

Abstract
Adoptive T-cell therapy with CD19-specific chimeric antigen receptors (CARs) is promising for treatment of advanced B-cell malignancies. Tumor targeting of CAR-modified T-cells is likely to contribute therapeutic potency; therefore we examined the relationship between the ability of CD19-specific CAR (CD19-CAR)-transduced T-cells to accumulate at CD19+ tumor lesions, and their ability to provide antitumor effects in xenograft mouse models. Normal human peripheral blood lymphocytes, activated with immobilized RetroNectin and anti-CD3 antibodies, were transduced with retroviral vectors that encode CD19-CAR. Expanded CD19-CAR T-cells with a high transgene expression level of about 75% produced IL-2 and IFN-γ in response to CD19, and lysed both Raji and Daudi CD19+ human B-cell lymphoma cell lines. Furthermore, these cells efficiently accumulated at Raji tumor lesions where they suppressed tumor progression and prolonged survival in tumor-bearing Rag2−/− γc−/− immunodeficient mice compared to control cohorts. These results show that the ability of CD19-CAR T-cells to home in on tumor lesions is pivotal for their anti-tumor effects in our xenograft models, and therefore may enhance the efficacy of adoptive T-cell therapy for refractory B-cell lymphoma.
Keywords: Adoptive T-cell therapy, CD19, Tumor targeting, B-cell lymphoma, Chimeric antigen receptor
1. Introduction
Chimeric antigen receptor (CAR)-based T-cell therapy is a promising approach to targeted cancer immunotherapy. CARs are hybrid proteins consisting of an extracellular single chain fragment of variable region (scFv) fused to intracellular lymphocyte signaling domains CD28 or 4-1BB (CD137), coupled with CD3ξ, to mediate T-cell activation. This particular configuration of CARs is widely used in current preclinical and clinical studies [1]. Independent of the human leukocyte antigen system, CAR-transduced T-cells can directly recognize and kill tumor cells that express specific cell surface antigens.
CD19 antigen is an attractive target for CAR-based T-cell therapy since it is a B-cell lineage-specific surface molecule, which is expressed on normal and most malignant B-cells but not on hematopoietic stem cells [2]. Preclinical studies have demonstrated that CD19-specific CAR (CD19-CAR) T-cells eliminated CD19+ B-cell malignancies [3–5]. Recent phase I clinical trials using CD19-CAR T-cells have been shown to be safe and exhibit impressive anti-tumor effects for advanced CD19+ chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), and B-cell lymphoma patients [6–11]. However, the optimal CAR-based T-cell therapy for maximizing therapeutic potency is yet to be realized.
Both persistence and tumor targeting of adoptive transferred CAR-modified T-cells are likely to contribute to therapeutic potency. Preclinical and early clinical trial results have shown that persistence of CD19-CAR T-cells in vivo was correlated with anti-tumor effects for B-cell malignancies [4,5,12]. Thus, CAR designs which involve a signaling feature, that activates and prolongs T-cell survival in vivo, have been extensively pursued [13–16]. In contrast, much less is known regarding the correlation between the anti-tumor effects of CD19-CAR T-cells and their tumor targeting ability.
In the present study, we generated large numbers of primary T-cells that expressed CD19-CAR with a CD28 signaling domain. These expanded T-cells exhibited CD19-dependent redirected effector functions in vitro. We also observed that they accumulated at the site of tumors and exhibited anti-tumor effects at these sites in human B-cell lymphoma xenograft immunodeficient mice. Our results show that efficient accumulation at tumors of CD19-CAR T-cells can enhance CAR-based T-cell therapy for B-cell lymphoma.
2. Materials and methods
2.1. Plasmids
The gamma retroviral vector SFG-1928z that encodes CD19-specific scFv fused to CD28- and CD3ξ intracellular signal domains has been described [3] (Fig. 1A). A luciferase expression plasmid pEF-Luc was generated by ligating the luciferase gene from pGL3 (Promega, Madison, WI) into a pEF-PGKneo vector containing the EF1α promoter and Neomycin resistance gene cassette [17].
Fig. 1.
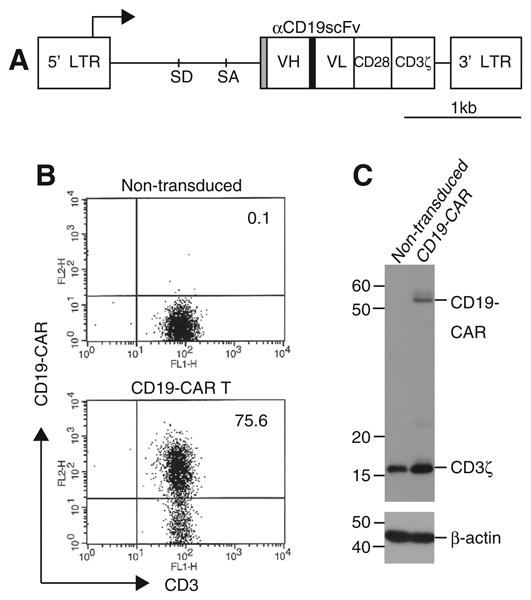
Generation of CD19-CAR modified T-cells using RetroNectin/anti-CD3 stimulation. (A) Schematic structure of SFG-1928z retroviral vector. VH and VL: variable heavy- and light chain; SD: splice donor; SA: splice acceptor; LTR: long terminal repeat; Gray box: CD8α signal peptide; Black box: (GGGGS)3 linker. PBLs were stimulated with RetroNectin/anti-CD3 and transduced twice with SFG-1928z retroviral vectors. They were propagated on CD19+ feeder cells. (B) Surface expression of CD19-CAR on transduced or non-transduced T-cells was examined by flow cytometry. All numbers show the percentages of CD3+ CD19-CAR+ populations. Representative data was shown in three independent experiments. (C) CD19-CAR expression in cell lysates from transduced T-cells or the control non-transduced T-cells was detected by Western blotting with anti-CD3ξ; β-actin is a loading control.
2.2. Peripheral blood lymphocytes and cell lines
Peripheral blood lymphocytes (PBLs) from three healthy donors were obtained with the approval of the Jichi Medical University Institutional Review Board, and written consent of each donor. PBLs were cultured with X-VIVO 15 (Takara Bio, Shiga, Japan), supplemented with 5% human AB serum (NOVA Biologics, Oceanside, CA) and 1 nanomolar recombinant human interleukin-2 (IL-2) (Life technologies, Carlsbad, CA). The retrovirus packaging cells PG13 [18] and mouse fibroblast NIH3T3 cells that expresses human CD19 (3T3/CD19) [19] were cultured with DMEM, supplemented with 10% FBS. Human Burkitt lymphoma cell lines Raji and Daudi (Health Science Research Resources Bank, Osaka, Japan) were maintained in RPMI1640 supplemented with 10% FBS. Raji cells expressing luciferase (Raji/Luc) were established by transfection with the pEF-Luc plasmid, followed by G418 selection.
2.3. Retroviral transduction and T-cell expansion
For retroviral transduction, PBLs were stimulated with 20 μg/ml of immobilized RetroNectin (Takara Bio) and 10 μg/ml anti-CD3e antibodies (R&D Systems, Minneapolis, MN) for 3 days and transduced with retroviral particles using a preloading method [20]. Briefly, PG13 viral producer cells were established by stable transduction of SFG-1928z retroviral vectors (Fig. 1A). RetroNectin-coated plates were loaded with retroviral particles obtained from the PG13 cells, and centrifuged (1000g, 2 h, 32 °C). Stimulated PBMCs were added to the preloaded plates, and incubated overnight. The procedure was repeated on the next day. For selective ex vivo expansion, transduced T-cells were co-cultured with γ-irradiated (50 Gy) 3T3/CD19 cells at 1:1 ratio. On days 5 and 10, the 3T3/CD19 cells were added to the T-cell cultures.
2.4. Flow cytometry
We analyzed cell surface expression of CD19-CAR on transduced T-cells by flow cytometry using a BD LSR with CellQuest software (BD Biosciences, San Jose, CA). Antibodies used for CD19-CAR detection were biotin goat anti-mouse F (ab'), PE Strep-tavidin (Jackson Immunoresearch, West Grove, PA), and FITC anti-human CD3 (Biolegend, San Jose, CA). Isotype-matched antibodies were used as controls.
2.5. Western blotting
CD19-CAR cellular protein expression was examined by Western blotting. Cell lysate prepared from CD19-CAR T-cells was separated on 12% polyacrylamide gels and then transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). The membranes were incubated with a mouse monoclonal anti-human CD3ξ antibody (BD Biosciences) or a rabbit anti-human β-actin antibody (Cell Signaling, Danvers, MA) as a control, followed by anti-mouse immunoglobulin conjugated with horseradish peroxidase. Proteins recognized by antibodies were visualized with an enhanced chemiluminescent detection system (GE Healthcare, Buckinghamshire, UK).
2.6. Elisa
Supernatants from the duplicate wells of co-cultures of 3T3/CD19 cells with CD19-CAR T-cells at 1:1 ratio were harvested after 48 h incubation. Human IL-2 and IFN-γ levels in supernatants were measured by ELISA kits (Biolegend).
2.7. Cell lytic activity
Cell lytic activity of CD19-CAR T-cells was examined by standard 4-h chromium release assays [21].
2.8. Mouse tumor model
To determine if modified T-cells accumulated at tumor lesions in vivo, 10- to 12-week-old Balb/c Rag2−/−γc−/− (Rag2−/γ−) immunodeficient mice [22-24] were injected subcutaneously with 5 × 106 Raji/Luc cells. Two weeks after inoculation, 107 CD19-CAR T-cells were injected intravenously into tumor-bearing mice. Tumors were removed 24 h after T-cell infusion, and analyzed by immunohistochemistry with an anti-human CD3 antibody (Dako, Glostrup, Denmark). The localization of human CD3+ T-cells was examined using a BX50 microscope (Olympus, Tokyo, Japan). To evaluate accumulation at tumor sites of CAR-modified T-cells and their anti-tumor effects, Rag2−/γ− mice injected intravenously with 5 × 104 Raji/Luc cells on day 0, were adoptively transferred with a single intravenous infusion of 107 CD19-CAR T-cells on day 3. To monitor the progression of Raji/Luc cells in vivo, the substrate of luciferase, D-luciferin was injected intraperitonealy into mice (75 mg/kg body weight), and bioluminescence imaging was performed using an IVIS imaging system (Xenogen, Hopkinton, MA) with Living Image software (Xenogen). In this therapeutic experiment, accumulations at tumor lesions of CD19-CAR T-cells were determined by immunohistochemistry as described above. All mouse experiments were carried out in a humane manner after receiving approval from the Institutional Animal Experiment Committee of Jichi Medical University.
2.9. Statistics
Student's t test was used to evaluate in vitro experimental differences. Survival data was assessed by the log-rank test. GraphPad Prism 5 (GraphPad Software, San Diego, CA) was used for the statistical calculations. p < 0.05 was considered statistically significant.
3. Results and discussion
3.1. Generation of CD19-CAR T-cells and transgene expression
To generate CD19-CAR T-cells, we utilized RetroNectin/anti-CD3 stimulation to activate PBLs from healthy donors, since this method allows efficient proliferation and high-level transgene expression in retrovirally modified T-cells, with less differentiated phenotypes as described previously [20]. Comparative analysis showed that RetroNectin/anti-CD3 treatment mediated ex vivo expansion of transduced T-cells to levels comparable to treatment with anti-CD3 and anti-CD28 beads, but was significantly higher than treatment with anti-CD3 alone [20]. Activated PBLs were transduced with SFG-1928z retroviral vectors that encode the 2nd generation of CD19-CAR with the CD28 gene (Fig. 1A), and were selectively propagated on 3T3/CD19 cells. Transduced T-cell numbers increased about 200-fold within two weeks (data not shown). Expression of CD19-CAR+ CD3+ in transduced T-cells was approximately 75%, as assessed by flow cytometry (Fig. 1B). Cellular protein expression of CD19-CAR in transduced T-cells was also confirmed by Western blotting using an anti-CD3ξ antibody (Fig. 1C). We then examined immunophenotypes of CD19-CAR T-cells by flow cytometry. At day 14 of culture, CD19-CAR T-cells predominantly expressed CD8 (75%), compared to the control PBLs without culture (35%) (Fig. S1) as described previously [20]; whereas, anti-CD3 and anti-CD28 beads preferentially induced the CD4+ population (63%), under the same experimental setting (data not shown). While both CD4+ and CD8+ T-cell subsets are targets for CD19-CAR gene transduction, it is not known which subset is suitable for T-cell therapy. Compared to control PBLs without culture, CD19-CAR T-cells cultured with antigen contained primarily central or effector memory subpopulations as exemplified by the co-expression of either CD62L or CCR7 along with CD45RA, or CD27 with CD28 (Fig. S1). Especially noteworthy is the fact that many of the CD19-CAR T-cells were less differentiated (i.e., exhibited a more primitive phenotype), and co-expressed both CD27+ and CD28+, an expression phenotype thought to be suitable for adoptive T-cell transfer [25]. In addition, the lack of CD279 (PD-1) expression (<1%) supports the absence of exhaustion among the CD19-CAR T-cells (data not shown). These results show that large numbers of these cells with high-level transgene expression were generated in our culture conditions.
3.2. CD19-dependent cytokine production and cell lytic activity
We examined the ability of CD19-CAR T-cells to produce cytokines by ELISA and found that they selectively produced IL-2 and IFN-γ in response to CD19+ target cells (Fig. 2A). Cytotoxic assays revealed that they also effectively lysed CD19+ Raji and Daudi cells at increasing effector/target ratios compared to non-transduced T-cells (Fig. 2B). To address the specific killing ability of CD19-CAR T-cells, K562 cells expressing CD19 (K562/CD19) and parental K562 cells (CD19 negative) were used as target cells. As expected, CD19-CAR T-cells lysed CD19+ cells more effectively than CD19− cells (Fig. S2). Taken together, these results show that expanded CD19-CAR T-cells exhibited CD19-dependent redirected effector functions in vitro.
Fig. 2.

Antigen-specific cytokine production and cell lytic activity of CD19-CAR T-cells. (A) IL-2 and IFN-γ secretion by CD19-CAR T-cells after co-culture with 3T3/CD19 or the control 3T3 cells (p < 0.05). N.D., not detectable. (B) Cell lytic activity of CD19-CAR T-cells (black circles) or the control non-transduced T-cells (white circles) against Raji and Daudi cells in 51Cr release assays. Data show mean ± SD of triplicate wells. CD19-CAR T-cells lysed well compared to non-transduced T-cells at all effector/target ratios (p < 0.01).
3.3. Accumulation at tumors and anti-tumor activity
To investigate that modified T-cells were able to accumulate at tumor lesions, Rag2−/γ− mice with subcutaneous Raji tumors were injected with either CD19-CAR T-cells or non-transduced T-cells. Immunohistochemistry showed that robust accumulation of human CD3+ T-cells occurred at tumor lesions 24 h after administration of CD19-CAR T-cells. A few CD3+ T-cells were detected in control mice (Fig. 3A). Engineered T-cells effectively localized to tumor lesions compared with control T-cells (Fig. 3B). These results are similar to those found for infused T-cells that accumulated at tumors induced by the subcutaneous injection of NALM-6 cells (an ALL cell line) into SCID/beige mice 48 h after T-cell infusion with first generation CD19-CARs [26]. We examined the relationship between accumulation at tumor sites of CD19-CAR T-cells and their anti-tumor effects using a systemic Raji tumor mouse model that we established. When Rag2−/γ− mice were injected intravenously with Raji/Luc cells, these tumors grew progressively as assessed by bioluminescent imaging. We further confirmed CD19+ tumor infiltration in the spleen, liver and bone marrow by immunohistochemistry (Fig. S3). These infiltrates resembled an aggressive form of human B-cell lymphoma.
Fig. 3.

Accumulation at tumor lesions of CD19-CAR T-cells in subcutaneous xenograft mouse models. (A) Immunohistochemical staining for human CD3 (brown) in subcutaneous Raji tumors from Rag2−/γ− mice at 24 h after infusion of either CD19-CAR T-cells or the control non-transduced T-cells. Sections from representative mice are shown. (B) CD3+ cell counts in tumor sections from mice receiving CD19-CAR T-cell were higher than those of control sections (p < 0.01). Data show mean ± SD of four different fields of view per section. N.D., not detectable. Scale lines, 50 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
For adoptive T-cell therapy, Rag2−/γ− mice injected intravenously with Raji/Luc cells were subsequently infused with CD19-CAR T-cells. Bioluminescent imaging revealed that the infused cells successfully suppressed Raji tumor progression; whereas, infusion of non-transduced T-cells did not (Fig. 4A), thereby excluding alloreactivity as a mechanism of tumor suppression. Furthermore, mice treated with CD19-CAR T-cells exhibited significantly prolonged survival compared to control cohorts (Fig. 4B). We also examined the ability of infused T-cells to accumulate at tumors in each cohort. Immunohistochemistry revealed more efficient accumulation of human CD3+ T-cells at spleen lesions in mice receiving CD19-CAR T-cells, compared to mice receiving non-transduced T-cells at day 29 (Fig. 4C and D). These results suggest that such cells survive for at least 29 days in tumor bearing-mice, and that the ability of CD19-CAR T-cells to accumulate at tumor lesions is likely associated with the anti-tumor effects observed in human B-cell lymphoma xenograft mouse models.
Fig. 4.

(A) Bioluminescent imaging of systemic Raji tumor progression in Rag2−/γ− mice following T-cell infusion. Mice inoculated intravenously with Raji/Luc cells on day 0, were injected intravenously with either CD19-CAR T-cells or control non-transduced T-cells on day 3. Bioluminescent imaging was performed at days 15, 21 and 29. (B) Enhanced survival of CD19-CART-cell-treated mice when compared with the control cohorts (p < 0.01). (C) Immunohistochemical staining for human CD3 (brown) in spleen lesions from tumor-bearing mice at day 29 after T-cell administration. (D) CD3+ cell counts in tumor sections from mice receiving CD19-CAR T-cell were higher than those of control sections (p < 0.01). Data show mean ± SD of four different fields of view per section. Scale lines, 50 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
It is important to note that other parameters, in addition to the accumulation of CD19-CAR T-cells at tumor sites, may also contribute to tumoricidal effects. In CLL patients for example, the localization of CD19-CAR T-cells to tumor lesions was detected by immunohistochemistry up to two months post T-cell infusion, but the accumulation of CAR-modified T-cells at tumor sites was not always consistent with clinical outcomes [7].
In any event, tumor targeting of infused effector T-cells is likely an important prerequisite for CD19-CAR anti-tumor effects. Parameters such as a CAR's structure, scFv antibody region, specific target epitope, and affinity for a given epitope, may be important factors which influence CAR-mediated tumor targeting. In current clinical trials using CD19-CAR T-cells for advanced B-cell malignancies, two types of CD19-CARs expressing either FMC63 or SJ25C1 monoclonal antibody-derived scFvs recognize distinct epitopes with different binding affinities that may play an important role in tumor targeting, anti-tumor effects, and subsequent clinical outcomes [27]. However, there is no direct functional comparison between two CD19-CARs, and critical determinants to select the optimal scFv remain unknown. Recently, Haso et. al. demonstrated that epitope specificity has a major impact on CAR efficacy, since CD22 antigen-specific CARs (CD22-CAR) incorporating an scFv directed to a more membrane proximal epitope, mediated more potent anti-tumor effects in an ALL tumor mouse model than other CD22-CARs of similar affinity, but which targeted distinct epitopes [28]. Thus, the careful selection of a specified CAR antigen epitope is an important consideration towards the improvement of CAR-modified T-cells in eliciting anti-tumor effects. As an alternative approach to efficient tumor targeting of CARs, Craddock et. al. showed that the transfer of CCR2b-chemokine-receptor-expressing CAR-modified T-cells (targeted to GD2 antigen) could enforce preferential migration to tumor lesions, and boost anti-tumor effects in a neuroblastoma mouse model [29]. Therefore, refinement with regard to tumor targeting of CAR-modified T-cells may confer enhanced therapeutic potency to CAR-based T-cell therapy.
In conclusion, we have successfully generated large numbers of CD19-CAR T-cells that exhibited antigen-specific cytokine production and cell lytic activity in vitro. We also showed that adoptive transfer of these cells strongly suppressed human B-cell lymphoma progression, and this activity correlated with the ability of infused T-cells to accumulate at tumor sites in the xenograft mouse model. These studies expand the repertoire of more potent CD19-CAR-based T-cell therapies for refractory B-cell lymphoma.
Based on our findings here and in other clinical trials [6,7], we will conduct a phase I/II clinical trial, whereby patients with chemorefractory non-Hodgkin B-cell lymphoma will be treated with autologous CD19-CAR T-cells.
Supplementary Material
Acknowledgments
This work was supported by JSPS KAKENHI Grant Nos. 22700923 to T. Tsukahara; 24390247 to K. Ozawa and Jichi Medical University Young Investigator Award (to T. Tsukahara). This publication was subsidized by JKA through its promotion funds from KEIRIN RACE. We thank Dr. M. Ito (Central Institute for Experimental Animals) for generously providing Balb/c Rag2−/−γc−/− mice. We also thank Gary Baley for critical editing as academic editor.
Abbreviations
- CAR
chimeric antigen receptor
- CD19-CAR T-cells
CD19-specific CAR-transduced T-cells
Footnotes
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bbrc.2013.07.030.
References
- 1.Davila ML, Brentjens R, Wang X, Riviere I, Sadelain M. How do CARs work?: early insights from recent clinical studies targeting CD19. Oncoimmunology. 2012;1:1577–1583. doi: 10.4161/onci.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li YS, Wasserman R, Hayakawa K, Hardy RR. Identification of the earliest B lineage stage in mouse bone marrow. Immunity. 1996;5:527–535. doi: 10.1016/s1074-7613(00)80268-x. [DOI] [PubMed] [Google Scholar]
- 3.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, Quintas-Cardama A, Larson SM, Sadelain M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13:5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 4.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, Smith DD, Forman SJ, Jensen MC, Cooper LJ. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 5.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, Sadelain M. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 14.Tammana S, Huang X, Wong M, Milone MC, Ma L, Levine BL, June CH, Wagner JE, Blazar BR, Zhou X. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum Gene Ther. 2010;21:75–86. doi: 10.1089/hum.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, Till B, Raubitschek A, Forman SJ, Qian X, James S, Greenberg P, Riddell S, Press OW. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18:712–725. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 16.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhen L, King AA, Xiao Y, Chanock SJ, Orkin SH, Dinauer MC. Gene targeting of X chromosome-linked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox. Proc Natl Acad Sci USA. 1993:9832–9836. doi: 10.1073/pnas.90.21.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller AD, Garcia JV, von Suhr N, Lynch CM, Wilson C, Eiden MV. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol. 1991;65:2220–2224. doi: 10.1128/jvi.65.5.2220-2224.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Latouche JB, Sadelain M. Induction of human cytotoxic T lymphocytes by artificial antigen-presenting cells. Nat Biotechnol. 2000;18:405–409. doi: 10.1038/74455. [DOI] [PubMed] [Google Scholar]
- 20.Yu SS, Nukaya I, Enoki T, Chatani E, Kato A, Goto Y, Dan K, Sasaki M, Tomita K, Tanabe M, Chono H, Mineno J, Kato I. In vivo persistence of genetically modified T cells generated ex vivo using the fibronectin CH296 stimulation method. Cancer Gene Ther. 2008;15:508–516. doi: 10.1038/cgt.2008.21. [DOI] [PubMed] [Google Scholar]
- 21.Kannagi M, Sugamura K, Sato H, Okochi K, Uchino H, Hinuma Y. Establishment of human cytotoxic T cell lines specific for human adult T cell leukemia virus-bearing cells. J Immunol. 1983;130:2942–2946. [PubMed] [Google Scholar]
- 22.Ohbo K, Suda T, Hashiyama M, Mantani A, Ikebe M, Miyakawa K, Moriyama M, Nakamura M, Katsuki M, Takahashi K, Yamamura K, Sugamura K. Modulation of hematopoiesis in mice with a truncated mutant of the interleukin-2 receptor gamma chain. Blood. 1996;87:956–967. [PubMed] [Google Scholar]
- 23.Ohteki T, Fukao T, Suzue K, Maki C, Ito M, Nakamura M, Koyasu S. Interleukin 12-dependent interferon gamma production by CD8alpha+ lymphoid dendritic cells. J Exp Med. 1999;189:1981–1986. doi: 10.1084/jem.189.12.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, Alt FW. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 25.Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, Phan GQ, Kammula US, Hughes MS, Citrin DE, Restifo NP, Wunderlich JR, Prieto PA, Hong JJ, Langan RC, Zlott DA, Morton KE, White DE, Laurencot CM, Rosenberg SA. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. 2010;16:6122–6131. doi: 10.1158/1078-0432.CCR-10-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santos EB, Yeh R, Lee J, Nikhamin Y, Punzalan B, La Perle K, Larson SM, Sadelain M, Brentjens RJ. Sensitive in vivo imaging of T cells using a membrane-bound Gaussia princeps luciferase. Nat Med. 2009;15:338–344. doi: 10.1038/nm.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brentjens RJ, Curran KJ. Novel cellular therapies for leukemia: CAR-modified T cells targeted to the CD19 antigen. Hematol Am Soc Hematol Educ Program. 2012;2012:143–151. doi: 10.1182/asheducation-2012.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM, Wayne AS, Mackall CL, Orentas RJ. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.