

Abstract
The regulators of the Rho-family GTPases, GTPase-activating proteins (GAPs) and guanine exchange factors (GEFs), play important roles in axon guidance. By means of a functional genomic study of the Rho-family GEFs and GAPs in Drosophila, we have identified a Rho-family GAP, CrossGAP (CrGAP), which is involved in Roundabout (Robo) receptor-mediated repulsive axon guidance. CrGAP physically associates with the Robo receptor. Too much or too little CrGAP activity leads to defects in Robo-mediated repulsion at the midline choice point. The CrGAP gain-of-function phenotype mimics the loss-of-function phenotypes of both Robo and Rac. Dosage-sensitive genetic interactions among CrGAP, Robo, and Rac support a model in which CrGAP transduces signals downstream of Robo receptor to regulate Rac-dependent cytoskeletal changes.
Keywords: axon guidance, GTPase-activating protein, guanine nucleotide exchange factor, Slit, Roundabout
Axonal repulsion is a major force guiding the formation of the neural network during development, and it is thought to restrict the ability of axons to regenerate after injury (1, 2). In the developing CNS, most growth cones confront the midline during their journey and make the decision of whether or not to cross (3). An important family of receptors that controls midline crossing is the Roundabouts (Robos), which respond to their repellent ligand Slit (4). In Drosophila robo mutants, too many axons cross and recross the midline (5). Several molecules have been implicated in the Robo signaling pathway, including the actin binding protein Enabled (6, 7), the tyrosine kinase Abelson (Abl) (6, 8, 9), and the Ras/Rho GEF Son of Sevenless (10, 11). Nevertheless, our understanding of the signal-transduction mechanism leading from Slit/Robo to cytoskeleton rearrangement during repulsive axon guidance remains incomplete.
The Rho family of small GTPases (including Rac, Rho, and Cdc42) are key regulators of the actin cytoskeleton in neurons (12, 13). Analysis of mutants of the three Drosophila rac genes (rac1, rac2, and mtl) has suggested an important role for rac in midline axon guidance (14). In rac1, mtl double mutants, axons are misrouted across the midline (14). Biochemical and genetic studies have revealed an important role of Cdc42 and Rac in Robo repulsion (15, 16). In contrast to the Slit-dependent negative regulation of Cdc42 in vertebrates, where the Slit/Robo GAPs (srGAPs) link the Robo cytoplasmic domain directly to Cdc42 (16), how Slit stimulation leads to Rac activation in both vertebrates and Drosophila is unknown.
The small GTPases are activated by the guanine nucleotide exchange factors (GEFs) and inactivated by the GTPase-activating proteins (GAPs). Increasing evidence suggests that guidance receptors can regulate growth cone or cell motility by regulating the Rho-family GTPases through these regulatory proteins (16). Therefore, we began with a systematic search to screen for Rho GEFs and GAPs that could be involved in CNS axon guidance and, in particular, the Robo-mediated repulsive guidance. We carried out an expression analysis of all of the Rho family GEFs and GAPs in Drosophila and performed a genetic screen by using transgenic RNA interference (RNAi). Our study has led to the discovery of CrossGAP (CrGAP), which functions in Robo-mediated repulsion. We show CrGAP directly interacts with Robo both biochemically and genetically and acts as a GAP specifically for Rac to regulate midline crossing.
During the preparation of our manuscript, CrGAP was independently discovered as a mediator of Robo repulsion in tracheal cells and axons, and was named Vilse (17). Intriguingly, our studies revealed that CrGAP/Vilse may function differently during signaling downstream of Robo in midline neurons versus tracheal cells.
Materials and Methods
Molecular Constructs. Drosophila CrGAP was PCR amplified from LD10379 and subcloned into the pUAST vector. (At the time that we made this construct, the genome annotation predicted a sequence with the start codon at amino acid 22 of the gene. Therefore, our construct lacks the N-terminal 21 aa). To make the CrGAP WW domain GST-fusion construct, amino acids 22–154 of CrGAP were PCR amplified and cloned into pGEX4T-2 vector. For the WW-domain-containing CrGAP construct, amino acids 22–507 of the gene were fused with a N-terminal 6-myc tag, and cloned into the pcDNA3 vector. Robo constructs have been published in ref. 6.
Genetics. Upstream activating sequence (UAS)-CrGAP was transformed into W1118 flies by using standard procedures. Three independent insertions (lines 28, 33 and 37) were generated. Dose-dependent gain-of-function phenotypes are consistently seen in all these three transgenic lines. The following stocks were generated: (i) UAS-CrGAP28, UAS-CrGAP33/CyOWgβgal; (ii) UAS-CrGAP37, elav GAL4 3A/TM3Ubxβgal; (iii) UAS-CrGAP28/CyO; elav GAL43A/TM2; (iv) UAS-Rac1, GMR-GAL4/CyOGAL-80; (v) UAS-RhoA, GMR-GAL4/CyO; and (vi) UAS-CrGAP28; UAS-CrGAPRNAi-1/CyOWgβgal. The rac1J10mtlΔ mutant flies were provided by B. Dickson (Institute of Molecular Biotechnology, Vienna). GFP/CyoTubGal80 was a gift from J. Thomas (The Salk Institute, La Jolla, CA).
Ab Generation. The 6xHis-tagged CrGAP fusion protein (amino acids 147–297 in pQE-31 (Qiagen, Valencia, CA) was expressed in Escherichia coli, purified with Ni-NTA agarose beads (Qiagen), and used for mouse immunization at the Hybridoma Facility at the University of Pennsylvania. Polyclonal antiserum was used for immunoprecipitation (IP) (2 μl per IP reaction), and mAb was used for Western blotting (1:100) and immunohistochemistry (1:200).
Biochemistry. GST pull-down assays were performed as described in ref. 6. For coimmunoprecipitation (Co-IP), HEK 293T cells were transfected by using Effectene (Qiagen). For Co-IP of HA-Robo and myc-WWGAP, cells were washed once with PBS and lysed in lysis buffer [1× PBS/0.5% Triton X-100/1× protease inhibitor (Complete, Roche)], with 0.1 mM sodium orthovanadate. IPs were performed for 2 h at 4°C, washed three times with lysis buffer, and then analyzed by 8% SDS/PAGE and immunoblotted with Abs to visualize precipitated proteins. The following Abs were used for IPs and Western blot analyses: mouse anti-HA mAb (12CA5), mouse anti-myc mAb (9E10), and anti-Robo Ab (13C9). In vivo Co-IP was performed as described in Fan et al. (15). Supernatants from embryos expressing UAS-CrGAP driven by elavGAL4 were incubated with polyclonal anti-CrGAP (or anti-myc mAb as a negative control) and protein A Sepharose 4B beads (Zymed) for 4 h at 4°C. Beads were washed three times with lysis buffer. IP complexes were analyzed by 8% SDS/PAGE and immunoblotted with anti-CrGAP mAb or anti-Robo mAb (13C9).
Results
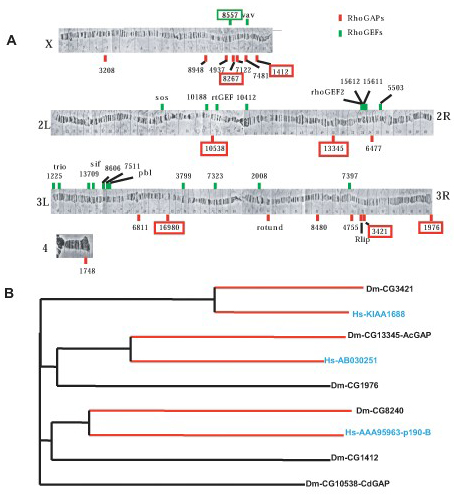
We surveyed the Drosophila genome and identified 20 Rho GAPs and 22 Rho GEFs (Fig. 6, which is published as supporting information on the PNAS web site) by using the tblastn program. We then examined the mRNA expression patterns of these genes (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site). Neuronal differentiation and axon guidance begin at stage 13 and are nearly complete by stage 17 in Drosophila embryos. Therefore, we focused our analysis on these embryonic stages. Excluding previously characterized genes, 17 of the 20 Rho GAPs and 14 of the 22 Rho GEFs were analyzed by RNA in situ. (Fig. 6). Six previously uncharacterized Rho GAPs and one Rho GEF are strongly expressed in the developing embryonic CNS (Fig. 7, which is published as supporting information on the PNAS web site), suggesting possible roles during axon guidance.
Genetic Analysis Implicates CrGAP in Midline Repulsion. To study the function of these Rho regulators in embryonic axon guidance, we performed a loss-of-function analysis by using transgenic double-stranded RNAi (18, 19), with a focus on the six CNS expressed Rho GAP genes. First, we evaluated the efficiency and the specificity of the transgenic RNAi method by a genetic test in the eye. Overexpression of RhoA using the GAL4/UAS system (20) causes a rough eye phenotype in adult flies because of a disruption of ommatidial cell development (Fig. 1c) (21). When an RNAi construct for RhoA is coexpressed in the eye, it largely restores the eye morphology to normal (Fig. 1f), indicating that the RNAi transgene can efficiently suppress the expression of the corresponding gene. In contrast, RhoA RNAi does not suppress the phenotype caused by rac1 overexpression (Fig. 1 b and e), even though rac1 shares 72% sequence similarity with RhoA. This observation indicates that the effect of the transgenic RNAi is highly specific.
Fig. 1.

Efficiency of the RNAi method and the specificity of CrGAP. Scanning-electron photomicrographs of adult Drosophila eyes are shown. The pictures are representative of >20 flies examined for each genotype. (a) A WT eye. (b and c) UAS-Rac1, GMR-GAL4/+ (b) and UAS-RhoA,GMR-GAL4/+ (c) flies have rough eye phenotypes. (e) An eye from a UAS-Rac1,GMR-GAL4/+; UAS-RhoARNAi/+ fly. The phenotype caused by overexpressing Rac is unaffected by expression of RhoRNAi. (f) UAS-RhoA,GMR-GAL4/+; UAS-RhoARNAi/+ eyes show a marked suppression of the RhoA rough eye phenotype. (g and h) The gain-of-function phenotype of Rac (b) is strongly suppressed by the coexpression of UAS-CrGAP (g), but the gain-of-function phenotype of RhoA is not (h). Expression of UAS-CrGAP alone does not cause an observable phenotype (data not shown). The flies were raised at 25°C.
To investigate potential roles in axon guidance, multiple independent RNAi lines of each of the six CNS-enriched GAPs were expressed in all postmitotic neurons, and the resulting embryos were examined with Abs that label all axons (mAb BP102) or a subset of longitudinal axons (mAb FasII). Somewhat surprisingly, none of the RhoGAP RNAi lines revealed significant defects in embryonic axon guidance when examined in this way (data not shown), suggesting that either the single RhoGAP genes do not have indispensable roles in axon guidance at these stages or, alternatively, that the RNAi technique does not completely eliminate gene function when expressed with elavGal4.
To further assess the potential function of the six RhoGAP genes during axon guidance, we examined the effects of GAP RNAi expression in a sensitized genetic background, in which slit and robo gene dose are reduced by one-half. slit, robo/+ transheterozygous embryos show mild midline guidance defects, with the medial-most longitudinal pathway occasionally crossing the midline (Fig. 2b). CNS expression of multiple independent RNAi transgenes for one of the six GAP genes (CrGAP), but none of the other five, resulted in a significant enhancement of the slit, robo/+ phenotype (Fig. 2 b–d). This dominant enhancement suggests that CrGAP may be involved in midline repulsion and that it may function in the Robo pathway.
Fig. 2.

CrGAP RNAi enhances guidance defects in slit, robo/+ embryos. Stage 16 embryos are stained with the mAb1D4 to reveal the three FasII-positive longitudinal axon pathways. SRE indicates slit1,robo5/+; elav-GAL4/+.(a) In WT embryos, the FasII-positive axon pathways never cross the midline. (b) In slit1, robo5/+ transheterozygous embryos, the medial (innermost) longitudinal pathway occasionally crosses at the midline (arrows). (c) In slit1,robo5/+ transheterozygotes, reducing CrGAP gene dose with the RNAi transgene significantly enhances the ectopic crossing defects. (d) Histogram showing the quantification of the midline crossing phenotypes in each of the indicated genotypes. The percentages of segments that showed ectopic crossing are shown. The number of segments scored in each genotype (n) was 275, 173, 297, 126, 136, 96, 159, and 136. *, P < 0.01, in Fisher's exact test. A second line of CrGAPRNAi, UAS-CrGAPRNAi-2/TM3 also shows significant enhancement of the crossing phenotype (53.3% crossing, n = 137) (data not shown).
To demonstrate that expression of CrGAP RNAi specifically targets the CrGAP gene, we generated flies that carry a WT UAS-CrGAP transgene (encoding all but the first 21 aa) and attempted to rescue the genetic interaction between slit, robo/+, and CrGAPRNAi. Pan-neural expression of UAS-CrGAP strongly suppressed the effects of CrGAPRNAi and restored the ectopic crossing phenotype to a level similar to that seen in slit, robo/+alone (Fig. 2d). This result supports the interpretation that the excessive midline crossing observed in slit, robo/+; CrGAPRNAi embryos are, in fact, due to reduction of the endogenous CrGAP gene expression, and it suggests that overexpression of our UAS transgene provides WT CrGAP activity. Furthermore, the rescuing effect of our UAS transgene suggests that the first 21 aa of CrGAP encoding part of the first WW domain is dispensable for function. Immunostaining of embryos overexpressing CrGAP with a CrGAP mAb reveals high-level overexpression of CrGAP in the CNS, indicating that our UAS transgenes efficiently express CrGAP (data not shown).
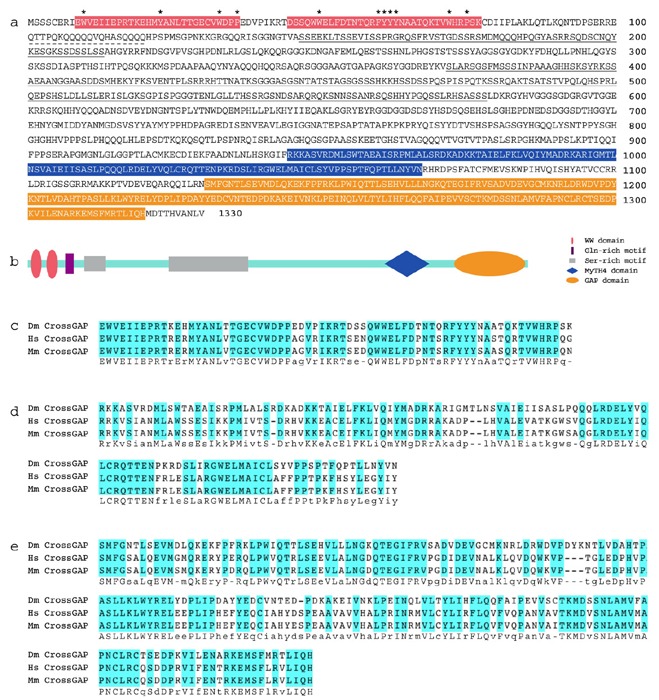
CrGAP Is a Conserved Rac-Specific GAP. CrGAP encodes a protein with at least one highly conserved homologue in human and in mouse. The human homologue of CrGAP, KIAA1688, was identified as a cDNA with large protein product from the brain (22), and shares 54.4% sequence similarity with Drosophila CrGAP. The gene contains at least three highly conserved signaling motifs: two WW domains, a MyTh4 (myosin tail homology) domain, and a Rho GAP domain (Fig. 8, which is published as supporting information on the PNAS web site). Interestingly, the MyTh4 domain is also present in MAX-1, a protein implicated in Netrin-mediated axon repulsion (23), suggesting that there might be a functional conservation of this domain in mediating output from repulsive guidance receptors. To determine which Rho GTPase(s) is the likely in vivo target of CrGAP, we again used the ectopic expression system in the Drosophila eye. Our results show that CrGAP can strongly and specifically down-regulate the activity of Rac1, but not that of RhoA (Fig. 1 g and h), and Cdc42 (data not shown). Together, these data suggest that the primary in vivo target (at least in the eye) of CrGAP is Rac, consistent with the sequence analysis in which the GAP domain in CrGAP is found to be most homologous to the GAP domain in Rac GAP proteins (data not shown).
CrGAP Overexpression Antagonizes the Repulsive Function of Robo. Because reducing CrGAP function enhances the defects observed in slit, robo/+ transheterozygous embryos (Fig. 2c), one might have predicted that increasing CrGAP function would show a reciprocal effect and suppress the slit, robo/+ defects. However, overexpressing CrGAP by using either EPCrGAP or UASCrGAP (missing only the first 21 aa) led to an exacerbation of the phenotypes of the slit, robo/+ transheterozygous embryos, with strongly increased crossing defects of longitudinal axons (Fig. 3 f and h, and data not shown), as well as a dramatically altered axon scaffold (Fig. 3 b and d). Similar expression of another RacGAP, rotundGAP, did not result in any enhancement of the slit, robo/+ phenotype, indicating that the effect is specific for CrGAP (Fig. 3h).
Fig. 3.

Dose-sensitive genetic interactions among CrGAP, robo, and rac. All shown embryos are at stage 16 and have been stained with either the BP102 (a–c) or 1D4 (e–g and i–k) mAb. (a–h) Antagonistic genetic interaction between CrGAP and Slit/Robo. In embryos that overexpress CrGAP in the SRE background, the ladder-like WT-looking axon scaffold (a and c) is severely disrupted and shows characteristics of the robo mutant phenotype (b). The ectopic crossing defect in SRE embryos (e) is also significantly enhanced (f). (g and i–l) Antagonistic genetic interaction between CrGAP and rac. Note the ectopic crossing defect is present only in embryos overexpressing both CrGAP and RacN17 (j) but not in embryos of the other genotypes (g, i, and k). The stars in l indicate the following complication in obtaining UAS-CrGAP28, UAS-CrGAP33/+; UAS-RacN17/elav-GAL4 embryos. UAS-CrGAP28, UAS-CrGAP33/+; elav-GAL4/+ flies were mated to UAS-RacN17/UAS-RacN17 flies. Statistically, only 25% of the progeny from these parents are expected to have the genotype of RacN17; UCrGAP shown in j. Therefore, the fact that close to 20% of the segments showed the crossing defect indicates that the penetrance of the crossing phenotype in embryos with the RacN17; UCrGAP genotype is close to 80% (l). A similar scheme was used to generate the Cdc42N17; UCrGAP embryos for the test with Cdc42. The number of segments scored in each genotype was 165, 164, and 98 (d); 275, 120, 243, and 165 (h); and 103, 220, 98, and 124 (l), respectively.
Although CrGAP overexpression does not appear to result in a reciprocal phenotype to that observed when CrGAP function is reduced, the apparent antagonism of Robo repulsion is consistent with the observed positive role for rac in regulating midline crossing (14, 15, 24). To test directly whether CrGAP antagonizes rac, we increased CrGAP levels in embryos expressing the dominant negative Rac, N17Rac. The N17Rac-expressing embryos display mild midline axon outgrowth defects, with small breaks in the outermost longitudinal pathways, but rarely show axons ectopically crossing the midline (Fig. 3 i and l) (25). Moreover, N17Rac inactivates endogenous rac function only partially and, therefore, provides a sensitive background for testing genes that regulate the remaining rac activity (25, 26). When the level of CrGAP is increased in the N17Rac expressing embryos, multiple ectopic crossings are observed in most of the embryos (Fig. 3 j and l). In contrast, no phenotype is observed when CrGAP is increased in embryos expressing the dominant negative version of another Rho GTPase, Cdc42 (Fig. 3 k and l). It is important to note that the phenotypic enhancement observed in both experiments is achieved with relatively low levels of CrGAP overexpression, which by itself does not generate any observable phenotype in the midline axons (Fig. 3 c and g). Thus, the observed guidance defects are likely not a result of simple additive effects of the two genotypes but, rather, reflect a genuine genetic interaction between these genes. Together, these results suggest that Robo signaling may down-regulate CrGAP to prevent down-regulation of Rac. These findings are consistent with previous work indicating that Slit stimulation leads to the activation of Rac and that reducing rac disrupts Robo repulsion (15).
To further investigate the antagonistic relationship between CrGAP and Robo, we generated embryos expressing even higher levels of CrGAP. In contrast to the rather modest effects of CrGAP loss of function, high-level overexpression of CrGAP results in defects that mimic the phenotypes observed in robo and rac loss-of-function embryos (Fig. 4). With low to medium levels of CrGAP overexpression (up to two copies of UAS and one copy of elav or one copy of UAS and two copies of elav), the midline axon projection pattern is identical to WT (Fig. 4 a and f). With higher expression levels of CrGAP (two copies of UAS and two copies of elav or three copies of UAS and one copy of elav), a range of defects is observed. The milder class of defects involves multiple ectopic crosses of the longitudinal axons at the midline (Fig. 4c). These guidance errors are remarkably similar to those observed in robo mutants (Fig. 4b), or the double rac mutant, rac1J10mtlΔ (Fig. 4d, and previously published in ref. 14). Indeed, the defect is severe enough to alter the whole CNS axon scaffold, causing a significant thickening of the commissures and thinning of the longitudinal connectives because of too many axons crossing and recrossing the midline (Fig. 4h). In addition to this axon guidance defect, some embryos also show an axon outgrowth defect, in which multiple longitudinal axon pathways fail to extend (Fig. 4e). This phenotype is reminiscent of the axon outgrowth defect observed in triple loss-of-function rac mutants (14).
Fig. 4.

CrGAP gain-of-function mimics Robo and Rac loss-of-function. Stage 16 filleted embryos of the indicated genotype stained with Abs that label a subset of noncrossing axons, anti-FasII (a–e), or all CNS axons, anti-BP102 (f–h). Anterior is at the top in all images. The genotypes of the embryos are UAS-CrGAP37/elav (a and f); robo1 (b and g); UAS-CrGAP28/UAS-CrGAP28; elav-GAL4/elav-GAL4 (c, e, and h); and rac1J10mtlΔ (d). (a and f) Embryos with low level of CrGAP overexpression show axon patterns identical to WT embryos. The longitudinal axon pathways run parallel to the midline and never cross (a). The axon scaffold has a characteristic ladder-like appearance with commissural axon bundles crossing the midline forming two commissures in each segment (f). (b and g) In the robo mutant, many axons cross and recross the midline. Regions that show ectopic midline crossing are indicated by arrows (b). The axon scaffold is severely altered with much thicker and fused commissures and thinner longitudinal connectives (g). (c and h) Embryos with a high level of CrGAP overexpression show defects similar to robo mutant in terms of both ectopic crossing (c) and altered axon scaffold (h). (d) The rac, mtl double mutant show multiple midline crossings reminiscent of robo mutants. (e) The axon outgrowth defects in CrGAP overexpression embryos. Arrows with asterisk indicate the sites where all of the three longitudinal pathways fail to extend and appear broken. (i) Percentage of segments in UAS-CrGAP28/UAS-CrGAP28; elav-GAL4/elav-GAL4 embryos showing the three classes of phenotypes. The “1D4 guidance defect” refers to the ectopic crossing as indicated by arrow heads in c. The “1D4 outgrowth defect” refers to the phenotype where all of the three longitudinal axon pathways show stalling, as indicated by the arrowheads with asterisk in e. The “BP102 guidance defect” refers to the robo-like axon pattern as shown in all of the four segments in h.
In agreement with the eye-suppression experiments, the similarity between the CrGAP gain-of-function and the rac loss-of-function phenotypes argues that CrGAP can act as a Rac GAP in CNS neurons. When expressed at high enough levels, CrGAP appears to be able to inactivate all three rac genes in axons. The similarity between the CrGAP gain-of-function and robo loss-of-function further supports a connection between Robo signaling and CrGAP regulation. To determine whether the midline guidance effects of manipulating CrGAP are specific for robo repulsion, we tested whether increasing and decreasing CrGAP function would interact with other genes known to influence midline crossing, including the frazzled attractive Netrin receptor and the Receptor tyrosine phosphatases 10D and 69D. No dose-dependent interactions were observed, suggesting that the effects of CrGAP are specific for robo (see Table 1, which is published as supporting information on the PNAS web site).
CrGAP and Robo Physically Interact in Vitro and in Vivo. To test whether the genetic interactions observed between CrGAP and robo reflect a direct physical association between the two proteins, we performed both in vitro and in vivo biochemical assays for protein–protein interactions. The WW domains in CrGAP are protein-interaction modules that are known to interact with proline-rich sequences (27), which are abundant in the Robo cytoplasmic domain (5). By using an in vitro GST pull-down assay, we were able to detect a direct interaction between the cytoplasmic domain of Robo and an N-terminal fragment of CrGAP that contains one of the two WW domains (Fig. 5a). Co-IP of full-length Robo and an N-terminal myc tag (myc-WWGAP) from transfected HEK 293T cells suggests that CrGAP and Robo can interact in intact cells (Fig. 5b). Last, further support for the significance of the interaction between CrGAP and Robo comes from the finding that the two proteins can be detected by Co-IP from embryonic extracts (Fig. 5c).
Fig. 5.

Biochemical Interactions between CrGAP and Robo. (a) The Robo cytoplasmic domain binds to the N-terminal part of CrGAP that contains the second WW domain. 35S-labeled Robo was incubated with either GST-CrGAP or GST protein alone to test for binding. (b) Myc-tagged CrGAP amino acids 22–154 (myc-WWGAP) associates with HA-tagged Robo (HA-Robo) when coexpressed in HEK 293T cells. Cells expressing HA-Robo alone or in combination with myc-WWGAP were subjected to IP with anti-Robo Ab. To visualize protein levels, aliquots of the same lysates were directly run on a separate SDS/PAGE and probed with anti-HA to visualize Robo or anti-myc for CrGAP. (c) In vivo Co-IP of CrGAP and Robo. Embryos overexpressing CrGAP in the CNS were lysed. Soluble extract (left lane) was assessed by IP with anti-CrGAP Ab. The precipitated CrGAP and Robo were detected by using anti-CrGAP and anti-Robo, respectively (middle lane). Embryonic extract was also assessed by IP with anti-myc or preimmune serum (data not shown) as a negative control (right lane).
Discussion
The Drosophila genome contains 22 Rho GEFs and 20 GAPs, which far outnumbers the six Rho GTPases (28). In the human genome, there are also a large number of GEFs (>60) and GAPs (>70) for the Rho family. The large number of regulators suggests that Rho GTPases may achieve signaling specificity and diversity by coupling to various upstream receptor pathways through these different regulators. Here, through sequence and expression analysis, we identified six CNS Rho GAPs and one CNS Rho GEF. Probing the individual function of these CNS-expressed regulators should help to understand the specific roles Rho GTPases play in the multiple morphological processes in the nervous system, ranging from axon growth and guidance to dendritic elaboration and stabilization (12).
In particular, our study has led to the identification of a Rho-family GAP gene, CrGAP, involved in Robo receptor signaling. CrGAP is present at the right developmental time and place to potentially interact with the Robo receptor. Biochemical experiments showed that CrGAP can directly associate with the cytoplasmic domain of Robo in vitro and protein–protein interactions are also detected in vivo in embryonic extracts. CrGAP displays antagonistic genetic interactions with robo and rac, and its gain-of-function phenotype mimics the loss-of-function phenotypes of both robo and rac. Further, by using genetic tests in the eye as well as in the nervous system, we demonstrated that CrGAP is a GAP specific for Rac. Together, these results support a model in which CrGAP regulation of Rac activity is important to mediate efficient repulsion during Robo signaling.
Paradoxically, our genetic manipulations with both loss-of-function (RNAi) and gain-of-function of CrGAP have led to a similar qualitative effect: enhancing the inappropriate midline crossing defects in sensitized slit, robo/+ embryos. Also, both CrGAP RNAi and UAS CrGAP enhance the midline crossing defects in embryos overexpressing Commissureless, a negative regulator of Robo (G.J.B., unpublished data). One possible explanation, among others, is that both abnormally high Rac activity (consequence of CrGAP loss-of-function) and abnormally low Rac activity (consequence of CrGAP gain-of-function) are disruptive to Robo signaling. This idea is consistent with the observation that constitutive-active and dominant-negative forms of the Rho GTPases sometimes result in similar phenotypic consequences (see ref. 12 for references); indeed, in some neurons both constitutive-active and dominant-negative Rac appear to antagonize Robo repulsion (G.J.B., unpublished data). The observation that only CrGAP gain of function, and not CrGAP loss of function, can generate strong midline guidance defects in WT embryos (where slit and robo levels are not reduced) supports the idea that Robo might normally function to down-regulate CrGAP to control midline crossing.
Recently, Lundström et al. (17) reported the identification of Vilse through a genetic screen for embryonic tracheal phenotypes. Although both of our studies point to a role of CrGAP/Vilse in Robo signaling, it appears that the exact mechanism might differ in midline neurons versus in trachea. Overexpression of Vilse in the trachea of robo mutants ameliorates the phenotypes of robo (17); in contrast, overexpression of CrGAP/Vilse in midline axons exacerbates the robo phenotype (this study). This intriguing difference calls for further investigation of how precisely Robo activation regulates CrGAP function in neurons.
High levels of CrGAP overexpression lead to phenotypes ranging from axon guidance errors to axon outgrowth defects. With an even higher level of CrGAP overexpression, we observed the phenotype shifting predominantly toward axon outgrowth defects (data not shown). This observation echoes the previously raised notion that a low level of Rac activity is essential to maintain axon outgrowth, whereas a higher level of Rac activity is required for accurate guidance decisions of the growth cone (14, 29). Our finding that the phenotype of CrGAP overexpression can approach that of triple rac mutants supports the idea that CrGAP can down-regulate the activity of all three rac genes. Thus, modulating the level or activity of CrGAP could potentially overcome the problem of redundancy of multiple rac genes and provide an efficient way to alter Rac-dependent cytoskeletal dynamics in the growth cone, making CrGAP an exciting molecular target for promoting regrowth of injured axons in the adult CNS.
Supplementary Material
Acknowledgments
We thank L. Luo (Stanford University, Stanford, CA) for the gift of the transgenic GAP RNAi flies; M. Fauvarque (Commissariat à l'Energie Atomique, Grenoble, France) for UAS rotund flies; A. Gontang, D. Soper, S. Riley, C. Uemura, and B. Blankemeier for superb technical assistance; and G. Liao and P. Tomancak for helpful discussions on the genome information. This work was supported by a Howard Hughes Medical Institute predoctoral fellowship (to H.H.); a Human Frontier Science Program postdoctoral fellowship (to J.-P.L.); a Damon Runyon postdoctoral fellowship (to E.C.L.); National Institutes of Health Grant NS18366 (to C.S.G.); and a Burroughs Welcome Career award, a Whitehall Foundation Research grant, and National Institutes of Health Grant NS046333 (to G.J.B.). C.S.G. was an Investigator of the Howard Hughes Medical Institute.
Author contributions: H.H., M.L., J.P.L., J.M., C.S.G., and G.J.B. designed research; H.H., M.L., J.P.L., E.C.L., and G.J.B. performed research; H.H., M.L., and G.J.B. contributed new reagents/analytic tools; H.H., M.L., J.P.L., J.M., E.C.L., and G.J.B. analyzed data; and H.H. and G.J.B. wrote the paper.
Abbreviations: GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; CrGAP, CrossGAP; Robo, Roundabout; IP, immunoprecipitation; Co-IP, coimmunoprecipitation; RNAi, RNA interference; UAS, upstream activating sequence.
References
- 1.Tessier-Lavigne, M. & Goodman, C. S. (2000) Science 287, 813–814. [DOI] [PubMed] [Google Scholar]
- 2.Tessier-Lavigne, M. & Goodman, C. S. (1996) Science 274, 1123–1133. [DOI] [PubMed] [Google Scholar]
- 3.Seeger, M., Tear, G., Ferres-Marco, D. & Goodman, C. S. (1993) Neuron 10, 409–426. [DOI] [PubMed] [Google Scholar]
- 4.Brose, K. & Tessier-Lavigne, M. (2000) Curr. Opin. Neurobiol. 10, 95–102. [DOI] [PubMed] [Google Scholar]
- 5.Kidd, T., Brose, K., Mitchell, K. J., Fetter, R. D., Tessier-Lavigne, M., Goodman, C. S. & Tear, G. (1998a) Cell 92, 205–215. [DOI] [PubMed] [Google Scholar]
- 6.Bashaw, G. J., Kidd, T., Murray, D., Pawson, T. & Goodman, C. S. (2000) Cell 101, 703–715. [DOI] [PubMed] [Google Scholar]
- 7.Yu, T. W., Hao, J. C., Lim, W., Tessier-Lavigne, M. & Bargmann, C. I. (2002) Nat. Neurosci. 5, 1147–1154. [DOI] [PubMed] [Google Scholar]
- 8.Hsouna, A., Kim, Y. S. & VanBerkum, M. F. (2003) J. Neurobiol. 57, 15–30. [DOI] [PubMed] [Google Scholar]
- 9.Wills, Z., Emerson, M., Rusch, J., Bikoff, J., Baum, B., Perrimon, N. & Van Vactor, D. (2002) Neuron 36, 611–622. [DOI] [PubMed] [Google Scholar]
- 10.Fritz, J. L. & VanBerkum, M. F. (2000) Development (Cambridge, U.K.) 127, 1991–2000. [DOI] [PubMed] [Google Scholar]
- 11.Fritz, J. L. & VanBerkum, M. F. (2002) Dev. Biol. 252, 46–58. [DOI] [PubMed] [Google Scholar]
- 12.Luo, L. (2000) Nat. Rev. Neurosci. 1, 173–180. [DOI] [PubMed] [Google Scholar]
- 13.Dickson, B. J. (2001) Curr. Opin. Neurobiol. 11, 103–110. [DOI] [PubMed] [Google Scholar]
- 14.Hakeda-Suzuki, S., Ng, J., Tzu, J., Dietzl, G., Sun, Y., Harms, M., Nardine, T., Luo, L. & Dickson, B. J. (2002) Nature 416, 438–442. [DOI] [PubMed] [Google Scholar]
- 15.Fan, X., Labrador, J. P., Hing, H. & Bashaw, G. J. (2003) Neuron 40, 113–127. [DOI] [PubMed] [Google Scholar]
- 16.Wong, K., Ren, X. R., Huang, Y. Z., Xie, Y., Liu, G., Saito, H., Tang, H., Wen, L., Brady-Kalnay, S. M., Mei, L., et al. (2001) Cell 107, 209–221. [DOI] [PubMed] [Google Scholar]
- 17.Lundström, A., Gallio, M., Englund, C., Steneberg, P., Hemphala, J., Aspenstrom, P., Keleman, K., Falileeva, L., Dickson, B. J. & Samakovlis, C. (2004) Genes Dev. 18, 2161–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavernarakis, N., Wang, S. L., Dorovkov, M., Ryazanov, A. & Driscoll, M. (2000) Nat. Genet. 24, 180–183. [DOI] [PubMed] [Google Scholar]
- 19.Billuart, P., Winter, C. G., Maresh, A., Zhao, X. & Luo, L. (2001) Cell 107, 195–207. [DOI] [PubMed] [Google Scholar]
- 20.Brand, A. H. & Perrimon, N. (1993) Development (Cambridge, U.K.) 118, 401–415. [DOI] [PubMed] [Google Scholar]
- 21.Hariharan, I. K., Hu, K. Q., Asha, H., Quintanilla, A., Ezzell, R. M. & Settleman, J. (1995) EMBO J. 14, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagase, T., Kikuno, R., Hattori, A., Kondo, Y., Okumura, K. & Ohara, O. (2000) DNA Res. 7, 347–355. [DOI] [PubMed] [Google Scholar]
- 23.Huang, X., Cheng, H. J., Tessier-Lavigne, M. & Jin, Y. (2002) Neuron 34, 563–576. [DOI] [PubMed] [Google Scholar]
- 24.Matsuura, R., Tanaka, H. & Go, M. J. (2004) Eur. J. Neurosci. 19, 21–31. [DOI] [PubMed] [Google Scholar]
- 25.Kaufmann, N., Wills, Z. P. & Van Vactor, D. (1998) Development (Cambridge, U.K.) 125, 453–461. [DOI] [PubMed] [Google Scholar]
- 26.Hu, H., Marton, T. F. & Goodman, C. S. (2001) Neuron 32, 39–51. [DOI] [PubMed] [Google Scholar]
- 27.Chen, H. I., Einbond, A., Kwak, S. J., Linn, H., Koepf, E., Peterson, S., Kelly, J. W. & Sudol, M. (1997) J. Biol. Chem. 272, 17070–17077. [DOI] [PubMed] [Google Scholar]
- 28.Rubin, G. M. (2000) Novartis Found. Symp. 229, 79–83. [PubMed] [Google Scholar]
- 29.Ng, J., Nardine, T., Harms, M., Tzu, J., Goldstein, A., Sun, Y., Dietzl, G., Dickson, B. J. & Luo, L. (2002) Nature 416, 442–447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}