

Abstract
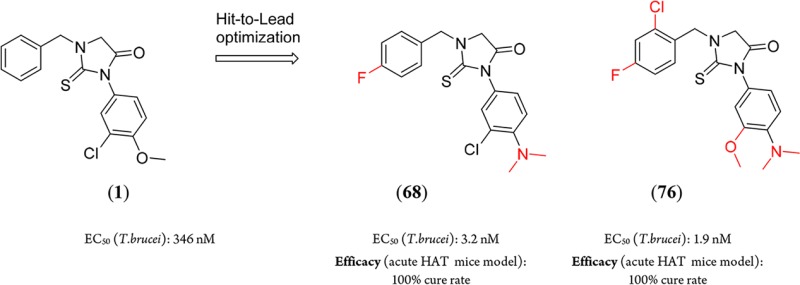
A high throughput screening and subsequent hit validation identified compound 1 as an inhibitor of Trypanosoma brucei parasite growth. Extensive structure–activity relationship optimization based on antiparasitic activity led to the highly potent compounds, 1-(4-fluorobenzyl)-3-(4-dimethylamino-3-chlorophenyl)-2-thiohydantoin (68) and 1-(2-chloro-4-fluorobenzyl)-3-(4-dimethylamino-3-methoxyphenyl)-2-thiohydantoin (76), with a T. brucei EC50 of 3 and 2 nM, respectively. This represents >100-fold improvement in potency compared to compound 1. In vivo efficacy experiments of 68 and 76 in an acute mouse model of Human African Trypanosomiasis showed a 100% cure rate after 4 days of oral treatment at 50 mg/kg twice per day.
Keywords: Human African Trypanosomiasis, “sleeping sickness”, Trypanosoma brucei inhibitor, thiohydantoins, hit-to-lead optimization
HAT (Human African Trypanosomiasis) or sleeping sickness is one of the most neglected diseases of human, caused by parasites of Trypanosoma brucei species, and spread through the bite of infected tsetse flies. Most of the morbidity is due to the late neurological stage when parasites cross the blood–brain barrier. Without treatment, the disease is invariably fatal. The WHO approved treatment for late-stage HAT is nifurtimox–eflornithine combination therapy (NECT), which requires intravenous administration, skilled staff, and inpatient monitoring.1 A new drug, effective for late stage disease, which is nontoxic and orally administered, is urgently needed. Moreover, this drug should be affordable by national health systems in the disease-endemic countries located in sub-Saharan Africa.
In the course of a high throughput phenotypic screen for compounds that inhibit T. brucei growth in vitro,2,3 we discovered the substituted 2-thiohydantoin (1) (Figure 1).
Figure 1.

Screening hit, a 2-thiohydantoin (compound 1).
Compounds with the 2-thiohydantoin moiety have been investigated for a variety of applications, including hypolipidemics,4 anticarcinogenics,5 antimutagenics,6 antithyroidals,7 antivirals,8−10 antimicrobials,11,12 antiulcer, and anti-inflammatories,13 as well as herbicides.14 Enzalutamide is an FDA approved anticancer drug with a 2-thiohydantoin pharmacophore and is an androgenic receptor antagonist.15 Despite their use in multiple disease indications, 2-thiohydantoins should not be regarded as promiscuous inhibitors, but rather as a scaffold with a distinct intermolecular interaction profile that can be usefully exploited for drug development.16
Different synthetic methods to prepare 2-thiohydantoin and its derivatives have been described. The most commonly used methods are the treatment of α-amino acids with acetic anhydride followed by ammonium thiocyanate17 or the coupling reaction between α-amino acid derivatives and isothiocyanate.18,19 We have used the isothiocyanate route to prepare our thiohydantoin library, as it is the most suitable for our purposes. N-Benzyl substituted glycine esters and isothioocyanates were prepared as reagents for the synthesis of 1,3-disubstituted 2-thiohydantions (Schemes 1 and 2). Depending on the commercial availability of starting materials and chemical tractability, N-substituted glycine esters were prepared by reductive amination of aromatic aldehydes with glycine esters (Scheme 1) or by nucleophilic substitution of bromoacetic esters with benzyl amines (Scheme 2). For synthesis of isothiocyanates, not only the commercial availability and chemical tractability but also structural features, such as the presence of a halogen in the aromatic ring, determined the synthetic route. The 3-chloro-4-N,N-dimethylphenyl isothiocyanate (7) was prepared by aromatic nucleophilic substitution of fluorine with dimethylamine, reduction of the nitro group in the presence of halogens (chlorine) with tin chloride, and finally the conversion of the amino group to the isothiocyanate by treatment with thiophosgene in a heterogeneous mixture (Scheme 1). The preparation of 3-methoxy-4-N,N-dimethylphenyl isothiocyanate (13) started with reductive alkylation of 2-methoxy-4-nitroaniline, followed by reduction of the nitro group with hydrogen/palladium catalyst and conversation of the amino group to the isothiocyanate (Scheme 2). The final condensation and subsequent cyclization reaction of the N-substituted glycine ester with isothiocyanates were done in ethanol at room temperature. The corresponding 2-thiohydantoin derivatives were precipitated from the reaction mixture or purified by column chromatography.
Scheme 1. Representative Synthesis of 2-Thiohydantoin Derivative 68.

Reagents and conditions: (a) Et3N, mol. sieves 3 Å, NaCNBH3, CHCl3, rt, yield 48%; (b) dimethylamine HCl salt, K2CO3, DMSO, 80 °C, yield 95%; (c) SnCl2, EtOAc/EtOH, 80 °C, yield 64%; (d) CSCl2, DCM-aq·NaHCO3, 4 °C–rt, yield 84%; (e) EtOH, rt, yield 70%.
Scheme 2. Representative Synthesis of 2-Thiohydantoin Derivative 76.

Reagents and conditions: (a) THF, 4 °C–rt, yield 62%; (b) paraformaldehyde, AcOH, NaCNBH3, yield 94%; (c) Pd/C, H2, MeOH, yield 60%; (d) CSCl2, DCM-aq·NaHCO3, 4 °C–rt, yield 94%; (e) EtOH, rt, yield 84%.
Hit compound 1 showed submicromolar activity in T. brucei growth inhibition assays, low CYP inhibition activity, and no detected cytotoxicity on the mammalian CRL-8155 or Hep G2 cell lines. Compound 1 displayed moderately rapid degradation in the presence of murine microsomes (Figure 1). Improving T. brucei activity and microsomal stability were the first prerequisites for advancing this scaffold to efficacy testing in mouse models of T. brucei infection.
We started SAR investigation of this scaffold by modification of the 3-chloro-4-methoxyphenyl moiety. Replacing 3-Cl and/or 4-OMe with hydrogen(s) led to inactive compounds 14, 15, and 16 (Table 1). Changing the substitution position of chlorine or methoxy group in monoderivatives 17, 18, and 19 did not show any positive effects on the compound’s activity. The relative position of chlorine and methoxy group was important for the activity as seen by the inactive 5-chloro-2-methoxy analogue (20). The results indicated that both substituents had synergetic effects on activity, and their positions in the ring were important. When the methoxy group in 1 was replaced with the trifluoromethyl 21 or the fluoro 22 group, the potency decreased. The combination of chlorine and fluorine groups in compound 23 as well as monofluoro substitution in the compound 24 phenyl ring led to inactive compounds. Bis-trifluoromethyl derivative 25 was also inactive.
Table 1. In Vitro Antitrypanosomal Evaluation of N-Benzyl-2-thiohydantoins Derivatives (SAR R1–R2).
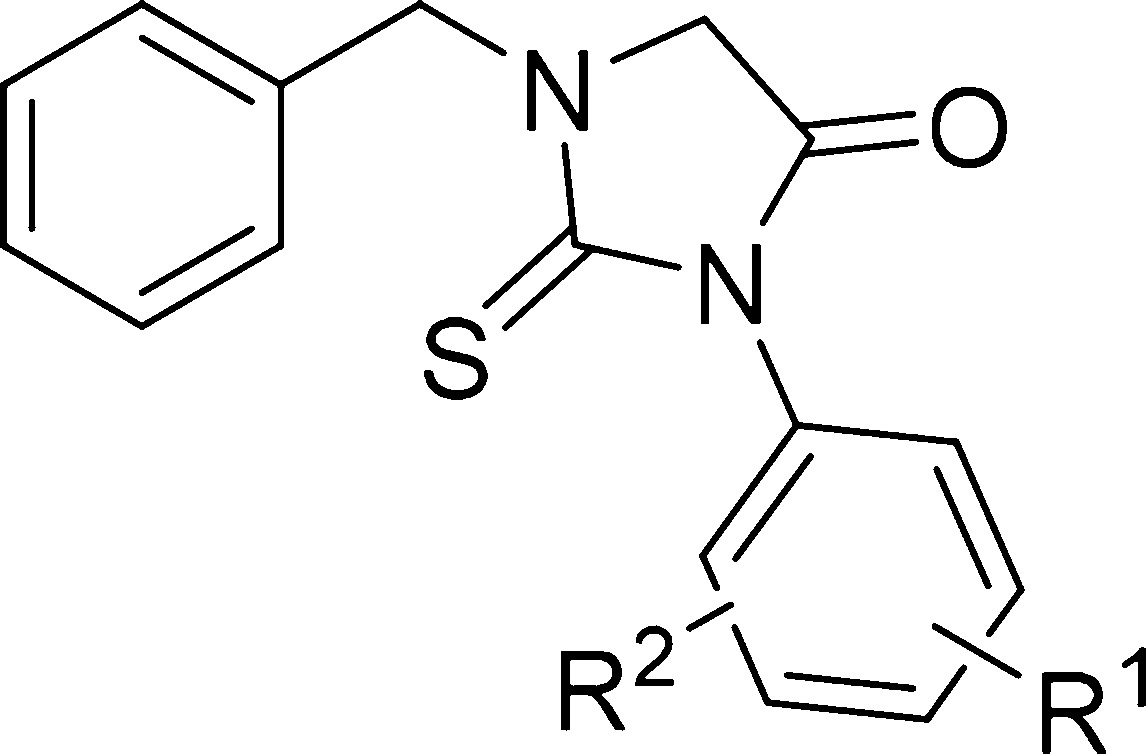
| compd | R1 | R2 | T.brucei EC50 (nM)a |
|---|---|---|---|
| 1 | 4-OMe | 3-Cl | 346 |
| 14 | 4-OMe | H | >10000 |
| 15 | 3-Cl | H | >20000 |
| 16 | H | H | >10000 |
| 17 | 3-OMe | H | >10000 |
| 18 | 4-Cl | H | >10000 |
| 19 | 2-Cl | H | >20000 |
| 20 | 5-Cl | 2-OMe | >10000 |
| 21 | 4-CF3 | 3-Cl | >10000 |
| 22 | 4-F | 3-Cl | >10000 |
| 23 | 5-Cl | 2-F | >10000 |
| 24 | 4-F | H | >10000 |
| 25 | 3-CF3 | 5-CF3 | >10000 |
| 26 | 4-N(Me)2 | H | 1500 |
| 27 | 4-isoPr | H | >10000 |
| 28 | 4-OH | H | >10000 |
| 29 | 4–C(O)Me | H | >10000 |
| 30 | 4-COOH | H | >10000 |
| 31 | 4-OMe | 2-OMe | 637 |
| 32 | 4-OMe | 3-OMe | 161 |
| 33 | 4-OMe, 5-OMe | 3-OMe | >10000 |
| 34 | 4-N(Me)2 | 3-Cl | 16 |
| 35 | 4-N(Me)2 | 3-F | 125 |
| 36 | 4-N(Me)2 | 3-OMe | 13 |
| 37 | 4-OMe | 3-N(Me)2 | 431 |
Concentration of compound required to inhibit growth by 50% (EC50) in T. bruceibrucei strain BF427. Pentamidine isethionate was included as a control for all of the T. brucei EC50 assays with average ± SEM: 1.15 ± 0.09 nM (n = 32).
The breakthrough came with the 4-N,N-dimethylamino analogue (26), with an EC50 of 1500 nM, five times less active than 1, but at least six times more potent than 4-methoxy analogue (14), which indicated a higher influence on activity from the N,N-dimethylamino group compared to the methoxy-group. 4-Iso-propyl 27, 4-phenol 28, 4-acetyl 29, and 4-carboxylic 30 derivatives were all inactive. Substitution of chlorine in 1 with a methoxy group increased the potency of the 3,4-dimethoxy derivative 31 two-fold, while 2,4-dimethoxy derivative 32 showed a decrease in antitrypanosomal activity (EC50 of 161 and 637 nM, respectively), confirming the preferable substitution position as the para–meta combination. Addition of the third methoxy group at position 5 to compound 31 was detrimental to activity, as the corresponding compound 33 was completely inactive. By combining the best groups and positions mentioned above, we synthesized the 4-N,N-dimentylamino-3-chloro derivative 34 with T. brucei EC50 = 16 nM, 4-N,N-dimethylamino-3-methoxy analogue 36 with EC50 = 13 nM, and 4-N,N-dimethylamino-3-fluoro analogue 35 with EC50 = 125 nM. Switching the substitution position of dimethylamino and methoxy group in 36 led to a 30-fold drop in activity in compound 37, which provided further support of the importance of para–meta positions for activity.
The next region that has been explored was the substitution on the benzyl ring in (3-chloro-4-methoxyphenyl)-2-thiohydantoin, starting with introduction of substituents at position 4 (Table 2). The 4-chloro derivative (38) was less potent than unsubstituted benzyl compound 1. Compounds with activating or deactivating groups such as 4-methyl (41), 4-methoxy (42), 4-trifluoro (43), 4-cyano (44), 4-ethylcarboxylate (45), and 3-pyridyl analogue (46) had low or no activity. Improvement of antiparasitic potency was observed with the 4-fluoro derivative (39), which is seven times more potent against T. brucei parasites (EC50 = 52 nM) relative to 1. Changing the position of the fluorine from para to meta (compound 40) decreased potency 18-fold. Based on these results, we speculate that there is not enough space in the binding pocket for a large substituent near the para position and/or that there is specific hydrogen bonding to the fluorine at the para position. Nevertheless, we kept the 4-fluorine substitution in the benzyl ring and added substituents at the ortho and meta positions. The addition of the second fluorine at the meta position (47) decreased the potency 6-fold compared to 39, while addition at the ortho position (48) retained the activity. The relative position of both fluorines was important as seen by inactive 2,5-difluoro analogue (49). Introducing a larger (compared with H or F) substituent like cyano (50) at the meta position eliminated the activity. The ortho position in compound 39 tolerated changes to the size and nature of the substituents, with 4-fluoro-2-chloro derivative (51) as the most active analogue with EC50 = 46 nM, and 4-fluoro-2-methoxy (52), 4-fluoro-2-nitro (53), 4-fluoro-2-trifluoromethyl (54) analogues were considered for further optimization to increase potency and stability.
Table 2. In Vitro Antitrypanosomal Evaluation of 3-Chloro-4-(dimethylamino)phenyl-2-thiohydantoin Derivatives (SAR of R3–R4).
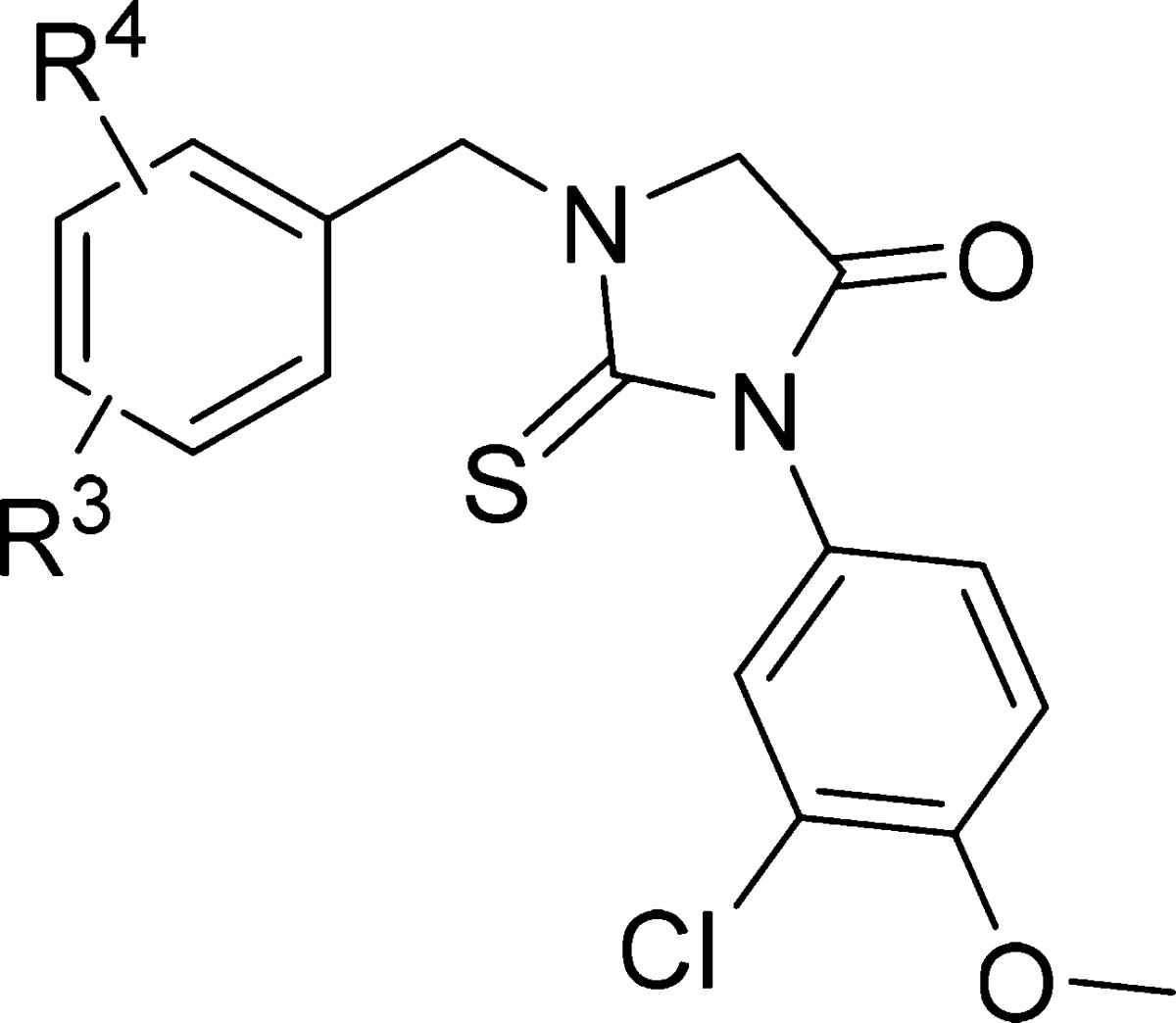
| compd | R3 | R4 | T.brucei EC50 (nM)a |
|---|---|---|---|
| 1 | H | H | 346 |
| 38 | 4-Cl | H | 853 |
| 39 | 4-F | H | 52 |
| 40 | 3-F | H | 968 |
| 41 | 4-Me | H | 1553 |
| 42 | 4-OMe | H | >20000 |
| 43 | 4-CF3 | H | >20000 |
| 44 | 4-CN | H | 17818 |
| 45 | 4–C(O)OEt | H | >20000 |
| 46 | 3-pyridyl | H | 3000 |
| 47 | 4-F | 3-F | 313 |
| 48 | 4-F | 2-F | 63 |
| 49 | 5-F | 2-F | 9260 |
| 50 | 4-F | 3-CN | >20000 |
| 51 | 4-F | 2-Cl | 46 |
| 52 | 4-F | 2-OMe | 191 |
| 53 | 4-F | 2-NO2 | 350 |
| 54 | 4-F | 2-CF3 | 924 |
Concentration of compounds required to inhibit growth by 50% (EC50) of T. brucei brucei strain BF427. Pentamidine isethionate was included as a control for all of the T. brucei EC50 assays with average ± SEM: 1.15 ± 0.09 nM (n = 32).
To determine the importance of the 2-thiohydantoin core, we modified the 2-thiohydantoin cycle by conversion to hydantoin (S to O change) (55) or substitution of the glycine α-hydrogen with ethoxy (56), methyl (57), or carbonyl (58) groups (Figure 2). All changes to the 2-thiohydantoin core were highly detrimental to antitrypanozomal activity. Methylation of the α-benzyl position of 39 resulted in loss of activity for compound 59. It was determined that the 2-thiohydantoin cycle should remain unmodified in further optimization. All compounds with a chiral center were prepared as racemates.
Figure 2.

2-Thiohydantoin ring modifications.
Based on the SAR described above, the next step was to combine the substituents with the highest influence on activity into one molecule in order to optimize the compounds’ antitrypanosomal properties. We chose more than one pair of substituents (four for the phenyl ring and seven for the benzyl ring) in order to have a large pool to select the best compounds from.
For substituents R1 and R2 on the phenyl group, we selected 4-methoxy and 3-methoxy groups (60–66) and three 4-dimethylamino pairs: 4-dimethylamino and 3-chloro (67–73), 4-dimethylamino and 3-fluoro (74–75), and 4-dimethylamino and 3-methoxy (76–82) groups (Table 3). 4-Pyrrolidino-3-chloro analogues (83–85) were prepared with the aims discussed below. As for substituents R3 and R4 at the benzyl moiety, we selected the seven most active 4-fluoro analogues: 4-fluoro and 2-chloro, 4-fluoro with H at R4, 4-fluoro and 2-fluoro, 4-fluoro and 2-methoxy, 4-fluoro and 3-fluoro, 4-fluoro and 2-nitro, and 4-fluoro and 2-trifluoromethyl groups (Table 3). The influence of each pair of substituents on the anti-T. brucei activity can be evaluated as follows (R1 and R2): (4-N(CH3)2 and 3-Cl) ≥ (4-N(CH3)2 and 3-OMe) > (4-pyrrolidino and 3-Cl) > (4-N(CH3)2 and 3-F) ≥ (4-OMe and 3-OMe). The R3 and R4 substituents are in the order (4-F and 2-Cl) > (4-F and H) ≥ (4-F and 2-F) > (4-F and 2-OMe) > (4-F and 3-F) > (4-F and 2-CF3) ≥ (4-F and 2-NO2) and are ordered in Table 3 accordingly. Out of 26 compounds, 23 had T. brucei EC50 values below 50 nM and showed no cytotoxicity up to 50 μM. The most active compounds 68 and 76, with EC50 of 3 and 2 nM, respectively, were assessed for plasma protein binding, solubility, and metabolic stability against pooled mouse liver microsomes. They were also tested in mice for oral pharmacokinetics and brain concentrations (at 60 min postdose) to assess the brain to plasma ratios (Table 4).
Table 3. In Vitro Activity and Cytotoxicity for Select 2-Thiohydantoins.

| compd | R1 | R2 | R3 | R4 | T. brucei EC50 (nM)a | cytotoxycity CC50 (× 103 nM)b |
|---|---|---|---|---|---|---|
| 60 | OMe | OMe | F | 2-Cl | 14 | 36.8 |
| 61 | F | H | 49 | >50 | ||
| 62 | F | 2-F | 33 | >50 | ||
| 63 | F | 2-OMe | 107 | >50 | ||
| 64 | F | 3-F | 115 | >50 | ||
| 65 | F | 2-NO2 | 555 | >50 | ||
| 66 | F | 2-CF3 | 37 | >50 | ||
| 67 | N(Me)2 | Cl | F | 2-Cl | 2 | >50 |
| 68 | F | H | 3 | 39 | ||
| 69 | F | 2-F | 4 | >50 | ||
| 70 | F | 2-OMe | 11 | 12.6 | ||
| 71 | F | 3-F | 13 | >50 | ||
| 72 | F | 2-NO2 | 19 | >50 | ||
| 73 | F | 2-CF3 | 22 | >50 | ||
| 74 | N(Me)2 | F | F | 2-Cl | 15 | >50 |
| 75 | F | 2-F | 28 | >50 | ||
| 76 | N(Me)2 | OMe | F | 2-Cl | 2 | >50 |
| 77 | F | H | 7 | >50 | ||
| 78 | F | 2-F | 6 | >50 | ||
| 79 | F | 2-OMe | 11 | 39.6 | ||
| 80 | F | 3-F | 28 | >50 | ||
| 81 | F | 2-NO2 | 44 | >50 | ||
| 82 | F | 2-CF3 | 28 | >50 | ||
| 83 | PYR | Cl | F | 2-Cl | 9 | >50 |
| 84 | F | H | 27 | >50 | ||
| 85 | F | 2-F | 10 | >50 |
Concentration of compound required to inhibit growth by 50% (EC50) of T. brucei brucei strain BF427. Pentamidine isethionate was included as a control for all of the T. brucei EC50 assays with average ± SEM: 1.15 ± 0.09 nM (n = 32).
Concentration of compound required to inhibit growth by 50% (CC50) of human lymphoblasts (CRL-8155). Quinacrine was included as a control for all CRL-8155 assays, with an average ± SEM: 2.34 ± 0.31 μM (n = 5).
Table 4. Activity, CYP Inhibition, Metabolic Stability, Oral Pharmacokinetics, Brain Penetration, Protein Binding, and Solubility for Compounds 68 and 76.
| compd | 68 | 76 |
|---|---|---|
| T. brucei EC50 (nM)a | 3.2 | 1.9 |
| CYP3A4 IC50 (nM)b | 12798 | 7760 |
| microsome T1/2 (min)c | 15 | 11 |
| oral PK Cmax (μM)d | 2.1 ± 0.1 | 2.5 ± 0.3 |
| oral PK AUC (min·μmol/L)d | 1376 ± 187 | 750 ± 130 |
| brain/plasma ratioe | 0.74 ± 0.30 | 1.68 ± 0.35 |
| protein binding (%)f | 88.8 ± 1.5 | 96.3 ± 0.5 |
| solubility at pH 7.4 (μM)g | 2.1 | 4.1 |
Concentration of compound required to inhibit growth by 50% (EC50) of T. brucei brucei strain BF427. Pentamidine isethionate was included as a control for all of the T. brucei EC50 assays with average ± SEM: 1.15 ± 0.09 nM (n = 32).
Concentration to inhibit CYP3A4 by 50%. Ketoconazole was run as a control with a value of 29.6 nM, which aligned with other assays (25.62 ± 3.48 nM (n = 29)).
Compound half-life time in mouse microsomes. Testosterone (T1/2 = 3.97 min) and dextromethorphan HBr (T1/2 = 9.51 min) were included as controls and fell into acceptable ranges. Average ± SEM of testosterone, 4.6 ± 0.6 min (n = 47), and dextromethorphan HBr, 10.2 ± 0.9 min (n = 51)
Average values ± SEM of three mice each given a single dose at 50 mg/kg by oral gavage.
Average values ± SEM of three mice each given a single ip dose at 5 mg/kg.
Protein binding was assessed for each compound in triplicate at 2 μM solution in 50% mouse plasma. Propranolol was included as a binding control with 91.7 ± 1.0% total binding.
Solubility was assessed in phosphate buffered saline.
With rather moderate metabolic stability, high brain plasma ratios, a reasonable PK profile, and low protein binding, compounds 68 and 76 were selected for in vivo mouse efficacy experiments. In a model of the acute phase of HAT, five mice were infected with T. brucei rhodesiense at day 0 and were given the test compounds for 4 days, beginning 2 days postinfection. Compounds 68 and 76 attained 5/5 cures, as determined by the absence of detectable parasitemia in any of the treated mice for 60 days postinfection (Figure 3). All mice that received only vehicle showed high parasitemia on the last day of dosing, with all concentrations >1.5 × 107 parasites/mL of blood, and were euthanized.
Figure 3.
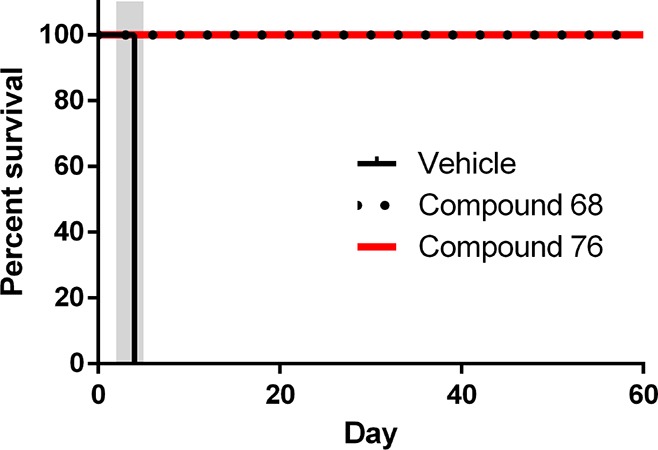
Mouse efficacy model of acute T. brucei infection. All mice were infected with T. brucei rhodesiense STIB900 on day 0. Groups of five mice each were treated with compound 68 and compound 76 (50 mg/kg by oral gavage b.i.d.) or vehicle from days 2–5 (gray-shaded area). Mice were monitored for parasitemia in tail-blood samples through day 60 postinfection.
The 100% cure rate in the murine model demonstrates that in vitro activity of these compounds translates to an in vivo model, clearing parasites from mice blood and lymphatic systems.
In summary, we have discovered 1-benzyl-3-aryl-2-thiohydantoins as potent antitrypanosomal agents. Extensive SAR studies identified 4-fluorobenzyl/4-fluoro-2-chloro benzyl and 4-dimethylamino-3-chloro/(methoxy)phenyl substituents at 2-thiohydantion core with the highest improvement in antiparasitic activity. Compounds 68 and 76 completely cleared parasites from mice in an acute T. brucei infection model. Investigation of more thiohydantion analogues is needed to find compounds optimized for metabolic stability, oral PK properties, and brain exposure, at which point we can advance the scaffold to efficacy studies in a chronic T. brucei infection model, where the parasites have crossed the blood–brain barrier.
Acknowledgments
Authors would like to thank Uyen T. T. Nguyen for her contributions in collecting cytotoxicity data and Martin Sadilek for recording high-resolution mass spectra.
Glossary
ABBREVIATIONS
- HAT
Human African Trypanosomiasis
- SAR
structure–activity relationship
- WHO
World Health Organization
- PYR
pyrrolidine
- SEM
standard error of the mean
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00230.
Biochemical assays, pharmacokinetic and in vivo experiments, experimental procedures for the synthesis of compounds 68 and 76, general experimental procedure and characterization of compounds 1 and 14–85 (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the National Institutes of Health (Grant 5R01AI106850).
All rodent experiments were done in compliance with the University of Washington Institutional Animal Care and Use Committee (IACUC) approved protocol.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. Human African Trypanosomiasis. http://www.who.int/trypanosomiasis_african/disease/diagnosis/en/.
- Tatipaka H. B.; Gillespie J. R.; Chatterjee A. K.; Norcross N. R.; Hulverson M. A.; Ranade R. M.; Nagendar P.; Creason S. A.; McQueen J.; Duster N. A.; Nagle A.; Supek F.; Molteni V.; Wenzler T.; Brun R.; Glynne R.; Buckner F. S.; Gelb M. H. Substituted 2-phenylimidazopyridines: a new class of drug leads for human African trypanosomiasis. J. Med. Chem. 2014, 57, 828–35. 10.1021/jm401178t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchynskyy A.; Gillespie J. R.; Hulverson M.; McQueen J.; Creason A. S.; Ranade M. R.; Duster A. N.; Gelb H. M.; Buckner S. F. Discovery of N-(2-aminoethyl)-N-benzyloxyphenyl benzamides: New potent Trypanosoma brucei inhibitors. Bioorg. Med. Chem. 2017, 25, 1571–1584. 10.1016/j.bmc.2016.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins E. J. 5,5-Diaryl-2-thiohydantoins and 5,5-diaryl N3-substituted 2-thiohydantoins as potential hypolipidemic agents. J. Med. Chem. 1986, 29, 855–859. 10.1021/jm00155a042. [DOI] [PubMed] [Google Scholar]
- Al-Obaid A. M.; El-Subbagh H. I.; Khodair A. I.; Elmazar M. M. 5-Substituted-2-thiohydantoin analogs as a novel class of antitumor agents. Anti-Cancer Drugs 1996, 7, 873–880. 10.1097/00001813-199611000-00009. [DOI] [PubMed] [Google Scholar]
- Takahashi A.; Matsuoka H.; Yamada K.; Uda Y. Characterization of antimutagenic mechanism of 3-allyl-5-substituted 2-thiohydantoins against 2-amino-3-methylimidazo[4,5-f]quinolone. Food Chem. Toxicol. 2005, 43, 521–528. 10.1016/j.fct.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Marx J. V.; Richert D. A.; Westerfeld W. W. Peripheral inhibition of thyroxine by thiohydantoins derived from amino acids. J. Med. Chem. 1970, 13, 1179–1181. 10.1021/jm00300a036. [DOI] [PubMed] [Google Scholar]
- WO 2013/171281 Al, 21 November 2013.
- El-Barbary A. A.; Khodair A. I.; Pedersen E. B.; Nielsen C. S-Glucosylated hydantoins as new antiviral agents. J. Med. Chem. 1994, 37, 73–77. 10.1021/jm00027a009. [DOI] [PubMed] [Google Scholar]
- Chérouvrier J. R.; Carreaux F.; Bazureau J. P. Reactivity of 2-Thiohydantoins towards various electrophilic reagents: applications to the synthesis of new 2-ylidene-3,5-dihydro-4H-imidazol-4-ones. Molecules 2004, 9, 867–875. and references cited therein 10.3390/91000867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S.; Unser M. J.; Froelich E. Some 5-(oxoalkyl)-2-thiohydantoins and their derivatives. J. Am. Chem. Soc. 1956, 78, 6182–6185. 10.1021/ja01604a064. [DOI] [Google Scholar]
- Marton J.; Enisz J.; Hosztafi S.; Timar T. Preparation and fungicidal activity of 5-substituted hydantoins and their 2-thio analogs. J. Agric. Food Chem. 1993, 41, 148–152. 10.1021/jf00025a031. [DOI] [Google Scholar]
- Curran A. C. W. U.S. Pat. 3,984,430, 1976.
- Han J.; Wang J.; Dong H.; Lei J.; Wang M.; Fang J. Synthesis and herbicidal activity of 5-(4-hydroxybenzyl)-2- thioxoimidazolidin-4-one esters. Molecules 2011, 16, 2833–2845. 10.3390/molecules16042833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo M.; Xu X.; Xie Z.; Ge R.; Zhang Z.; Li Z.; Bian J. Design and synthesis of indoline thiohydantoin derivatives based on enzalutamide as antiproliferative agents against prostate cancer. Eur. J. Med. Chem. 2017, 125, 1002–1022. 10.1016/j.ejmech.2016.10.049. [DOI] [PubMed] [Google Scholar]
- Mendgen Th; Steuer Ch.; Klein D. Ch. Privileged scaffolds or promiscuous binders: a comparative study on rhodanines and related heterocycles in medicinal chemistry. J. Med. Chem. 2012, 55, 743–753. 10.1021/jm201243p. [DOI] [PubMed] [Google Scholar]
- Johnson T. B.; Bengis R. Hydantoins: synthesis of hydantoin of 3-amino-tyrosine. J. Am. Chem. Soc. 1912, 34, 1054–1061. 10.1021/ja02209a013. [DOI] [Google Scholar]
- Innocenti A.; Casini A.; Alcaro M. C.; Papini A. M.; Scozzafava A.; Supuran C. T. Carbonic anhydrase inhibitors: the first on-resin screening of a 4-sulfamoylphenylthiourea library. J. Med. Chem. 2004, 47, 5224–5229. 10.1021/jm049692i. [DOI] [PubMed] [Google Scholar]
- Elokdah H.; Sulkowski T. S.; Abou-Gharbia M.; Butera J. A.; Chai S.-Y.; McFarlane G. R.; McKean M. L.; Babiak J. L.; Adelman S. J.; Quinet E. M. Design, synthesis, and biological evaluation of thio-containing compounds with serum HDL-cholesterol-elevating properties. J. Med. Chem. 2004, 47, 681–695. 10.1021/jm030219z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.