

Abstract
Phospholipase C-γ1 (PLC-γ1) contains two tandem Src homology 2 (SH2) domains. The NH2-terminal SH2 domain has been known to mediate the binding of PLC-γ1 to receptor protein tyrosine kinases, which then activate PLC-γ1 via phosphorylation at Y783. We now show that the phosphorylated Y783 residue (pY783) associates with the COOH-terminal SH2 domain [SH2(C)] within the same molecule of PLC-γ1. The specificity of this intramolecular interaction is demonstrated in several ways. The mutation of SH2(C), but not of the NH2-terminal SH2 domain, exposes pY783 and makes it available for binding by anti-pY783 antibodies, for intermolecular association with a GST fusion protein containing the tandem SH2 domains of PLC-γ1 and for dephosphorylation by phosphatases. The intramolecular interaction between pY783 and SH2(C) induces a rearrangement of surface charge such that PLC-γ1 molecules phosphorylated at Y783 are retained more strongly by heparin resins than are unphosphorylated molecules. Finally, the intramolecular interaction of pY783 with SH2(C) results in activation of phospholipase activity. Our results thus clarify the molecular mechanism of PLC-γ1 activation, revealing the specific function of pY783 and the distinct roles of the two SH2 domains in this process.
Keywords: affinity chromatography, platelet-derived growth factor
Phospholipase C (PLC) hydrolyzes phosphatidylinositol 4,5-bisphosphate to generate the second messengers inositol 1,4,5-trisphosphate and diacylglycerol. Mammalian cells express 12 different PLC gene products, which are grouped into five subfamilies: β (β1–β4), γ (γ1 and γ2), δ (δ1–δ4), and ε and ζ (1, 2). All PLC isoforms are subject to strict regulation: They are inactive in quiescent cells but are rapidly activated in response to cell stimulation via various surface receptors.
Among the various isozymes of PLC, members of the γ subfamily are structurally distinct, in that the insert sequence between the catalytic X and Y domains is much longer than those in other class of isozymes and contains two tandem Src homology 2 (SH2) domains and an SH3 domain, both flanked by the split halves of a pleckstrin homology domain (Fig. 1A). The X and Y domains together form an inseparable catalytic unit, whereas the insert sequence has been proposed to serve as an autoinhibitory domain based on the fact that PLC-γ1 proteins, either genetically or proteolytically disrupted in the insertion sequence, exhibit substantially higher activities than the intact protein (reviewed in ref. 3). Expression of PLC-γ activity upon cell stimulation, therefore, should occur through release of autoinhibition.
Fig. 1.

Effect of mutation of Y783 or Y1253 on PDGF-induced PLC-γ1 activation. (A) Schematic representation of the domain organization of rat PLC-γ1. (B) Null TV-1 cells expressing wild-type (WT) PLC-γ1 or its Y783F or Y1253F mutants were left unstimulated or stimulated for 10 min with PDGF (50 ng/ml), and cell lysates were then subjected to immunoblot analysis with antibodies to PLC-γ1 (α-PLC-γ1) or with antibodies specific for PLC-γ1 phosphorylated either on Y783 (α-pY783) or on Y1253 (α-pY1253). (C) Null TV-1 cells expressing wild-type PLC-γ1 or the Y783F or Y1253F mutant were metabolically labeled with myo-[3H]inositol and then stimulated with PDGF (50 ng/ml) for the indicated times. The amount of [3H]inositol phosphates in cell lysates was measured. Data are the means of two independent experiments.
Activation of PLC-γ isozymes in vivo depends on tyrosine phosphorylation in two critical steps. First, PLC-γ associates either with a receptor protein tyrosine kinase (PTK), such as the receptor for platelet-derived growth factor (PDGF), or with an adapter protein in supramolecular complex containing receptor and nonreceptor PTK, such as B and T cell antigen receptor complexes. Such association is mediated by the recognition of a phosphorylated tyrosine residue of the receptor or the adapter by the NH2-terminal SH2 [SH2(N)] domain of PLC-γ (4–8). Second, the receptor PTK or the nonreceptor PTK bound to the adapter phosphorylates PLC-γ.
The PDGF and EGF receptors phosphorylate rat PLC-γ1 on three tyrosine residues: Y771, Y783, and Y1253 (9–11). PLC-γ1 phosphorylated on tyrosines exhibits increased enzymatic activity in vitro (12, 13), and phosphorylation of Y783 is shown to be essential for PLC-γ1 activation in vivo (11). Moreover, Y783 is the major site of phosphorylation; the extent of phosphorylation at Y1253 is about one-half of that apparent at Y783 in PDGF- or EGF-stimulated cells and is virtually undetectable in lymphocytes stimulated via their immunoreceptors, whereas the extent of phosphorylation at Y771 is less than one-tenth of that at Y783 (14). The molecular events that link Y783 phosphorylation and PLC-γ1 activation, however, have remained unknown. Here we provide evidence that the phosphorylated Y783 residue participates in an intramolecular interaction with the COOH-terminal SH2 [SH2(C)] domain of PLC-γ1 and thereby converts the phospholipase to the active state.
Experimental Procedures
Antibodies and Other Materials. A mixture of mouse monoclonal antibodies to PLC-γ1 (15) and both rabbit polyclonal anti-pY783 and -pY1253 (14) were described previously. Recombinant rat PLC-γ1 produced with a baculovirus expression system was purified as described (16). ZAP-70 was obtained from Upstate Biotechnology (Lake Placid, NY), and PDGF-BB (rat recombinant) was from R & D Systems. Heparin-Sepharose CL-6B, protein G-Sepharose, and GSH-Sepharose were from Amersham Pharmacia Biotech.
Construction of Vaccinia Virus Vectors. The Tyr → Phe mutants (11) and SH2-domain mutants (8) of rat PLC-γ1 were described previously. In the case of the SH2(C) domain, we introduced an Arg → Ala substitution at position 696 in addition to that at position 694 originally described (8). The coding sequences of wild-type and mutant PLC-γ1 cDNAs were transferred into the plasmid pSC11, and recombinant vaccinia viruses for protein expression were prepared as described (17).
Measurement of in Vivo Production of Inositol Phosphates. Null TV-1 cells (18) were kindly provided by G. Carpenter (Vanderbilt University School of Medicine, Nashville, TN). The cells were cultured in six-well plates to ≈80% confluency and then incubated for 24 h with 2 ml of myo-inositol-free DMEM supplemented with 1% FBS myo-[2-3H]inositol [1 μCi/ml (1 Ci = 37 GBq)/25 Ci/mol; NEN Life Science] and vaccinia viruses. The cells were then washed once with standard DMEM, incubated for 10 min in DMEM supplemented with 20 mM LiCl and 20 mM Hepes-NaOH (pH 7.4), and then exposed to PDGF for various times. [3H]Inositol phosphates in the cells were then analyzed by HPLC equipped with a radiometric detector, as described (8).
Purification of PLC-γ1. Null TV-1 cells were grown to confluency in 150-mm dishes and then incubated for 24 h with vaccinia viruses in serum-free medium. The medium was then replaced with fresh DMEM supplemented with 1 mM sodium vanadate, and the cells were incubated for 10 min at 37°C before stimulation with PDGF (50 ng/ml) plus H2O2 (2 mM) for an additional 10 min. The cells were lysed in 5 ml of an ice-cold solution containing 20 mM Hepes-NaOH (pH 7.0), 1 mM EDTA, 1 mM EGTA, 1 mM sodium vanadate, 0.1% Triton X-100, and a mixture of protease inhibitors [0.5 mM 4-(2-aminoethyl)benzenesulfonylfluoride/leupeptin (25 μg/ml)/aprotinin (25 μg/ml)], and the dishes were immediately transferred to a –70°C freezer.
The frozen cells were thawed and scraped off the dishes, and the lysates were centrifuged. The resulting supernatants were mixed with heparin-Sepharose CL-6B (2 ml of packed resin for samples pooled from 10 dishes) that had been equilibrated with a solution containing 20 mM Hepes-NaOH (pH 7.0), 1 mM EDTA, 1 mM EGTA, and 1 mM DTT. After incubation for 1 h at 4°C with rocking, the mixture was transferred to a column and washed extensively with equilibration buffer. Bound material was then eluted with equilibration buffer supplemented with 1 M NaCl (5 ml per 2 ml of resin).
The column eluates were subjected to HPLC on a heparin-5PW column (7.5-mm i.d. × 7.5 cm; TosoHaas, Montgomeryville, PA) that had been equilibrated with a solution containing 20 mM Hepes-NaOH (pH 7.0), 0.1 mM EDTA, and 1 mM DTT. The samples were injected after 10-fold dilution with equilibration buffer. The column was washed to remove unbound material, and bound proteins were then eluted with a linear gradient of NaCl (0–1 M) in equilibration buffer over 1 h at a flow rate of 1 ml/min. PLC-γ1 was detected in the fractions by immunoblot analysis.
For determination of the monomeric or dimeric state of PLC-γ1, the partially purified PLC-γ1 preparations obtained by batch chromatography on heparin-Sepharose were subjected to gel filtration on a G3000SW column (7.5-mm i.d. × 60 cm; TosoHaas). The column was equilibrated and subjected to elution with a solution containing 20 mM Tris·HCl (pH 7.5), 0.1 M NaCl, 1 mM DTT, and 1 mM EDTA at a flow rate of 0.5 ml/min, and 0.5-ml fractions were collected.
Immunoblot Analysis, Immunoprecipitation, and GST Precipitation Analysis. Cells cultured in six-well plates were lysed in 1 ml of an ice-cold lysis buffer containing 20 mM Hepes-NaOH (pH 7.0), 50 mM NaF, 10% glycerol, 1% Triton X-100, 1 mM sodium vanadate, and a mixture of protease inhibitors. After incubation for 30 min on ice, the lysates were centrifuged, and the resulting supernatants were fractionated by SDS/PAGE on a 6% gel (NOVEX, San Diego). The separated proteins were transferred to a nitrocellulose membrane and probed with primary antibodies. Immune complexes were detected with alkaline phosphatase-conjugated secondary antibodies (Kirkegaard & Perry Laboratories) and the chemiluminescent reagent CDP-Star (Tropix, Bedford, MA). Chemiluminescence images were captured, digitized, and analyzed with a Kodak Image Station 440.
For immunoprecipitation, cell lysates were first subjected to batch chromatography with heparin-Sepharose CL-6B, as described above. Portions (100 μl) of the partially purified PLC-γ1 preparations were diluted to 1 ml with PBS supplemented with 0.1% Triton X-100/1 mM EDTA/1 mM sodium vanadate and were incubated with rocking, first for 1 h at 4°C with excess amounts of antibodies and then for 2 h in the additional presence of 50 μl of a 50% slurry of protein G-Sepharose. The beads were then washed twice with the incubation buffer and once with water, suspended in the original volume of SDS sample buffer, and boiled. The samples were then subjected to immunoblot analysis.
For the GST precipitation assay, cell lysates were subjected to batch chromatography on heparin-Sepharose as described above, and portions of the partially purified PLC-γ1 preparations were incubated for 1 h at room temperature with 1 μg of a GST fusion protein containing amino acids 533–851 of rat PLC-γ1 (19) in a total volume of 0.5 ml of the lysis buffer supplemented with 1 mM EDTA/1 mM DTT/0.1 mg/ml BSA. The incubation was continued for an additional 1 h after the addition of 50 μl of a 50% slurry of GSH-Sepharose. The beads were then washed twice with the incubation buffer, after which bound proteins were subjected to immunoblot analysis.
Phosphorylation of PLC-γ1 in Vitro. Purified PLC-γ1(2 mg/ml) was incubated for 4 h at 30°C with ZAP-70 (10 μg/ml) in a solution containing 20 mM Hepes-NaOH (pH 7.0), 100 mM NaCl, 1 mM EGTA, 1 mM DTT, 10 mM MgCl2, 5 mM MnCl2, 0.1 mM sodium vanadate, 1 mM ATP, and 0.1 mg/ml BSA. The reaction mixture was then immediately subjected to HPLC on a heparin-5PW column, as described above.
Measurement of Phospholipase Activity in Vitro. For preparation of substrates, a solution of phosphatidylinositol (PI) (bovine liver; Avanti Polar Lipids) in chloroform was mixed with (2-[3H]-myo-inositol) PI (17 Ci/mmol; NEN) and then dried under a stream of N2. The dried lipid was suspended in a solution containing 50 mM Hepes-NaOH (pH 7.0), 100 mM NaCl, 0.1 mM EDTA, and either 0.2% sodium deoxycholate or 0.1% Triton X-100 plus 0.3% octyl glucoside.
Enzyme samples (50 μl) diluted in a solution containing 50 mM Hepes-NaOH (pH 7.0), 4 mM EGTA, 4.6 mM CaCl2, 0.6 mM MgCl2, and 100 mM NaCl were mixed with 50 μl of substrate suspension. The resulting mixture thus contained 50 mM Hepes-NaOH (pH 7.0), 100 mM NaCl, 2 mM EGTA, 2.3 mM CaCl2 (free Ca2+ ≈ 0.3 mM), 0.3 mM MgCl2, 0.05 mM EDTA, 0.4 mM PI, 30,000 cpm of [3H]PI, and either 0.1% sodium deoxycholate or 0.05% Triton X-100 plus 0.15% octyl glucoside. After incubation at 30°C for 10–20 min, the reaction was terminated by the addition of 100 μl of 10% BSA and 200 μl of 10% trichloroacetic acid followed by centrifugation. The amount of radioactivity in the supernatant was measured by liquid scintillation spectroscopy.
Results
Y783 Is the Only Phosphorylation Site Necessary for Activation of PLC-γ1 in Cells. We have previously shown that the PDGF-induced activation of PLC-γ1 is completely blocked by the Y783 → F mutation, reduced by ≈40% by the Y1253 → F mutation, and not affected by the Y771 → F mutation (11). However, these observations were made with NIH 3T3 cell lines that had been selected for stable overexpression of the mutants and that also expressed endogenous PLC-γ1. Moreover, the expression levels of the mutant proteins in these cell lines were not identical. We therefore reexamined the Y → F mutant enzymes by inducing their transient expression in Null TV-1 cells, which are derived from embryonic fibroblasts of a PLC-γ1-deficient mouse (18). We chose a recombinant vaccinia virus as the expression vector because of its high efficiency of gene transduction and the fact that it allows the level of expression to be controlled readily. The expression levels of the Y783F and Y1253F mutants of PLC-γ1 and of the wild-type protein were thus adjusted to be approximately equal. Phosphorylation on Y783 and on Y1253 induced by exposure of the infected cells to PDGF appeared to occur independently, given that phosphorylation at one site was not affected by mutation of the other site (Fig. 1B). Mutation of Y783 completely blocked the PDGF-induced accumulation of inositol phosphates; however, mutation of Y1253 and Y771 had no effect on PLC-γ1 activation (Fig. 1C and data not shown). We therefore conclude that phosphorylation of Y783 is necessary and sufficient for induction of phospholipase activity in response to growth factor stimulation.
Phosphorylation on Y783 Induces a Conformational Change in PLC-γ1. We attempted to isolate phosphorylated PLC-γ1. High levels of PLC-γ1 expression in Null TV-1 cells were achieved by infection with large doses of recombinant vaccinia viruses. Infected cells were then stimulated with a saturated concentration of PDGF plus pervanadate (sodium vanadate plus H2O2) to induce high stoichiometry of PLC-γ1 phosphorylation. It was found that affinity chromatography on heparin-5PW could effectively separate phosphorylated PLC-γ1 from the nonphosphorylated protein.
Lysates of Null TV-1 cells expressing wild-type PLC-γ1 were subjected to heparin-5PW HPLC, which was then developed with an NaCl gradient. PLC-γ1 molecules from unstimulated cells were present in a peak centered at fraction 21 (Fig. 2A). In contrast, although this peak was still detected with stimulated cells, a second peak of PLC-γ1 molecules centered around fractions 29–31 emerged. Immunoblot analysis with PLC-γ1 phosphospecific antibodies revealed that PLC-γ1 molecules in the first peak were phosphorylated neither at Y783 nor at Y1253, whereas those in the second peak were phosphorylated on Y783, Y1253, or both sites.
Fig. 2.

Phosphorylation of Y783, but not of Y1253, increases the retention time of PLC-γ1 on a heparin column. (A) Null TV-1 cells expressing wild-type PLC-γ1 or its Y783F or Y1253F mutants were left unstimulated or stimulated for 10 min with PDGF (50 ng/ml) plus pervanadate (2 mM H2O2 + 1 mM sodium vanadate). Soluble proteins were then extracted from the cells and subjected to fractionation by HPLC on a heparin-5PW column. Bound proteins were eluted with a linear NaCl gradient (0–1 M) at a flow rate of 1 ml/min over 60 min. Fractions (1 ml) were collected and subjected to immunoblot analysis with indicated antibodies. (B) Purified PLC-γ1 was phosphorylated with ZAP-70 in vitro and subjected to HPLC on a Heparin-5PW column as in A.
Given that heparin is a highly anionic sulfated polysaccharide and that tyrosine phosphorylation introduces negative charges to PLC-γ1, the unexpected higher affinity of phosphorylated PLC-γ1 molecules for the heparin column suggested that phosphorylation might induce a major conformational change in PLC-γ1 that results in a rearrangement of surface charge. Similar analysis of a lysate of NIH 3T3 cells that had been stimulated with PDGF in the absence of pervanadate revealed that a fraction of PLC-γ1 molecules became phosphorylated, and that the phosphorylated molecules, which solely recovered in the second peak, reacted with both anti-pY783 and -pY1253 (data not shown). This result indicated that the phosphorylation-induced conformational change was not an artifact due to viral infection or of cell stimulation in the presence of pervanadate, an inhibitor of protein tyrosine phosphatases.
Lysates of the Null TV-1 cells expressing the Y783F or Y1253F mutants of PLC-γ1 were analyzed to determine which phosphorylation site was responsible for the conformational change. The Y1253F mutant that had been phosphorylated on Y783 in response to cell stimulation eluted in the second peak (Fig. 2 A). Similarly, stimulation of cells expressing the Y771F mutant resulted in the elution of PLC-γ1 molecules phosphorylated on Y783 and Y1253 in the second peak (data not shown). In contrast, molecules of the Y783F mutant phosphorylated on Y1253 were all recovered in the first peak, and the second peak was no longer detected with this mutant (Fig. 2 A). These results indicate that phosphorylation of PLC-γ1 on Y783 is necessary and sufficient for the shift in the position of the elution peak.
We also analyzed the elution pattern of purified PLC-γ1 that had been phosphorylated by a nonreceptor PTK (ZAP-70) in vitro. Whereas the nonphosphorylated enzyme eluted from the heparin column in a single peak centered at fraction 21 (data not shown), phosphorylation resulted in the appearance of the second peak; all of the Y783-phosphorylated PLC-γ1 molecules were detected in the second peak, but Y1253-phosphorylated molecules were present in both peaks (Fig. 2B). These data further support the conclusion that phosphorylation onY783, not that on Y1253, is responsible for the shift in elution profile. They further rule out the possibility that the shift is caused by the association of an unknown protein with Y783-phosphorylated PLC-γ1.
Intramolecular Interaction of Phosphorylated Y783 with the SH2(C) Domain of PLC-γ1. To examine whether the conformational change induced in PLC-γ1 by the phosphorylation of Y783 resulted from the interaction of pY783 with one of the two SH2 domains of the protein, we introduced an R586→A mutation into SH2(N) and both R694→A and R696→A mutations into SH2(C). The resulting mutant proteins were designated N– and C–, respectively. Arginine residues equivalent to R586 and R694 in PLC-γ1 are conserved in all SH2 domains and contribute to a binding site for the phosphoryl group of phosphotyrosine. R696 is not present in other SH2 domains, but in the SH2(C) domains of PLC-γ1 (and presumably in that of PLC-γ2), it also interacts with the phosphate moiety (20). The binding of pY783 to an SH2 domain would be expected to shield pY783 from interaction with other proteins.
Null TV-1 cells expressing wild-type PLC-γ1 or the N– or C– mutants were stimulated with PDGF plus pervanadate, and PLC-γ1 molecules were then enriched by batch purification with heparin-Sepharose from cell lysates. The mutation of the SH2(N) domain impairs the binding of the N– protein to the activated PDGF receptor, and its phosphorylation by the receptor is therefore nearly undetectable (8). Nevertheless, stimulation of cells with a saturating concentration of PDGF together with pervanadate ensured that the phosphorylation level of N– was similar to those of the wild-type and C– proteins (see Fig. 4). The partially purified fractions containing the wild-type, N–, or C– proteins were subjected to immunoprecipitation with anti-PLC-γ1, anti-pY783, or anti-pY1253, and the resulting precipitates were analyzed by immunoblotting.
Fig. 4.

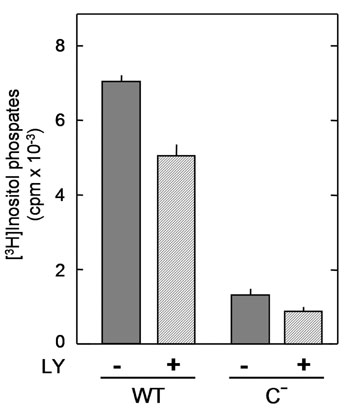
Effect of SH2 domain mutation on the conformation and activity of PLC-γ1 measured in vitro.(A) Null TV-1 cells expressing wild-type PLC-γ1orthe N– or C– mutants thereof were stimulated for 10 min with PDGF plus pervanadate. Soluble proteins were then extracted and subjected to fractionation by HPLC on a heparin-5PW column as in Fig. 2, with the exception that a shallower NaCl gradient (0–0.6 M over 1 h) was used, and that sodium vanadate (0.1 mM) was included in the elution buffer to prevent dephosphorylation of the C– mutant. The fractions were subjected to immunoblot analysis with anti-PLC-γ1 (Upper), and the amount of PLC-γ1 in each fraction was quantified (Lower). (B) The column fractions corresponding to peaks 1 and 2 from A were concentrated, their concentrations were adjusted on the basis of their phospholipase activity measured with PI-deoxycholate micelles, and the adjusted preparations were subjected to immunoblot analysis with the indicated antibodies. (C) The adjusted protein solutions from B were assayed for phospholipase activity with micelles prepared from PI and Triton X-100. Data are expressed relative to the value for peak 1 of the wild-type enzyme and are means ± SE of values from three independent experiments.
Anti-pY1253 precipitated Y1253-phosphorylated protein from all three lysates as efficiently as did anti-PLC-γ1 (Fig. 3A), indicating that pY1253 in all three PLC-γ1 proteins was unobstructed. In contrast, the amount of Y783-phosphorylated wild-type or N– proteins precipitated by anti-pY783 was greatly reduced compared with that precipitated by anti-PLC-γ1, suggesting that pY783 was shielded. Mutation of SH2(C) appeared to largely prevent such shielding, given that the efficiency of immunoprecipitation of the Y783-phosphorylated protein by anti-pY783 was increased. This result suggested that pY783 binds to SH2(C).
Fig. 3.

Intramolecular association of pY783 with the SH2(C) domain in PLC-γ1. (A) Null TV-1 cells expressing wild-type, N–,orC– forms of PLC-γ1 were stimulated for 10 min with PDGF plus pervanadate, after which the PLC-γ1 proteins were partially purified from cell lysates by batch chromatography on heparin-Sepharose. The partially purified proteins were then subjected to immunoprecipitation with anti-PLC-γ1 (α-γ1), -pY783, or -pY1253, and the resulting precipitates were subjected to immunoblot analysis with the same antibodies. (B) Partially purified PLC-γ1 proteins prepared as in A from non-stimulated or cells stimulated with PDGF plus pervanadate were incubated with a GST fusion protein containing the SH2(N)-SH2(C)-SH3 domains of PLC-γ1. Proteins that bound to the GST fusion protein were precipitated with GSH-Sepharose and subjected to immunoblot analysis with the indicated antibodies (Bound). The partially purified PLC-γ1 proteins equivalent to one-fourth of the amount used for the binding assay were also subjected directly to immunoblot analysis (Input). (C) Null TV-1 cells were stimulated as in A and then lysed in the absence of phosphatase inhibitors (NaF, vanadate). The lysates were incubated in the presence of 5 mM DTT and 5 mM MgCl2 to activate phosphatases at 30°C for the indicated times and then subjected to immunoblot analysis with the indicated antibodies. (D) Null TV-1 cells expressing wild-type PLC-γ1 were left unstimulated or stimulated for 10 min with PDGF plus pervanadate. PLC-γ1 was then partially purified from cell lysates as in A and subjected to gel filtration by HPLC. The fractions from unstimulated (filled circles) and stimulated (open circles) cells were subjected to immunoblot analysis with anti-PLC-γ1 and anti-pY783, respectively. The elution positions of marker proteins are also shown: 670 kDa, thyroglobulin; 138 kDa, BSA dimer; 69 kDa, BSA monomer; 44 kDa, ovalbumin; and 17 kDa, myoglobin.
The partially purified fractions containing wild-type, N–, or C– forms of PLC-γ1 were also subjected to precipitation with a GST fusion protein containing the SH2(N)-SH2(C)-SH3 region of rat PLC-γ1 (amino acids 533–851). The GST fusion protein did not bind PLC-γ1 present in fractions prepared from non-stimulated cells (Fig. 3B). Even after induction of tyrosine phosphorylation, neither wild-type nor N– proteins bound the GST fusion protein. In contrast, phosphorylated C– protein was precipitated from fractions prepared from stimulated cells, indicating again that mutation of SH2(C) results in the exposure of pY783. These observations also indicated that pY1253 is unable to interact with either of the two SH2 domains in the GST fusion protein.
During purification, we noticed that the phosphate moiety of pY783 in the C– mutant was readily lost when phosphatase inhibitors were not included in the buffer, whereas pY783 in the wild-type and N– proteins was stable. This increased susceptibility of pY783 of the C– mutant to dephosphorylation was also apparent in crude cell lysates, which had been prepared in the absence of phosphatase inhibitors (Fig. 3C). This observation thus provided additional support for the interaction of pY783 with SH2(C).
To determine whether this interaction occurs intra- or intermolecularly, we subjected the lysates of Null TV-1 cells expressing wild-type PLC-γ1 prepared either before or after stimulation to gel filtration. The elution profile of the phosphorylated protein obtained from stimulated cells revealed by anti-pY783 was similar to that of the nonphosphorylated protein in unstimulated cells revealed by anti-PLC-γ1 (Fig. 3D). A purified preparation of PLC-γ1 was also eluted in the same position (data not shown). The apparent molecular size of PLC-γ1 was estimated to be ≈70 kDa, which is smaller than the molecular mass of 149 kDa deduced from the cDNA sequence, possibly because of the aglobular shape of the molecule. These results thus suggested that the interaction between pY783 and SH2(C) is intramolecular.
Role of the pY783-SH2(C) Interaction in Phospholipase Activation. Null TV-1 cells expressing wild-type, N–, or C– PLC-γ1 proteins were stimulated with PDGF and pervanadate, lysed, and subjected to HPLC on the heparin column by using a buffer containing sodium vanadate (phosphatase inhibitor). Like the wild-type protein, N– yielded two well separated peaks corresponding to nonphosphorylated (peak 1) and phosphorylated (peak 2) forms of the protein (Fig. 4A). In the case of C–, however, peak 2 had a broader appearance and eluted much earlier than did those for the wild-type and N– proteins. That peaks 1 and 2 of C– did not merge fully indicates that phosphorylation of Y783 alone can change the distribution of surface charge in the absence of interaction with SH2(C).
We next measured the phospholipase activity associated with peaks 1 and 2. The activity of phosphorylated PLC-γ1 measured with a mixed micellar substrate containing PI and Triton X-100 is much greater than that of the nonphosphorylated enzyme, whereas both phosphorylated and nonphosphorylated enzymes show similar activities toward PI-containing micelles prepared with deoxycholate (12, 13). We therefore first equalized the PLC-γ1 concentrations of peaks 1 and 2 on the basis of phospholipase activity measured with the deoxycholate-based substrate, and the presence of similar amounts of PLC-γ1 protein in the normalized preparations was confirmed by immunoblot analysis with anti-PLC-γ1 (Fig. 4B). A similar extent of phosphorylation at Y783 of peak 2 preparations was also confirmed. Phospholipase activity of the normalized preparations was then measured with the Triton X-100-based substrate (Fig. 4C). For both the wild-type and N– proteins, the activity of the phosphorylated form (peak 2) was ≈20 times that of the nonphosphorylated form (peak 1). In contrast, phosphorylation of the C– mutant increased its activity only 2-fold. These results suggested that the conformational change required for full expression of phospholipase activity in vitro requires the interaction of pY783 with the SH2(C) domain, and phosphorylation of Y783 alone appears capable of inducing this change only to a much smaller extent.
Discussion
Stimulation of cells via various surface receptors induces the phosphorylation of PLC-γ1 on Y771, Y783, and Y1253. Of these three sites, phosphorylation of Y771 and Y1253 occurs at a low level or depends on cell type (14), and phosphorylation of neither of these residues is required for phospholipase activation. In contrast, Y783 is the major site of phosphorylation, and its phosphorylation is indispensable for phospholipase activation both in vitro and in vivo (ref. 11 and this study). The mechanistic basis of this requirement, however, has not been known. We now provide evidence that phosphorylation of Y783, the residue located in the middle of the linker region, induces a major conformational change in PLC-γ1 through the intramolecular interaction between pY783 and SH2(C). If in fact the uniquely long linker region between the X and Y domains is an intramolecular inhibitor, which keeps PLC-γ1 activity low in quiescent cells (3), the phosphorylation of Y783 may be a molecular switch that reverses this inhibition. The closely related isozyme, PLC-γ2, might also undergo a similar structural rearrangement after phosphorylation of the site equivalent to Y783 in PLC-γ1 (21). Our results also suggest that phosphorylation of Y783 causes a conformational change to some extent in the absence of its binding to SH2(C), but phosphorylation alone yields only an intermediate weakly active form. It is the intramolecular interaction that transforms PLC-γ1 into its fully active form.
The two SH2 domains of PLC-γ1 are arranged in a tandem manner. Such an arrangement of SH2 domains is sometimes necessary to ensure high specificity and affinity for interaction with bisphosphorylated target proteins, as exemplified by the case of binding of p85α regulatory subunit of PI 3-kinase to the β-PDGF receptor phosphorylated both at Y740 and at Y751 (ref. 22 and references therein). The two SH2 domains of PLC-γ1 were initially suggested to be necessary to bind multiply phosphorylated receptor PTKs more efficiently (22, 23). More recently, however, the SH2(N), but not the SH2(C), domain has been shown to be sufficient to mediate the association of PLC-γ1 with either the PDGF or EGF receptor (5, 7, 8) or with adapter proteins responsible for the recruitment of PLC-γ1 to the vicinity of nonreceptor PTKs in cells stimulated via immune receptors (4, 6). The two SH2 domains of PLC-γ1 thus appear to play distinct roles, with SH2(N) first mediating the positioning of PLC-γ1 close to a PTK for phosphorylation and then SH2(C) contributing to a conformational change of the phosphorylated enzyme that results in phospholipase activation.
Despite the important role of SH2(C) in the activation demonstrated in the present study, however, the effects of SH2(C) mutation have ranged from a slight (8) to a moderate decrease (24) to a near total loss in the induction of activity (6, 7). This inconsistency is likely attributable to the facts that these effects were evaluated in various cell types expressing the mutant enzymes at different levels, and that pY783 alone in the absence of its interaction with SH2(C) is able to increase catalytic activity, albeit weakly, as seen with the C– mutant in the present study. We reevaluated in vivo activity of the C– protein; when expressed at a low level in Null cells, C–-expressing cells exhibited only weak phosphoinositide hydrolysis after stimulation with PDGF (≈20% of those expressing wild type), as opposed to the case of overexpression (8) (see Supporting Text and Fig. 6, which are published as supporting information on the PNAS web site).
Based on our present results, we propose a model for the activation of PLC-γ1 in cells stimulated with PDGF (Fig. 5). The SH2(N) domain first recognizes the phosphorylated receptor PTK, which then phosphorylates PLC-γ1 on Y783 and two other minor sites. Phosphorylation of Y783 itself induces an initial conformational change in PLC-γ1 that results in induction of a low level of phospholipase activity. The intramolecular association of pY783 with the SH2(C) domain, however, promotes a major conformational change and thereby increases phospholipase activity to a much higher level. Nevertheless, phosphorylation of Y783 is not sufficient for full activation of PLC-γ1 in cells. Additional cellular factors, such as phosphatidylinositol 3,4,5-trisphosphate, further increase the activity of PLC-γ1 to maximal levels (14, 19, 25).
Fig. 5.

Model for the mechanism of PLC-γ1 activation by tyrosine phosphorylation. See Discussion for details.
Supplementary Material
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PLC, phospholipase C; PDGF, platelet-derived growth factor; PI, phosphatidylinositol; PTK, protein tyrosine kinase; SH2, Src homology 2; SH2(N), NH2-terminal SH2; SH2(C), COOH-terminal SH2.
References
- 1.Rhee, S. G. (2001) Annu. Rev. Biochem. 70, 281–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saunders, C. M., Larman, M. G., Parrington, J., Cox, L. J., Royse, J., Blayney, L. M., Swann, K. & Lai, F. A. (2002) Development (Cambridge, U.K.) 129, 3533–3544. [DOI] [PubMed] [Google Scholar]
- 3.Carpenter, G. & Ji, Q. (1999) Exp. Cell. Res. 253, 15–24. [DOI] [PubMed] [Google Scholar]
- 4.Stoica, B., DeBell, K. E., Graham, L., Rellahan, B. L., Alava, M. A., Laborda, J. & Bonvini, E. (1998) J. Immunol. 160, 1059–1066. [PubMed] [Google Scholar]
- 5.Chattopadhyay, A., Vecchi, M., Ji, Q., Mernaugh, R. & Carpenter, G. (1999) J. Biol. Chem. 274, 26091–26097. [DOI] [PubMed] [Google Scholar]
- 6.DeBell, K. E., Stoica, B. A., Veri, M. C., Di Baldassarre, A., Miscia, S., Graham, L. J., Rellahan, B. L., Ishiai, M., Kurosaki, T. & Bonvini, E. (1999) Mol. Cell. Biol. 19, 7388–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji, Q. S., Chattopadhyay, A., Vecchi, M. & Carpenter, G. (1999) Mol. Cell. Biol. 19, 4961–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poulin, B., Sekiya, F. & Rhee, S. G. (2000) J. Biol. Chem. 275, 6411–6416. [DOI] [PubMed] [Google Scholar]
- 9.Kim, J. W., Sim, S. S., Kim, U. H., Nishibe, S., Wahl, M. I., Carpenter, G. & Rhee, S. G. (1990) J. Biol. Chem. 265, 3940–3943. [PubMed] [Google Scholar]
- 10.Wahl, M. I., Nishibe, S., Kim, J. W., Kim, H., Rhee, S. G. & Carpenter, G. (1990) J. Biol. Chem. 265, 3944–3948. [PubMed] [Google Scholar]
- 11.Kim, H. K., Kim, J. W., Zilberstein, A., Margolis, B., Kim, J. G., Schlessinger, J. & Rhee, S. G. (1991) Cell 65, 435–441. [DOI] [PubMed] [Google Scholar]
- 12.Nishibe, S., Wahl, M. I., Hernandez-Sotomayor, S. M., Tonks, N. K., Rhee, S. G. & Carpenter, G. (1990) Science 250, 1253–1256. [DOI] [PubMed] [Google Scholar]
- 13.Wahl, M. I., Jones, G. A., Nishibe, S., Rhee, S. G. & Carpenter, G. (1992) J. Biol. Chem. 267, 10447–10456. [PubMed] [Google Scholar]
- 14.Sekiya, F., Poulin, B., Kim, Y. J. & Rhee, S. G. (2004) J. Biol. Chem. 279, 32181–90. [DOI] [PubMed] [Google Scholar]
- 15.Suh, P. G., Ryu, S. H., Choi, W. C., Lee, K. Y. & Rhee, S. G. (1988) J. Biol. Chem. 263, 14497–14504. [PubMed] [Google Scholar]
- 16.Park, D., Jhon, D. Y., Kriz, R., Knopf, J. & Rhee, S. G. (1992) J. Biol. Chem. 267, 16048–16055. [PubMed] [Google Scholar]
- 17.Chakrabarti, S., Brechling, K. & Moss, B. (1985) Mol. Cell. Biol. 5, 3403–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji, Q. S., Winnier, G. E., Niswender, K. D., Horstman, D., Wisdom, R., Magnuson, M. A. & Carpenter, G. (1997) Proc. Natl. Acad. Sci. USA 94, 2999–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bae, Y. S., Cantley, L. G., Chen, C. S., Kim, S. R., Kwon, K. S. & Rhee, S. G. (1998) J. Biol. Chem. 273, 4465–4469. [DOI] [PubMed] [Google Scholar]
- 20.Pascal, S. M., Singer, A. U., Gish, G., Yamazaki, T., Shoelson, S. E., Pawson, T., Kay, L. E. & Forman-Kay, J. D. (1994) Cell 77, 461–472. [DOI] [PubMed] [Google Scholar]
- 21.Kim, Y. J., Sekiya, F., Poulin, B., Bae, Y. S. & Rhee, S. G. (2004) Mol. Cell. Biol. 24, 9986–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ottinger, E. A., Botfield, M. C. & Shoelson, S. E. (1998) J. Biol. Chem. 273, 729–735. [DOI] [PubMed] [Google Scholar]
- 23.Rotin, D., Honegger, A. M., Margolis, B. L., Ullrich, A. & Schlessinger, J. (1992) J. Biol. Chem. 267, 9678–9683. [PubMed] [Google Scholar]
- 24.Ishiai, M., Sugawara, H., Kurosaki, M. & Kurosaki, T. (1999) J. Immunol. 163, 1746–1749. [PubMed] [Google Scholar]
- 25.Falasca, M., Logan, S. K., Lehto, V. P., Baccante, G., Lemmon, M. A. & Schlessinger, J. (1998) EMBO J. 17, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}