

Abstract
Increased protein kinase C (PKC) activity is associated with heart failure, and can target multiple cardiac troponin I (cTnI) residues in myocytes, including S23/24, S43/45 and T144. In earlier studies, cTnI-S43D and/or -S45D augmented S23/24 and T144 phosphorylation, which suggested there is communication between clusters. This communication is now explored by evaluating the impact of phospho-mimetic cTnI S43/45D combined with S23/24D (cTnIS4D) or T144D (cTnISDTD). Gene transfer of epitope-tagged cTnIS4D and cTnISDTD into adult cardiac myocytes progressively replaced endogenous cTnI. Partial replacement with cTnISDTD or cTnIS4D accelerated the time to peak (TTP) shortening and time to 50% re-lengthening (TTR50%) on day 2, but peak shortening was only diminished by cTnIS4D. Extensive cTnIS4D replacement continued to accelerate TTP, and decrease shortening amplitude, while TTR50% returned to baseline levels on day 4. In contrast, cTnISDTD modestly reduced shortening amplitude and continued to accelerate myocyte TTP and TTR50%. These results indicate cTnIS43/45 communicates with S23/24 and T144, with S23/24 exacerbating and T144 attenuating the S43/45D-dependent functional deficit. In addition, more severe functional alterations in cTnIS4D myocytes were accompanied by higher levels of secondary phosphorylation compared to cTnISDTD. These results suggest that secondary phosphorylation helps to maintain steady-state contractile function during chronic cTnI phosphorylation at PKC sites.
Keywords: troponin I, cardiac myocyte, myofilament, phosphorylation, contractile function
INTRODUCTION
Elevated protein kinase C (PKC) expression and activity is associated with cardiac dysfunction in human and animal models of heart failure (HF; (2,4,11)). One of the downstream targets for PKC is the thin filament molecular switch, cardiac troponin I (cTnI), which is phosphorylated at residues S23/24, S43/45 and T144 (29,34,35). Cardiac dysfunction also is linked to PKC-targeted cTnI phosphorylation of S43/45 and T144 (11,47,50). However, questions remain about the contribution of these PKC-targeted cTnI residues to the modulation of contractile function and/or dysfunction.
In biophysical and biochemical studies on myofilaments, phosphorylation of each cTnI cluster modulates contractile function (6,26,34,51). Extensive studies also resulted in a signal transduction mechanism to explain the accelerated in vivo relaxation produced by phosphorylation of or phosphomimetic substitutions in cTnI S23/24 (10,44,49,51). In contrast, the in vivo modulatory role(s) and mechanism(s) produced by cTnI-S43/45 and -T144 phosphorylation are less clear. Phospho-mimetic cTnIS43/45 substitutions increase the TnC Ca2+ off rate in reconstituted thin filaments (26), and reduce actomyosin ATPase Ca2+ sensitivity and peak activity (34). In permeabilized papillary muscles, S43/45 phospho-mimetics reduce Ca2+ sensitivity and peak tension while sliding speed is slowed in motility assays (6). Biochemical studies show T144 has a different influence than either S23/24 or S43/45, with phospho-mimetic T144 having little influence or decreasing thin filament Ca2+ off rate (26,27), reducing cross-bridge-activated actomyosin ATPase activity, and decreasing the Ca2+ sensitivity of in vitro sliding speed without significantly modifying Ca2+-activated isometric tension (6,27). There are no animal models expressing phospho-mimetic cTnIS43/45 or T144 substitutions alone. However, there are mice which express cTnIS43/45 in combination with T144 or S23/24 phospho-mimetics and they develop a range of systolic dysfunction and variable changes in diastolic function (3,20,37).
Studies on intact myocytes serve as an important bridge for understanding and integrating earlier in vitro and in vivo studies. The reduced shortening and slowed contraction rate observed in myocytes expressing phospho-mimetic cTnIS43D and/or S45D is consistent with earlier in vitro results. However, the cellular responses are more complex than the in vitro results, as they continue to change over time (24). For example, the initial reduction in shortening rate returned toward baseline over time, and coincided with secondary increases in the phosphorylation of other cTnI residues and additional myofilament proteins (24). Increases in one or more of these secondary phosphorylation sites appear to restore myocyte function back to a steady state or “setpoint”. These data also suggest function may deteriorate more rapidly if there is a loss of secondary or “compensating” myofilament protein phosphorylation. The enhanced S43/45 and diminished S23/24 phosphorylation of cTnI associated with human HF is consistent with this idea (8,47,50). The ongoing changes in cellular function produced by phospho-mimetic S43/45 also are consistent with systolic dysfunction accompanied by variable changes in diastolic function reported in existing animal models expressing phospho-mimetic substitutions at S43/45 combined with S23/24 and/or T144 (3,20,24,37). However, the diverse range of cardiac phenotypes observed in existing mouse models expressing multiple phospho-mimetic substitutions at PKC-targeted cTnI residues (3,20,37) indicates in vivo animal models alone are not easily able to determine whether PKC-targeted S23/24 or T144 compensate for and/or communicate with S43/45 to modify contractile function.
The present study examines whether cTnI with phospho-mimetic S43/45 combined with S23/24 (cTnIS23/24/43/45D or cTnIS4D) or T144 (cTnIS43/45D/T144D or cTnISDTD) can attenuate the influence of S43/45 on cardiac myocyte contractile function. The results indicate phospho-mimetic S23/24 and T144 independently modify the functional response to S43/45D in myocytes. Based on the in vitro impact of each cluster, the combined cTnI phospho-mimetics do not simply produce an additive response. Instead, the results suggest communication between S43/45D and S23/24D or T144D contributes to the contractile response. Moreover, greater dysfunction is associated with more targets and a higher level of secondary phosphorylation. This communication together with the secondary phosphorylation patterns observed in myocytes provide insight into the role these PKC-targeted sites play in modulating function and their long-term contribution in the development of contractile dysfunction.
METHODS
Site-directed mutagenesis and construction of adenoviral vectors for gene transfer
The cTnISDTD and cTnIS4D were produced by sequentially replacing cTnI-S43/45 followed by -T144 or -S23/24, respectively with negatively charged D in full-length, wild type cTnI cDNA (rat) via site-directed mutagenesis (QuikChange, Agilent Tech, Inc., Santa Clara, CA) (24). Both FLAG-tagged and non-tagged versions of cTnISDTD and cTnIS4D were prepared in pGEM3Z (24), and then individually subcloned into a pDC315 shuttle vector. Recombinant adenovirus was produced by homologous recombination of each shuttle vector with pBHGLoxΔE1,3Cre in HEK293 cells (18,24). The mutagenesis primers for cTnIS23/24D were (mutations underlined) 5′-CTGCTCCTG-TCCGACGTCGCGATGATGCCAACTACCGAGCCTATG-3′ (sense) and 5′-CATAGGCTCGGT-AGTTGGCATCATCGCGACGTCGGACAGGAGCAG-3′ (anti-sense), and for cTnIT144D were 5′-GTGGCAAGTTTAAGCGGCCAGATCTCCGAAGAGTGAGAATC-3′ (sense) and 5′-GATTC-TCACTCTTCGGAGATCTGGCCGCTTAAACTTGCCAC-3′ (anti-sense). Virus containing wild type cTnI (±FLAG), cTnI-S43D, -S45D, or -S43/45D were prepared as described earlier (24).
Cardiac myocyte isolation and culture
Animal work was carried out according to handling protocols and procedures developed by the University Committee for the Use and Care of Animals (UCUCA) at the University of Michigan. Isolated Ca2+-tolerant adult rat myocytes were re-suspended in DMEM containing 5% fetal bovine serum (FBS), penicillin (50 U/ml) and streptomycin (50μg/ml; P/S), and plated on laminin-coated coverslips at 37°C for 2 hrs, as described earlier (24,25). Recombinant adenovirus diluted in serum-free M199 + P/S was added to myocytes at 37°C and after one hour an 2 mL M199 + P/S aliquot was added to cells (25). Media was changed 24 hr after gene transfer and then every other day.
Expression analysis: Western blotting and immunohistochemistry
Western blot analysis
Replacement of endogenous cTnI with tagged or non-tagged wild type cTnI, cTnISDTD, or cTnIS4D, determination of cTnI/tropomyosin ratio to monitor myofilament stoichiometry, and protein phosphorylation were each monitored by Western blot analysis 2–4 days after gene transfer (24). Prior to Western detection, cells were scraped into ice-cold sample buffer, and proteins were separated on 12% SDS polyacrylamide gels, electrophoretically transferred to PVDF membranes, and probed with primary and secondary antibody (Ab) pairs (24). Primary antibodies (Abs) included TnI (MAB1691; Millipore; Billerica, CA), tropomyosin (Tm; T2780; Sigma-Aldrich; St.Louis, MO), plus phospho-specific cTnI Abs for S23/24 (p-S23/24; Cell Signaling Technology; Boston, MA), T144 (p-T144; Abcam), and S150 (p-S150; (32)). In addition, cardiac myosin binding protein C (cMyBP-C) levels were analyzed with an Ab directed to the N-terminus of cMyBP-C (Santa Cruz Biotechnology, Inc.; Santa Cruz, CA), along with phospho-Abs for S273 (p-S273), S282 (p-S282), and S302 (p-S302; (36)). Western analysis also analyzed PP2 primary Abs for methylated (mPP2; Abcam) and catalytic PP2 (cPP2; EMD Millipore) as well as alpha-4 (a4; EMD Millipore) expression. Each primary Ab-stained blot was then incubated with appropriate goat anti-mouse (GAM) or goat anti-rabbit (GAR) horseradish-peroxidase (HRP)-conjugated secondary Ab followed by enhanced chemilumenscence (ECL), which was detected with a ChemiDoc MP Imager (BioRad, Hercules, CA). A portion of each gel and blot was silver stained and Sypro-stained, respectively. Quantitative analysis of Western blots, Sypro-stained blots, and silver (Ag)-stained gels was performed using Quantity One® software (24).
The influence of phosphatase inhibition on myofilament phosphorylation was compared by Western analysis using a subset of myocytes treated with calyculin A (calA; 10nM; EMD-Millipore) or media, as described earlier (24). Changes in the phosphorylation of cTnIS23/24 (p-S23/24) and cMyBP-C S282 (p-S282) with (+calA) and without calA (media) were calculated using the following equation: ([p-S+CalA]-[p-Smedia])/(p-Smedia)*100.
Immunohistochemistry
Paraformaldehye-fixed, detergent-permeabilized myocytes were dual immunostained with the TnI Ab (MAB1691) plus fluorescein isothiocyanate (FITC)-conjugated secondary Ab pair followed by the FLAG Ab (Sigma) plus Texas Red (TR)-conjugated secondary Ab (TR) pair, 4 days after gene transfer, as described earlier (24). To establish sarcomere localization of exogenous cTnI, immunostained myocytes were analyzed using a Fluoview 500 laser scanning confocal microscope (Olympus; Center Valley, PA) for confocal projection imaging and de-convolution with Autoquant X software (Media Cybernetics; Rockville, MD).
Cellular function: sarcomeric shortening and Ca2+ transient measurements
For functional measurements, coverslips with myocytes were transferred to stimulation chambers and paced at 0.2 Hz starting 24hr after gene transfer. Media was changed every 12 hr for these experiments. Myocyte shortening was measured at 37°C in paced myocytes 2 and 4 days after gene transfer under basal conditions and in response to 10–15 min perfusion with endothelin-1 (ET, 10 nM; (24,48)). Resting sarcomere length (SL, μm), peak shortening amplitude (% baseline), time to peak (TTP; ms), shortening rate (μm/s), re-lengthening rate (μm/s), and time to 50% and 75% re-lengthening (TTR50%, TTR75%; ms) were measured on signal-averaged traces collected with a videobased CCD camera system (Ionoptix), as previously earlier (24). Sarcomere shortening and Ca2+ transients were simultaneously measured in 4 day, Fura-2AM-loaded myocytes. Signal averaged recordings from these experiments were analyzed for basal and peak Ca2+ ratios, rates of Ca2+ transient rise and decay (Ratio/s), the time to 50% and 75% decay (TTD50% ;TTD75%; ms; (24)).
Data analysis and statistics
Results are presented as mean±SEM, and statistical significance was set to p<0.05 (*). Results were analyzed using a one- or two-way analysis of variance (ANOVA) and post-hoc Dunnett or Tukey multiple comparison test, respectively unless otherwise noted.
RESULTS
Expression and sarcomere incorporation of cTnISDTD and cTnIS4D after gene transfer
Protein expression and replacement was analyzed by Western blot 2 and 4 days after gene transfer of cTnI, cTnI-SDTD, and -S4D with and without FLAG (Fig. 1A–C). In agreement with earlier work on cTnIS43/45D (24), replacement of FLAG-tagged constructs increased over time, with similar levels of partial replacement (35–40%) observed for cTnI-FLAG, -SDTDFLAG and - S4DFLAG 2 days after gene transfer. By day 4, more extensive replacement developed in each group (65–80%; Fig. 1A,B), although cTnISDTDFLAG lagged slightly behind the other constructs (Fig. 1A,B). Analysis of total cTnI and tropomyosin (Tm) protein expression also indicated thin filament stoichiometry is preserved in myocytes expressing cTnI, cTnI-SDTD, or -S4D with or without the epitope tag (Fig. 1A,C). In addition, a striated staining pattern consistent with sarcomere incorporation of each construct was detected for cTnI and FLAG by confocal image analysis of immunostained myocytes expressing cTnI-FLAG, -SDTDFLAG, and -S4DFLAG on day 4 (Fig. 1D). Together, these results show gene transfer results in cTnI replacement in the sarcomere of adult myocytes without a change in thin filament stoichiometry for each construct.
FIGURE 1. Protein expression, thin filament stoichiometry and sarcomere incorporation of cTnISDTD and cTnIS4D after gene transfer.
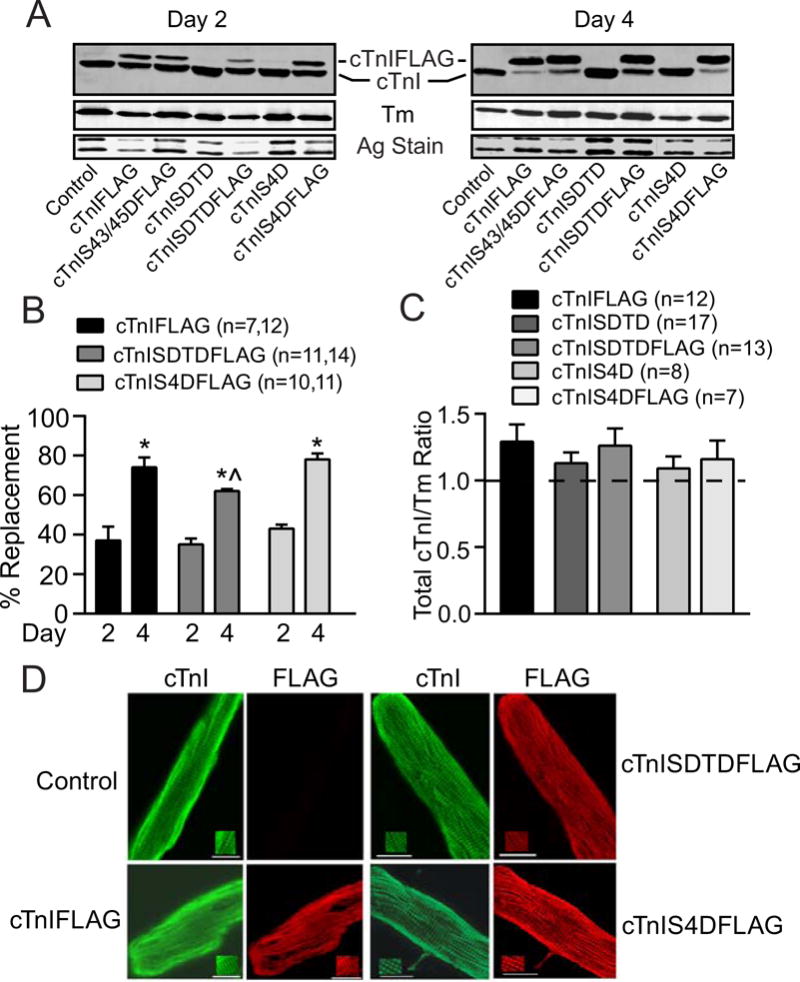
A. Representative Western blots show cTnI (upper panel) and tropomyosin (Tm; middle panel) expression 2 and 4 days after gene transfer. A silver-stained (Ag stain) portion of gel also is included to indicate protein loading (lower panel). Gene transfer of FLAG-tagged cTnI, cTnI-S43/45D (30), -SDTD, and –S4D resulted in partial (left panel) and more extensive (right panel) replacement of endogenous cTnI 2 and 4 days after gene transfer, respectively. B. Quantitative analysis of FLAG-tagged cTnI, cTnISDTD, and cTnIS4D replacement of endogenous cTnI at 2 and 4 days post-gene transfer. Replacement was calculated as the percent FLAG-tagged/total cTnI ratio. FLAG-tagged expression increased significantly between days 2 and 4 for each construct (*p<0.05, 2-way ANOVA plus post-hoc Tukey), although cTnISDTDFLAG expression lagged behind cTnIFLAG on day 4 (^p<0.05). C. Quantitative analysis of thin filament stoichiometry based on total cTnI/Tm ratios in controls, myocytes expressing FLAG-tagged cTnI, and myocytes expressing either FLAG- or non-tagged cTnISDTD or cTnIS4D 4 days after gene transfer. The non-treated, time-matched control cTnI/Tm ratio is set to 1.0 for each blot and is indicated by a dotted line in this graph. The cTnI/Tm ratios observed for each construct are normalized to this control value and compared using a one-way ANOVA (p>0.05). The n in panels C and D indicate the number of rat samples analyzed in each group. D. Immunohistochemical staining for TnI (left; FITC) and FLAG (right; TR) in controls (upper left panels), and myocytes expressing cTnI-FLAG (lower left panels), -SDTDFLAG (upper right panels) and -S4DFLAG (lower right panels). Insets in each panel emphasize the striated patterns detected in each myocyte (bar =10μm). The detection of a comparable striated pattern in myocytes stained for TnI and FLAG Abs confirms there is sarcomere incorporation of each construct.
Myocyte contractile performance
Partial replacement produced changes in contractile function 2 days after gene transfer, although the responses differed in myocytes expressing cTnI-SDTD versus -S4D (Fig. 2). The reduction in peak shortening amplitude observed in cTnIS4D myocytes is comparable to the cTnI-S43D and – S45D responses at the same time point, and is consistent with phospho-mimetic S43/45-induced reductions in maximum force, peak shortening, sliding motility and actomyosin ATPase activity (6,24). Both cTnI-SDTD and –S4D also accelerated TTP and TTR50%, and –SDTD increased maximum re-lengthening rate compared to control myocytes on day 2 (Fig. 2). Earlier, cTnIS23/24D accelerated re-lengthening alone (49), which is consistent with this cluster acting to accelerate re-lengthening in cTnIS4D myocytes. However, the acceleration of both re-lengthening rate and TTR50% in cTnISDTD myocytes is less easily explained by the T144D substitution, which slows thin filament Ca2+ off rates, and reduces the Ca2+ sensitivity and peak cross-bridge cycling rate in motility assays (6, 26). The current cTnISDTD response also differs from the decrease in tension and cross-bridge cycling found after extensive replacement with phospho-mimetic cTnIS43/45/T144 in reconstituted myofilaments (6). In addition, the accelerated TTP plus TTR50% detected on day 2 in cTnI-SDTD and -S4D myocytes (Fig. 2) was absent at the same time point for cTnI-S43D and/or -S45D expressing myocytes (24). To test whether secondary phosphorylation responses developed earlier in cTnISDTD myocytes, p-S23/24 levels were analyzed in day 2 myocytes but were not different in cTnISDTD versus control myocytes (Fig. 2B). Collectively, the contractile function observed in day 2 myocytes indicates S23/24D and T144D each modulate contraction (e.g. TTP) and relaxation (TTR50%) responses produced by S43/45D alone, although S23/24D in cTnIS4D is less able to preserve peak shortening than T144D in cTnISDTD.
FIGURE 2. Myocyte contractile function and phosphorylation 2 days after gene transfer of cTnI-FLAG, -SDTD, and –S4D compared to non-treated controls.
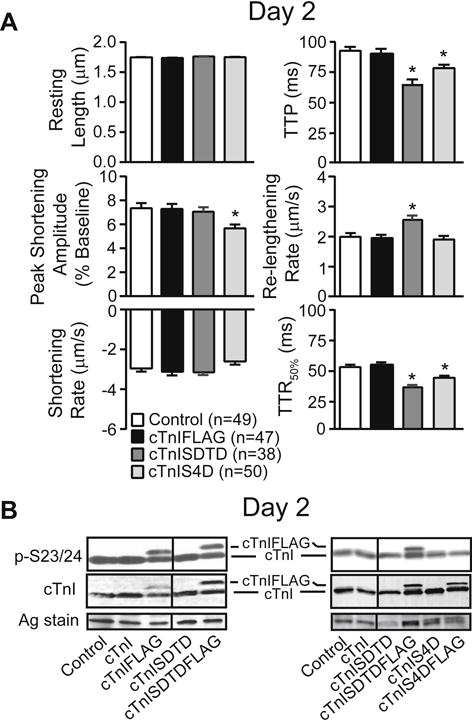
A. Analysis of signal averaged resting sarcomere length (SL, μm), peak shortening amplitude (expressed as a percent of resting length), shortening and re-lengthening rates (μm/s), along with the time to peak (TTP), and time to 50% re-lengthening (TTR50%) in 38–50 myocytes per group (6–8 hearts/group), as described earlier (24). Results were compared to control values using 1-way ANOVA and post-hoc Newman-Keuls tests, with statistical significance set at p<0.05 (*). B. Representative Westerns show results for cTnI p-S23/24 and total cTnI in control, cTnI, cTnISDTD ± FLAG, and cTnIS4D ± FLAG expressing myocytes 2 days after gene transfer. The response for cTnISDTDFLAG is compared to cTnIFLAG in the left panel, and the responses for cTnISDTDFLAG and cTnIS4DFLAG are shown in the right panel. The relative p-S23/24 levels remain similar to control values for myocytes expressing each of the phospho-mimetics 2 days after gene transfer. A silver (Ag) stained portion of each gel also is shown to indicate protein loading in each lane. Vertical black lines in each blot indicate a separation between cTnI or cTnI-FLAG and -SDTD on the same blot.
Contractile function continued to change after more extensive replacement with cTnI-SDTD or - S4D 4 days after gene transfer (Fig. 3A). Extensive cTnIS4D replacement further reduced shortening amplitude and the maximum rates of shortening and re-lengthening, although accelerated TTP continued to be present in these myocytes (Fig. 3A). This response is similar to the pattern observed in day 4 myocytes expressing cTnI-S43D and -S45D (24), and the reduced contraction is consistent with the systolic dysfunction reported in transgenic (tg) mice with S43/45 and S23/24 phospho-mimietic substitutions (3,37). A small, but significant increase in resting sarcomere length developed in the cTnIS4D myocytes. The shortening amplitude also decreased and re-lengthening rate returned to baseline levels after more extensive cTnISDTD replacement (Fig. 3A), although TTP and TTR50% continued to be accelerated in these myocytes (Fig. 3A). Taken together, the functional responses observed 2–4 days after gene transfer indicate S23/24 and T144 individually modulate the S43/45D response in cTnI. The earlier onset of reduced shortening observed with cTnI-S4D versus -SDTD (Fig. 2,3A) or -S43/45D alone (24), suggests S23/24D creates a permissive environment for S43/45D to reduce the rate and amplitude of shortening compared to T144.
FIGURE 3. Analysis of basal contractile function (A), Ca2+ transients (B), and the contractile response to endothelin (ET; C) 4 days after gene transfer.
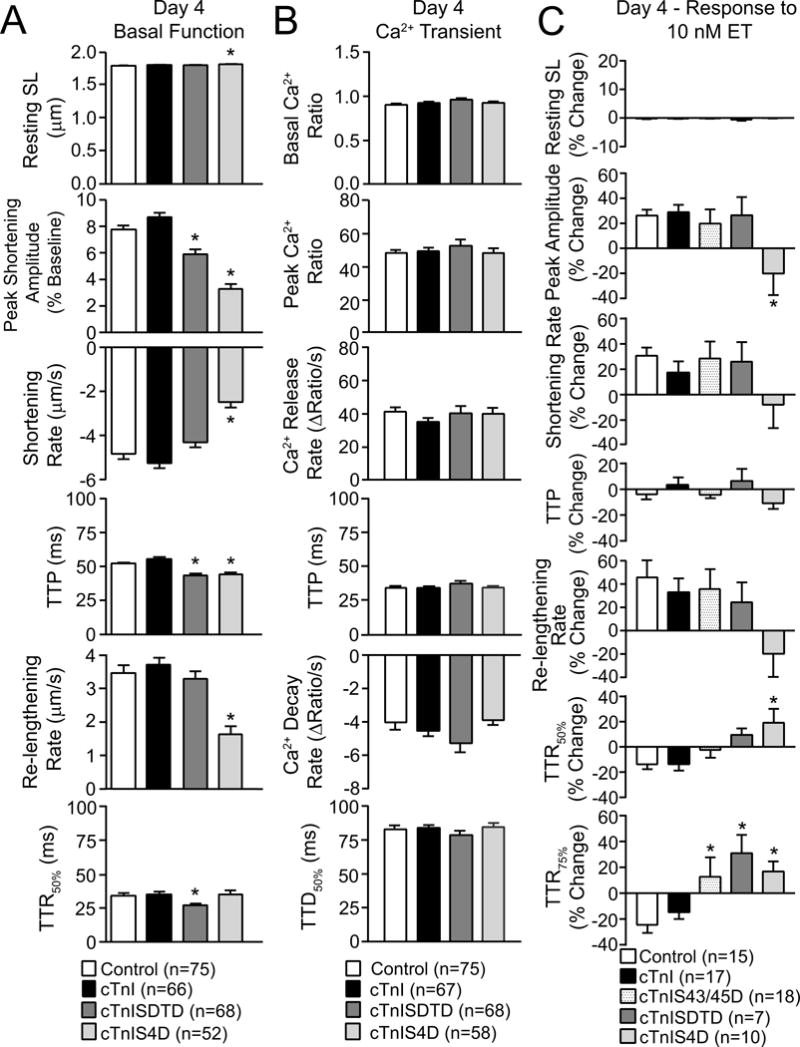
Adult myocytes from control, cTnI, cTnISDTD and cTnIS4D expressing myocytes were loaded with Fura-2AM 4 days after gene transfer and analyzed for basal contractile function (A), as described in Figure 2, plus Ca2+ transients (B). For the Ca2+ transient analysis, basal and peak Ca2+ ratios, Ca2+ release and decay rates (ΔRatio/s), and the time to 50% Ca2+ decay (TTD50%; ms) were measured in each myocyte. C. Contractile function responses before and after ET also were evaluated in the same groups plus myocytes expressing cTnIS43/45D. ET responses are expressed as the percent change in resting SL, peak amplitude, rates of shortening and re-lengthening, and TTP, TTR50% and TTR75% after ET. The TTR75% response is included in this panel due to the significant differences detected for all 3 phospho-mimetic substitutions compared to controls. In each panel, responses in cTnI-, cTnISDTD- and cTnIS4D-expressing myocytes were compared to controls, with significance set at *p<0.05.
Functional adaptations and secondary signaling in cTnI-SDTD- and -S4D-expressing myocytes
In earlier work, adaptive changes in the Ca2+ transient and myofilament phosphorylation accompanied the contractile function responses in myocytes after extensive replacement with phospho-mimetic cTnI-S43 and/or -S45 (24). Although Ca2+ transients were measured in Fura- 2AM-loaded myocytes on day 4 to evaluate secondary changes in Ca2+ cycling, transients were not different between cTnI, cTnI-SDTD or -S4D expressing myocytes (Fig. 3B). In contrast, divergent contractile responses to the PKC agonist ET hint that there may be secondary adaptations present by day 4. Previously, ET acceleration of re-lengthening was blocked in day 4 myocytes expressing a similar level of phospho-null cTnIT144P, but not cTnIS23/24A (48). Based on this earlier work, an attenuated ET-mediated acceleration of TTR75% was expected and observed with higher level cTnISDTD replacement in myocytes (Fig. 3C). The unexpected attenuation of the ET-induced acceleration of TTR75% by cTnI-S43/45D and -S4D myocytes (Fig. 3C) may result from the secondary p-T144, which tended to increase in myocytes expressing S43/45 phospho-mimetics (24). Secondary modifications also may contribute to the inability of ET to increase peak shortening amplitude in the cTnIS4D myocytes (Fig. 3C).
Phosphorylation of other cTnI residues and cardiac myosin binding protein C (cMyBP-C), another representative myofilament protein targeted for phosphorylation, were analyzed to evaluate secondary adaptations in myofilament phosphorylation. The cTnISDTD myocytes developed modest increases in cTnI p-S23/24, and this increase was distributed in the same ratio as FLAG-tagged cTnISDTD/endogenous cTnI (see Data in Brief), in agreement with earlier results in cTnIS43/45D (24). For cTnIS4D myocytes, Although earlier work showed a non-significant trend for increased p-T144 and p-S273 in myocytes expressing phospho-mimetic cTnI-S43 and/or -S45 (24), secondary phosphorylation of nearly all monitored sites, including p-T144 and p-S150 in cTnI and p-S273 and p-S282 in cMyBP-C increased in cTnIS4D myocytes on day 4 (Fig. 4A–F). In contrast to cTnIS4D, p-S150 also remained comparable to controls in myocytes expressing cTnISDTD- (Fig. 4C), or cTnI-S43D and/or -S45D (see Data in Brief). Most importantly, cTnIS4D produces more severe dysfunction than cTnISDTD in myocytes (Fig. 3A), and yet a higher magnitude of secondary phosphorylation and greater number of residues are targeted for secondary phosphorylation in cTnIS4D compared to cTnISDTD myocytes (Fig. 4). This association between dysfunction and secondary myofilament phosphorylation could indicate more phosphorylation is necessary to return S43/45D-induced dysfunction to the basal steady state in cTnI-S4D compared to -SDTD myocytes (6,24). Alternatively, separate, unrelated mechanisms may cause dysfunction and secondary phosphorylation, and both mechanisms may be triggered by cTnI-S4D but not –SDTD.
FIGURE 4. Phosphorylation of myofilament cTnI and cMyBP-C residues 4 days after gene transfer.
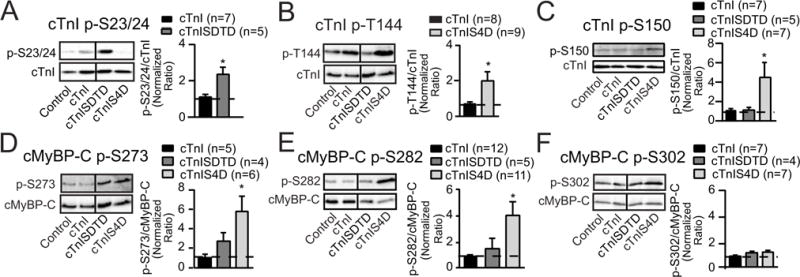
Representative immunoblots (left panels) and quantitative analysis (right panels) of phosphorylated cTnI -S23/24 (p-S23/24; A), -T144 (p-T144, B) and -S150 (p-S150, C) relative to total cTnI expression (lower panels) in controls and myocytes expressing cTnI, cTnISDTD or cTnIS4D 4 days post-gene transfer. The ratios of phosphorylated/total cTnI in each panel are normalized to control ratios, which are set to 1.0 (dotted line in each graph) for each quantitative analysis (30). The ratios in cTnISDTD- and/or cTnIS4D-expressing myocytes were compared to myocytes expressing cTnI after gene transfer, with statistical significance set at p<0.05 (*). This Western analysis also shows the expected reduction in p-S23/24 and p-T144 after replacement with cTnIS4D and cTnISDTD, respectively. Due to this reduction, p-S23/24 in cTnIS4D- and p-T144 in cTnISDTD- expressing myocytes were not quantitatively analyzed in their respective panels. Representative immunoblots shown in panels D–F show site-specific phosphorylation of cMyBP-C in the same groups of day 4 myocytes shown in A–C. Immunoblots (left panels) and quantitative analysis (right panels) are shown for phosphorylated cMyBP-C -S273 (p-S273, D), -S282 (p-S282, E), and -S302 (p-S302, F). Detection of the specific phosphorylated residue (left panels) is shown in the upper blot and total cMyBP-C in the lower panel. Quantitative analysis was performed using the normalized phosphorylated/total cMyBP-C ratio in each group, with the control ratio set to 1.0 (indicated by dashed line). Results in each panel were compared to cTnI expressing myocytes, with statistical significance set at p<0.05 (*). Vertical black lines shown in Western blots in this figure and in Figures 5 and 6 indicate a separation between cTnI and cTnISDTD on the same blot.
Enhanced secondary myofilament phosphorylation in myocytes could result from increased kinase activity and/or reduced phosphatase activity. Previously, reduced phosphatase rather than increased kinase activity coincided with the elevated myofilament phosphorylation detected in myocytes expressing cTnI-S43D and/or -S45D (24). To evaluate whether changes in phosphatase also coincide with the enhanced secondary phosphorylation in cTnISDTD or cTnIS4D myocytes, cTnI p-S23/24 and cMyBP-C p-S282 levels were measured in the presence and absence of the phosphatase inhibitor, calyculin A (calA). In contrast to basal phosphorylation, p-S23/24 in cTnISDTD myocytes (Fig. 5A) and p-S282 in cTnIS4D-expressing myocytes (Fig. 5B) treated with calA remained comparable to similarly treated controls. As a result, the percent change in calA- induced phosphorylation was significantly blunted in the cTnISDTD and cTnIS4D myocytes compared to myocytes expressing cTnI (Fig. 5A,B). Loss of mPP2 reduces regulated PP2 activity, and is associated with cell stress and cardiac dysfunction (9,40,46). Western analysis detected a reduced mPP2/cPP2 ratio in cTnI-SDTD and -S4D myocytes compared to controls (Fig. 6A), although cPP2 expression remained comparable in these myocyte groups (see Fig. 6A legend). A loss of α-4 protein compromises PP2 stability (21), and reduced α-4 protein levels can be found in failing ischemic vs non-failing human hearts (19). In the present study, α-4 protein expression decreased in cTnIS4D, but not cTnI or cTnISDTD myocytes compared to controls (Fig. 6B). In summary, calA, mPP2/cPP2, and α-4 expression results each point to lower PP2 activity in cTnI-S4D, and to a lesser extent –SDTD myocytes versus controls (Fig. 5,6), and reduced PP2 activity could result in the enhanced secondary myofilament phosphorylation detected in the same myocytes.
FIGURE 5. Secondary myofilament phosphorylation in the presence and absence of phosphatase inhibition by calyculin A (calA).
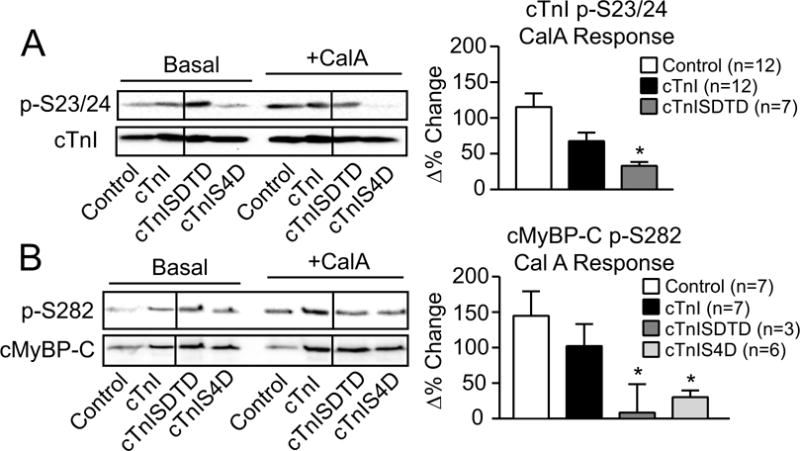
Representative Western (left panel) and quantitative (right panel) analyses of cTnI p-S23/24 (A) and cMyBP-C p-S282 (B) in the absence and presence of the phosphatase inhibitor calA (10 nM) and expressed relative to total cTnI and cMyBP-C, respectively. The ratio of phosphorylated/total protein is normalized to control values (set to 1.0) in the absence of calyculin A (CalA) for the quantitative analysis. Then, control responses to calA (Δ% change) on day 4 are compared to responses in cTnI-, cTnISDTD- and cTnIS4D-expressing myocytes, with the Δ% response shown in each right panel graph. The p-S23/24 and p-S282 levels after calA treatment are not different among groups. The Δ% change in the p-S23/24/cTnI and the p-S282/cMyBP-C ratios were analyzed by ANOVA (see Methods) with statistical significance set at p<0.05 (*). Note the absence of p-S23/24 detection in the cTnIS4D response (panel A). Any p-S23/24 detection in cTnIS4D myocytes is due to residual endogenous cTnI expression and not explained by calA treatment.
FIGURE 6. Analysis of phosphatase activity and stability in myocytes expressing cTnISDTD and cTnIS4D compared to controls and cTnI.
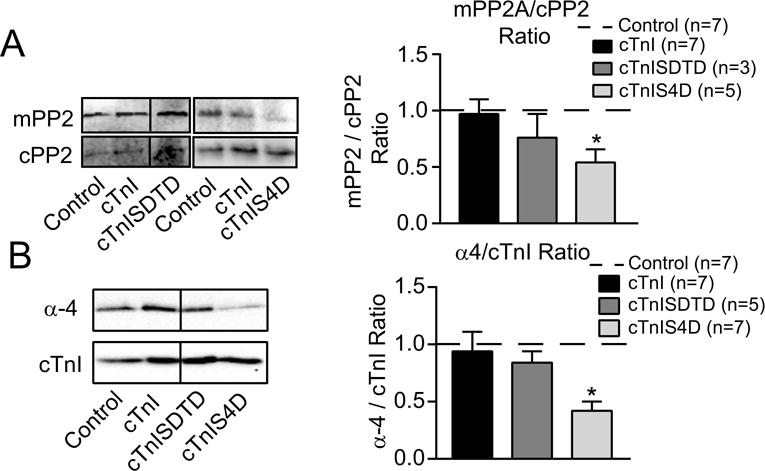
A. Western analysis of methylated protein phosphatase 2A (mPP2; upper left panel) and PP2A catalytic subunit (cPP2; lower left panel) along with the quantitative analysis of the mPP2/cPP2 ratio (right panel) for cTnISDTD and cTnIS4D. Blots were initially probed for mPP2, stripped and re-probed for cPP2, and then stained with Sypro stain. Then, the cPP2/Sypro band ratios were normalized to control values on the same blot (set to 1.0, n=7) and these ratios were not different between groups (cPP2/Sypro ratio: cTnI 1.11+0.15, n = 7; cTnISDTD 1.30+0.86, n = 3; cTnIS4D 1.15+0.23, n = 5). The ratio of mPP2/normalized cPP2 ratio was determined for the quantitative analysis shown in the right panel. B. Representative Western analysis of alpha-4 (a-4; left upper panel) relative to cTnI (lower left panel), plus the quantitative analysis of a-4/cTnI ratio (right panel) in day 4 myocytes. Each set of results was statistically analyzed by ANOVA (see Methods) with significance set at p<0.05 (*).
DISCUSSION
Phospho-mimetic cTnI-S43 and/or -S45 act as a functionally dominant brake in myocytes (24), and the current results show additional PKC-targeted residues can fine tune this brake. Specifically, T144D in cTnISDTD and S23/24D in cTnIS4D individually modify S43/45D-induced contractile function responses on day 2 (Figs. 2), and continue to produce separate functional responses as well as secondary myofilament phosphorylation patterns on day 4 (Fig. 3,4). The secondary phosphorylation is postulated to serve an early and functionally important role by helping to restore S43/45D-induced contractile dysfunction back toward a basal steady state or “setpoint”. Interestingly, severe contractile and relaxation dysfunction was associated with a greater magnitude and number of residues targeted for secondary phosphorylation in cTnIS4D compared to cTnISDTD-expressing myocytes (Fig. 3,4). Thus, even short term secondary myofilament phosphorylation in the cTnIS4D myocytes proved to be insufficient to restore contractile function toward this postulated basal setpoint. The distinct functional responses and secondary phosphorylation patterns produced by cTnI-SDTD and -S4D also indicate S23/24 and T144 may utilize different transduction mechanisms to modify myofilament function. Most importantly, the results suggest PKC phosphorylates multiple cTnI residues to efficiently counteract a dynamic range of cardiac performance produced by sympathetic activation (39,41). As a result, this signaling may play a critical role in preserving steady state function during sustained or chronic sympathetic drive.
Functional communication between cTnI phosphorylation targets
The independent and sometimes unexpected functional responses observed in cTnI-SDTD and – S4D myocytes could result from communication between S43/45 and the other cTnI residues. For example, reduced peak shortening and accelerated TTR50% were expected to be comparable in myocytes expressing cTnIS4D and cTnIS43/45D (Figs. 2,3). A similar response is predicted based on the reduction in peak shortening produced by cTnIS43/45D alone, the accelerated relaxation produced by p-S23/24 or S23/24 phospho-mimetics alone (44,49), and the enhanced p-S23/24 associated with accelerated TTR50% in myocytes expressing cTnIS43D and/or S45D (24). Indeed, the accelerated TTR50% response in cTnIS4D myocytes agrees with this prediction (Figs. 2,3), and can be attributed to S23/24D. However, the earlier appearance and greater magnitude of impaired shortening in cTnI-S4D compared to -S43/45D myocytes (Figs. 2,3; (24)) suggests S23/24 directly and/or indirectly communicates to enhance the functional impact produced by S43/45.
Communication also could accelerate TTP in day 2 cTnIS4D myocytes, which is absent from cTnIS43/45D myocytes on day 2 (Fig. 2; (24)). Although this idea differs from an earlier conclusion that phospho-mimetic S43/45 merely dominates over S23/24 in the functional response (14), the divergent conclusions may stem from a more complex environment in intact myocytes and/or external load differences among cardiac preparations.
The specific mechanism and/or interaction responsible for the exacerbated dysfunction in cTnI-S4D versus -S43/45D myocytes is not known, but could involve one or more of the transduction mechanisms proposed for S43/45 and/or S23/24 signal transduction. Phosphorylation of S23/24 reduces thin filament Ca2+ sensitivity by reducing the interaction between the cardiac-specific N- terminus and the N-lobe of cTnC, and also may depend on increased intra-domain interactions within cTnI (15,17). A variety of mechanisms also are proposed to explain the actions of p-S43/45, including altered thin filament TnC Ca2+ dissociation and/or association, stabilization of the thin filament inactive state, and associated changes in cross-bridge formation or detachment (6,12,26,28, 37). The greater dysfunction observed with cTnIS4D compared to cTnISDTD suggests that at least some portion of the S43/45D transduction mechanism(s) lies downstream from Ca2+ binding to cTnC. This prediction is based in part on the functionally important sites S43/45 occupy in the highly conserved H1 a-helix (43), and the increased helix rigidity caused by phosphorylation of residues at the same location in other a-helices (1). In turn, H1 a-helix alterations could modify the I-T arm and downstream switch domains in cTnI and/or upstream TnT domains to modulate the troponin switch. The S23/24D in cTnIS4D could further modify the S43/45D a-helix rigidity and/or position or work indirectly via intra-domain interactions to modify conformational changes in troponin, and its influence on thin filament activation state and/or cross-bridge cycling.
The cTnISDTD results also are physiologically important because phospho-mimetic cTnI- S43/45 combined with -T144 most closely mimic the influence of PKC treatment on myofilament function (6). The acceleration of both TTP and TTR50% in cTnISDTD myocytes does not develop in cTnIS43/45D myocytes (Figs. 2, 3; (24)), and may be due to T144D alone. Phospho-mimetic cTnIT144 alone slows cTn-Ca2+ off rates and reduces Ca2+ sensitivity of unloaded sliding speed in myofilament (6,26,27). Thus, slowed Ca2+ dissociation could lead to faster shortening while the sliding speed response predicts an accelerated relaxation in cTnISDTD myocytes, but only if each response is mediated by spatially and temporally separate transduction mechanisms within the thin filament. This interpretation suggests that PKC could mediate a range of thin filament functional responses. However, shortening rate remained constant and only the TTR50% changed intact myocytes with cTnIT144 substitutions ((48), Figs. 2,3). Moreover, the accentuated shift in the Ca2+ sensitivity of tension and sliding speed produced by phospho-mimetic cTnI S43/45 plus T144 suggests communication between residues in cTnI, rather than T144D alone contributes to the functional myocyte response produced by cTnISDTD (6). This communication also may contribute to the attenuated decrease in amplitude and accelerated contraction rate produced by cTnISDTD (Figs. 2,3), which remains difficult to explain based on the individual roles for S43/45 or T144 defined in earlier work (6,26,27).
Although further studies are needed to establish the underlying mechanism(s) and/or cTn domains contributing to each response, the cTnISDTD-specific responses along with the accentuated functional response observed in cTnIS4D myocytes support the idea that PKC-targeted cTnI residues communicate to modulate contractile function. Communication between phosphorylation sites could differentially influence more than one cTn transduction mechanisms to produce a range of functional responses to PKC. Altered transduction is anticipated to result from changes in the relative position or conformation of functional regions or domains within the cTn complex, which is likely to include the flexible amino terminus, H1 helix, I-T arm and inhibitory peptide (IP) region within cTnI, as well as additional domains responsible for transduction of the Ca2+ signal within the larger cTn complex. To gain insight into this communication, studies to evaluate in vivo dose-dependent cardiac responses over time are needed in animal models with individual and combined cTnI phospho-mimetic substitutions.
Secondary myofilament phosphorylation in response to cTnI-SDTD and -S4D expression
The secondary phosphorylation and PP2 alterations observed in cTnI-S4D and -SDTD myocytes by day 4 is consistent with earlier work in myocytes expressing cTnIS43D and/or S45D (24), although the secondary patterns are specific for each phospho-mimetic construct (Fig. 4–6). As anticipated, similar secondary changes in mPP2 and cMyBP-C pS282 developed in response to cTnIS4D and -S43/45 phospho-mimetics (24). However, cTnIS43D and/or S45D alone did not increase the cMyBP-C p-S273, cTnI p-T144 or p-S150 observed in cTnIS4D myocytes (24). These different secondary phosphorylation patterns could be due to the permanent substitutions in cTnIS4D compared to a more stochastic distribution of p-S23/24 in phospho-mimetic cTnIS43/45 myocytes (24). Alternatively, divergent phosphorylation turnover rates in myocytes expressing cTnI with multiple phospho-mimetic substitutions also could contribute to specific phosphorylation patterns and downstream functional responses. The phosphatase inhibition studies are consistent with a role for phosphorylation turnover (Fig. 5). Phosphorylation turnover changes in other signaling pathways plus evidence cardiac PKC phosphorylates downstream targets and activates phosphatases also indirectly support this idea (5,7,16).
The representative residues evaluated for secondary phosphorylation in cTnI and cMyBP-C were chosen because they are PKC phosphorylation targets (e.g. cTnI p-S23/24, p-S43/45, p-T144 (35); cMyBP-C p-S273, p-S282, p-S302 (30)), are associated with stress responses (e.g. cTnI p-S150; (32, 33)), and/or their phosphorylation changes during heart failure (22,50). The individual role of each residue in this response may fine tune the S43/45 modulatory brake in an effort to maintain or restore steady state contractile function. For example, the secondary cMyBP-C phosphorylation could increase cross-bridge cycling rate to attenuate the reductions in shortening rate (Fig. 4D,E; (45)). Enhanced p-S23/24 may preserve relaxation in cTnISDTD myocytes, while p-T144 counteracts changes in TnC Ca2+ binding/release and/or thin filament activation state in cTnIS4D myocytes (Fig. 4B; (6,14,26,27,37,42)). The ability of phospho-mimetic cTnIS150 to attenuate S23/24D-mediated decreases in myofilament TnC Ca2+ binding while also accelerating Ca2+ dissociation also suggests secondary p-S150 may help restore or minimize reductions in contractile function (Fig. 4C; (32,33)).
More than 15 different residues are targeted for phosphorylation in cTnI, and many of these sites undergo changes in phosphorylation levels under pathophysiological conditions (32,46,50). Thus, secondary phosphorylation of other cTnI residues, as well as other myofilament proteins, may modulate function during sustained myofilament phosphorylation (13,31,38). The dynamic myofilament signaling process detected in the current study is postulated to play a significant role during the early compensatory phase of cardiac dysfunction. An inability of this myofilament network to restore function and/or a loss of this response could set the stage for progressive structural remodeling and/or a transition to end-stage heart failure.
Insights into cTnI phospho-mimetic animal models
The current results also provide some insight into the range of cardiac phenotypes reported in earlier mouse models (Fig. 2,3;(3,20,37)), which has led to divergent interpretations about the modulatory role(s) of PKC-targeted cTnI phosphorylation. Sustained cTnIS43/45D expression plays a dominant role in causing contractile dysfunction in myocytes (Fig. 3; (24)). However, the present work also indicates phenotypic variability is possible when S43/45D is combined with S23/24D or T144D (Figs. 3,4,6) in mouse models (3,20,37). Communication between cTnI residues may cause subtle differences in thin filament conformation plus specific secondary phosphorylation patterns to produce a specific cardiac phenotype that differs from a simple additive response. Secondary myofilament responses may precede and/or develop in tandem with Ca2+ cycling proteins alterations reported in at least one phospho-mimetic cTnI mouse model (3).
In summary, our results demonstrate that cTnIS43/45 acts as a dominant brake on function, which is modified by other phospho-mimetic cTnI residues and/or via communication with other cTnI and cMyBP-C residues. As a result, both direct and secondary phosphorylation responses contribute to function in cTnISDTD and cTnIS4D myocytes. Based on these results, S43/45 could be a clinical target to attenuate or delay cardiac remodeling and improve early stage dysfunction.
Highlights.
-
-
PKC phosphorylates cardiac troponin I (cTnI) S23/24, S43/45 and T144 to fine tune myocyte function.
-
-
Function differs in myocytes expressing phospho-mimetic cTnI-S23/24/43/45 versus - S43/45/T144.
-
-
Primary alterations and secondary phosphorylation produce each myocyte functional response.
-
-
Secondary phosphorylation coincides with a return toward steady state cardiac performance.
Acknowledgments
The cMyBP-C pS273, pS282, and pS302 primary Abs were kind gifts of S. Sadayappan. This work was supported by National Institutes of Health Grant R01-HL-067254 (MVW) and R01-HL-114940 (BJB), NIGMS T32 GM007315 (SEL), and American Heart Association pre-doctoral award 12PRE8830022 (SEL). Studies presented here utilized the Core Facilities (Morphology and Image Analysis, and Cell and Molecular Biology Cores) of the Michigan Diabetes Research and Training Center (supported by National Institutes of Health Grant P60-DK20572).
Abbreviations
- α-4
alpha-4
- ANOVA
analysis of variance
- Ab
antibody
- Abs
antibodies
- calA
calyculin A
- cMyBP-C
cardiac myosin binding protein C
- cTnI
cardiac troponin I
- cPP2
catalytic protein phosphatase 2A
- ET
endothelin-1
- ECL
enhanced chemiluminescence
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- GAM
goat anti-mouse
- GAR
goat anti-rabbit
- HF
heart failure
- HRP
horseradish peroxidase
- mPP2
methylated protein phosphatase 2A
- P/S
penicillin and streptomycin
- PKC
protein kinase C
- p-
phosphorylated
- SL
sarcomere length
- Ag
silver
- TR
texas red
- TTP
time to peak
- TTD50%
time to 50% decay
- TTD75%
time to 75% decay
- TTR50%
time to 50% re-lengthening
- TTR75%
time to 75% re-lengthening
- tg
transgenic
- Tm
tropomyosin
- TnC
troponin C
- TnT
troponin T
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- 1.Andrew CD, Warwicker J, Jones GR, Doig AJ. Effect of phosphorylation on alpha-helix stability as a function of position. Biochemistry. 2002;41:1897–1905. doi: 10.1021/bi0113216. [DOI] [PubMed] [Google Scholar]
- 2.Bayer AL, Heidkamp MC, Patel N, Porter M, Engman S, Samarel AM. Alterations in protein kinase C isoenzyme expression and autophosphorylation during the progression of pressure overload-induced left ventricular hypertrophy. Mol Cell Biochem. 2003;242:145–152. [PubMed] [Google Scholar]
- 3.Bilchick KC, Duncan JG, Ravi R, Takimoto E, Champion HC, Gao WD, Stull LB, Kass DA, Murphy AM. Heart failure-associated alterations in troponin I phosphorylation impair ventricular relaxation-afterload and force-frequency responses and systolic function. Am J Physiol Heart Circ Physiol. 2007;292:H318–325. doi: 10.1152/ajpheart.00283.2006. [DOI] [PubMed] [Google Scholar]
- 4.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- 5.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 6.Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003;278:11265–11272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
- 7.Chaudhri VK, Kumar D, Misra M, Dua R, Rao KV. Integration of a phosphatase cascade with the mitogen-activated protein kinase pathway provides for a novel signal processing function. J Biol Chem. 2010;285:1296–1310. doi: 10.1074/jbc.M109.055863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christopher B, Pizarro GO, Nicholson B, Yuen S, Hoit BD, Ogut O. Reduced force production during low blood flow to the heart correlates with altered troponin I phosphorylation. J Muscle Res Cell Motil. 2009;30:111–123. doi: 10.1007/s10974-009-9180-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeGrande ST, Little SC, Nixon DJ, Wright P, Snyder J, Dun W, Murphy N, Kilic A, Higgins R, Binkley PF, Boyden PA, Carnes CA, Anderson ME, Hund TJ, Mohler PJ. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J Biol Chem. 2013;288:1032–1046. doi: 10.1074/jbc.M112.426957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong WJ, Jayasundar JJ, An J, Xing J, Cheung HC. Effects of PKA phosphorylation of cardiac troponin I and strong crossbridge on conformational transitions of the N-domain of cardiac troponin C in regulated thin filaments. Biochemistry. 2007;46:9752–9761. doi: 10.1021/bi700574n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong X, Sumandea CA, Chen YC, Garcia-Cazarin ML, Zhang J, Balke CW, Sumandea MP, Ge Y. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J Biol Chem. 2012;287:848–857. doi: 10.1074/jbc.M111.293258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finley NL, Rosevear PR. Introduction of negative charge mimicking protein kinase C phosphorylation of cardiac troponin I. Effects on cardiac troponin C. J Biol Chem. 2004;279:54833–54840. doi: 10.1074/jbc.M408304200. [DOI] [PubMed] [Google Scholar]
- 13.Hidalgo C, Granzier H. Tuning the molecular giant titin through phosphorylation: role in health and disease. Trends Cardiovasc Med. 2013;23:165–171. doi: 10.1016/j.tcm.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinken AC, Hanft LM, Scruggs SB, Sadayappan S, Robbins J, Solaro RJ, McDonald KS. Protein kinase C depresses cardiac myocyte power output and attenuates myofilament responses induced by protein kinase A. J Muscle Res Cell Motil. 2012;33:439–448. doi: 10.1007/s10974-012-9294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howarth JW, Meller J, Solaro RJ, Trewhella J, Rosevear PR. Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J Mol Biol. 2007;373:706–722. doi: 10.1016/j.jmb.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 16.Hwang H, Robinson D, Rogers JB, Stevenson TK, Lang SE, Sadayappan S, Day SM, Sivaramakrishnan S, Westfall MV. Agonist activated PKCbetaII translocation and modulation of cardiac myocyte contractile function. Sci Rep. 2013;3:1971. doi: 10.1038/srep01971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang PM, Cai F, Pineda-Sanabria SE, Corson DC, Sykes BD. The cardiac-specific N-terminal region of troponin I positions the regulatory domain of troponin C. Proc Nat Acad Sci. 2014;111:14412–14417. doi: 10.1073/pnas.1410775111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kampert SE, Devaney E, Westfall MV. Gene Transfer and Expression in Animal Cells. In: Cseke LJ, Kirakosayan A, Kaufman PB, Westfall MV, editors. Handbook of Molecular and Cellular Methods in Biology and Medicine. CRC Press; Boca Raton, Florida: 2011. pp. 557–578. [Google Scholar]
- 19.Kim EH, Galchev VI, Kim JY, Misek SA, Stevenson TK, Campbell MD, Pagani FD, Day SM, Johnson TC, Washburn JG, Vikstrom KL, Michele DE, Misek DE, Westfall MV. Differential protein expression and basal lamina remodeling in human heart failure. Proteomics Clin Appl. 2016;10:585–596. doi: 10.1002/prca.201500099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirk JA, MacGowan GA, Evans C, Smith SH, Warren CM, Mamidi R, Chandra M, Stewart AF, Solaro RJ, Shroff SG. Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ Res. 2009;105:1232–1239. doi: 10.1161/CIRCRESAHA.109.205427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong M, Ditsworth D, Lindsten T, Thompson CB. Alpha4 is an essential regulator of PP2A phosphatase activity. Mol Cell. 2009;36:51–60. doi: 10.1016/j.molcel.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kooij V, Holewinski RJ, Murphy AM, Van Eyk JE. Characterization of the cardiac myosin binding protein-C phosphoproteome in healthy and failing human hearts. J Mol Cell Cardiol. 2013;60:116–120. doi: 10.1016/j.yjmcc.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kooij V, Zhang P, Piersma SR, Sequeira V, Boontje NM, Wijnker PJ, Jimenez CR, Jaquet KE, dos Remedios C, Murphy AM, Van Eyk JE, van der Velden J, Stienen GJ. PKCalpha-specific phosphorylation of the troponin complex in human myocardium: a functional and proteomics analysis. PLoS One. 2013;8:e74847. doi: 10.1371/journal.pone.0074847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang SE, Schwank J, Stevenson TK, Jensen MA, Westfall MV. Independent modulation of contractile performance by cardiac troponin I Ser43 and Ser45 in the dynamic sarcomere. J Mol Cell Cardiol. 2015;79:264–274. doi: 10.1016/j.yjmcc.2014.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lang SE, Westfall MV. Gene transfer into cardiac myocytes. Methods Mol Biol. 2015;1299:177–190. doi: 10.1007/978-1-4939-2572-8_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Lopez JJ, Biesiadecki BJ, Davis JP. Protein kinase C phosphomimetics alter thin filament Ca2+ binding properties. PLoS One. 2014;9:e86279. doi: 10.1371/journal.pone.0086279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu QW, Hinken AC, Patrick SE, Solaro RJ, Kobayashi T. Phosphorylation of cardiac troponin I at protein kinase C site threonine 144 depresses cooperative activation of thin filaments. J Biol Chem. 2010;285:11810–11817. doi: 10.1074/jbc.M109.055657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathur MC, Kobayashi T, Chalovich JM. Negative charges at protein kinase C sites of troponin I stabilize the inactive state of actin. Biophys J. 2008;94:542–549. doi: 10.1529/biophysj.107.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metzger JM, Westfall MV. Covalent and noncovalent modification of thin filament action: the essential role of troponin in cardiac muscle regulation. Circ Res. 2004;94:146–158. doi: 10.1161/01.RES.0000110083.17024.60. [DOI] [PubMed] [Google Scholar]
- 30.Mohamed AS, Dignam JD, Schlender KK. Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch Biochem Biophys. 1998;358:313–319. doi: 10.1006/abbi.1998.0857. [DOI] [PubMed] [Google Scholar]
- 31.Montgomery DE, Wolska BM, Pyle WG, Roman BB, Dowell JC, Buttrick PM, Koretsky AP, Del Nido P, Solaro RJ. a-Adrenergic response and myofilament activity in mouse hearts lacking PKC phosphorylation sites on cardiac TnI. Am J Physiol Heart Circ Physiol. 2002;282:H2397–2405H. doi: 10.1152/ajpheart.00714.2001. [DOI] [PubMed] [Google Scholar]
- 32.Nixon BR, Thawornkaiwong A, Jin J, Brundage EA, Little SC, Davis JP, Solaro RJ, Biesiadecki BJ. AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. J Biol Chem. 2012;287:19136–19147. doi: 10.1074/jbc.M111.323048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nixon BR, Walton SD, Zhang B, Brundage EA, Little SC, Ziolo MT, Davis JP, Biesiadecki BJ. Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca(2+) sensitivity and accelerates deactivation in an acidic environment. J Mol Cell Cardiol. 2014;72:177–185. doi: 10.1016/j.yjmcc.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noland TA, Jr, Raynor RL, Jideama NM, Guo X, Kazanietz MG, Blumberg PM, Solaro RJ, Kuo JF. Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry. 1996;35:14923–14931. doi: 10.1021/bi9616357. [DOI] [PubMed] [Google Scholar]
- 35.Noland TA, Jr, Raynor RL, Kuo JF. Identification of sites phosphorylated in bovine cardiac troponin I and troponin T by protein kinase C and comparative substrate activity of synthetic peptides containing the phosphorylation sites. J Biol Chem. 1989;264:20778–20785. [PubMed] [Google Scholar]
- 36.Sadayappan S, Gulick J, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Robbins J. Cardiac myosin binding protein-C phosphorylation in a (beta)-myosin heavy chain background. Circulation. 2009;119:1253–1262. doi: 10.1161/CIRCULATIONAHA.108.798983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, VanBuren P, Martin LA, Robbins J. In vivo and in vitro analysis of cardiac troponin I phosphorylation. J Biol Chem. 2005;280:703–714. doi: 10.1074/jbc.M409513200. [DOI] [PubMed] [Google Scholar]
- 38.Scruggs SB, Hinken AC, Thawornkaiwong A, Robbins J, Walker LA, de Tombe PP, Geenen DL, Buttrick PM, Solaro RJ. Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. J Biol Chem. 2009;284:5097–5106. doi: 10.1074/jbc.M807414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solaro RJ, Westfall MV. Physiology of the Myocardium. In: Selke FW, del Nido P, Swanson SJ, editors. Sabiston & Spencer Surgery of the Chest Saunders. Elsevier; Philadelphia: 2010. pp. 725–737. [Google Scholar]
- 40.Stanevich V, Jiang L, Satyshur KA, Li Y, Jeffrey PD, Li Z, Menden P, Semmelhack MF, Xing Y. The structural basis for tight control of PP2A methylation and function by LCMT-1. Mol Cell. 2011;41:331–342. doi: 10.1016/j.molcel.2010.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sumandea MP, Rybin VO, Hinken AC, Wang C, Kobayashi T, Harleton E, Sievert G, Balke CW, Feinmark SJ, Solaro RJ, Steinberg SF. Tyrosine phosphorylation modifies protein kinase C delta-dependent phosphorylation of cardiac troponin I. J Biol Chem. 2008;283:22680–22689. doi: 10.1074/jbc.M802396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 44.Takimoto E, Soergel DG, Janssen PM, Stull LB, Kass DA, Murphy AM. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ Res. 2004;94:496–504. doi: 10.1161/01.RES.0000117307.57798.F5. [DOI] [PubMed] [Google Scholar]
- 45.Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walker LA, Fullerton DA, Buttrick PM. Contractile protein phosphorylation predicts human heart disease phenotypes. Am J Physiol Heart Circ Physiol. 2013;304:H1644–H1650. doi: 10.1152/ajpheart.00957.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker LA, Walker JS, Ambler SK, Buttrick PM. Stage-specific changes in myofilament protein phosphorylation following myocardial infarction in mice. J Mol Cell Cardiol. 2010;48:1180–1186. doi: 10.1016/j.yjmcc.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westfall MV, Lee AM, Robinson DA. Differential contribution of troponin I phosphorylation sites to the endothelin-modulated contractile response. J Biol Chem. 2005;280:41324–41331. doi: 10.1074/jbc.M506043200. [DOI] [PubMed] [Google Scholar]
- 49.Yasuda S, Coutu P, Sadayappan S, Robbins J, Metzger JM. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ Res. 2007;101:377–386. doi: 10.1161/CIRCRESAHA.106.145557. [DOI] [PubMed] [Google Scholar]
- 50.Zhang P, Kirk JA, Ji W, dos Remedios CG, Kass DA, Van Eyk JE, Murphy AM. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang R, Zhao J, Mandveno A, Potter JD. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995;76:1028–1035. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]