

Abstract

Enterovirus 71 (EV71) is a major causative agent of hand, foot, and mouth disease (HFMD), which can spread its infections to the central nervous and other systems with severe consequences. The viral caspid protein VP1 is a well-known target for antiviral efficacy because its occupancy by suitable compounds could stabilize the virus capsid, thus preventing uncoating of virus for RNA release. In this Letter, design, synthesis, and biological evaluation of novel anti-EV71 agents (aminopyridyl 1,2,5-thiadiazolidine 1,1-dioxides) are described. One of the most promising compounds (14) showed excellent antiviral activity against EV71 (EC50 = 4 nM) and exhibited excellent in vivo efficacy in the EV71 infected mouse model.
Keywords: HFMD, enterovirus 71, capsid inhibitor
Hand, foot, and mouth disease (HFMD) is a common infectious disease mainly occurring in young children. Clinical symptoms of HMFD are characterized by fever, sore throat, and vesicular rash on the hands and feet as well as exanthema on oral mucosa and tongue. For several decades, HFMD has been a very common infection in children all over the world, especially in the Asia-Pacific region. HFMD is caused by a spectrum of pathogens in the enterovirus (EV) family; enterovirus 71 (EV71) and coxsackievirus A16 (Cox A16) are regarded as the major causative agents. In particular, EV71 is more often associated with neurological diseases such as meningitis, encephalitis, monoplegia, acute flaccid paralysis, and even death, especially among young children (≤5 years old).1,2 So far, no marketed antienterovirus drugs are available for the treatment of EV71-related disease, though the first EV71 vaccine is now approved by cFDA to prevent this disease.3 Supportive therapy is still the primary treatment for severe cases of EV71 infection. Ribavirin was used for treating mild HFMD in clinic. However, ribavirin showed no anti-EV71 activity with EC50 > 100 μM in the cytopathogenic effect inhibition assay (CPE) in-house. Considering the danger of severe EV71 infection in children, there is an urgent need to develop effective antiviral treatment to prevent or treat EV71-related disease before the next EV71 outbreak.
EV71 has nonenveloped, positive-sense, single-stranded RNA encoding capsid proteins (VP1, VP2, VP3, and VP4) and nonstructural proteins (2A, 2B, 2C, 3A, VPg, 3C, and 3D) that are involved in the life cycle of viruses including absorption, invasion, shelling, and replication. All the functional proteins involved in the life cycle of viruses can be targets for the development of antiviral drugs.4 We have previously disclosed the discovery of a series of peptidomimetic 3Cpro inhibitors.5
The viral protein VP1 is a well-known target for antiviral design because its hydrophobic pocket can be occupied by suitable compounds, which lead to stabilizing the virus capsid and preventing uncoating of virus for RNA release.6 WIN series from Sterling-Winthrop is a prominent series of capsid-binding compounds. Pleconaril in this series is a typical small molecule inhibitor, which has been shown to have a broad spectrum of activities against enteroviruses and rhinoviruses and has been tested in clinical trials.7 According to what Shia and co-workers reported, Pleconaril showed no antiviral activity against EV71. Nevertheless, using skeletons of WIN compounds as templates, they identified a novel series of imidazolidinone derivatives with significant antiviral activity against enterovirus 71. Among this series of compounds, DBPR103 was identified as a very potent enterovirus 71 inhibitor (EC50 = 54 nM) with no apparent cytotoxic effect toward RD (rhabdomyosarcoma) cell lines (CC50 > 25 μM).8,9
During our research, Rao and co-workers reported using structural analysis of the complete virus to guide the design of EV71 inhibitors. Using pyridyl imidazolidinones as a template, they discovered a potent EV71 capsid inhibitor-NLD, which was able to protect cells from EV71 infection at a concentration of 25 pM.10,11 Based on these studies, here we report the chemical synthesis and biological evaluation of novel aminopyridyl 1,2,5-thiadiazolidine 1,1-dioxides as EV71 caspid inhibitors to treat HMFD.
Result and Discussion
Pleconaril and DBPR-103 were tested in the cytopathogenic effect inhibition assay (CPE) in-house. Pleconaril showed no anti-EV71 activity with EC50 > 100 μM, and DBPR-103 was a potent EV71 inhibitor with EC50 = 91 nM. Data were consistent with those reported in the literature.8,9 Our research was focused on structure modifications of pyridyl imidazolidone compound DBPR-103. Therefore, the primary task of the project is to identify a novel skeleton based on pyridyl imidazolidone series that could maintain potent antiviral activity. As reported by Chern, the five-carbon alkyl linker and terminal pyridine play a very important role in influencing anti-EV71 activity.9 We keep the two moieties unchanged.
First, imidazolidone modification was designed and evaluated by the CPE assay. Among this series, 2-ketopiperazines 1 and 1,2,5-thiadiazolidine 1,1-dioxide 5(12) kept antiviral activity with EC50 of 180 and 110 nM, respectively. The other modifications all resulted in loss of antiviral activity. Compound 1 (CC50 = 3.2 μM) has moderate cytotoxicity, while compound 5 (CC50 > 100 μM) has no such issue (Table 1).
Table 1. Structure–Activity Relationship of Imidazolidone Modification.

The compound concentration required to reduce the virus-induced cell death by 50% was defined as EC50.
The compound concentration required to reduce cell viability to 50% of the tested control culture was defined as CC50.
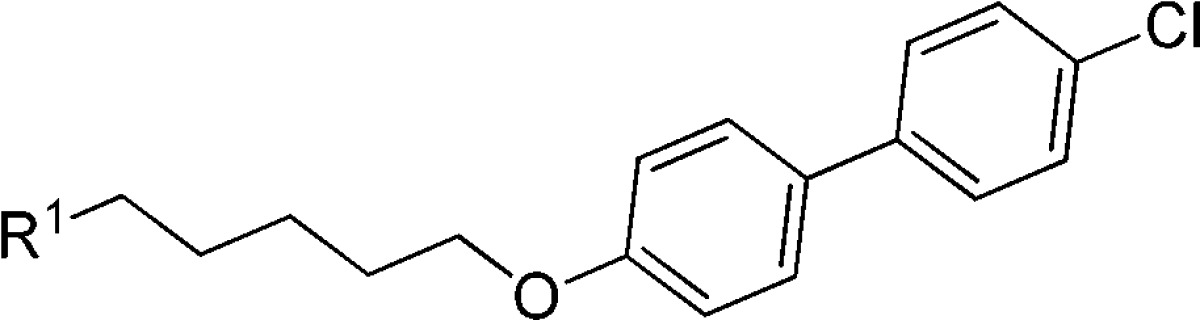
Another structure modification strategy was the cyclization of phenyl alkyl ether. According to this strategy, several benzoheterocycle compounds were designed and evaluated, as shown in Table 2. Most of these compounds lost activity, except the benzodihydropyran 6 (EC50 = 48 nM), which is 2-fold more potent than DBPR-103. Compound 6 also showed moderate cytotoxicity (CC50 = 3 μM) like 1.
Table 2. Structure–Activity Relationship of Phenyl Alkyl Ether Cyclization.

The compound concentration required to reduce the virus-induced cell death by 50% was defined as EC50.
The compound concentration required to reduce cell viability to 50% of the tested control culture was defined as CC50.
The three potent compounds 1, 5, and 6 were further evaluated and compared with DBPR103 through in vitro ADME and in vivo PK profiles. The mouse liver microsomal stability of 5 (<10 μL/min/mg) indicated slower metabolism than the other three compounds (40–50 μL/min/mg). The PK data with 2 mg/kg IV and 5 mg/kg IP are summarized in Table 3. Following IV/IP administration, the elimination half-life (T1/2) of 1, 5, and 6 ranged from 5 to 6 h, which is better than DBPR103, and would be suitable for once daily dosing. Compound 6 exhibited the lowest clearance and best exposure in these compounds. Compound 5 also showed good exposure, which was 2-fold more than that of DBPR103, both in intraperitoneal and intravenous injections. However, 1 and 6 showed hERG inhibition issues (IC50 = 0.37 and 1.75 μM, respectively), compared with 5 (IC50 > 30 μM). Compound 5 was selected for further tests in oral PK. The oral exposure of 5 indicated it is also suitable for oral dosing. Taking all factors into consideration, compound 5 is believed to be more suitable as a lead compound.
Table 3. Single Dose Mouse in Vivo PK Propertiesa.
| Compd | DBPR103 | 1 | 5 | 6 | |
|---|---|---|---|---|---|
| iv@2mpk | |||||
| Cl (mL/min/kg) | 28 | 21 | 12 | 6 | |
| T1/2 (h) | 4 | 6 | 6 | 5 | |
| Vd (L/kg) | 9 | 10 | 5 | 3 | |
| AUC (nM·h) | 2731 | 3344 | 5710 | 10999 | |
| ip@5mpk | ip@5mpk | po@5mpk | |||
| Cmax (nM) | 453 | 1255 | 682 | 617 | 3362 |
| Tmax (h) | 2 | 2 | 2 | 2 | 1 |
| AUC (nM·h) | 4275 | 8947 | 9011 | 6166 | 23240 |
| %F | 64 | 112 | 63 | 43 | 86 |
Data reported are the means from the dosing cohorts (Male CD-1 mouse, n = 3/dose). Dosed at 2 mg/kg iv (20% solutol in water), 5 mg/kg ip (20% solutol in water), and 5 mg/kg po (20% solutol in water). Cl is clearance. Vd is volume of distribution. Cmax represents the highest observed concentration. Tmax is the time taken to reach the maximum concentration. T1/2 is the half-life of the compound exposure in plasma. AUC is the area under the curve. %F is percent bioavailability.
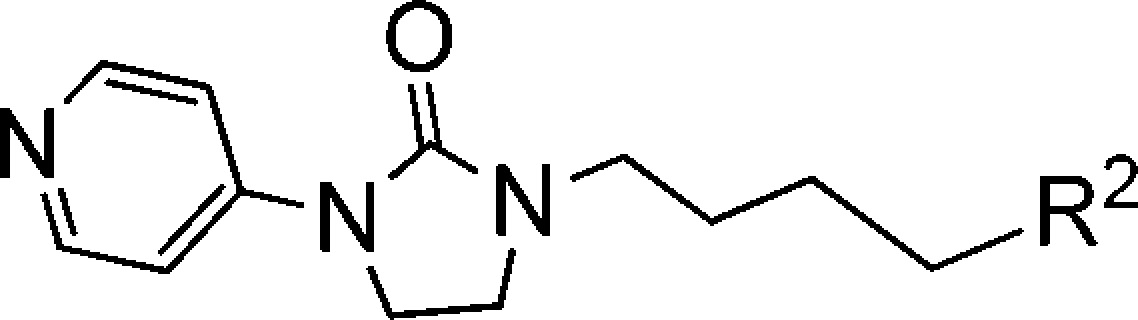
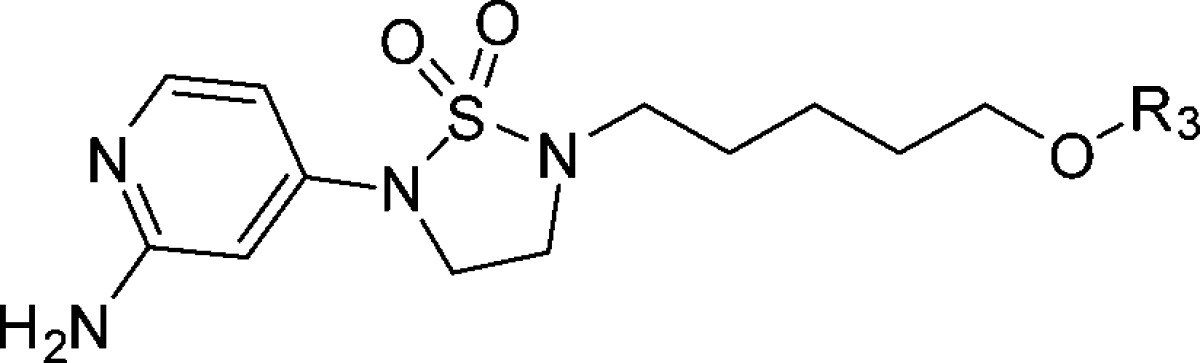
Since the PK profile of 5 is good, the next goal of the project is to further increase activity of compounds based on 5 (EC50 = 110 nM). According to the Chern report, antiviral activity of DBPR compounds could be improved by replacing chlorobenzene with oxime ether.13,14 So, we planned to introduce these moieties into 1,2,5-thiadiazolidine 1,1-dioxides to improve the activity. Coincidently, Rao and co-workers reported a series of EV71 capsid inhibitors (NLD as a representative) during our research process. As shown in the NLD structure (Scheme 1), the methyl group in the linker and oxime ether have been confirmed to make contribution to activity. The main difference between NLD and DBPR series is the amine group in the 2-position of pyridine. With such slight alteration, NLD was able to protect cells from EV71 infection at a concentration of 25 pM, which is an amazing improvement in activity. To confirm this, NLD was synthesized and tested in CPE assay. NLD exhibited excellent antiviral activity (EC50 = 0.03 nM), as previously reported.10 However, NLD shows moderate cytotoxicity (CC50 = 5 μM) and also suffers from severe hERG inhibition (0.8 μM). We planned to introduce an amine group into our 1,2,5-thiadiazolidine 1,1-dioxide serials to check whether this modification can improve activity.
Scheme 1. Chemical Structures of Pleconaril, DBPR-103, and NLD.

The amine group was added to compound 5 to generate 10,12 which was 10-fold more potent than 5 (Scheme 2). This indicated the amine made a great contribution to the activity. Compound 11 (EC50 = 0.9 nM), which replaced the chlorobenzene of 10 with oxime ether, is 10-fold more potent than 10. In total, compound 11 exhibited 100-fold improvement in activity compared to 5 with this two-step modification. Furthermore, 10 and 11 have no cytotoxicity (CC50 > 100 μM).
Scheme 2. Chemical Structures of 5, 10, and 11.

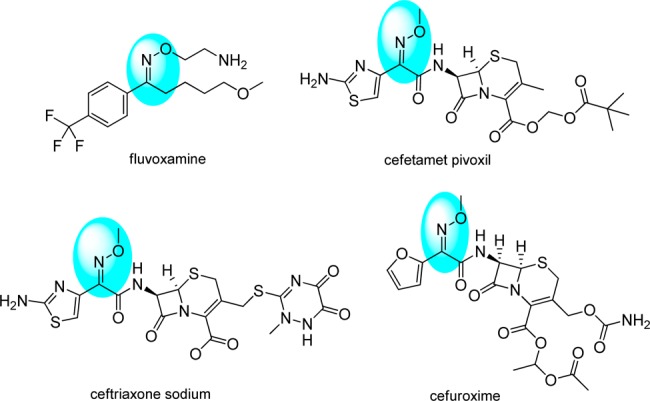
To the best of our knowledge, no aldoxime ether structures were tested in clinical therapies, while ketoxime ether structures were widely used in pharmaceutical molecules in the market, like fluvoxamine, ceftriaxone sodium, cefetamet pivoxil, and cefuroxime (Scheme 3). This might be because aldoxime ether is more easily metabolized to aldehydes, which are potential genotoxic agents. In this respect, we speculate it will be more drug-like and safer to replace aldoxime ether of 11 to ketoxime ether. So we designed several compounds on the basis of 11, including acetophenone oxime ether (12), propiophenone oxime ether (13), benzocyclopentanone oxime ether (14), benzocyclohexanone oxime ether (15), and some heterocyclic-fused ring as bioisosteres of oxime ether (16, 17, and 18).
Scheme 3. Marketed Drugs Contain Ketoxime Ether Structure.
As shown in Table 4, most of these compounds lost antiviral activity in CPE assay. Fortunately, benzocyclopentanone oxime ether (14) kept excellent anti-EV71 activity (EC50 = 4 nM).
Table 4. Modifications of Compound 11.

The compound concentration required to reduce the virus-induced cell death by 50% was defined as EC50.
The compound concentration required to reduce cell viability to 50% of the tested control culture was defined as CC50.
The molecular modeling method was used to illustrate the binding mode of compounds 12, 14, and 16. The results indicated that the overall binding mode of compound 14 was similar to that of the NLD on EV71 capsid. The amino-pyridine group could form hydrogen bonding with Asp112. The sulphonyl group could form hydrogen bonding to the backbone NH of residue Ile113, as did the carbonyl oxygen of the imidazole of NLD. The phenoxyl group was sandwiched between Phe135 and Phe155, and the binding mode of the tail oxime group of compound 14 could overlay well with that of NLD in the cocrystal structure (pdb code: 4CEY) (Figure 1). The aminopyridine group of compound 12 could overlay with NLD, but its phenoxyl group could not overlay well with NLD (see SI Figure S2). Both the aminopyridine group and phenoxyl group of compound 16 could not overlay well with NLD (see SI Figure S3). The binding results are consistent with the cell-based bioactivities of 12, 14, and 16. This implies that the inhibition mechanism of compound 14 is targeting the EV71 capsid and blocking viral uncoating as NLD does.
Figure 1.
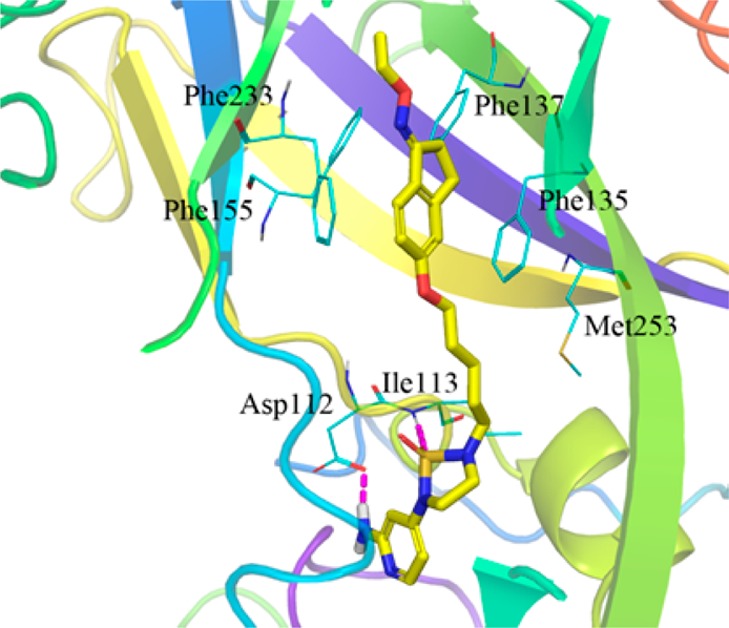
Predicted binding mode of compound 14 on EV71 capsid. X-ray structure of NLD (PDB code 4CEY) was used as the docking template. Pink dashes are hydrogen bonds. The figure was generated with PyMOL Molecular Graphics System, version 1.5.0.4, Schrödinger, LLC.
Since NLD suffers from severe hERG inhibition (0.8 μM), we tested the hERG inhibition of two potent compounds, 11 and 14. The hERG activities of 11 (9 μM) and 14 (6 μM) are much better than NLD. Compounds 11 and 14 and NLD were further tested and compared by PK characterization (Table 5). Pharmacokinetic studies were conducted in mouse to assess the intraperitoneal and intravenous exposure for 11, 14, and NLD. The elimination half-life (T1/2) of 11 and 14 is 1.6 and 1.5 h, respectively. NLD showed high clearance, and its half-life (T1/2) is only 0.3 h. The IV/IP exposure of 14 is better than that of 11 and NLD. Also considering the poor drug-like property of aldoxime ether in compound 11, compound 14 was selected for further exploration in an in vivo efficacy model.
Table 5. Single Dose Mouse in Vivo PK Propertiesa.
| Compd | 11 | 14 | NLD |
|---|---|---|---|
| iv@1 mpk | |||
| Cl (mL/min/kg) | 36 | 34 | 105 |
| T1/2 (h) | 1.6 | 1.5 | 0.3 |
| Vd (L/kg) | 3.2 | 2.8 | 2.4 |
| AUC (nM·h) | 592 | 821 | 371 |
| ip@5 mpk | |||
| Cmax (nM) | 1443 | 2152 | 1558 |
| Tmax (h) | 0.5 | 0.7 | 0.3 |
| AUC (nM·h) | 2449 | 3868 | 1711 |
| %F | 79 | 88 | 92 |
Data reported are the means from the dosing cohorts (Male CD-1 mouse, n = 3/dose). Dosed at 2 mg/kg iv (20% solutol in water) and 5 mg/kg ip (20% solutol in water). Cl is clearance. Vd is volume of distribution. Cmax represents the highest observed concentration. Tmax is the time taken to reach the maximum concentration. T1/2 is the half-life of the compound exposure in plasma. AUC is the area under the curve. %F is percent bioavailability.
The ICR mouse model (3 days old) with lethal-dose (1 × 106 PFU) EV71 strain 695F infection was used to imitate the severe complications of EV71 infected children. Lycorine, as reported in an EV71 infected model, was used as a positive control.15,16 Ribavirin is a broad-spectrum antiviral agent and was used for treating mild HFMD in clinic. Compound 14 was dosed 10 mg/kg/d via intraperitoneal injection route at different times. One is dosing before EV71 infection (group 3), the other is dosing after EV71 infection (group 4). The vehicle, lycorine, and ribavirin groups were dosed before EV71 infection. The results showed that mice in vehicle group all died at day 8. Ribavirin group mice all died at day 5 (100 mg/kg) and day 6 (50 mg/kg), due to toxicity of ribavirin. Lycorine group mice protection was only 30% after 14 days of dosing. Compound 14 showed excellent protection of EV71 infected mouse, which was 100% in two groups (Figure 2). The body weight and clinical score also showed 14 was much better than lycorine.
Figure 2.
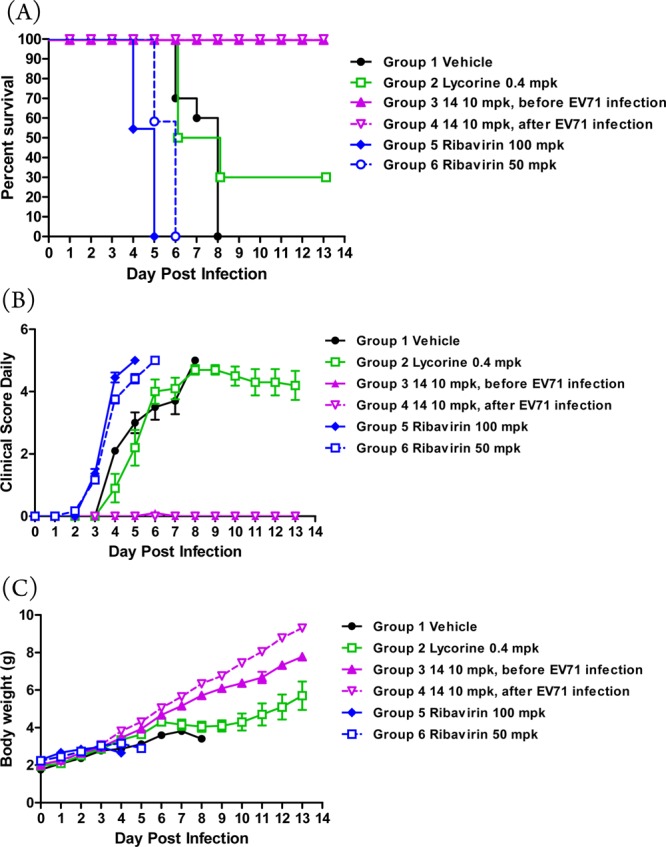
In vivo antiviral activity of 14. (A) Survival rate. (B) Mice body weight. (C) Clinical score. Three-day-old ICR mice are i.p. inoculated with EV71 and administrated with vehicle (N = 10), lycorine (0.4 mpk, N = 10), 14 before EV71 infection (10 mpk, N = 10), 14 after EV71 infection (10 mpk, N = 9), and ribavirin (100 mpk, N = 11, or 50 mpk, N = 12).
Chemistry
The chemical synthesis of 14 is shown in Scheme 4. Starting from 5-hydroxy-1-indanone 14a, condensation of 14a with ethoxyamine gave oxime ether 14b. Monosubstitution of 14b with 1,5-dibromopentane afforded intermediate 14c, which then reacted with C to give 14d. Final product 14 was obtained by the reaction of chloropyridine intermediate 14d with BocNH2 and Boc deprotection in situ. The synthesis of other compounds can be found in the Supporting Information.
Scheme 4. Synthesis of 14.

Conclusion
Considering the danger of severe EV71 infection in children and that no marketed antiviral drugs are available, there is an unmet medical need to develop effective antiviral treatment. The novel aminopyridyl 1,2,5-thiadiazolidine 1,1-dioxides were first synthesized and their anti-EV71 activities were evaluated by CPE assay. In this series, compound 14 was identified to have excellent antiviral activity against EV71 (EC50 = 4 nM) and is more potent than DBPR103. Furthermore, 14 showed excellent in vivo efficacy in the EV71 infected mouse model. Thus, 14 is a promising drug candidate for potential application in the clinical therapy of HFMD.
Glossary
ABBREVIATIONS
- EV71
Enterovirus 71
- HFMD
hand, foot, and mouth disease
- Cox A16
coxsackievirus A16
- CPE
cytopathogenic effect
- RD
rhabdomyosarcoma
- ADME
absorption, distribution, metabolism, and excretion
- PK
pharmacokinetics
- CYP
Cytochrome P450
- hERG
human ether-a-go-go related gene
- MMS
microsomes metabolic stability
- IV
intravenous
- IP
intraperitoneal
- PO
per oral
- AUC
area under curve
- CL
clearance
- T1/2
half-life
- Vd
volume of distribution
- PFU
plaqueforming unit
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00188.
Synthetic procedures, analytical data, and assay protocol (PDF)
Author Contributions
§ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Yang Y.; Wang H.; Gong E.; Du J.; Zhao X.; McNutt M. A.; Wang S.; Zhong Y.; Gao Z.; Zheng J. N. Neuropathology in 2 cases of fatal enterovirus type 71 infection from a recent pidemic in the People’s Republic of China: a histopathologic, immunohistochemical, and reverse transcription polymerase chain reaction study. Hum. Pathol. 2009, 40, 1288. 10.1016/j.humpath.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhu Z.; Yang W.; Ren J.; Tan X.; Wang Y.; Mao N.; Xu S.; Zhu S.; Cui A.; Zhang Y.; Yan D.; Li Q.; Dong X.; Zhang J.; Zhao Y.; Wan J.; Feng Z.; Sun J.; Wang S.; Li D.; Xu W. An emerging recombinant human enterovirus 71 responsible for the 2008 outbreak of Hand Foot and Mouth Disease in Fuyang city of China. Virol. J. 2010, 7, 94. 10.1186/1743-422X-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Yang B.; Zhai Y.; Yin Z.; Sun Y.; Rao Z. Peptidyl Aldehyde NK-1.8k Suppresses Enterovirus 71 and Enterovirus 68 Infection by Targeting Protease 3C. Antimicrob. Agents Chemother. 2015, 59, 2636. 10.1128/AAC.00049-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; He Y. Q.; Yi L. N.; Zan H.; Kung H. F.; He M. L. Viral kinetics of Enterovirus 71 in human abdomyosarcoma cells. World J. Gastroenterol. 2011, 17, 4135. 10.3748/wjg.v17.i36.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Yang B.; Hao F.; Wang P.; He H.; Huang L.; Zhang X.; Zhang S.; Peng X.; Yin K.; Hu J.; Chen X.; Gu Z.; Wang L.; Shen L.; Hu G.; Li N.; Li J.; Chen S.; Xiao W.; Wang Z.; Guo G.; Chang X.; Zhang L.; Cai Q.; Lin T. Design, Synthesis, and Biological Evaluation of anti-EV71 Agents. Bioorg. Med. Chem. Lett. 2016, 26, 3346. 10.1016/j.bmcl.2016.05.036. [DOI] [PubMed] [Google Scholar]
- Smyth M. S.; Martin J. H. Picornavirus uncoating. Mol. Pathol. 2002, 55, 214. 10.1136/mp.55.4.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevear D. C.; Tina M. T.; Martin E. S.; James M. G. Activity of pleconaril against enteroviruses. Antimicrob. Agents Chemother. 1999, 43, 2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shia K. S.; Li W. T.; Chang C. M.; Hsu M. C.; Chern J. H.; Leong M. K.; Tseng S. N.; Lee C. C.; Lee Y. C.; Chen S. J.; Peng K. C.; Tseng H. Y.; Chang Y. L.; Tai C. L.; Shih S. R. Design, Synthesis, and Structure-Activity Relationship of Pyridyl Imidazolidinones: A Novel Class of Potent and Selective Human Enterovirus 71 Inhibitors. J. Med. Chem. 2002, 45, 1644. 10.1021/jm010536a. [DOI] [PubMed] [Google Scholar]
- Chern J. H.; Chang C. S.; Tai C. L.; Lee Y. C.; Lee C. C.; Kang I. J.; Lee C. Y.; Shih S. R. Synthesis and antipicornavirus activity of (R)- and (S)-1-[5-(40-chlorobiphenyl-4-yloxy)-3-methylpentyl]-3-pyridin-4-yl-imidazolidin-2-one. Bioorg. Med. Chem. Lett. 2005, 15, 4206–4211. 10.1016/j.bmcl.2005.06.069. [DOI] [PubMed] [Google Scholar]
- De Colibus L.; Wang X.; John A. B. S.; James K.; Ren J.; Jonathan G.; Gerhard P.; Nicola S.; Thomas S. W.; Hu Z.; Wang J.; Li X.; Peng W.; David J. R.; Elizabeth E. F.; Rao Z.; David I. S. More-powerful virus inhibitors from structure-based analysis of HEV71 capsid-binding molecules. Nat. Struct. Mol. Biol. 2014, 21, 282. 10.1038/nsmb.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Peng W.; Ren J.; Hu Z.; Xu J.; Lou Z.; Li X.; Yin W.; Shen X.; Claudine P.; Thomas S.; Walter; Gwyndaf E.; Danny A.; Robin O.; David J. R.; Wang J.; David I. S.; Elizabeth E. F.; Rao Z. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 2012, 19, 424. 10.1038/nsmb.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compounds 5 and 10–18 were reported in WO2015165340.
- Chang C. S.; Lin Y. T.; Shih S. R.; Lee C. C.; Lee Y. C.; Tai C. L.; Tseng S. N.; Chern J. H. Design, Synthesis, and Antipicornavirus Activity of 1-[5-(4-Arylphenoxy)alkyl]-3-pyridin-4-ylimidazolidin-2-one Derivatives. J. Med. Chem. 2005, 48, 3522. 10.1021/jm050033v. [DOI] [PubMed] [Google Scholar]
- Chern J. H.; Lee C. C.; Chang C. S.; Lee Y. C.; Tai C. L.; Lin Y. T.; Shia K. S.; Lee C. Y.; Shih S. R. Synthesis and antienteroviral activity of a series of novel, oxime ether-containing pyridyl imidazolidinones. Bioorg. Med. Chem. Lett. 2004, 14, 5051. 10.1016/j.bmcl.2004.07.084. [DOI] [PubMed] [Google Scholar]
- Liu J.; Yang Y.; Xu Y.; Ma C.; Qin C.; Zhang L. Lycorine reduces mortality of human enterovirus 71-infected mice by inhibiting virus replication. Virol. J. 2011, 8, 483. 10.1186/1743-422X-8-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Yu C. Animal models of enterovirus 71 infection: applications and limitations. J. Biomed. Sci. 2014, 21, 31. 10.1186/1423-0127-21-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.