

Abstract
The ubiquitination of proteins can signal their degradation, modify their activity or target them to specific membranes or cellular organelles. Here, we show that monoubiquitination regulates the plasma membrane abundance and function of the potassium channel, ROMK. Immunoprecipitation of proteins obtained from renal cortex and outer medulla with ROMK antibody revealed that this channel was monoubiquitinated. To determine the ubiquitin binding site on ROMK1, all intracellular lysine (Lys) residues of ROMK1 were individually mutated to arginine (Arg), and a two-electrode voltage clamp was used to measure the ROMK1 channel activity in Xenopus oocytes. ROMK1 channel activity increased from 8.1 to 27.2 μA only when Lys-22 was mutated to Arg. Furthermore, Western blotting failed to detect the ubiquitinated ROMK1 in oocytes injected with R1K22R. Patch-clamp experiments showed that biophysical properties of R1K22R were identical to those of wild-type ROMK1. Although total protein expression levels of GFP-ROMK1 and GFP-R1K22R in oocytes were similar, confocal microscopy showed that the surface fluorescence intensity in oocytes injected with GFP-R1K22R was higher than that of GFP-ROMK1. In addition, biotin labeling of ROMK1 and R1K22R proteins expressed in HEK293 cells showed increased surface expression of the Lys-22 mutant channel. Finally, expression of R1K22R in COS7 cells significantly stimulated the surface expression of ROMK1. We conclude that ROMK1 can be monoubiquitinated and that Lys-22 is an ubiquitin-binding site. Thus, monoubiquitination of ROMK1 regulates channel activity by reducing the surface expression of channel protein. This finding implicates the linking of a single ubiquitin molecule to channels as an important posttranslational regulatory signal.
Keywords: ubiquitination, inwardly rectifying K channel (Kir.11), K recycling, collecting duct, thick ascending limb
ROMK (KCNJ1) channel is an inwardly rectifying K channel located in the apical membrane of the thick ascending limb (TAL) and of the cortical collecting duct (CCD) (1). ROMK channels in the TAL are responsible for K recycling across the apical membrane and K recycling is essential for maintaining normal function of Na/Cl/K cotransporter (2). The importance of ROMK in K recycling in the TAL has been best demonstrated by the finding that loss-function mutations in ROMK cause salt wasting and metabolic alkalosis (Barter's syndrome) (3). ROMK is also responsible for K secretion in the CCD. Although the Ca2+-dependent maxi K channel has been shown to be involved in K secretion when urinary flow rate in the CCD is high (4, 5), ROMK channels play a principal role in K secretion under normal tubule flow (1, 2, 6).
ROMK channel activity is regulated by hormones and dietary K intake by modulating the number of channels in the apical membrane (1, 7, 8). We and others have demonstrated that stimulation of V2 receptor with vasopressin or an increase in dietary K content increases the apical ROMK channel number in the CCD (9, 10). In contrast, low K intake decreases the apical ROMK channel number in the CCD. Furthermore, we have shown that decreases in ROMK channel activity induced by low K intake are the result of stimulating endocytosis (11). Although the mechanism of ROMK internalization is not completely understood, it is possible that ROMK channels are internalized by a clathrin-dependent mechanism (12, 13). Recently, ubiquitination has been shown to be involved in the regulation of endocytosis by attaching one or two ubiquitin molecules to lysine residue of substrate protein (14). Given the importance of endocytosis in regulating ROMK channel activity and that ROMK channels have 20 lysine residues in the intracellular segment, we examined the possibility that the monoubiquitination is involved in the regulation of ROMK trafficking.
Materials and Methods
Tissue Preparation. The renal cortex and outer medulla were separated under a dissecting microscope and suspended in RIPA solution (1:8 ratio, wt/vol) containing 50 mM Tris·HCl (pH 7.4), 10 mM NaCl, 1% Nonidet P-40, 1% Triton X-100, 0.1% SDS, 1 mM sodium molybdate, 1 mM para-nitrophenyl-phosphate, and 1 mM EDTA. For every 125-mg tissue sample, we added a 25-μl mixture of protease and phosphatase inhibitors containing aprotinin (1 μg/ml), leupeptin (1 μg/ml), pepstatin A (1 μg/ml), sodium vanadate (Na3VO4) (1.5 mM), and sodium fluoride (1 mM). The samples were left on ice for 15 min and homogenized with a mortar and pestle. The protein concentrations were measured twice by using the Pierce BSA protein assay. The homogenized tissue samples were incubated in the presence of DNase (5 μg/ml) and rabbit IgG serum at 4°C for 60 min. The mixture was centrifuged at 500 × g for 10 min at 4°C and the resultant supernatant was collected.
Transfection of HEK293 and COS7 Cells. HEK293 and COS7 cells were plated in 35-mm dishes and transfected with ROMK channels or other interested cDNA by using 7 μl of LT1 reagent (PanVera, Madison, WI) according to instructions provided by the manufacturer. The experiments were carried out 2 days after the transfection. The successful rate for the cell transfection was usually between 60 and 70%.
Immunoprecipitation with ROMK Antibody Cross-Linked to the Immobilized Protein G. We used seize the X Immunoprecipitation kit (Pierce) to perform immunoprecipitation. To attach the ROMK antibody (10 μg) to the immobilized protein G, the column containing antibody and protein G was rotated for 30 min followed by centrifuging and washing with 0.5 ml of binding/wash buffer three times. To crosslink the antibody, 2 mg of disuccinimidyl suberate (DSS) was dissolved in 80 μl of DMSO, and 12.5 μl of DSS containing solution was added to the column containing ROMK antibody and protein G. The column was equilibrated by gently mixing for 60 min at room temperature followed by centrifuge and washing three times by adding 500 μl of ImmunoPure Elution buffer provided in the kit. The column was held upside-down briefly, centrifuged, and washed with 500 μl of binding/wash buffer twice. After preparation of the column, the lysate was diluted with binding/wash buffer in 1:1 ratio and added to the column. The mixture was gently rotated for 1 h at room temperature, centrifuged, and washed three times. The immunoprecipitated ROMK protein was eluted by adding 50 μl of ImmunoPure Elution buffer. The sample was mixed and centrifuged, and the resultant flow was collected to harvest the precipitated ROMK protein. The advantage of this procedure is that it removes the heavy chain of the antibody.
Immunoprecipitation and Western Blot. The corresponding antibody was added to the protein samples (500 μg) harvested from oocytes or cell cultures with a ratio of (4 μg per 1 mg of protein). The mixture was gently rotated at 4°C overnight, followed by incubation with 25 μl of protein A/G plus agarose (Santa Cruz Biotechnology) for an additional 2 h at 4°C. The tube containing the mixture was centrifuged at 500 × g, and the agarose bead pellet was mixed with 25 μlof2× SDS sample buffer containing 4% SDS, 100 mM Tris·HCl (pH 6.8), 20% glycerol, 200 mM DTT, and 0.2% bromophenol blue. After boiling the sample for 5 min, we loaded the supernatant to separate the proteins by electrophoresis on 10% SDS-polyacrylamide gels and transferred it to the nitrocellulose membrane. The membranes were blocked with 5% nonfat dry milk in TBS, rinsed, and washed with 0.05% Tween 20/TBS buffer. We used enhanced chemiluminescence (Amersham Pharmacia) to detect the protein bands, and the intensity of the corresponding band was determined with alpha digidoc 1000 (Alpha Innotech, San Leandro, CA).
Biotinylation. Changes in surface ROMK were quantitated by labeling the cells with cell impermeant Sulfo-NHS-biotin (Pierce) according to the protocol provided by the manufacturer. After biotinylation, the cells were washed twice with PBS and trypsinized with trypsin-EDTA. They were pelleted by centrifugation for 5 min at 7,000 × g, washed twice with PBS, and lysed with cold RIPA buffer (1× PBS/1% Igepal CA-630/0.1% SDS/0.5% deoxycholate) supplemented with 1 mM sodium molybdate, 1 mM sodium fluoride, 1 μΜ PMSF, and 100 μl of protease inhibitor mixture (Sigma) per ml of lysis buffer. Aliquots of lysates containing equal amounts of protein were immunoprecipitated overnight with 1 μg of anti-GFP (Clontech) monoclonal antibody and 20 μl of protein A/G agarose. The biotin-labeled GFP-ROMK1 proteins were detected by using NeutrAvidin horseradish peroxidase (HRP) (Pierce). Changes in biotin-labeled surface ROMK proteins were normalized with the corresponding total ROMK1 protein.
Preparation of Xenopus Oocytes. Xenopus laevis females were obtained from NASCO (Fort Atkinson, WI). The method for obtaining oocytes has been described (15) Viable oocytes were selected for injection with different cRNA. The oocytes were incubated at 19°C in a 66% DMEM/F12 medium with freshly added 2.5 mM sodium pyruvate and 50 μg/ml gentamycin. Experiments were performed on days 1 and 2 after injection with a two-electrode voltage clamp.
Two-Electrode Whole Cell Voltage-Clamping. A Warner oocyte clamp OC-725C was used to measure the whole cell K current. Voltage and current microelectrodes were filled with 1,000 mM KCl and had resistance of <2MΩ. The current was recorded on a chart recorder (Gould TA240). To exclude the leaky current, 2mMBa2+ was used to determine the Ba2+-sensitive K+ current.
Confocal Microscope. Surface fluorescence detected by confocal microscopy at the equatorial plane of oocytes expressing GFP-tagged ROMK correlates with channel activity and has been used by us to assess channel expression in the plasma membrane (16). Briefly, GFP fluorescence was excited at 488 nM with an argon laser beam and viewed with an inverted Olympus FV300 confocal system equipped with a ×60 oil lens. All images were acquired and processed with identical parameters.
Purification of ROMK1 Protein. HEK 293 cells were transfected with hemagglutinin–ubiquitin and His-ROMK1 and were lysed 24 h after transfection. The cell lysate was centrifuged and the resultant supernatant was collected. The sample was added to 5% Ni2+-nitrilotriacetic acid magnetic agarose beads (Qiagen, Valencia, CA) containing solution in 1:0.2 ratio and mixed gently for 30 min at room temperature. The tube containing beads was placed on a magnetic separator to remove the fluid inside the tube. The beads were rinsed several times with 500 μl of wash buffer (50 mM NaH2PO4/300 mM NaCl/20 mM imidazole, pH 8.0). Finally, 100 μl of elution buffer (50 mM NaH2PO4/300 mM NaCl/250 mM imidazole/0.005% Tween 20, pH 8.0) was added to the tube to harvest His-tagged ROMK channels.
Patch Clamping. An Axon200A patch-clamp amplifier was used to record channel current. The current was low-pass filtered at 1 KHz by an eight-pole Bessel filter (902LPF, Frequency Devices, Haverhill, MA) and digitized by an Axon interface (Digidata 1200). Data were analyzed by using the pclamp software system 7 (Axon). Channel activity defined as NPo was calculated from data samples of 60-s duration in the steady state as follows:
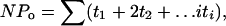 |
[1] |
where ti is the fractional open time spent at each of the observed current levels.
The pipette solution for the patch clamp study contained 140 mM KCl, 1.8 mM MgCl2, and 10 mM Hepes (pH 7.4). The bath solution was composed of 140 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1.8 mM MgCl2, 5 mM glucose, and 10 mM Hepes (pH 7.4).
Immunocytochemistry. Cos7 cells transfected with pEYFP-Mem and ROMK1/mutants were washed with PBS (pH 7.2) and fixed in 4% polyformaldehyde in PBS for 15 min at room temperature. The cell membrane was permeabilized in 0.4% Triton in PBS for 30 min and the slide was blocked in 2% goat serum in PBS for 1 h, followed by 1-h incubation with 1:100 ROMK antibody at room temperature.
Materials and Statistics. Site-directed mutagenesis was carried out to generate ROMK1 mutants, and each mutation was confirmed by nucleotide sequencing. We used T7 RNA polymerase to synthesize cRNA of ROMK1 and ROMK1 mutants. ROMK antibody was obtained from Alomone (Jerusalem), whereas ubiquitin antibody was purchased from Covance (Berkeley, CA) and Santa Cruz Biotechnology. Also, antibody for His-probe and Flag-tag antibody were from Santa Cruz Biotechnology and Sigma, respectively. The GFP antibody was from Upstate Biotechnology. Ubiquitin construct was a gift from D. Bohmann (University of Rochester, Rochester, NY), and pEYFP-mem construct was obtained from Clontech.
Data are shown as mean ± SEM, and paired or unpaired Student's t test was used to determine the significance between the two groups. Statistical significance was taken as P < 0.05.
Results
ROMK Is Regulated by Monoubiquitination. To determine whether ROMK channels can be monoubiquitinated in the kidney, we immunoprecipitated ROMK from renal cortex and outer medulla and probed the immunoprecipitated with ubiquitin antibody. Fig. 1 is a representative Western blot from six such experiments (four rats) showing that ROMK antibody recognized three protein bands of 42, 50, and 80 kDa (Fig. 1 Left). The 42-kDa protein is ROMK, whereas the 80-kDa protein has been considered to be a dimmer of ROMK (17) or a cross-reacting protein unrelated to ROMK (18–20). To explore the possibility that the 50-kDa protein represents monoubiquitinated ROMK, we stripped the membrane and exposed the membrane to ubiquitin antibody. From inspection of Fig. 1 Right, it is apparent that ubiquitin antibody recognized a 50-kDa protein band. This finding suggests that one ubiquitin molecule (8 kDa) has been added to ROMK (42 kDa).
Fig. 1.

A Western blot showing ROMK (Left) and ubiquitinated ROMK (Right) from cortex and outer medulla of the rat kidney. ROMK proteins were harvested by immunoprecipitation of tissue lysates with ROMK antibody cross-linked to the immobilized protein G.
To further demonstrate that ROMK channels are monoubiquitinated, we cotransfected HEK293 cells with hemagglutinin–ubiquitin and His-tagged ROMK1 constructs. The cells were lysed 24 h after transfection and the lysates were incubated with Ni2+ nitrilotriacetic acid agarose beads to precipitate His-tagged ROMK1. The proteins were resolved by SDS gel, and ROMK and ubiquitinated ROMK were detected by His and ubiquitin antibody, respectively. Fig. 2 is a representative Western blot from five such experiments showing that His-antibody recognized a major 42-kDa protein band, presumably ROMK1, and a light 50-kDa protein band that was also recognized by ubiquitin antibody. Thus, our data obtained from rat kidney and transfected HEK293 cells have demonstrated that ROMK channel can be monoubiquitinated.
Fig. 2.

A Western blot showing the purified His-tagged ROMK1 (Right) and ubiquitinated ROMK1 (Left) from HEK293 cells transfected with His-Tagged ROMK1 and ubuquitin. His-tagged ROMK1 proteins were harvested by precipitation of the cell lysate with Ni2+ nitrilotriacetic acid agarose beads.
Monoubiquitination of proteins is mediated by adding one ubiquitin molecule to Lys of the intracellular sequence of the substrate protein (21). Thus, we next determined which lysine residue in ROMK1 is responsible for the ubiquitin binding. We individually mutated every Lys on the N and C termini of ROMK1 except Lys-80, which has been shown to be a pH sensor for ROMK1 (22): R1K22A, R1K45R, R1K76R, R1K181R, R1K186R, R1K187R, R1K190R, R1K196R, R1K202R, R1K206R, R1K218R, R1K229R, R1K232R, R1K 331R, R1K333R, R1K336R, R1K346R, R1K363R, and R1K370R. A two-electrode voltage clamp was used to measure K current in oocytes injected with ROMK1 and mutants. Results summarized in Fig. 3 demonstrate that mutation of Lys-22 in the N terminus significantly increased K current from 8.1 ± 0.5 μA (ROMK1) (n = 67 measurements from 6 frogs) to 27.2 ± 1.2 μA (R1K22R) (n = 122 measurements from 12 frogs). This observation suggested that Lys-22 could be the ubiquitin binding site. This notion was also confirmed by experiments in which ubiquitination of ROMK1 was examined in oocytes injected with either GFP-Flag-tagged-ROMK1 or GFP-Flag-tagged-R1K22R (Fig. 4). From inspection of Fig. 4, it is apparent that ROMK antibody detected both a 72- and 80-kDa protein band in oocytes injected with GFP-Flag-tagged-ROMK1 (Fig. 4 Lower, left lane). Moreover, when the membrane was stripped and exposed to ubiquitin antibody, the 80-kDa protein band was recognized by ubiquitin antibody (Fig. 4 Upper, left lane). This finding indicates that ROMK1 is monoubiquitinated in oocytes. In contrast, there is only a 72-kDa protein band (Fig. 4 Lower, center lane), and ubiquitin antibody failed to recognize this band in oocytes injected with GFP-Flag-tagged R1K22R.
Fig. 3.

K currents in oocytes injected with ROMK1 (10 ng) and Lys to Arg mutants of ROMK1 (10 ng). The K current was measured with two-electrode voltage clamp. The data represent mean value ± SEM, and the experimental numbers ranged from 10 to 122.
Fig. 4.

A Western blot showing the ubiquitinated ROMK1 (Upper) and the whole ROMK1 (Lower). The ROMK1 channels were expressed in Xenopus oocytes and harvested by immunoprecipitation. ROMK1 is indicated by a short arrow.
Inhibition of Ubiquitination Increases the Surface Expression of ROMK1. Stimulation of the ROMK1 channel activity induced by mutation of Lys-22 could result from an increased channel open probability, a higher channel conductance, a higher protein expression, or an augmentation of ROMK1 channel number at the cell surface. Because the channel open probability of ROMK1 under control conditions is >0.9, it is unlikely that mutation of Lys 22-induced stimulation of ROMK1 channel activity is the result of an increase in channel open probability. Nevertheless we examined the single-channel properties of the R1K22R mutant expressed in oocytes. Fig. 5 is a single-channel recording showing that the channel open probability is ≈0.93 ± 0.1 (n = 6). Furthermore, the channel conductance is 38 ± 1 pS in the range of –40 to 0 mV (n = 6). Thus, neither channel open probability nor single-channel conductance of R1K22R is significantly different from that of wild-type ROMK1.
Fig. 5.

A single-channel recording in an inside-out patch from oocytes injected with R1K22R. The channel closed level is indicated by C, and a short bar indicates channel levels.
We next examined whether mutation of Lys-22 alters the protein expression level in Xenopus oocytes. We first measured the K current in oocytes injected with GFP-ROMK1 and R1K22R and confirmed that the K current in oocytes injected with GFP-R1K22R was 220 ± 20% (n = 12) higher than that with GFP-ROMK1. After measurement of K currents, the oocytes were immediately lysed and the ROMK1 channel was detected with ROMK antibody. Fig. 6A is a Western blot demonstrating that the protein expression level of wild-type ROMK1 and R1K22R were similar. Thus, the high ROMK1 channel activity in oocytes injected with R1K22R is not the result of an increase in protein expression levels.
Fig. 6.

Mutation of Lys-22 increases ROMK expression in plasma membrane. (A) Western blot demonstrating ROMK1 expression in oocytes injected with ROMK1 or R1K22R, respectively. (B) The fluorescence intensity in oocytes expressing R1K22R and ROMK1.
To explore the possibility that mutation of Lys-22 increases the ROMK1 channel number in the cell membrane, we examined the fluorescence intensity in oocytes injected with GFP-ROMK1 or GFP-R1K22R. Fig. 6B is a typical confocal image showing the fluorescence intensity of oocytes injected with ROMK1 or R1K22R. It is apparent that the fluorescence intensity in the cell membrane of oocytes injected with R1K22R is 180 ± 20% (n = 12) higher than that with ROMK1. We also used biotin labeling to examine the ROMK1 expression in cell plasma membrane of HEK293 cells transfected with GFP-ROMK1 and GFP-R1K22R (Fig. 7). The ROMK1 channels were harvested by immunoprecipitation of cell lysate with GFP antibody. The surface-located ROMK1 (labeled with biotin) was detected with neutraavidin (Fig. 7 Upper), and total ROMK proteins were indicated by ROMK antibody (Fig. 7 Lower). The level of surface-located ROMK1 channels was normalized in comparison with total ROMK protein. Although the total ROMK protein levels were almost the same between ROMK1 and R1K22R, mutation of Lys-22 significantly increased the surface ROMK1 channel by 190 ± 20% (n = 4) compared to that with ROMK1.
Fig. 7.

Mutation of Lys-22 to Arg increases the biotin-labeled ROMK1. (Upper) The biotin-label ROMK1. (Lower) The whole ROMK1 expression in HEK293 cells. Note that the 80-kDa band is present only in cells transfected with ROMK1.
We also extended the study by examining the expression pattern of ROMK1 in COS7 cells transfected with either ROMK1 or R1K22R. We and others have observed that only a small fraction of ROMK1 is actually expressed in the plasma membrane of COS7 cells transfected with ROMK1 (23, 24). Fig. 8 is a confocal image showing that ROMK1 is highly expressed in the cell plasma membrane in COS7 cells transfected with R1K22R as evidenced by the fact that ROMK1 staining (red) is highly overlapped with membrane marker, pEYFP-Mem (green). In contrast, colocalization between ROMK1 and membrane marker is low in the cell transfected with ROMK1.
Fig. 8.

A confocal image showing the expression of ROMK1 in COS cells transfected with R1K22R/ROMK1 (red) and pEYFP-Mem, a membrane marker.
Discussion
Here, we have demonstrated a mechanism by which ROMK channel activity is regulated by monoubiquitination. Three lines of evidence strongly suggest that ROMK channels can be monoubiquitinated. First, ubiquitin antibody reacts with a 50-kDa protein that is also recognized by ROMK antibody from the tissue lysates of renal cortex and outer medulla. Second, HEK cells transfected with ubiquitin and His-tagged ROMK1 also yields a 50-kDa protein recognized by both ROMK and ubiquitin antibodies. Third, in oocytes injected with GFP-ROMK1, ubiquitin antibody detected an 80-kDa protein band that was also recognized by the ROMK antibody.
Monoubiquitination (linking of a single or no more than two ubiquitin molecules to a substrate protein) has been reported to regulate protein metabolism (21). The fate of the monoubiquitinated plasma membrane protein is different from that of polyubiquitinated proteins which is achieved by forming a polyubiquitin chain (four or more ubiquitin molecules) through Lys-48. The monoubiquitinated protein is internalized to the endosome and may be recycled to the plasma membrane or delivered to a late endosome (21, 25, 26), whereas polyubiquitinated proteins are a signal for protein degradation via the 26S proteasome (21, 25, 26). Polyubiquitination has been shown to play a key role in controlling a variety of signal transduction pathways such as c-Jun (27–29). Polyubiquitination is also responsible for the degradation of epithelial Na channel (ENaC) and defective regulation of polyubiquitination of ENaC results in Liddle's syndrome, a dominant hereditary form of hypertension (30). In contrast, monoubiquitination can act as a regulatory signal similar to phosphorylation and affect the activity, trafficking and endosomal sorting of the substrate proteins (25, 26, 31, 32).
Like polyubiquitination, monoubiquitination of substrate protein is achieved through forming a covalent bond between the glysine residue in the C terminus of 76-aa ubiquitin and lysine residues in substrate proteins (25). The covalent attachment of ubiquitin to a substrate protein is a three-step process: (i) the ubiquitin-activating enzyme (E1) forms a thiol ester bond with ubiquitin by an ATP-dependent reaction; (ii) the activated ubiquitin is then transferred to a ubiquitin-conjugated enzyme (E2); and (iii) the ubiquitin ligase (E3) catalyzes the formation of an isopeptide bond with a lysine residue in substrate protein (21). It has been reported that monoubiquitination on a single lysine residue is sufficient to cause internalization of G protein-coupled receptor (14) and yeast plasma membrane protein (33).
The present study also demonstrated that Lys-22 is the ubiquitin-binding site in ROMK1. This conclusion is supported by two lines of evidence: (i) mutation of Lys-22 to Arg increased ROMK1 channel activity by >2-fold; and (ii) no 8-kDa band shift has been observed from the cell lysate obtained from oocytes injected with R1K22R. Increases in ROMK1 channel activity induced by mutation of Lys-22 could be the result of stimulation of protein expression, changes in channel open probability, and augmentation of channel number in the plasma membrane. Three lines of evidence suggest that inhibition of ubiquitination increases the surface expression of ROMK1. First, neither the channel expression level nor the biophysical properties of RIK22R were altered in comparison to that of wild-type ROMK1. Second, confocal microscopy showed higher surface fluorescence intensity in oocytes injected with R1K22R than that with ROMK1. Third, surface biotinylation in HEK cells transfected with R1K22R or wild-type ROMK1 demonstrated a higher surface expression of the mutant channel lacking the monoubiquitination site. Although we did not detect a strong signal for polyubiquitinated ROMK, it is conceivable that polyubiquitination of ROMK takes place also in the kidney for the ROMK metabolism. However, it is possible that polyubiquitinated ROMK channels were rapidly degraded. Thus, it would be difficult to detect the signal of polyubiquitinated ROMK with our methodology.
Both mono- and polyubiquitination are catalyzed by E3 ubiquitin ligase. However, we do not know which E3 ubiquitin ligase is responsible for mediating ubiquitination of ROMK1. Nedd4, A HECT-domain containing E3 ubiquitin ligase, has been shown to be expressed in the CCD (34). Moreover, Nedd4 is responsible to catalyzing polyubiquitination of ENaC (35). Interestingly, Nedd4 has been also reported to perform the monoubiquitination of EPS15, a ubiquitin-binding protein (36). Thus, it is possible that Nedd4 may also be responsible for conducting monoubiquitination of ROMK1.
Although it is not known whether the increase in ROMK1 surface expression induced by mutation of Lys-22 is the result of stimulating export or inhibiting internalization, we suggest that monoubiquitination of ROMK1 likely enhances endocytosis. Monoubiquitination has been implicated in the endocytosis of receptors (37). The monoubiquitinated protein is recognized by ubiquitin binding proteins that contain ubiquitin-binding domains such as the ubiquitin-interacting motif (38–40). One function of these ubiquitin-binding proteins is to link the monoubiquitinated protein to clathrin-coated pits, which are then pinched off to form clathrin-coated endosomes. Because the endocytosis of ROMK has been shown to be mediated by clathrin-coated pits (12), it is possible that monoubiquitination of ROMK may be involved in mediating the clathrin-dependent endocytosis. In addition, it is conceivable that monoubiquitination of ROMK channels may be involved in the modulation of ROMK channel number in plasma membranes in response to hormones and dietary K intake (7). We have demonstrated that stimulation of PTK enhanced the internalization of ROMK channels in the CCD (7). It is possible that monoubiquitination of ROMK channels may be required for initiating endocytosis induced by stimulation of PTK activity. Interestingly, it has been demonstrated that activity of c-Cbl, an E3 ubiquitin ligase with RING finger domain, is stimulated by tyrosine phosphorylation (41). Fig. 9 shows a model illustrating the potential physiological role of monoubiquitination in the regulation of ROMK1 channel activity. Low K intake stimulates PTK activity, which could phosphorylate ROMK1 and E3 ubiquitin ligase. As consequence, the activated E3 ubiquitin ligase facilitates monoubiquitination of ROMK1, which in turn leads to the internalization.
Fig. 9.

A model illustrating the possible physiological role of monoubiquitination in the regulation of the effect of low K intake on ROMK1 channel activity.
Acknowledgments
We thank Dr. V. Fried for stimulating discussion and technical support. The work was supported by National Institutes of Health Grants DK54983 (to W.-H.W.), DK47402 (to W.-H.W.), DK-54999 (to S.C.H.), and DK54998 (to G.G.). D.-H.L. was also supported by Chinese National Science Foundation Project 30370734.
Author contributions: D.L., S.C.H., G.G., and W.-H.W. designed research; D.L., H.S., Z.W., E.B., K.D., and W.-H.W. performed research; B.Y. contributed new reagents/analytic tools; D.L. and W.-H.W. analyzed data; and D.L., S.C.H., W.-H.W., and G.G. wrote the paper.
Abbreviation: CCD, cortical collecting duct.
References
- 1.Wang, W. H., Hebert, S. C. & Giebisch, G. (1997) Annu. Rev. Physiol. 59, 413–436. [DOI] [PubMed] [Google Scholar]
- 2.Giebisch, G. (1998) Am. J. Physiol. 274, F817–F833. [DOI] [PubMed] [Google Scholar]
- 3.Simon, D. B., Karet, F. E., Rodriguez, J., Hamdan, J. H., DiPietro, A., Trachtman, H., Sanjad, S. A. & Lifton, R. P. (1996) Nat. Genet. 14, 152–156. [DOI] [PubMed] [Google Scholar]
- 4.Woda, C. B., Bragin, A., Kleyman, T. & Satlin, L. M. (2001) Am. J. Physiol. 280, F786–F793. [DOI] [PubMed] [Google Scholar]
- 5.Woda, C. B., Leite, M., Jr., Rohatgi, R. & Satlin, L. M. (2002) Am. J. Physiol. 283, F437–F446. [DOI] [PubMed] [Google Scholar]
- 6.Palmer, L. G. (1999) Am. J. Physiol. 277, F821–F825. [DOI] [PubMed] [Google Scholar]
- 7.Wang, W. H. (2004) Annu. Rev. Physiol. 66, 547–569. [DOI] [PubMed] [Google Scholar]
- 8.Palmer, L. G. & Frindt, G. (1999) Am. J. Physiol. 277, F805–F812. [DOI] [PubMed] [Google Scholar]
- 9.Cassola, A. C., Giebisch, G. & Wang, W. H. (1993) Am. J. Physiol. 264, F502–F509. [DOI] [PubMed] [Google Scholar]
- 10.Palmer, L. G., Antonian, L. & Frindt, G. (1994) J. Gen. Physiol. 105, 693–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei, Y., Bloom, P., Lin, D. H., Gu, R. M. & Wang, W. H. (2001) Am. J. Physiol. 281, F206–F212. [DOI] [PubMed] [Google Scholar]
- 12.Zeng, W. Z., Babich, V., Ortega, B., Quigley, R., White, S. J., Welling, P. A. & Huang, C. L. (2002) Am. J. Physiol. 283, F630–F639. [DOI] [PubMed] [Google Scholar]
- 13.Kahle, K. T., Wilson, F. H., Leng, Q., Lalioti, M. D., Connell, A. D., Dong, K., Rapson, A. K., MacGregor, G. G., Giebisch, G., Hebert, S. C., et al. (2003) Nat. Genet. 35, 372–376. [DOI] [PubMed] [Google Scholar]
- 14.Terrell, J., Shih, S., Dunn, R. & Hicke, L. (1998) Mol. Cell 1, 193–202. [DOI] [PubMed] [Google Scholar]
- 15.Macica, C. M., Yang, Y. H., Hebert, S. C. & Wang, W. H. (1996) Am. J. Physiol. 271, F588–F594. [DOI] [PubMed] [Google Scholar]
- 16.Moral, Z., Deng, K., Wei, Y., Sterling, H., Deng H., Ali, S., Gu, R. M., Huang, X. Y., Hebert, S. C., Giebisch, G., et al. (2001) J. Biol. Chem. 276, 7156–7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu, J. Z., Hall, A. E., Peterson, L. N., Bienkowski, M. J., Eessalu, T. E. & Hebert, S. C. (1997) Am. J. Physiol. 273, F739–F748. [DOI] [PubMed] [Google Scholar]
- 18.Mennitt, P. A., Wade, J. B., Ecelbarger, C. A., Palmer, L. G. & Frindt, G. (1997) J. Am. Soc. Nephrol. 8, 1823–1830. [DOI] [PubMed] [Google Scholar]
- 19.Ecelbarger, C. A., Kim, C. H., Knepper, M. A., Liu, J., Tate, M., Welling, P. A. & Wade, J. B. (2001) J. Am. Soc. Nephrol. 12, 10–18. [DOI] [PubMed] [Google Scholar]
- 20.Kohda, Y., Ding, W., Phan, E., Housini, I., Wang, J., Star, R. A. & Huang, C. L. (1998) Kidney Int. 54, 1214–1223. [DOI] [PubMed] [Google Scholar]
- 21.Hicke, L. & Dunn, R. (2003) Annu. Rev. Cell Dev. Biol. 19, 141–172. [DOI] [PubMed] [Google Scholar]
- 22.Fakler, B., Schultz, J. H., Yang, J., Schulte, U., Braendle, U., Zenner, H. P., Jan, L. Y. & Ruppersberg, J. (1996) EMBO J. 15, 4093–4099. [PMC free article] [PubMed] [Google Scholar]
- 23.Lin, D. H., Sterling, H., Lerea, K. M., Giebisch, G. & Wang, W. H. (2002) J. Biol. Chem. 277, 44332–44338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoo, D., Kim, B. Y., Campo, C., Nance, L., King, A., Maouya, D. & Welling, P. A. (2003) J. Biol. Chem. 278, 23066–23075. [DOI] [PubMed] [Google Scholar]
- 25.DiFiore, P. P. D., Polo, S. & Hofmann, K. (2003) Nat. Rev. Mol. Cell. Biol. 4, 491–497. [DOI] [PubMed] [Google Scholar]
- 26.Schnell, J. D. & Hicke, L. (2003) J. Biol. Chem. 278, 35857–35860. [DOI] [PubMed] [Google Scholar]
- 27.Ciechanover, A., Finley, D. & Varshavsky, A. (1984) Cell 37, 57–66. [DOI] [PubMed] [Google Scholar]
- 28.Reed, S. I. (2003) Nat. Rev. Mol. Cell. Biol. 4, 855–864. [DOI] [PubMed] [Google Scholar]
- 29.Treier, M., Staszewski, L. M. & Bohmann, D. (1994) Cell 78, 787–798. [DOI] [PubMed] [Google Scholar]
- 30.Shimkets, R. A., Warnock, D. G., Bositis, C. M., Nelson-Williams, C., Hansson, J. H., Schambelan, M., Gill, J. R., Jr., Ulick, S., Milora, R. V., Findling, J. W., et al. (1994) Cell 79, 407–414. [DOI] [PubMed] [Google Scholar]
- 31.Shih, S. C., Sloper-Mould, K. E. & Hicke, L. (2000) EMBO J. 19, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Umebayashi, K. (2003) Cell Struct. Funct. 28, 443–453. [DOI] [PubMed] [Google Scholar]
- 33.Galan, J. M. & Haguenauer-Tsapis, R. (1997) EMBO J. 16, 5847–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staub, O., Yeger, H., Plant, P. J., Kim, H., Ernst, S. A. & Rotin, D. (1997) Am. J. Physiol. 272, C1871–C1880. [DOI] [PubMed] [Google Scholar]
- 35.Staub, O., Gautschi, I., Ishikawa, T., Breitschopf, K., Ciechanover, A., Schild, L. & Rotin, D. (1997) EMBO J. 16, 6325–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polo, S., Sigismund, S., Faretta, M., Guidi, M., Capua, M. R., Bossi, G., Chen, H., Camilli, P. D. & Fiore, P. P. D. (2002) Nature 416, 451–455. [DOI] [PubMed] [Google Scholar]
- 37.Haglund, K., Sigismund, S., Polo, S., Szymkiewicz, I., Fiore, P. P. D. & Dikic, I. (2003) Nat. Cell Biol. 5, 461–466. [DOI] [PubMed] [Google Scholar]
- 38.Shih, S. C., Prag, G., Francis, S. A., Sutanto, M. A., Hurley, J. H. & Hicke, L. (2003) EMBO J. 22, 1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swanson, K. A., Kang, R. S., Stamenova, S. D., Hicke, L. & Radhakrishnan, I. (2003) EMBO J. 22, 4597–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raiborg, C., Bache, K. G., Gillooly, D. J., Madshus, I. H., Stang, E. & Stenmark, H. (2002) Nat. Cell Biol. 4, 394–398. [DOI] [PubMed] [Google Scholar]
- 41.Kassenbrock, C. K. & Anderson, S. M. (2004) J. Biol. Chem. 279, 28017–28027. [DOI] [PubMed] [Google Scholar]