

SUMMARY
Abetalipoproteinemia (ABL) is an inherited disorder of lipoprotein metabolism resulting from mutations in Microsomal Triglyceride Transfer Protein (MTTP). In addition to the liver and intestine, MTTP is expressed in cardiomyocytes and cardiomyopathy has been reported in several ABL cases. Using induced pluripotent stem cells (iPSCs) generated from an ABL patient homozygous for a missense mutation (MTTPR46G), we show that human hepatocytes and cardiomyocytes exhibit defects associated with ABL disease including loss of apoB secretion and intracellular accumulation of lipid. MTTPR46G iPSC-derived cardiomyocytes failed to secrete apoB, accumulated intracellular lipid, and displayed increased cell death suggesting intrinsic defects in lipid metabolism due to loss of MTTP function. Importantly, these phenotypes were reversed after the correction of the MTTPR46G mutation by CRISPR/Cas9 gene editing. Together, these data reveal clear cellular defects in iPSC-derived hepatocytes and cardiomyocytes lacking MTTP activity including a cardiomyocyte-specific regulated stress response to elevated lipids.
Keywords: Abetaliproteinemia, induced pluripotent stem cells, iPSC-derived hepatocytes and cardiomyocytes, lipid accumulation, apoB, cardiac stress
Graphical abstract
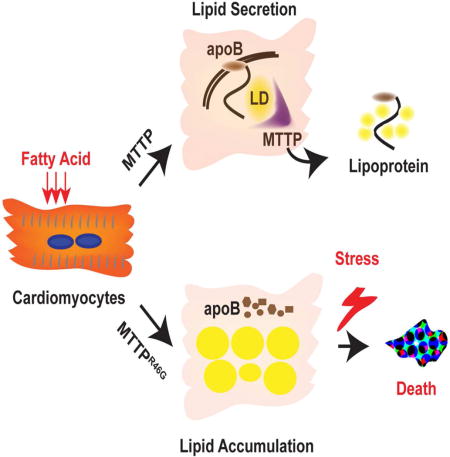
INTRODUCTION
Abetalipoproteinemia (ABL) is an autosomal recessive disorder caused by mutations in the gene MTTP, which encodes the endoplasmic reticulum (ER)-resident lipid transfer protein microsomal triglyceride transfer protein (MTTP). MTTP functions to promote lipid transfer to apolipoprotein B (apoB) within the ER, facilitating its secretion by the enterocyte and the hepatocyte (Wetterau et al., 1991; Wetterau et al., 1990). Genetic deficiency in MTTP results in undetectable apoB-containing lipoproteins in plasma due to the inability to secrete them. While hepatic steatosis is a known complication of ABL, primary hepatocytes from patients with ABL have never been characterized.
Both MTTP and apoB are expressed in human and mouse heart tissue, suggesting that the heart has the capacity to secrete lipoproteins (Boren et al., 1998; Nielsen et al., 1999; Nielsen et al., 1998). Mttp null mice have elevated triglyceride stores in the heart (Bjorkegren et al., 2001). However, it is not known whether cardiac lipid metabolism is affected in patients with ABL. Interestingly, some patients with ABL disease exhibit cardiac arrhythmias and heart failure (Dische and Porro, 1970; Gregg and Wetterau, 1994; Ledmyr et al., 2004; Sobrevilla et al., 1964; Zamel et al., 2008). These findings are consistent with the concept that MTTP-mediated secretion of apoB is a possible mechanism for protecting against cardiomyocyte lipid overload.
In order to define the cell intrinsic roles for MTTP in hepatocytes and cardiomyocytes, induced pluripotent stem cells were generated from an ABL patient and control subjects and differentiated into hepatocytes and cardiomyocytes. As expected, the ABL iPSC-hepatocytes failed to secrete apoB and accumulated lipid. Interestingly, compared with control iPSC-cardiomyocytes, iPSC-cardiomyocytes from the ABL patient failed to secrete apoB, accumulated intracellular lipid, and responded poorly after stress induction through increased apoptosis. These data provide the evidence that MTTP in human cardiomyocytes facilitates lipid export leading to protection from cellular stress in the setting of lipid overload.
RESULTS
Generation of MTTPR46G specific and control iPSCs
We generated iPSCs from an ABL patient homozygous for a rare missense mutation in the MTTP gene (136C>G) and matched control subjects using standard procedures(Yang et al., 2012). This missense mutation occurs in the N-terminal region of MTTP, causing an amino acid switch from arginine to glycine at residue 46 (MTTPR46G)(Fig. S1A and B). Clinically, the patient with the MTTPR46G mutation had undetectable plasma apoB, very low plasma cholesterol and triglyceride (Table S1), steatorrhea, microvesicular steatosis, spinocerebellar degeneration, and retinopathy, all consistent with classic ABL (Miller et al., 2014; Walsh et al., 2015; Zeissig et al., 2010). Three different iPSC lines were established from this patient and control subjects (Table S2). These iPSCs expressed high levels of pluripotency markers including NANOG, OCT4, and SOX2 (Fig. S1C). Quantitative flow cytometry demonstrated over 90% of the MTTPR46G iPSCs express the cell surface pluripotency markers SSEA-4 and TRA-1-60 (Fig. S1D).
Recapitulation of the ABL phenotype in mutant MTTPR46G hepatocytes
To determine the effect of the MTTPR46G mutation on apoB expression in hepatocytes, we differentiated MTTPR46G and control iPSCs into hepatocytes using a standard protocol (Si-Tayeb et al., 2010). After twenty days, the majority of the differentiated cells from control and MTTPR46G iPSCs were positive for the hepatocyte markers HNF4α and ASGPR1 (Fig. S2A). Expression of hepatic genes including ALB, AFP, HNF4α and ASGPR1 were at similar levels to the hepatic cell line Huh7 (Fig. S2B). Additionally, functional synthesis and secretion of albumin were equivalent between control and MTTPR46G hepatocytes (Fig. S2C and D).
Hepatocytes derived from MTTPR46G iPSCs expressed comparable levels of the MTTP gene and MTTP protein as differentiated hepatocytes from control iPSCs (Fig. 1A and B). However, as shown in Figure 1C, lysate from control hepatocytes displayed triglyceride transfer in an MTTP activity assay, whereas lysate from the MTTPR46G iPSC-hepatocytes had no detectable MTTP activity (Fig. 1C). Thus, the MTTPR46G mutation abolished MTTP triglyceride transfer activity, consistent with the phenotypes of ABL in the patient. The apoB mRNA level was normal in MTTPR46G hepatocytes, but there was an almost complete absence of intracellular and extracellular secreted apoB protein (Fig. 1D–F). Because poorly lipidated apoB is known to undergo proteasomal degradation (Fisher et al., 1997; Yeung et al., 1996), we inhibited this process with N-acetyl-leucyl-leucyl-norleucinal (ALLN) (Sakata and Dixon, 1999). After one hour pretreatment with ALLN followed by 20 minutes pulse with 35S methionine/cysteine, a significant amount of new apoB protein was synthesized in MTTPR46G hepatocytes, whereas the control cells exhibited only a moderate elevation in newly synthesized apoB, indicating substantial proteasomal degradation of apoB in the mutant MTTPR46G hepatocytes (Fig. 1G and H). When these cells were chased in label free media, control cells exhibited a slower decrease in newly synthesized cellular apoB levels that reached 30% of initial levels after a 120 minute chase, whereas MTTPR46G hepatocytes had a dramatic reduction in apoB after just a 30 minute chase with only 9% of the initially labeled protein remaining after a 60 minute chase (Fig. 1I).
Figure 1. Abolished apoB secretion in mutant MTTPR46G differentiated hepatocytes.

(A and B) Quantification of messenger RNA MTTP and MTTP protein by real-time PCR and Western blot, respectively. (C) Microsomal triglyceride transfer activity was measured. (D and E) Quantification of messenger RNA and protein of apoB 100 by real-time PCR and Western blot, respectively. (F) ApoB protein secreted into media after a 16 hour incubation was measured by ELISA. (G) Newly synthesized cellular apoB was analyzed by immunoprecipitation and SDS-PAGE after a 20 minute label with [35S]methionine/cysteine in the presence or absence of ALLN. (H) Quantification of newly synthesized apoB protein with normalization to albumin after 20 min pulse with [35S]methionine/cysteine. (I) Pulse-chase of newly synthesized apoB for 30min, 60min, and 120min. Graph shows the percentage relative to initial amount of apoB at the end of the label. ±S.D. *P<0.05 **P<0.01. Values are means for three independent experiments.
Hepatosteatosis is commonly observed in both ABL and mouse models representing loss of MTTP (Chang et al., 1999; Raabe et al., 1999). We examined whether iPSC-derived hepatocytes from the MTTPR46G patient exhibited lipid accumulation as assessed by Oil Red O staining. A significant amount of large lipid droplets were observed in MTTPR46G hepatocytes whereas control cells were nearly free of large intracellular lipid droplets (Fig. 2A). This is supported by a quantitative increase in triglycerides and cholesterol in MTTPR46G iPSC-derived hepatocytes (Fig. 2B and C). Oleic acid (OA) treatment, which stimulates neutral lipid synthesis and secretion in hepatocytes, also resulted in a significant increase in cellular triglycerides and cholesterol in MTTPR46G derived hepatocytes compared to control cells (Fig. 2B and C). Labeling cells with [3H]-OA showed that the increase in TG from OA treatment was due to defective secretion from hepatocytes (Fig. 2D and E). Taken together, MTTPR46G iPSC-derived hepatocytes exhibit the hallmark features of ABL, including the absence of apoB secretion, excess intracellular lipid storage, reduced hepatic lipid secretion, and excess intracellular lipid storage, presumably due to loss of MTTP lipid transfer activity.
Figure 2. Accumulated lipid droplets in mutant MTTPR46G differentiated hepatocytes.

(A) Representative images of lipid droplets stained with Oil Red O. Yellow arrowheads indicate red positive lipid stains. Hematoxylin stains the nuclei blue. (B and C) Cellular triglyceride (TG) and total cholesterol (TC) content was determined by enzymatic assays and normalized to total cellular protein as measured by BCA assay. (D and E) Cells were incubated with [3H]-OA(5 uCi/ml)+0.8mM OA for 4 hours. Cellular and medium TG were extracted, separated by TLC, and quantified by liquid scintillation counting. TG counts are normalized to protein contents determined by Lowry protein assay. ± S.D. (n=4) *P<0.05 Values are means for three independent experiments. Scale bar: 400 μm
Correction of the C136G mutation in MTTP by gene editing rescues the ABL phenotype in MTTPR46G iPSC-derived hepatocytes
We corrected MTTP C136G mutation causing the R46G mutant using a CRISPR/Cas9 gene editing approach (Fig. 3A)(Ran et al., 2013). We obtained two clones bearing the corrected allele as confirmed by DNA sequencing (Fig. 3B). These corrected iPSC lines expressed similar levels of pluripotency markers as well as hepatocyte markers upon differentiation (Fig. 3C and D). Newly synthesized apoB was examined in the corrected lines using [35S] methionine/cysteine 2-hour labeling experiments. In contrast to the mutant MTTPR46G line, levels of cellular and secreted apoB were mostly or partially normalized by the gene correction, indicating that the MTTPR46G mutation caused the decrease in apoB stability in hepatocytes (Fig. 3E,F, and G). Moreover, Oil Red O staining revealed a decrease in lipid droplet accumulation in the differentiated hepatocytes from the corrected line in comparison to the MTTPR46G line (Fig. 3H).
Figure 3. Correction of C136G in MTTP rescues the ABL phenotype.

(A) Schematic strategy for correction of C136G in MTTP by CRISPR/Cas9. (B) iPSC from ABL patient was transfected with plasmids containing guide RNA and Cas9. Genomic DNA was extracted from GFP+ colonies and subjected to PCR amplification. Subsequent DpnII digestion was applied to identify the positively targeted clones. (C) The corrected iPSC lines were tested for expression of pluripotency markers by real-time PCR. (D) Expression of hepatic genes was analyzed by real-time PCR in hepatocytes derived from the corrected iPSC lines. (E–G) Amount of newly synthesized apoB in the cell or secreted in the medium was measured at the end of a 2-hour label with [35S]methionine/cysteine. F is a Western blot for apoB in the media. (H) Cellular lipid accumulation by Oil Red O staining following rescue of the C136G MTTP mutation by CRISPR/Cas9. Scale bar: 400 μm ± S.D. *P<0.05. **P<0.01 Values are means for three independent experiments.
Cardiomyocyte intrinsic defects due to expression of the MTTPR46G protein
While liver and intestine are the major sites of MTTP expression, human and mouse heart also expresses MTTP proteins (Nielsen et al., 1998). However, what role MTTP plays in cardiac lipid metabolism remains unclear. To examine the role of MTTP in human cardiomyocytes, we differentiated both control and MTTPR46G iPSCs into cardiomyocytes using a previously published protocol (Laflamme et al., 2007; Shiba et al., 2012). Synchronic beating from induced cardiomyocytes was observed in both control, MTTPR46G, and corrected lines by day 14 of differentiation. All cells expressed equivalent levels of cardiomyocyte markers, including MYH6, MYH7, MLC2a, and MLC2v (Fig. S3).
Similar levels of MTTP mRNA and MTTP protein were observed in control, MTTPR46G, and correction cardiomyocytes (Fig S4A and S4B). We next examined the effect of lipid loading on all cell lines. Interestingly, both OA and palmitic acid (PA) induced apoB transcription in control, MTTPR46G, and corrected cells (Fig. 4A). However, apoB secretion was only detectable in control cardiomyocytes and the gene corrected line, whereas MTTPR46G cells had little to no detectable apoB secretion after OA or PA treatment (Fig. 4B). OA and PA treatment also induced increased neutral lipid accumulation in MTTPR46G compared to control cardiomyocytes (Fig. 4C,D and S4C), which is further supported by a significant increase in TG synthesis in MTTPR46G derived cardiomyocytes (Fig. 4E). This also led to a decrease in secreted TGs in the MTTPR46G derived cardiomyocytes (Fig. 4F). While the amounts of apoB and TG secreted are dramatically lower in cardiomyocytes as compared to hepatocytes, there was still a significant decrease observed in MTTPR46G relative to control cardiomyocytes. In addition, the TG:CE ratio of lipids secreted from cardiomyocytes and hepatocytes differs, suggesting less TG-rich particles produced by cardiomyocytes (Fig. S4D). This is consistent with the previous findings that the apoB-containing lipoproteins secreted from mouse and human hearts are not TG-rich but rather have a density consistent with a more cholesterol rich LDL particle (Boren et al., 1998). Correction of the R46G mutation restored apoB and TG secretion and lipid accumulation to those of control cardiomyocyte levels (Fig. 4B–F). These results demonstrate that human cardiomyocytes require MTTP for secretion of apoB and lipid and in the setting of genetic MTTP deficiency are vulnerable to lipid accumulation.
Figure 4. Abolished apoB and elevated lipid storage upon fatty acids treatment in cardiomyocytes was rescued by correction of MTTPR46G.
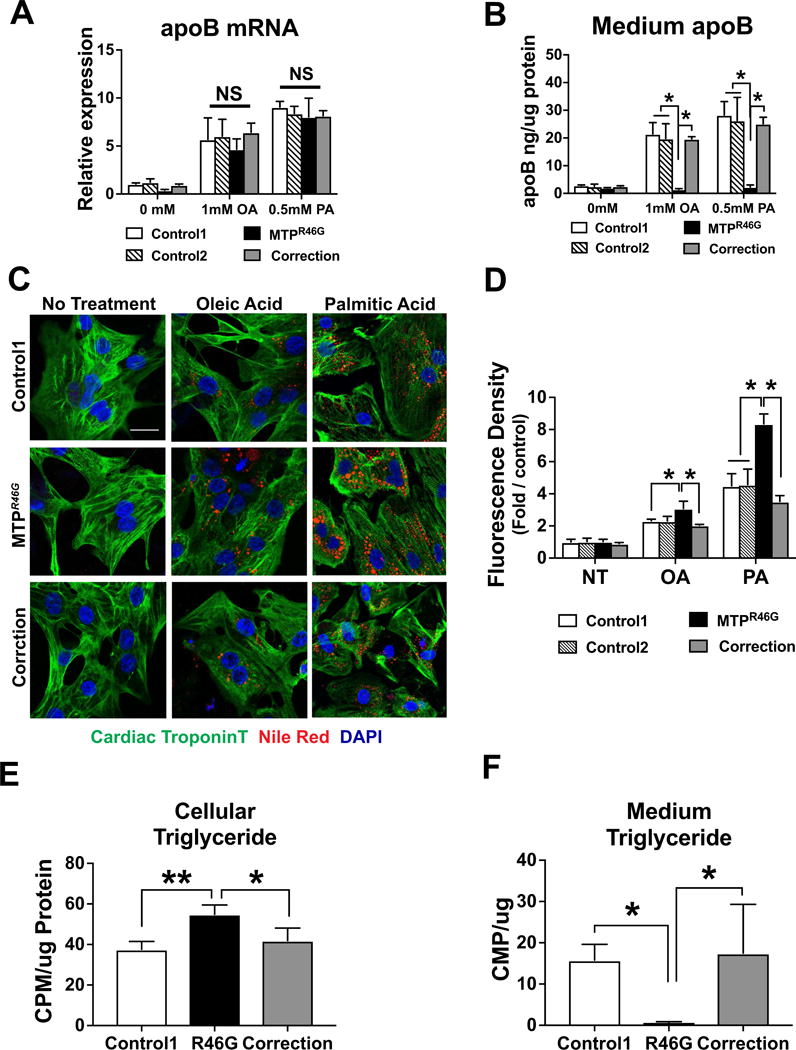
(A and B) ApoB transcript and medium apoB protein upon fatty acids treatment were quantified by real-time PCR and ELISA, respectively. Cells were incubated with Oleic Acid (OA: 1mM) or Palmitic Acid (PA: 0.5mM) for 48 hours. (C) Representative images of neutral lipid staining in cardiomyocytes. Cardiac Troponin T stains cardiac muscle fiber proteins as green. Nile Red stains lipid droplet as red. DAPI stains nuclei as blue. (D) Mean Fluorescence Intensity of Nile Red was analyzed by ImageJ. Fold relative to control is shown. (E and F) Newly synthesized cellular TG content is quantified in cardiomyocytes (E) and in the culture media (F) following a 24 hour label with [14C] oleic acid and lipid extraction. TG counts are normalized to protein contents determined by Lowry protein assay ± S.D. *P<0.05, Values are means for three independent experiments. Scale bar: 25 μm
Hypersensitivity of MTTPR46G cardiomyocytes to metabolic stresses
Altered metabolism of free fatty acids and lipid accumulation in the myocardium can cause myocardial dysfunction and cardiomyocyte apoptosis (Chiu et al., 2001; Christoffersen et al., 2003). Increased cardiac apoB secretion has been shown to ameliorate cardiac dysfunction in dietary and genetic mouse models of lipid overload (Bartels et al., 2009; Yokoyama et al., 2004). Therefore, we examined whether loss of MTTP function altered the response of mutant MTTPR46G cardiomyocytes to multiple metabolic stresses. MTTPR46G and control cells treated with sunitinib, a receptor tyrosine kinase inhibitor that has cardiac cytotoxicity (Force and Kolaja, 2011; Orphanos et al., 2009), exhibited a similar dose dependent increase in apoptosis (Fig. S5A and B). However, when sunitinib was added in the presence of PA to induce lipid synthesis, a significant increase in apoptosis, evaluated by TUNEL and cleaved caspase 3 staining, was noted in MTTPR46G cardiomyocytes relative to control cells (Fig. 5A–D). The increased apoptosis in MTTPR46G cardiomyocytes was normalized to control levels with correction of the MTTPR46G mutation (Fig. 5A–D). These results suggest an overload of intracellular lipid content reduces the tolerance towards the combined effects of multiple pharmacological stressors in MTTPR46G cells.
Figure 5. MTTPR46G cardiomyocytes are hypersensitive to metabolic stresses.

(A) Representative images showing sunitinib (15 μM) and PA (0.5mM) induced apoptosis as visualized by TUNEL positive cells (green). DAPI stains nuclei blue. (B) Percentage of sunitinib induced TUNEL positivity was quantified by cell counting using ImageJ (>1000 nuclei were counted per genotype). (C) Representative images showing expression of cleaved Caspase 3 in sunitinib and PA treated cardiomyocytes. Troponin T (green), Cleaved caspase 3 (red), and DAPI (blue). (D) Mean fluorescence density for cleaved Caspase 3 was quantified using ImageJ. Fold change relative to control is shown. (E) Quantification of TUNEL positivity in response to Hypoxia/Reoxygenation and PA. (F) Representative images showing expression of cleaved Caspase 3 in hypoxia/reoxygenation and PA treated cardiomyocytes. Troponin T (white), Cleaved caspase 3 (green), Nile Red (red), and DAPI (blue). Graph to the right shows mean fluorescence density for cleaved Caspase 3 was quantified using ImageJ. Fold change relative to control is shown. *P<0.05. Values are means for three independent experiments ± S.D. Scale bar: A=150 μm, C=40 μm.
We next tested cardiomyocytes stress tolerance with hypoxia followed by reoxygenation, another metabolic stressor that is used as an in vitro model of IR injury. Cells were subjected to PA, hypoxia/reoxygenation (H/R), or H/R+PA treatment. While H/R+PA triggered an apoptotic response in all cell lines, a more pronounced response was noted in MTTPR46G cells relative to controls (Fig. 5E and F). This differential sensitivity in MTTPR46G cardiomyocytes to H/R+PA was normalized in the gene corrected line (Fig. 5E and F). This suggests PA induced overload of intracellular lipid in cardiomyocytes sensitizes stress responses to multiple metabolic stresses. Expression of genes associated with cardiac dysfunction and failure was significantly higher in MTTPR46G than in control cardiomyocytes after treatment of sunitinib together with PA (Fig. 6A). However, correction of the MTTP C136G mutation normalized the expression of genes after PA treatment, suggesting a protective role of MTTP during lipid-induced stresses in cardiomyocytes. ANP and BNP expression was also significantly upregulated in MTTPR46G cells after hypoxic treatment, suggesting a protective role of MTTP in cardiomyocytes. Importantly, hypoxic stress was normalized by correction of the MTTP C136G mutation (Fig. 6B).
Figure 6. Increased expression of stress response genes in MTTPR46G cardiomyocytes.
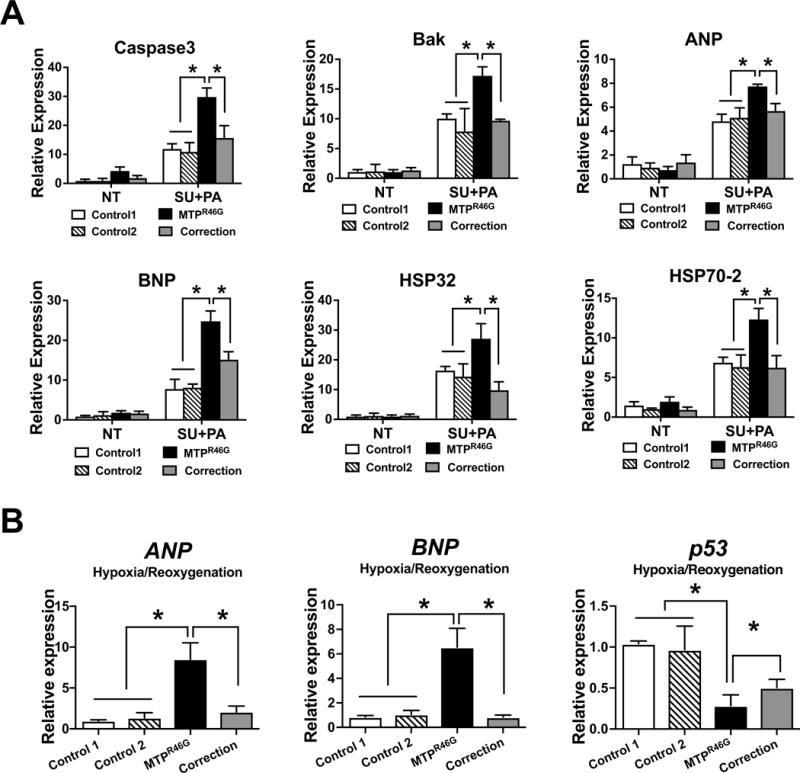
(A) Expression of stress associated genes, such as Caspase3, Caspase9, Bak, ANP, BNP, Hsp32, and HSP70-2 in response to sunitinib (SU) and PA by real-time PCR. (B) Expression of stress genes including Caspase3, p53, ANP, and BNP, in response to H/R and PA. ± S.D. *P<0.05. Values are means for three independent experiments.
DISCUSSION
The present study uses patient-specific iPSCs generated from a patient with ABL with a missense mutation (MTTPR46G) in both alleles of the MTTP gene. Differentiation of the iPSCs to hepatocytes produced the expected phenotype of abolished apoB production and accumulation of intracellular lipid, phenotypes that were corrected by genome editing of the causal MTTP mutation. Moreover, cardiomyocytes derived from the ABL iPSCs displayed impaired apoB secretion, lipid accumulation, and increased sensitivity to cellular stress. These results are consistent with a role for MTTP in promoting secretion of apoB in hepatocytes and cardiomyocytes and protecting cardiomyocytes from cellular stress.
A number of mutations in MTTP have been identified that result in the ABL phenotype (Miller et al., 2014; Shoulders et al., 1993; Walsh et al., 2015). Many of these mutations lead to premature stop codons or defective splicing and result in reduced or absent expression of a full length MTTP protein (Pons et al., 2011). Other missense mutations are not predicted to affect the triglyceride transfer function of MTTP and may act through different mechanisms to cause ABL phenotypes (Al-Shali et al., 2003; Ohashi et al., 2000; Wang and Hegele, 2000). While there are several mouse models to study the function of MTTP, none of these examine the effect of human missense mutations (Bartels et al., 2009; Chang et al., 1999; Liang et al., 2014; Raabe et al., 1999; Tietge et al., 1999). Our studies clearly establish that the missense mutation MTTPR46G abolishes MTTP activity and recapitulates the ABL phenotype, making the patient specific iPSCs in this report the human model for studying the cellular effects of MTTP deficiency.
MTTP plays a critical role in intracellular assembly of apoB-containing TG-rich lipoproteins in both liver and intestine (Young, 1990), The MTTP gene is expressed not only in the intestine and liver, but also in the heart (Berriot-Varoqueaux et al., 2000; Hussain et al., 2012). Expression and secretion of apoB lipoproteins has been demonstrated from human heart biopsies (Boren et al., 1998; Nielsen et al., 1999; Nielsen et al., 1998). However, the physiological role of this cardiac expression and secretion of lipoproteins remains to be determined. A role for MTTP in promoting secretion of apoB in the human heart has been previously proposed to protect the heart against toxic load of lipid accumulation (Bartels et al., 2009). Heart-specific MTTP knockout mice had elevated cardiac triglyceride levels (Bjorkegren et al., 2001). Although cardiac expression of MTTP is low (Aminoff et al., 2010), its expression increases in the ischemic myocardium. Interestingly, reduced myocardial expression of MTTP in hypoxic hearts is associated with increased myocardial lipid (Ledmyr et al., 2004; Nielsen et al., 2002). A genetic variant associated with reduced MTTP expression is associated in increased cardiac disease (Ledmyr et al., 2004). In addition, cardiomyopathy – arrhythmias, cardiomegaly, and cardiac failure has been described in ABL patients, though the patient in our study doesn’t present with obvious cardiomyopathy (Dische and Porro, 1970; Sobrevilla et al., 1964; Zamel et al., 2008). While the etiology of myopathy is unclear in these cases, it could be related to muscle weakness caused by vitamin E deficiency resulted from lack of absorption in intestine. Our studies reveal a cardiomyocyte intrinsic phenotype due to loss of MTTP activity, which may be critical in the setting of lipid overload. In particular, upon fatty acid treatment, apoB can be clearly detected in control cardiomyocytes and the conditioned media. Whereas, ABL cardiomyocytes had absent cellular apoB, lipid accumulated in the cell, and increased cellular triglycerides. This leads to increased sensitivity to cytotoxic stresses, ultimately resulting in increased apoptosis in lipid overloaded cardiomyocytes as well as increased sensitivity to hypoxia/rexoygenation. Together, our data suggest the low level secretion of lipidated apoB by cardiomyocytes serves a different physiological function—not bulk secretion of lipids but rather secretion of particular lipids. Furthermore, this pathway in cardiomyocytes may not be physiologically relevant except in times of stress and/or lipid overload. Importantly, the phenotypes we observed in both MTTPR46G hepatocytes and cardiomyocytes were rescued by CRISPR/Cas9 mediated gene correction. This strongly supports the contention that these phenotypes are due to the mutation in MTTP. Our studies provide new evidence to support a physiological role for MTTP expression in human cardiomyocytes to export apoB and excess lipid and protect the heart from stress.
Our studies reveal that coupling rigorous iPSC differentiation protocols for multiple cell lineages as well as gene correction allows for the discovery and analysis of new phenotypes caused by rare genetic mutations. Although our studies are limited to cells from one ABL patient, this human disease iPSC/CRISPR/Cas9 model allows for further resolution of the complex disease phenotype in ABL patients and favors for the development of therapies directed towards this rare genetic disorder as well as potentially other causes of cellular lipotoxicity.
EXPERIMENTAL PROCEDURES
Human Subjects
All human studies were approved by the University of Pennsylvania Human Subjects Research Institutional Review Board. The individuals were specifically recruited for this study and gave their informed written consent. The study was conducted at the Perelman School of Medicine at the University of Pennsylvania. Peripheral blood samples obtained from the subjects were used for general lipid measurements as well as generation of iPSC lines. See Table S1 for gender, age, and race of individuals used in this study.
Generation of subject-specific iPSCs and differentiation into hepatocytes/cardiomyocytes
Subject-specific PBMC-derived iPSCs were generated using Sendai viral vectors by the iPSC Core Facility at University of Pennsylvania as previously described (Yang et al., 2015; Yang et al., 2012). These cell lines have been deposited at WiCell Research Institute (http://www.wicell.org/home/stem-cell-lines/catalog-of-stem-cell-lines/collections/nhlbi-next-gen-rader.cmsx). The hepatocytes and cardiomyocytes were generated from iPSCs using standard protocols described previously (Cai et al., 2008; Laflamme et al., 2007; Mallanna and Duncan, 2013; Shiba et al., 2012).
ApoB labeling
Twenty days after initiation of differentiation, newly synthesized apoB in iPSC-derived hepatocytes was labeled and traced using methods described previously (Yamaguchi et al., 2006). In brief, immunoprecipitation of proteins from each sample was carried out using antibodies against apoB (Calbiochem) or albumin (Sigma). Cell lysates or conditioned medium was mixed with NET buffer (150 mM NaCl, 5 mM EDTA, 50 mM Tris (pH 7.4), 0.5% Triton X-100, and 0.1% SDS) and an excess amount of various antisera. The mixture was incubated at 4 °C for 1 h. Protein A-agarose was added to the reaction solution, and the incubation was continued for an additional 16 h. The beads were washed with NET buffer, and proteins were released with sample buffer (0.125 M Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, and 10% β-mercaptoethanol) by boiling for 5 min. Samples were resolved by gel electrophoresis followed by autoradiography. The total amount of apoB100 in the cell or media was normalized to the total amount of albumin protein in each sample.
ApoB pulse-chase assay
Newly synthesized apoB was traced using the method described previously (Yamaguchi et al., 2006). Briefly, iPSC-derived hepatocytes were preincubated in serum-free DMEM without methionine/cysteine containing 1.5% BSA with or without 40ug/ml ALLN (Calpain Inhibitor 1, Sigma) for 1 h and then labeled with DMEM without methionine/cysteine containing 1.5% BSA with or without ALLN and 200 μCi/ml [35S] methionine/cysteine for 20 min. After being washed, cells were incubated in serum-free DMEM plus 1.5% BSA containing 10 mM methionine and 3 mM cysteine for 10 or 120 min. The medium was collected and cells were lysed at 10, 40, 70, or 130 min. In other experiments, hepatocytes were preincubated for 1 h and labeled with 150 μCi/ml [35S]methionine/cysteine for 2 hours after which media was collected and cells were lysed. The lysis buffer contained 62.5 m M sucrose, 0.5% sodium deoxycholate, 0.5% Triton X-100, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 50 μg/ml leupeptin, 50 μg/ml pepstatin A, and 30 μl/ml protease inhibitor mixture (1 mM benzamidine, 5 mM EDTA, 100 units/ml aprotinin, and 10 mM HEPES (pH 8.0). Conditioned medium was mixed with protease inhibitor mixture and 0.86 mM freshly made phenylmethylsulfonyl fluoride. Cell lysates and conditioned medium were used for immunoprecipitations.
TG and TC measurement
Total TG and cholesterol contents were measured by enzymatic assays and normalized to protein as measured by BCA assay (Bi et al., 2014). Briefly, iPSC-derived hepatocytes and cardiomyocytes were pretreated with or without OA or PA for 24 hours. Cells were washed with PBS and pellets were obtained from centrifugation. Cell membrane was removed by sonication in RIPA buffer. Total TG and cholesterol contents were determined by enzymatic assays and normalized to protein as measured by BCA assay (Kohan et al., 2012). To measure newly synthesized triglycerides, cells were preincubated with [3H]-OA (5 uCi/ml) in the presence of OA (0.8mM) for 4 hours. To measure radiolabeled triglyceride, lipids were extracted from medium and cells, fractionated by TLC, and quantified by scintillation spectroscopy. TG counts in cell and medium were normalized to total cellular protein (Chung et al., 2010)
MTTP lipid transfer activity assay
MTTP lipid transfer activity in iPSC-derived hepatocytes was determined using method described previously (Athar et al., 2004). Details can be found in Supplementary Materials and Methods.
Genome Editing by CRISPR/Cas9
Precise gene correction was performed by CRISPR/Cas9 following the published protocol (Ran et al., 2013). Briefly, sgRNAs were designed using the MIT CRISPR Design Tool and cloned into the plasmid PX458 (Addgene plasmid ID: 48138). ssODNs were suspended in sterile H2O and transfected into iPSCs using 4D-Nucleofector (Lonza) together with PX458-sgRNA. The nucleofected iPSCs were plated with mTeSR1 supplemented with 2 μM Thiazovivin (Sigma). After 24 hours, GFP+ cells were sorted by FACSJazz (BD). Single GFP+ iPSCs were maintained in mTeSR1 and allowed to grow into colonies until manually picking for DNA extraction with Quick Extract - DNA Extraction Solution (Epicentre). Then DNA was subjected to PCR amplification around the cutting site and subsequent DpnII (NEB) digest to analyze successfully edited clones.
Lipid droplets staining
Oil Red O (Sigma O0625) and Nile Red (ThermoFisher) were used to label lipid droplets in iPSC-derived hepatocytes and cardiomyoyctes according to manufacturer’s instructions. IPSC-derived hepatocytes were fixed with 4% paraformaldehyde for 15 min, followed by incubation with distilled water and subsequently with 60% isopropanol for 2 min and stained with a filtered 0.35% Oil Red O (Sigma) solution in 60% isopropanol for 10 min at room temperature. Then cells were washed with sterile water and stained with Hematoxylin solution for 1 min at room temperature. Images were analyzed under a light microscope. Lipid droplets appear red and nuclei appear blue. To stain neutral lipids in iPSC-derived cardiomyocytes, cells at 20 days were treated with Oleic Acid or Palmitic Acid separately for 24 hours. Fixation and permeabilization were performed using the same method as described above. Cardiomyocytes were co-stained with cardiac TroponinT (ThermoFisher) and Nile Red (ThermoFisher), followed with Alexa 488 (Invitrogen). Images were acquired from Confocal Microscopy (Leica TCS SP8). Cardiac Troponin T protein stains green, lipid droplets appear red, nuclei stains blue.
TUNEL Assay
Cardiomyocytes derived from iPSCs were treated with Sunitinib (0, 1.5uM,15uM) with or without palmitate (0.5mM) for 18 hours. Cells were then fixed to determine apoptosis using In Situ Cell Death Detection Kit (Roche, 11684795910) according to the manufacturer’s protocol. Briefly, cells were fixed and permeabilized using the method described above. Cells were labeled with TUNEL enzyme mixture in a humidified environment at 37°C for 1h. DAPI solution was applied to stain total nuclei. Fluorescent microscopy was used to acquire the images. Apoptotic cell nuclei stain green. Total nuclei appear blue. Both nuclei were counted and calculated as the percentage of apoptotic index. Over 500 cells were counted in test samples.
Cleaved Caspase 3 assay
Cardiomyocytes derived from iPSCs were treated with Sunitinib (15uM) with or without palmitate (0.5mM) for 18 hours. Cells were then fixed and permeabalized for cleaved caspase 3. Cardiomyocytes were co-stained with cleaved caspase3 (Cell signaling) and cardiac TroponinT (ThermoFisher), followed by a 2nd antibody staining with Alexa 488 (Invitrogen) and Alexa 555. Images were acquired with Leica Microscope. Cardiac Troponin T protein stains green, cleaved caspase3 stains red, and nuclei stains blue.
Hypoxia/Reoxygenation stress assay
iPSC-derived cardiomyoyctes were maintained in serum free medium with or without palmitate followed by exposure to hypoxia (94% N2, 5%CO2, and 1%O2) for 18 hours. Cells were then moved back to an environment with 20% O2 (5%CO2) for reoxygenation. After 24 hours, cells were fixed for analysis of TUNEL or cleaved caspase 3 (Portal et al., 2013).
ELISA
Albumin (Bethyl Laboratories, Inc.) and apoB (Mabtech) in the medium were determined using commercial ELISA Kits.
Statistical Analysis
Data were analyzed for statistical significance using a two-tailed unpaired Student’s test (Graphpad Prism). P Values less than 0.05 were considered statistically significant. All quantitative data are presented as mean ±SEM.
Supplementary Material
Acknowledgments
We thank the study participants and administrative personnel at the Smilow Center for Translational Research. We also thank the support from the Induced Pluripotent Stem Cell Core Facility of the Institute for Regenerative Medicine at the University of Pennsylvania. This work was supported by NIH grant U01-HG006398. X.B is supported by AHA Postdoctoral Fellowship 15POST25160019.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
E.E.M and D.J. R conceived and designed the study. Y.L, D.M.C, and X.B designed and performed the experiments and analyzed the data. Y.L, D.J.R and E.E.M wrote the manuscript. Other authors also directly participated in the planning, execution, or analysis of the study. All authors read and approved the final version of the submitted manuscript.
References
- Al-Shali K, Wang J, Rosen F, Hegele RA. Ileal adenocarcinoma in a mild phenotype of abetalipoproteinemia. Clin Genet. 2003;63:135–138. doi: 10.1046/j.0009-9163.2002.00175.x. [DOI] [PubMed] [Google Scholar]
- Aminoff A, Ledmyr H, Thulin P, Lundell K, Nunez L, Strandhagen E, Murphy C, Lidberg U, Westerbacka J, Franco-Cereceda A, et al. Allele-specific regulation of MTTP expression influences the risk of ischemic heart disease. J Lipid Res. 2010;51:103–111. doi: 10.1194/jlr.M900195-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athar H, Iqbal J, Jiang XC, Hussain MM. A simple, rapid, and sensitive fluorescence assay for microsomal triglyceride transfer protein. J Lipid Res. 2004;45:764–772. doi: 10.1194/jlr.D300026-JLR200. [DOI] [PubMed] [Google Scholar]
- Bartels ED, Nielsen JM, Hellgren LI, Ploug T, Nielsen LB. Cardiac expression of microsomal triglyceride transfer protein is increased in obesity and serves to attenuate cardiac triglyceride accumulation. PLoS One. 2009;4:e5300. doi: 10.1371/journal.pone.0005300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, Wetterau JR. The role of the microsomal triglygeride transfer protein in abetalipoproteinemia. Annu Rev Nutr. 2000;20:663–697. doi: 10.1146/annurev.nutr.20.1.663. [DOI] [PubMed] [Google Scholar]
- Bi X, Zhu X, Gao C, Shewale S, Cao Q, Liu M, Boudyguina E, Gebre AK, Wilson MD, Brown AL, et al. Myeloid cell-specific ATP-binding cassette transporter A1 deletion has minimal impact on atherogenesis in atherogenic diet-fed low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2014;34:1888–1899. doi: 10.1161/ATVBAHA.114.303791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkegren J, Veniant M, Kim SK, Withycombe SK, Wood PA, Hellerstein MK, Neese RA, Young SG. Lipoprotein secretion and triglyceride stores in the heart. J Biol Chem. 2001;276:38511–38517. doi: 10.1074/jbc.M106839200. [DOI] [PubMed] [Google Scholar]
- Boren J, Veniant MM, Young SG. Apo B100-containing lipoproteins are secreted by the heart. J Clin Invest. 1998;101:1197–1202. doi: 10.1172/JCI1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, DeLaForest A, Fisher J, Urick A, Wagner T, Twaroski K, Cayo M, Nagaoka M, Duncan SA. Protocol for directed differentiation of human pluripotent stem cells toward a hepatocyte fate. StemBook; Cambridge (MA): 2008. [PubMed] [Google Scholar]
- Chang BH, Liao W, Li L, Nakamuta M, Mack D, Chan L. Liver-specific inactivation of the abetalipoproteinemia gene completely abrogates very low density lipoprotein/low density lipoprotein production in a viable conditional knockout mouse. J Biol Chem. 1999;274:6051–6055. doi: 10.1074/jbc.274.10.6051. [DOI] [PubMed] [Google Scholar]
- Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144:3483–3490. doi: 10.1210/en.2003-0242. [DOI] [PubMed] [Google Scholar]
- Chung S, Timmins JM, Duong M, Degirolamo C, Rong S, Sawyer JK, Singaraja RR, Hayden MR, Maeda N, Rudel LL, et al. Targeted deletion of hepatocyte ABCA1 leads to very low density lipoprotein triglyceride overproduction and low density lipoprotein hypercatabolism. J Biol Chem. 2010;285:12197–12209. doi: 10.1074/jbc.M109.096933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dische MR, Porro RS. The cardiac lesions in Bassen-Kornzweig syndrome. Report of a case, with autopsy findings. Am J Med. 1970;49:568–571. doi: 10.1016/s0002-9343(70)80053-5. [DOI] [PubMed] [Google Scholar]
- Fisher EA, Zhou M, Mitchell DM, Wu X, Omura S, Wang H, Goldberg AL, Ginsberg HN. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J Biol Chem. 1997;272:20427–20434. doi: 10.1074/jbc.272.33.20427. [DOI] [PubMed] [Google Scholar]
- Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov. 2011;10:111–126. doi: 10.1038/nrd3252. [DOI] [PubMed] [Google Scholar]
- Gregg RE, Wetterau JR. The molecular basis of abetalipoproteinemia. Curr Opin Lipidol. 1994;5:81–86. doi: 10.1097/00041433-199404000-00003. [DOI] [PubMed] [Google Scholar]
- Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. Multiple functions of microsomal triglyceride transfer protein. Nutr Metab (Lond) 2012;9:14. doi: 10.1186/1743-7075-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan AB, Wang F, Li X, Bradshaw S, Yang Q, Caldwell JL, Bullock TM, Tso P. Apolipoprotein A-IV regulates chylomicron metabolism-mechanism and function. Am J Physiol Gastrointest Liver Physiol. 2012;302:G628–636. doi: 10.1152/ajpgi.00225.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- Ledmyr H, McMahon AD, Ehrenborg E, Nielsen LB, Neville M, Lithell H, MacFarlane PW, Packard CJ, Karpe F, executive, W The microsomal triglyceride transfer protein gene-493T variant lowers cholesterol but increases the risk of coronary heart disease. Circulation. 2004;109:2279–2284. doi: 10.1161/01.CIR.0000130070.96758.7b. [DOI] [PubMed] [Google Scholar]
- Liang Z, Xie Y, Dominguez JA, Breed ER, Yoseph BP, Burd EM, Farris AB, Davidson NO, Coopersmith CM. Intestine-specific deletion of microsomal triglyceride transfer protein increases mortality in aged mice. PLoS One. 2014;9:e101828. doi: 10.1371/journal.pone.0101828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallanna SK, Duncan SA. Differentiation of hepatocytes from pluripotent stem cells. Curr Protoc Stem Cell Biol. 2013;26:4. doi: 10.1002/9780470151808.sc01g04s26. Unit 1G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Burnett JR, Leonis MA, McKnight CJ, van Bockxmeer FM, Hooper AJ. Novel missense MTTP gene mutations causing abetalipoproteinemia. Biochim Biophys Acta. 2014;1842:1548–1554. doi: 10.1016/j.bbalip.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Nielsen LB, Perko M, Arendrup H, Andersen CB. Microsomal triglyceride transfer protein gene expression and triglyceride accumulation in hypoxic human hearts. Arterioscler Thromb Vasc Biol. 2002;22:1489–1494. doi: 10.1161/01.atv.0000030199.06252.26. [DOI] [PubMed] [Google Scholar]
- Nielsen LB, Sullivan M, Vanni-Reyes T, Goldberg IJ, Young SG. The DNA sequences required for apolipoprotein B expression in the heart are distinct from those required for expression in the intestine. J Mol Cell Cardiol. 1999;31:695–703. doi: 10.1006/jmcc.1998.0918. [DOI] [PubMed] [Google Scholar]
- Nielsen LB, Veniant M, Boren J, Raabe M, Wong JS, Tam C, Flynn L, Vanni-Reyes T, Gunn MD, Goldberg IJ, et al. Genes for apolipoprotein B and microsomal triglyceride transfer protein are expressed in the heart: evidence that the heart has the capacity to synthesize and secrete lipoproteins. Circulation. 1998;98:13–16. doi: 10.1161/01.cir.98.1.13. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Ishibashi S, Osuga J, Tozawa R, Harada K, Yahagi N, Shionoiri F, Iizuka Y, Tamura Y, Nagai R, et al. Novel mutations in the microsomal triglyceride transfer protein gene causing abetalipoproteinemia. J Lipid Res. 2000;41:1199–1204. [PubMed] [Google Scholar]
- Orphanos GS, Ioannidis GN, Ardavanis AG. Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol. 2009;48:964–970. doi: 10.1080/02841860903229124. [DOI] [PubMed] [Google Scholar]
- Pons V, Rolland C, Nauze M, Danjoux M, Gaibelet G, Durandy A, Sassolas A, Levy E, Terce F, Collet X, et al. A severe form of abetalipoproteinemia caused by new splicing mutations of microsomal triglyceride transfer protein (MTTP) Hum Mutat. 2011;32:751–759. doi: 10.1002/humu.21494. [DOI] [PubMed] [Google Scholar]
- Portal L, Martin V, Assaly R, d’Anglemont de Tassigny A, Michineau S, Berdeaux A, Ghaleh B, Pons S. A model of hypoxia-reoxygenation on isolated adult mouse cardiomyocytes: characterization, comparison with ischemia-reperfusion, and application to the cardioprotective effect of regular treadmill exercise. J Cardiovasc Pharmacol Ther. 2013;18:367–375. doi: 10.1177/1074248412475158. [DOI] [PubMed] [Google Scholar]
- Raabe M, Veniant MM, Sullivan MA, Zlot CH, Bjorkegren J, Nielsen LB, Wong JS, Hamilton RL, Young SG. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest. 1999;103:1287–1298. doi: 10.1172/JCI6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata N, Dixon JL. Ubiquitin-proteasome-dependent degradation of apolipoprotein B100 in vitro. Biochim Biophys Acta. 1999;1437:71–79. doi: 10.1016/s1388-1981(98)00006-7. [DOI] [PubMed] [Google Scholar]
- Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J, Palpant NJ, Gantz J, Moyes KW, Reinecke H, et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012;489:322–325. doi: 10.1038/nature11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders CC, Brett DJ, Bayliss JD, Narcisi TM, Jarmuz A, Grantham TT, Leoni PR, Bhattacharya S, Pease RJ, Cullen PM, et al. Abetalipoproteinemia is caused by defects of the gene encoding the 97 kDa subunit of a microsomal triglyceride transfer protein. Hum Mol Genet. 1993;2:2109–2116. doi: 10.1093/hmg/2.12.2109. [DOI] [PubMed] [Google Scholar]
- Si-Tayeb K, Noto FK, Sepac A, Sedlic F, Bosnjak ZJ, Lough JW, Duncan SA. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev Biol. 2010;10:81. doi: 10.1186/1471-213X-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrevilla LA, Goodman ML, Kane CA. Demyelinating Central Nervous System Disease, Macular Atrophy and Acanthocytosis (Bassen-Kornzweig Syndrome) Am J Med. 1964;37:821–828. doi: 10.1016/0002-9343(64)90030-0. [DOI] [PubMed] [Google Scholar]
- Tietge UJ, Bakillah A, Maugeais C, Tsukamoto K, Hussain M, Rader DJ. Hepatic overexpression of microsomal triglyceride transfer protein (MTTP) results in increased in vivo secretion of VLDL triglycerides and apolipoprotein B. J Lipid Res. 1999;40:2134–2139. [PubMed] [Google Scholar]
- Walsh MT, Iqbal J, Josekutty J, Soh J, Di Leo E, Ozaydin E, Gunduz M, Tarugi P, Hussain MM. Novel Abetalipoproteinemia Missense Mutation Highlights the Importance of the N-Terminal beta-Barrel in Microsomal Triglyceride Transfer Protein Function. Circ Cardiovasc Genet. 2015;8:677–687. doi: 10.1161/CIRCGENETICS.115.001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hegele RA. Microsomal triglyceride transfer protein (MTTP) gene mutations in Canadian subjects with abetalipoproteinemia. Hum Mutat. 2000;15:294–295. doi: 10.1002/(SICI)1098-1004(200003)15:3<294::AID-HUMU14>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Wetterau JR, Combs KA, McLean LR, Spinner SN, Aggerbeck LP. Protein disulfide isomerase appears necessary to maintain the catalytically active structure of the microsomal triglyceride transfer protein. Biochemistry. 1991;30:9728–9735. doi: 10.1021/bi00104a023. [DOI] [PubMed] [Google Scholar]
- Wetterau JR, Combs KA, Spinner SN, Joiner BJ. Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex. J Biol Chem. 1990;265:9800–9807. [PubMed] [Google Scholar]
- Yamaguchi J, Conlon DM, Liang JJ, Fisher EA, Ginsberg HN. Translocation efficiency of apolipoprotein B is determined by the presence of beta-sheet domains, not pause transfer sequences. J Biol Chem. 2006;281:27063–27071. doi: 10.1074/jbc.M606809200. [DOI] [PubMed] [Google Scholar]
- Yang W, Liu Y, Slovik KJ, Wu JC, Duncan SA, Rader DJ, Morrisey EE. Generation of iPSCs as a Pooled Culture Using Magnetic Activated Cell Sorting of Newly Reprogrammed Cells. PLoS One. 2015;10:e0134995. doi: 10.1371/journal.pone.0134995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Mills JA, Sullivan S, Liu Y, French DL, Gadue P. iPSC Reprogramming from Human Peripheral Blood Using Sendai Virus Mediated Gene Transfer. StemBook; Cambridge (MA): 2012. [PubMed] [Google Scholar]
- Yeung SJ, Chen SH, Chan L. Ubiquitin-proteasome pathway mediates intracellular degradation of apolipoprotein B. Biochemistry. 1996;35:13843–13848. doi: 10.1021/bi9618777. [DOI] [PubMed] [Google Scholar]
- Yokoyama M, Yagyu H, Hu Y, Seo T, Hirata K, Homma S, Goldberg IJ. Apolipoprotein B production reduces lipotoxic cardiomyopathy: studies in heart-specific lipoprotein lipase transgenic mouse. J Biol Chem. 2004;279:4204–4211. doi: 10.1074/jbc.M311995200. [DOI] [PubMed] [Google Scholar]
- Young SG. Recent progress in understanding apolipoprotein B. Circulation. 1990;82:1574–1594. doi: 10.1161/01.cir.82.5.1574. [DOI] [PubMed] [Google Scholar]
- Zamel R, Khan R, Pollex RL, Hegele RA. Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis. 2008;3:19. doi: 10.1186/1750-1172-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeissig S, Dougan SK, Barral DC, Junker Y, Chen Z, Kaser A, Ho M, Mandel H, McIntyre A, Kennedy SM, et al. Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function. J Clin Invest. 2010;120:2889–2899. doi: 10.1172/JCI42703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.