

Abstract
Abnormal choline phospholipid metabolism is associated with oncogenesis and tumor progression. We have investigated the effects of targeting choline phospholipid metabolism by silencing two glycerophosphodiesterase genes, GDPD5 and GDPD6, using small interfering RNA (siRNA) in two breast cancer cell lines, MCF-7 and MDA-MB-231. Treatment with GDPD5 and GDPD6 siRNA resulted in significant increases in glycerophosphocholine (GPC) levels, and no change in the levels of phosphocholine (PC) and free choline, which further supports their role as GPC specific regulators in breast cancer. The GPC levels were more than twofold increased during GDPD6 silencing, and marginally increased during GDPD5 silencing. DNA laddering was negative in both cell lines treated with GDPD5 and GDPD6 siRNA, indicating absence of apoptosis. Treatment with GDPD5 siRNA caused a decrease in cell proliferation in both breast cancer cell lines at 72h, while GDPD6 siRNA treatment decreased cell proliferation in MCF-7 at 72h, but not in MDA-MB-231 cells. Decreased cell migration and invasion were observed in MDA-MB-231 cells treated with GDPD5 or GDPD6 siRNA, where a more pronounced reduction in cell migration and invasion was observed under GDPD5 siRNA treatment as compared to GDPD6 siRNA treatment. In conclusion, GDPD6 silencing increased the GPC levels in breast cancer cells more profoundly than GDPD5 silencing, while the effects of GDPD5 silencing on cell proliferation, migration, and invasion were more severe than those of GDPD6 silencing. Our results suggest that silencing GDPD5 and GDPD6 alone or in combination may have potential as new molecular targeting strategy for breast cancer treatment.
Keywords: breast cancer, choline phospholipid metabolism, gene targeted treatment, GDPD5, GDPD6
Introduction
Metabolic alterations have been suggested as an emerging hallmark of cancer and a possible therapeutic target (1, 2). Studies exploring the potential of key metabolic regulators as new targets for treatment of cancer are highly relevant and may help improve the effectiveness of currently available treatment strategies. Choline phospholipid metabolism regulates the synthesis and degradation of de novo phosphatidylcholine (PtdCho), which is a major lipid component of the cell membrane bilayer. The total choline (tCho) signal detected by in vivo magnetic resonance spectroscopy (MRS) can be resolved into single peaks consisting of phosphocholine (PC), glycerophosphocholine (GPC) and free choline signals using ex vivo high-resolution MRS applications, and thus enables the detection of changes in choline containing metabolites individually (3). As alterations in choline phospholipid metabolism are associated with oncogenic transformation and treatment response (4–6), the genes and enzymes regulating this metabolic pathway are potential targets for treatment of cancer, including breast cancer. Several genes and key regulatory enzymes have been identified in choline phospholipid metabolism of cancer, including choline kinase alpha (CHKA) and phosphatidylcholine-specific phospholipase D1 (PtdCho-PLD1) (7, 8). Down-regulation of CHKA, the gene regulating the conversion of free choline to PC, is associated with decreased cell proliferation (9), and increased effects of chemotherapy in ovarian (10) and breast (11) cancers, whereas CHKA overexpression was shown to increase drug resistance in breast cancer cells (12). Targeted therapy against CHKA using the small molecule CHKA inhibitor TCD-717 is currently being tested in a dose escalation study in a clinical phase I trial (https://clinicaltrials.gov/ct2/show/NCT01215864?term=TCD-717&rank=1). Although the trial closed in 2014, no final evaluation has been published yet.
Although GPC is a part of the tCho signal, which has been suggested as a biomarker for the diagnosis and treatment evaluation of breast cancer (13–17), relatively little effort has been made to identify the genes and proteins regulating the level of GPC, and to elucidate the molecular reasons for the changes in GPC observed in cancers. GPC is a membrane breakdown product generated from hydrolysis of PtdCho by phospholipase A2 (PLA2) and lysophospholipase A1 (Lyso-PLA1). In our previous study, we demonstrated that the expression of glycerophosphodiester phosphodiesterase domain containing 5 (GDPD5) negatively correlated with the GPC levels in human breast cancer cell lines and tumors from patients (18). Degradation of GPC to free choline and glycerol-3-phosphate is catalyzed by the glycerophosphodiesterase enzymatic unit of the GDPD5 protein (19). Subsequently free choline can be recycled to produce PC by CHKA. The expression of GDPD5 was also found to be positively correlated with CHKA and PtdCho-PLD1 mRNA levels which further support the involvement of GDPD5 in tumor progression (18).
Glycerophosphodiester phosphodiesterase domain containing 6 (GDPD6), also known as endometrial carcinoma differential 3 (EDI3), was also shown to be involved in the regulation of choline phospholipid metabolism (20). Silencing of GDPD6 resulted in an increased GPC/PC ratio and decreased cell migration in breast cancer cells. GDPD6 expression was also found to be associated with metastasis and survival in endometrial and ovarian cancers (20). In the same study, no significant changes were detected in the expression of GDPD5 during GDPD6 silencing, suggesting that GDPD5 did not contribute to the phenotypic changes observed by GDPD6 silencing (20). Furthermore, gene silencing and overexpression of GDPD6 resulted in changes of integrin ß1 expression, which were associated with disturbances of cell adhesion and spreading (21).
As both GDPD5 (18, 19, 22) and GDPD6 (20, 21) were shown to be involved in the regulation of GPC, it is of interest to systematically compare and evaluate their potential as molecular targets for breast cancer treatment. The purpose of this study was to investigate the potential of targeting choline phospholipid metabolism for treatment of breast cancer by comparing the effects of GDPD5 and GDPD6 silencing on choline metabolite profiles, apoptosis, cell proliferation, migration, and invasion in two different types of malignant breast cancer cell lines.
Materials and Methods
Human breast cancer cell lines
Two different types of malignant breast cancer cell lines were used in this study: MCF-7 (23), an estrogen-sensitive weakly metastatic cell line, and MDA-MB-231 (24), an estrogen-independent highly metastatic cell line. Both cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and cultured as previously described with 5% CO2 in air at 37°C (25). The cell lines were authenticated by ATCC using cytochrome C oxidase I polymerase chain reaction (PCR) assay and short tandem repeat profiling using multiplex PCR. The cells also tested negative for Mycoplasma.
Gene silencing by siRNA treatment
Transient gene silencing was mediated by transfection of custom-made targeted siRNA against GDPD5 or GDPD6 (Dharmacon) using Lipofectamine 2000 transfection agent (Invitrogen) following the manufacturer’s protocol. Non-targeted (scrambled) siRNA was used as a control (Dharmacon). For each transfection, the siRNA oligomer and Lipofectamine 2000 reagent were diluted in Opti-MEM Reduced Serum Medium (Invitrogen) before the complexes were mixed and transferred to the cells. To find the optimal treatment protocol, different siRNA dosages (25–100 nM) and short and long time effects (24–96 hours) of GDPD5 siRNA treatment were tested. The GDPD6 siRNA treatment protocol was performed using 75 nM siRNA for direct comparison with GDPD5 siRNA treatment, which was optimal at a concentration of 75 nM GDPD5 siRNA. The siRNA sequences for GDPD5 and GDPD6 siRNA used in this study are listed in Table 1. Each experiment was repeated three times.
Table 1.
siRNA and primer sequences used for GDPD5 and GDPD6
| Gene | Sequences |
|---|---|
| GDPD5 siRNA | ’GCTCTCCGTATGTTCAGACAA’ |
|
| |
| GDPD5-S primer | 5′-CTACAACCCTGAGCAGAT-3′ |
| GDPD5-AS primer | 5′-AACATACGGAGAGCACAT-3′ |
|
| |
| GDPD6 siRNA | ’GGAAAGAATCTGTGGTTCA’ |
|
| |
| GDPD6-S primer | 5 ′-TTT C A A AAT GCTGCAGGGTAAT-3′ |
| GDPD6-AS primer | 5′-ACCCACAAAGCAACAGTGTGTA-3′ |
|
| |
| Housekeeping gene | |
|
| |
| HPRT1-S primer | 5′-CCTGGCGTCGTGATTAGTGATG-3′ |
| HPRT1-AS primer | 5′-CAGAGGGCTACAATGTGATGGC-3′ |
All GDPD primers were designed to be gene specific and not transcript specific.
Assessment of silencing efficiency by qRT-PCR
Total cellular RNA was isolated using QIAshredder and RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. The quality of the RNA yield was determined by the A260/280 absorption ratio measured on a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific). For each sample, 1 μg RNA was used as template for preparing cDNA by SuperScript III First-Strand Synthesis SuperMix (Invitrogen). Gene expression based on the number of mRNA transcripts was measured by quantitative reverse transcription-PCR (qRT-PCR) using iCycler (Bio-Rad). Three technical PCR reactions were prepared for each sample using 2 μL of 1:10 diluted cDNA, 2 μM of the paired sense and antisense primers (Invitrogen, see Table 1), 10 μL iQ SYBR Green reagent (Quanta BioSciences), and diethyl pyrocarbonate-treated water. Relative fold changes of target mRNA were normalized to the housekeeping gene hypoxanthine phosphoribosyltransferase 1 (HPRT1) and calculated based on the threshold cycle (ct) using the ΔΔct method as previously described (18). Three technical repeats were performed for each sample.
MR spectroscopy and metabolite quantification
Cells were harvested by trypsinization with 0.25 % Trypsin-EDTA Solution (Sigma-Aldrich) and counted in a dilution of trypan blue to assess cell viability. To evaluate the efficiency of gene silencing in the same sample used for MRS analysis, a fraction of cells was used for RNA isolation and analyzed for gene expression, while the remaining cells were extracted for metabolite quantification. Water-soluble metabolites were obtained using a dual-phase extraction method based on methanol/chloroform/water (1:1:1; v/v/v) as previously described (18, 26, 27). The samples were dissolved in deuterium dioxide solvent containing 0.24×10-6 mol 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid (TSP; Sigma-Aldrich) for chemical shift referencing and as internal standard for metabolite quantification. Fully relaxed 1H high resolution MR spectra were measured on a Bruker Avance 500 (11.7 T) MR Spectrometer (Bruker BioSpin Corp.) as previously described (27). Quantification of free choline (3.209 ppm), PC (3.227 ppm), and GPC (3.236 ppm) was performed by peak integration using the MestReNova software (Mestrelab Research) and normalized to cell number as previously described (27). Experimental groups were repeated several times as follows. GDPD5 mRNA and metabolite measurements in MCF-7 cells were repeated five times for non-targeted controls and nine times for GDPD5 siRNA treatment. GDPD5 mRNA and metabolite measurements in MDA-MB-231 cells were repeated five times for non-targeted controls and eleven times for GDPD5 siRNA treatment. GDPD6 mRNA and metabolite measurements in MCF-7 cells were repeated three times for non-targeted controls as well as for GDPD6 siRNA treatment. GDPD6 mRNA and metabolite measurements in MDA-MB-231 cells were repeated four times for non-targeted controls as well as for GDPD6 siRNA treatment.
DNA ladder analysis for apoptosis assessment
DNA ladder analysis was performed using floating cells from the culture medium and adherent cells that were scraped from the culture dish. All cells were pooled and centrifuged to obtain a pellet containing both intact and apoptotic cells. After lysis of cells in Binding/Lysis buffer provided by the Apoptotic DNA Ladder Kit (Roche), DNA was isolated by passing through a column with a surface of glass fibers and chaotropic salts, and was eluted followed by two washing steps. The purity and concentration of the DNA yield was measured on a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific). For each sample, 2 μg DNA was transferred to an agarose gel (1%) containing ethidium bromide (5 μL). The gel was run for 1.5 hours at 75 V. Cellular DNA, DNA ladder, and positive control consisting of lyophilized apoptotic cells from the kit were separated and visualized with a UV light detector. Each full experiment was repeated twice.
Cell proliferation
Cell proliferation was monitored over a period of 24–96 hours during GDPD5 and GDPD6 siRNA treatment and was quantified using cell proliferation reagent WST-1 (Roche), as well as manual counting in a separate set of experiments. For WST-1 assays, each sample was measured against a background control containing medium and WST-1 reagent as blank. The optimal incubation time with WST-1 reagent was at 2 hours after its addition to the cells. Cell proliferation was normalized to the baseline at 0 hours and compared to non-targeted siRNA controls. Each treatment group and time point was repeated six times. For manual counting, MCF-7 or MDA-MB-231 cells were seeded in 48 well-plates at a density of 40,000 cells per well, and then transfected with 75 nM of non-target, GDPD5 or GDPD6 siRNA the next day. After 48h of siRNA transfection, viable cells were detached by trypsinization and the number of cells in each well was manually counted. Another set of wells were replenished with fresh siRNA-containing media and incubated for another 48h, and cell number was determined again in the same way for the 96h time point. Each sample was counted twice and each treatment time point was repeated three times. Both types of assays, WST-1 and cell counting, resulted in comparable data.
Migration and invasion assays
After treatment with siRNA, cell migration assays were performed using transwell inserts with polyethylene membrane and 8.0 μm size pores (Corning), while cell invasion assays were performed using the same transwell inserts coated with 15ug/cm2 Collagen-1. Cells were placed on the upper layer of the transwell containing serum free medium. The lower layer of the well was filled with medium containing 10% FBS, which served as chemoattractant. Cells were incubated at 37°C with 5% CO2 for 24 and 48 hours. After incubation, cells located on the upper layer of the membrane were removed using a cotton swab, while cells migrated/invaded through the lower side were fixed, stained, and counted at a magnification of 100× in five random fields of view. Each experimental group in the migration and invasion assays was repeated three times.
Cell migration by scratch assay
MCF-7 cells were seeded in a 24 well-plate at a density of 0.2 million cells per well, and then transfected with 75 nM non-target, GDPD5, or GDPD6 siRNA the next day. After 48h of siRNA transfection, siRNA was removed. Scratches in cell monolayers were made with P200 pipet tips, followed by washing away the removed cells with cell culture medium. Medium with reduced serum (5% FBS) was added to decrease cell proliferation. Phase contrast microscopic images were taken on a Nikon TS100 inverted microscope equipped with a Nikon Coolpix digital camera (Nikon Instruments, Inc., Melville, NY) at 10× magnification immediately following scratching at 0h and at 48h of incubation in a standard cell culture incubator with the microscope setting being kept the same for each measurement. The gap size of these scratches was measured using ImageJ software (http://rsb.info.nih.gov/ij/) at 5 different corresponding positions on images taken at 0h or 48h. Migration distance was calculated by gap size at 0h subtracted by that at 48h. Each siRNA treatment and scratch assay was repeated four times.
Statistical Analysis
All comparisons were calculated using the student’s t-test and considered significant if the p-values were ≤ 0.05. Each experiment was repeated several times as indicated in the end of each paragraph for each assay or measurement performed. Data are presented as mean ± standard deviation (SD) or mean ± standard error (SE) as indicated in each figure.
Results
Optimal siRNA treatment protocol
GDPD5 siRNA silencing efficiency was dose-dependent in both MCF-7 and MDA-MB-231 breast cancer cell lines when treated for 48 hours (Figure 1A). Cells treated for a shorter incubation time (24 hours) did not show an increased silencing with increased siRNA concentration. Higher concentrations of GDPD5 siRNA (100 nM) were also tested, however, an increase in cell death and debris was observed, which may have resulted from cell toxicity mediated by the high dose of Lipofectamine reagent applied with 100 nM GDPD5 siRNA. The optimal concentration of GDPD5 siRNA with the highest silencing efficiency was 75 nM for both cell lines. Best knockdown results were obtained with 75 nM siRNA and at the 48 and 72 hours incubation times in both cell lines (Figure 1B). The efficiency of GDPD6 treatment with 75 nM GDPD6 siRNA over time was also examined. GDPD6 knockdown efficiency did not change over time for MCF-7 cells, and was highest at 48 and 72 hours in MDA-MB-231 cells.
Figure 1.
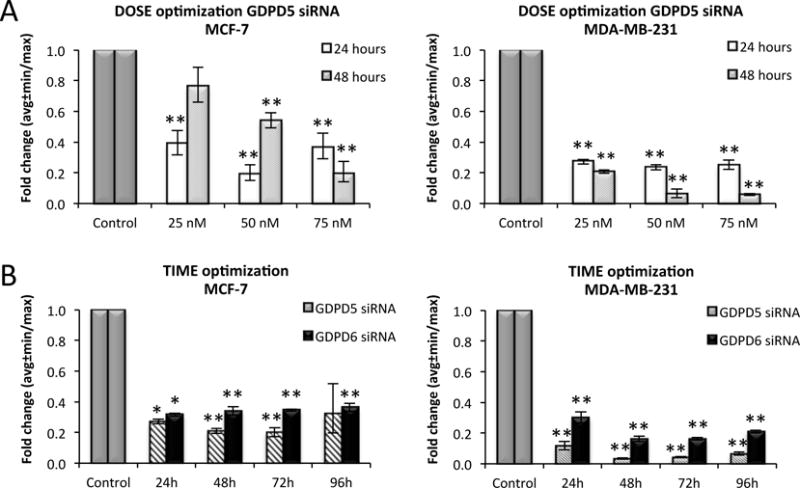
Optimization of siRNA A) dosage (25, 50 and 75 nM) for GDPD5 silencing and B) incubation time (24, 48, 72, 96 hours with 75 nM siRNA) for GDPD5 and GDPD6 silencing in MCF-7 and MDA-MB-231 breast cancer cells. Fold changes were normalized to control samples treated with non-targeted siRNA (gray scale). Each experiment was repeated three times. The optimal dosage and treatment time with the highest GDPD5 and GDPD6 silencing efficiency was 75 nM siRNA treated for 48/72 hours. Values are presented as mean ± standard error. *p≤0.05, **p≤0.01.
Gene silencing efficiency and metabolite quantification
GDPD5 was successfully down-regulated by 65% (p < 0.001) and 81% (p < 0.001) in MCF-7 and MDA-MB-231 cells, respectively (Figure 2A). We observed significant increases in GPC levels from 1.87 ± 0.19 fmol/cell in non-targeted controls to 2.63 ± 0.18 fmol/cell in GDPD5 siRNA treated MCF-7 cells (p = 0.016) and from 1.20 ± 0.12 fmol/cell in non-targeted controls to 1.68 ± 0.15 fmol/cell in GDPD5 siRNA treated MDA-MB-231 cells (p = 0.028). In this study, GDPD6 was successfully down-regulated by 67% in MCF-7 cells (p < 0.001) and 73% in MDA-MB-231 cells (p = 0.028) as shown in Figure 2B. MCF-7 cells treated with GDPD6 siRNA experienced a GPC increase from 3.88 ± 0.35 fmol/cell in controls to 8.19 ± 0.33 fmol/cell in GDPD6 siRNA treated cells, which is a more than twofold increase in GPC level (p < 0.001), and which led to a higher level of tCho (p = 0.031) and a decreased PC/GPC ratio (p = 0.017) compared to non-targeted controls. GPC significantly increased by threefold from 1.69 ± 0.18 fmol/cell in non-targeted controls to 6.21 ± 0.37 fmol/cell in MDA-MB-231 cells treated with GDPD6 siRNA (p = 0.048) which also led to a significant decrease in the PC/GPC ratio (p = 0.005). The PC and free choline levels did not change in GDPD5 or GDPD6 siRNA treated MCF-7 and MDA-MB-231 cells. Representative 1H MRS spectra of the choline metabolite profiles are shown in Figure 2C.
Figure 2.
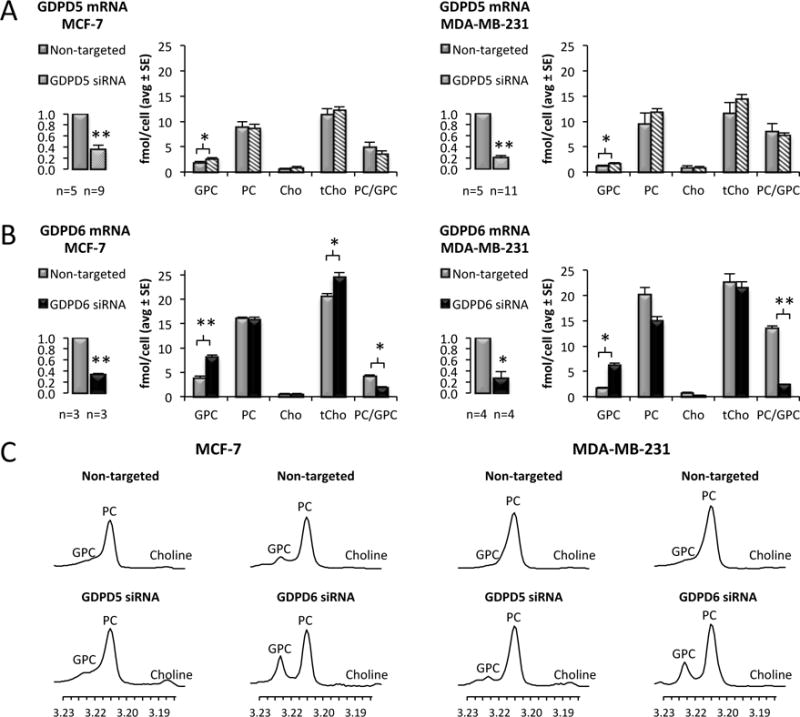
Effects of GDPD5 and GDPD6 silencing on choline metabolite profiles. A) GDPD5 was significantly down-regulated by 65% and 81% in MCF-7 and MDA-MB-231cells, respectively. A significant increase in GPC was observed in GDPD5 siRNA treated cells compared to non-targeted controls. B) GDPD6 was significantly down-regulated by 67% and 73% in MCF-7 and MDA-MB-231 cells, respectively. Increased GPC levels and decreased PC/GPC ratios were observed in GDPD6 treated cells compared to non-targeted controls. C) Representative 1H MRS choline metabolite profiles of GDPD5 and GDPD6 siRNA treated MCF-7 and MDA-MB-231 cells compared to non-targeted controls. Values are presented as mean ± standard error. *p≤0.05, **p≤0.01. The number of repeats (n) are given underneath each graph.
Apoptotic DNA laddering and cell proliferation
To test for apoptosis, a DNA laddering assay was carried out in extracts of MCF-7 and MDA-MB-231 cells treated with GDPD5 and GDPD6 siRNA. Apoptosis assays were performed on all cells, adherent and floating cells alike. In apoptotic cells, DNA is degraded and can be observed as multiple fragments with approximately 200 base pair length. As evident from Figure 3A, DNA laddering did not occur in either cell line treated with GDPD5 and GDPD6 siRNA. Cell proliferation was monitored for 24–96 hours with siRNA treatment (Figure 3B). In GDPD5 siRNA treated cells, a significant decrease in cell proliferation was observed in MCF-7 (p < 0.001) compared to non-targeted controls starting at 48h of treatment, which was not the case in MDA-MB-231 cells at any time point. In GDPD6 siRNA treated cells, neither MCF-7 nor MDA-MB-231 cells displayed significant changes in proliferation as compared to controls. Notably, only GDPD5 siRNA treatment in MCF-7 cells resulted in a pronounced effect on cell proliferation, leading to significantly lower proliferation rates upon treatment. By examining the cells under the microscope, we did not observe significant differences in the amount of floating cells between treatment groups, which indicates that the decrease in cell number in MCF-7 cells treated with GDPD5 is likely caused by a decrease in proliferation.
Figure 3.
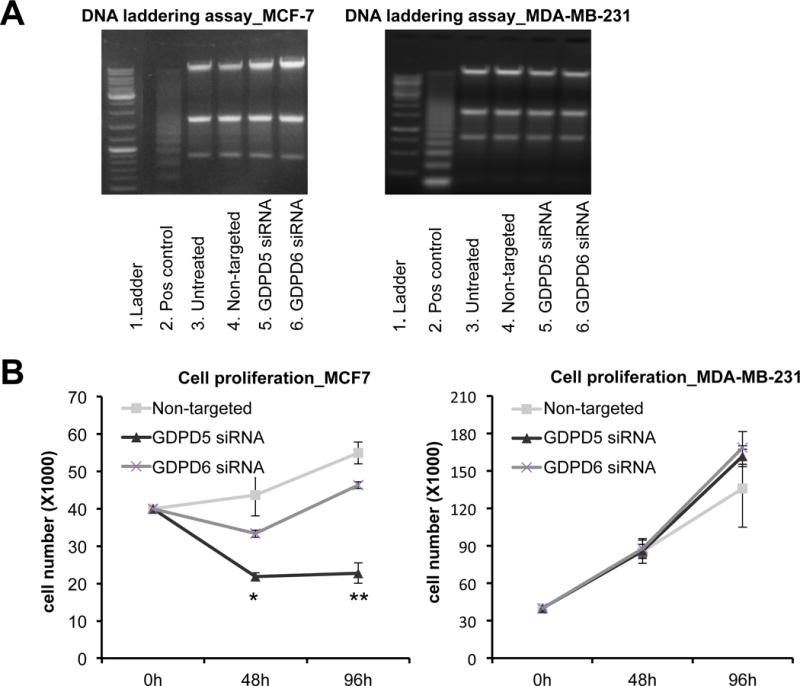
Effects of GDPD5 and GDPD6 silencing on apoptosis and cell proliferation. A) Apoptosis assay. DNA laddering was negative for both cell lines treated with GDPD5 and GDPD6 siRNA (n=2). B) Cell proliferation. GDPD5 silencing resulted in decreased cell proliferation in MCF-7 cells, but not MDA-MB-231 cells compared to non-targeted controls, while GDPD6 silencing had no significant effect on cell proliferation in MCF-7 and MDA-MB-231 cells. Values are presented as mean ± standard deviation. *p≤0.05, **p≤0.01, n=3 each.
Cell migration and invasion
MDA-MB-231 cells exhibited a significant decrease in cell migration when treated with GDPD5 (p < 0.001) and GDPD6 (p < 0.001) siRNA compared to non-targeted controls (Figure 4A) using transwell migration assay. In addition, decreased cell migration was observed in GDPD5 compared to GDPD6 siRNA treated cells (p = 0.002). Decreased cell invasion was also observed in GDPD5 siRNA treated MDA-MB-231 cells at 24h (p < 0.001) and 48h (p < 0.001) and in GDPD6 siRNA treated MDA-MB-231 cells at 24h (p = 0.056) and 48h (p < 0.001) compared to non-targeted controls (Figure 4B). We observed a more pronounced decrease in cell invasion in GDPD5 compared to GDPD6 siRNA treated MDA-MB-231 cells at 24h (p = 0.024) and 48h (p = 0.025). Cell migration and invasion assays were negative for MCF-7 cells. MCF-7 cells did not migrate or invade through the transwell using the same treatment protocol and conditions. To probe the effects of non-targeted, GDPD5, and GDPD6 siRNA treatment on MCF-7 cells, we performed scratch assays as shown in Figure 5. MCF-7 cell migration was significantly reduced following GDPD5 (p < 0.001) and GDPD6 (p < 0.01) siRNA treatment as compared to control.
Figure 4.
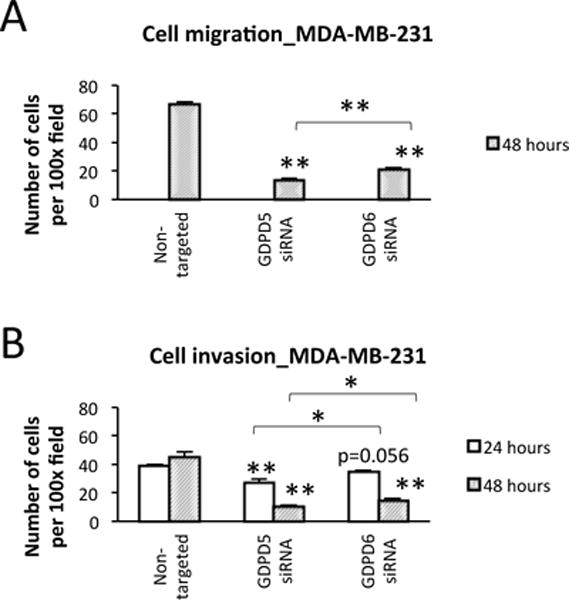
Effects of GDPD5 and GDPD6 silencing on cell migration and invasion. A) Cell migration. Decreased cell migration was observed in GDPD5 and GDPD6 treated MDA-MB-231 cells compared to non-targeted controls at 48h (n=3). Lower cell migration was observed in GDPD5 compared to GDPD6 siRNA treated cells. B) Cell invasion. GDPD5 and GDPD6 treated MDA-MB-231 cells showed significant decreases in cell invasion compared to non-targeted controls at 24h and 48h (n=3). Lower cell invasion was observed in GDPD5 compared to GDPD6 siRNA treated cells at both time points. Values are presented as mean ± standard error. *p≤0.05, **p≤0.01.
Figure 5.
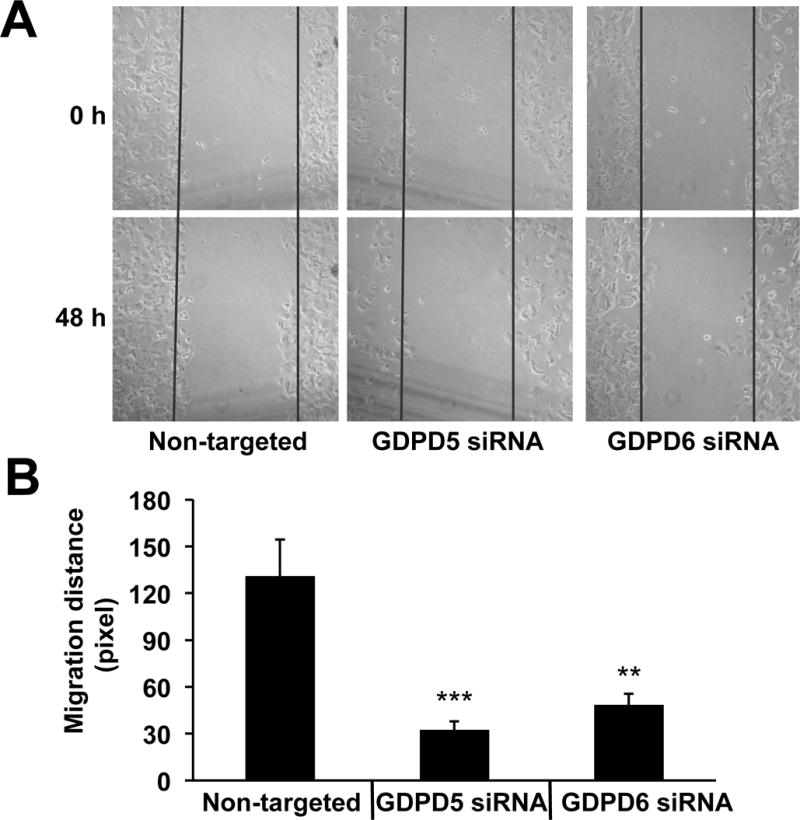
GDPD5 and GDPD6 silencing reduced MCF-7 cell migration. A) Representative phase contrast images taken immediately at 0h or at 48h after producing the scratch. B) Quantification of migration distance. Each assay was repeated four times. Values are presented as mean ± standard deviation. **P<0.01, ***P<0.001, n=4 each.
Discussion
We have investigated the effects of targeting two glycerophosphodiesterase genes, GDPD5 and GDPD6, which have shown to be involved in the regulation of choline phospholipid metabolism in breast cancer, in particularly the degradation of GPC to free choline and glycerol-3-phosphate (18, 20–22). Silencing of GDPD5 and GDPD6 in breast cancer cells increased the GPC levels, which further supports their roles as GPC specific regulators in breast cancer. GDPD6 probably has a more important role in regulating the cellular GPC levels in breast cancer cells than GDPD5, as the level of GPC was more dramatically increased upon GDPD6 silencing as compared to GDPD5 silencing. Future studies directly assessing the GDPD5-related GPC:Cho phosphodiesterase activity are needed to show the degree of GDPD5 involvement in degrading GPC. This is important because GDPD5 can also mediate glycosylphosphatidylinositol (GPI)-anchor hydrolysis (28), while GDPD6 is considered to be specific for GPC (20, 21). Accumulation of GPC mediated by decreased activity of GDPD5 and GDPD6 is postulated to affect the recycling of choline that is used as resource for the de novo synthesis of PtdCho, which is an important component in the cell membrane (29). However, we did not observe any changes in the levels of free choline and PC in response to treatment in both tested breast cancer cell lines. Apart from GPC degradation, choline can also be derived from the diet and transported into the cell by choline transporters, or it can be directly produced from hydrolysis of PtdCho by PtdCho-PLD1 (8, 30). Enhanced CHK activity and phosphatidylcholine-specific phospholipase C (PtdCho-PLC) were shown to increase the level of PC in breast and ovarian cancer (27, 31). It may therefore be possible that GDPD5 and GDPD6 siRNA treated cells compensate for their loss of free choline through enhanced uptake of extracellular choline and/or through breakdown of PtdCho directly. Also, GDPD5 and GDPD6 gene expressions were significantly down-regulated (63–81%), but not completely deleted and therefore minor gene expression and enzyme activity may still be possible.
Glycerol-3-phosphate is a precursor of several important intermediates involved in the biosynthesis of triacylglycerol and phospholipids, and can be derived from glycerol, GPC, and glycerophosphoethanolamine (GPE) in glycerophospholipid metabolism. The level of glycerol-3-phosphate was shown to be higher in breast cancer compared to normal breast tissue (32). In a recent study, overexpression of GDPD6 in NIH 3T3 cells resulted in a time dependent increase in glycerol-3-phosphate detected by 31P MRS (20), thus silencing of GDPD5 and GDPD6 and accumulation of GPC might affect the level of glycerol-3-phosphate in breast cancer cells. Synthesis of lysophosphatidic acid from glycerol-3-phosphate is mediated by the key regulator enzyme glycerol-3-phosphate acyltransferase (GPAM). Increased expression of GPAM is associated with an increased level of PtdCho and better survival in breast cancer patients (32). The glycerol-3-phosphate level might be reduced through GDPD5 and GDPD6 silencing, and it is possible that phospholipid biosynthesis is disturbed as a consequence, which could be a cause for the reduced cell proliferation that we observed in our study upon GDPD5 and GDPD6 silencing.
GDPD5 and GDPD6 siRNA treated MCF-7 and MDA-MB-231 cells did not undergo apoptosis, but did experience a decrease in cell proliferation in the case of GDPD5 siRNA silencing of MCF-7 cells only, while GDPD5 siRNA treatment of MDA-MB-231 cells, and GDPD6 siRNA treatment of MCF-7 and MDA-MB-231 cells did not have significant effects on cell proliferation. Stewart et al showed that, in agreement with our results, silencing or stably overexpressing GDPD6 did not affect proliferation in MCF-7 cells (20). The observed decrease in cell proliferation upon GDPD5 silencing in estrogen and progesterone receptor positive MCF-7 breast cancer cells is the first indication that GDPD5 is involved in pathways that mediate cell proliferation in these breast cancer cells. Prior to this study, GDPD5 had not been associated with cell proliferation. GDPD5 activity was previously shown to inactivate the Notch activator RECK (reversion-inducing cysteine-rich protein with kazal motifs) through GPI-anchor cleavage resulting in RECK release from the membrane (28). With previous reports showing that RECK acts as a metastasis suppressor (33), and that RECK increases proliferation, migration, and invasion in MDA-MB-231 cells (34), it is unlikely that GDPD5 silencing in our study resulted in reduced proliferation and migration through the RECK pathway. The pathways through which GDPD5 silencing reduces proliferation and migration in estrogen and progesterone receptor positive MCF-7 cells will be the subject of futures studies.
Serum response element (SRE) is a regulatory element for many important transcription factors in the MAPK pathway, which regulates a variety of cellular processes (35). GDPD5 is involved in the regulation of MAPK (36) and neuron differentiation (37). Overexpression of GDPD5 was shown to suppress the transcriptional activity of SRE in HEK-293 kidney cells, however, the biological effects and mechanisms causing this are still unclear (36). CHKA inhibitor treatment of breast cancer models has shown promising anti-proliferative effects and is currently being tested in clinical phase I trials (38–40). Inhibition of CHK resulted in decreased PtdCho and phosphatidic acid, which subsequently disrupts both the MAPK signaling and PI3K/AKT survival pathways (41). Interestingly, significantly positive correlations were found between the gene expression levels of GDPD5 and CHK in tumors from breast cancer patients and animal models, which further support the role of GDPD5 in oncogenic signaling and proliferation (18, 42). Based on our results, only GDPD5 siRNA treatment resulted in anti-proliferative effects, but not GDPD6 silencing. Treatment with GDPD5 and GDPD6 siRNA resulted in marginally increased GPC levels in the case of GDPD5, and dramatically increased GPC levels in the case of GDPD6. However, decreased GPC levels in response to neoadjuvant chemotherapy were shown to be associated with better response and survival in breast cancer patients (43, 44). The reasons behind these inconsistent observations are still unclear. Future studies exploring the mechanism by which GDPD5 silencing decreases cell proliferation in estrogen and progesterone receptor positive MCF-7 breast cancer cells are needed.
Cancer cells depend on migration and invasion to be able to move, enter the circulation, and metastasize to other organs (45). Decreased migration of MCF-7 cells caused by GDPD6 down-regulation was first observed by Stewart et al using scratch analysis (20), and was confirmed in our study along with showing decreased migration following GPDP5 silencing as well. In a recent study by the same group, silencing of GDPD6 resulted in a reduction of intergrin ß1 expression and disturbance of cell adhesion and spreading of MCF-7 cells (MDA-MB-231 cells had not yet been tested) (21). In our study, both GDPD5 and GDPD6 silencing reduced cell migration of MCF-7 cells using the scratch assay, with GDPD5 silencing leading to a more pronounced reduction in migration than GDPD6 silencing, while MCF-7 cells did not migrate or invade through the transwell. This latter observation might be due to the phenotypic characteristic of MCF-7 cells, which is a less aggressive and relatively non-invasive breast cancer cell line (46). Interestingly, not only did we find a significant decrease in cell migration and invasion in MDA-MB-231 breast cancer cells as a result of GDPD5 and GDPD6 silencing, we also observed that the inhibition of cell migration and invasion was more pronounced with GDPD5 as compared to GDPD6 siRNA treatment, which was consistent with our scratch assay results in MCF-7 cells. However, we need to further investigate the underlying molecular mechanisms to elucidate the roles of GDPD5 and GDPD6 in regulating these important cellular processes.
The tCho signal detected by in vivo 1H MRS was shown to provide beneficial additional information in the diagnosis of breast cancer (47). However, in a recent meta-analysis including 19 studies, the capability of tCho for detecting breast cancer was shown to have a relatively high specificity (~88%), but variable sensitivity (42–100%) (48). The spectral resolution of in vivo 1H MRS spectra acquired at 1.5 or 3.0 T clinical scanners nowadays cannot resolve the individual choline-containing metabolites. In our study, we observed an increased tCho level following GDPD6 siRNA treatment in MCF-7 cells. However, this was mainly due to the increased level of GPC, not PC, and free choline. As the tCho level is the sum of free choline, PC, and GPC, it depends on the expression of quite a large number of different regulatory genes and enzymes. Therefore interpreting the tCho signal as a general marker for choline phospholipid metabolism can be problematic.
A switch from low PC/high GPC to high PC/low GPC has been associated with malignant transformation of breast epithelial cells in vitro, where an increased PC/GPC ratio was shown to be correlated with increased malignancy (4). Significantly positive correlations between the PC/GPC ratio and GDPD5 have also been identified in breast tumors from patients (18). On the contrary, studies of in vivo animal models showed lower PC/GPC ratios in the most aggressive breast cancer subtypes (basal-like or triple negative) compared to the breast cancer subtype with a better survival (luminal) (42, 49). Similar to the in vivo animal studies, estrogen receptor negative breast cancers from patients have also been associated with low PC and high GPC compared to estrogen receptor positive tumors (50). These discrepancies that were observed between in vitro and in vivo studies might be due to the tumor microenvironment (42, 49).
We have previously demonstrated that a moderately negative correlation exists between GDPD5 expression and GPC levels in breast cancer cells and tumors (18). In contrast to our previous findings, GDPD5 was recently shown to have a positive, rather than negative, correlation with the GPC level in basal-like breast cancer and no significant correlation was observed between GDPD5 and GPC level in the luminal subtype (42). In the present study, GDPD6 silencing had a significantly more pronounced effect on the cellular GPC level and PC/GPC ratio compared to GDPD5 silencing. Additional studies are needed to investigate and validate the role of GDPD5 in regulating the cellular GPC level in breast cancer.
GDPD5 and GDPD6 are located in chromosomes 11q13.4-q13.5 and 20p12.3, respectively. The homology of these two genes was tested using NCBI blast (http://blast.ncbi.nlm.nih.gov/) and the results showed a total of 6% query subject alignments with 151 matched hits and highest percentage identity of 90%. This means that the genetic sequence of GDPD5 and GDPD6 are relatively different and might help explain the diverse effects observed in these two genes. The efficiency of GDPD5 siRNA knockdown was found to be dose-dependent and time-dependent in MCF-7 and MDA-MB-231 breast cancer cells. This information is critical for designing a robust and effective treatment protocol where repeated administration or combination of GDPD5 and GDPD6 siRNA treatment should be considered. Possible side effects of GDPD5 and GDPD6 silencing on non-tumoral tissues also need to be investigated, as it is for example well known that GPC:Cho phosphodiesterases play an important role in the control of osmotic stress in renal epithelial cells (51, 52).
RNA interference therapeutics has advanced in the last decade and has been shown to be well tolerated in pre-clinical animal models with the potential to suppress tumor growth and survival of tumor cells (53). The translation of siRNA treatment protocols to patients is currently being tested in clinical trials. Preliminary results show that this method may have a significant role in the development of future drugs for cancer therapy (54). Future studies investigating the interactions of GDPD5 and GDPD6 with other genes and proteins and their involvement in the regulation of other pathways besides choline phospholipid metabolism might be of interest.
Conclusions
By comparing the effects of GDPD5 and GDPD6 silencing, we observed that GDPD6 silencing resulted in a more pronounced increase in the GPC level of breast cancer cells, while GDPD5 silencing caused a more severe decrease in cell proliferation, migration, and invasion. Further investigations of the molecular and metabolic effects of GDPD5 and GDPD6 silencing alone or in combination are necessary to better understand their roles in choline phospholipid metabolism and to validate their potential as new targets for treatment of breast cancer.
Acknowledgments
Funding
This work was supported by NIH R01 CA134695.
References
- 1.Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Aboud OA, Weiss RH. New opportunities from the cancer metabolome. Clin Chem. 2013;59:138–46. doi: 10.1373/clinchem.2012.184598. [DOI] [PubMed] [Google Scholar]
- 3.Sitter B, Sonnewald U, Spraul M, Fjosne HE, Gribbestad IS. High-resolution magic angle spinning MRS of breast cancer tissue. NMR Biomed. 2002;15:327–37. doi: 10.1002/nbm.775. [DOI] [PubMed] [Google Scholar]
- 4.Aboagye EO, Bhujwalla ZM. Malignant Transformation Alters Membrane Choline Phospholipid Metabolism of Human Mammary Epithelial Cells. Cancer Res. 1999;59:80–4. [PubMed] [Google Scholar]
- 5.Ackerstaff E, Glunde K, Bhujwalla ZM. Choline phospholipid metabolism: a target in cancer cells? J Cell Biochem. 2003;90:525–33. doi: 10.1002/jcb.10659. [DOI] [PubMed] [Google Scholar]
- 6.Glunde K, Bhujwalla ZM, Ronen SM. Choline metabolism in malignant transformation. Nature reviews Cancer. 2011;11:835–48. doi: 10.1038/nrc3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramirez de Molina A, Gutierrez R, Ramos MA, Silva JM, Silva J, Bonilla F, Sanchez JJ, Lacal JC. Increased choline kinase activity in human breast carcinomas: clinical evidence for a potential novel antitumor strategy. Oncogene. 2002;21:4317–22. doi: 10.1038/sj.onc.1205556. [DOI] [PubMed] [Google Scholar]
- 8.Foster DA, Xu L. Phospholipase D in cell proliferation and cancer. Mol Cancer Res. 2003;1:789–800. [PubMed] [Google Scholar]
- 9.Glunde K, Raman V, Mori N, Bhujwalla ZM. RNA interference-mediated choline kinase suppression in breast cancer cells induces differentiation and reduces proliferation. Cancer Res. 2005;65:11034–43. doi: 10.1158/0008-5472.CAN-05-1807. [DOI] [PubMed] [Google Scholar]
- 10.Granata A, Nicoletti R, Tinaglia V, De Cecco L, Pisanu ME, Ricci A, Podo F, Canevari S, Iorio E, Bagnoli M, Mezzanzanica D. Choline kinase-alpha by regulating cell aggressiveness and drug sensitivity is a potential druggable target for ovarian cancer. British journal of cancer. 2014;110:330–40. doi: 10.1038/bjc.2013.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mori N, Glunde K, Takagi T, Raman V, Bhujwalla ZM. Choline kinase down-regulation increases the effect of 5-fluorouracil in breast cancer cells. Cancer Res. 2007;67:11284–90. doi: 10.1158/0008-5472.CAN-07-2728. [DOI] [PubMed] [Google Scholar]
- 12.Shah T, Wildes F, Penet MF, Winnard PT, Jr, Glunde K, Artemov D, Ackerstaff E, Gimi B, Kakkad S, Raman V, Bhujwalla ZM. Choline kinase overexpression increases invasiveness and drug resistance of human breast cancer cells. NMR Biomed. 2010;23:633–42. doi: 10.1002/nbm.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katz-Brull R, Lavin PT, Lenkinski RE. Clinical Utility of Proton Magnetic Resonance Spectroscopy in Characterizing Breast Lesions. J Natl Cancer Inst. 2002;94:1197–203. doi: 10.1093/jnci/94.16.1197. [DOI] [PubMed] [Google Scholar]
- 14.Meisamy S, Bolan PJ, Baker EH, Bliss RL, Gulbahce E, Everson LI, Nelson MT, Emory TH, Tuttle TM, Yee D, Garwood M. Neoadjuvant chemotherapy of locally advanced breast cancer: predicting response with in vivo (1)H MR spectroscopy–a pilot study at 4 T. Radiology. 2004;233:424–31. doi: 10.1148/radiol.2332031285. [DOI] [PubMed] [Google Scholar]
- 15.Shin HJ, Baek HM, Cha JH, Kim HH. Evaluation of breast cancer using proton MR spectroscopy: total choline peak integral and signal-to-noise ratio as prognostic indicators. AJR Am J Roentgenol. 2012;198:W488–97. doi: 10.2214/AJR.11.7292. [DOI] [PubMed] [Google Scholar]
- 16.Danishad KK, Sharma U, Sah RG, Seenu V, Parshad R, Jagannathan NR. Assessment of therapeutic response of locally advanced breast cancer (LABC) patients undergoing neoadjuvant chemotherapy (NACT) monitored using sequential magnetic resonance spectroscopic imaging (MRSI) NMR Biomed. 2010;23:233–41. doi: 10.1002/nbm.1436. [DOI] [PubMed] [Google Scholar]
- 17.Baek HM, Chen JH, Nalcioglu O, Su MY. Proton MR spectroscopy for monitoring early treatment response of breast cancer to neo-adjuvant chemotherapy. Ann Oncol. 2008;19:1022–4. doi: 10.1093/annonc/mdn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao MD, Dopkens M, Krishnamachary B, Vesuna F, Gadiya MM, Lonning PE, Bhujwalla ZM, Gribbestad IS, Glunde K. Glycerophosphodiester phosphodiesterase domain containing 5 (GDPD5) expression correlates with malignant choline phospholipid metabolite profiles in human breast cancer. NMR Biomed. 2012;25:1033–42. doi: 10.1002/nbm.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zablocki K, Miller SP, Garcia-Perez A, Burg MB. Accumulation of glycerophosphocholine (GPC) by renal cells: osmotic regulation of GPC:choline phosphodiesterase. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:7820–4. doi: 10.1073/pnas.88.17.7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stewart JD, Marchan R, Lesjak MS, Lambert J, Hergenroeder R, Ellis JK, Lau CH, Keun HC, Schmitz G, Schiller J, Eibisch M, Hedberg C, Waldmann H, Lausch E, Tanner B, Sehouli J, Sagemueller J, Staude H, Steiner E, Hengstler JG. Choline-releasing glycerophosphodiesterase EDI3 drives tumor cell migration and metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:8155–60. doi: 10.1073/pnas.1117654109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lesjak MS, Marchan R, Stewart JD, Rempel E, Rahnenfuhrer J, Hengstler JG. EDI3 links choline metabolism to integrin expression, cell adhesion and spreading. Cell adhesion & migration. 2014:8. doi: 10.4161/cam.29284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wijnen JP, Jiang L, Greenwood TR, Cheng M, Dopkens M, Cao MD, Bhujwalla ZM, Krishnamachary B, Klomp DW, Glunde K. Silencing of the glycerophosphocholine phosphodiesterase GDPD5 alters the phospholipid metabolite profile in a breast cancer model in vivo as monitored by (31) P MRS. NMR Biomed. 2014;27:692–9. doi: 10.1002/nbm.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soule HD, Vazguez J, Long A, Albert S, Brennan M. A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst. 1973;51:1409–16. doi: 10.1093/jnci/51.5.1409. [DOI] [PubMed] [Google Scholar]
- 24.Cailleau R, Young R, Olive M, Reeves WJ., Jr Breast tumor cell lines from pleural effusions. J Natl Cancer Inst. 1974;53:661–74. doi: 10.1093/jnci/53.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glunde K, Guggino SE, Solaiyappan M, Pathak AP, Ichikawa Y, Bhujwalla ZM. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia. 2003;5:533–45. doi: 10.1016/s1476-5586(03)80037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyagi RK, Azrad A, Degani H, Salomon Y. Simultaneous extraction of cellular lipids and water-soluble metabolites: evaluation by NMR spectroscopy. Magn Reson Med. 1996;35:194–200. doi: 10.1002/mrm.1910350210. [DOI] [PubMed] [Google Scholar]
- 27.Glunde K, Jie C, Bhujwalla ZM. Molecular causes of the aberrant choline phospholipid metabolism in breast cancer. Cancer Res. 2004;64:4270–6. doi: 10.1158/0008-5472.CAN-03-3829. [DOI] [PubMed] [Google Scholar]
- 28.Park S, Lee C, Sabharwal P, Zhang M, Meyers CL, Sockanathan S. GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science (New York, NY) 2013;339:324–8. doi: 10.1126/science.1231921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez-Murray JP, McMaster CR. Glycerophosphocholine catabolism as a new route for choline formation for phosphatidylcholine synthesis by the Kennedy pathway. J Biol Chem. 2005;280:38290–6. doi: 10.1074/jbc.M507700200. [DOI] [PubMed] [Google Scholar]
- 30.Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I. Identification and characterization of the high-affinity choline transporter. Nat Neurosci. 2000;3:120–5. doi: 10.1038/72059. [DOI] [PubMed] [Google Scholar]
- 31.Iorio E, Ricci A, Bagnoli M, Pisanu ME, Castellano G, Di Vito M, Venturini E, Glunde K, Bhujwalla ZM, Mezzanzanica D, Canevari S, Podo F. Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res. 2010;70:2126–35. doi: 10.1158/0008-5472.CAN-09-3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brockmoller SF, Bucher E, Muller BM, Budczies J, Hilvo M, Griffin JL, Oresic M, Kallioniemi O, Iljin K, Loibl S, Darb-Esfahani S, Sinn BV, Klauschen F, Prinzler J, Bangemann N, Ismaeel F, Fiehn O, Dietel M, Denkert C. Integration of metabolomics and expression of glycerol-3-phosphate acyltransferase (GPAM) in breast cancer-link to patient survival, hormone receptor status, and metabolic profiling. Journal of proteome research. 2012;11:850–60. doi: 10.1021/pr200685r. [DOI] [PubMed] [Google Scholar]
- 33.Walsh LA, Roy DM, Reyngold M, Giri D, Snyder A, Turcan S, Badwe CR, Lyman J, Bromberg J, King TA, Chan TA. RECK Controls Breast Cancer Metastasis by Modulating a Convergent, STAT3-dependent Neoangiogenic Switch. Oncogene. 2015;34:2189–203. doi: 10.1038/onc.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Kong X, Li J, Luo Q, Li X, Shen L, Chen L, Fang L. miR-96 promotes tumor proliferation and invasion by targeting RECK in breast cancer. Oncology reports. 2014;31:1357–63. doi: 10.3892/or.2013.2934. [DOI] [PubMed] [Google Scholar]
- 35.Treisman R. The serum response element. Trends Biochem Sci. 1992;17:423–6. doi: 10.1016/0968-0004(92)90013-y. [DOI] [PubMed] [Google Scholar]
- 36.Lang Q, Zhang H, Li J, Yin H, Zhang Y, Tang W, Wan B, Yu L. Cloning and characterization of a human GDPD domain-containing protein GDPD5. Mol Biol Rep. 2008;35:351–9. doi: 10.1007/s11033-007-9093-3. [DOI] [PubMed] [Google Scholar]
- 37.Rao M, Sockanathan S. Transmembrane protein GDE2 induces motor neuron differentiation in vivo. Science (New York, NY) 2005;309:2212–5. doi: 10.1126/science.1117156. [DOI] [PubMed] [Google Scholar]
- 38.Ramirez de Molina A, Banez-Coronel M, Gutierrez R, Rodriguez-Gonzalez A, Olmeda D, Megias D, Lacal JC. Choline kinase activation is a critical requirement for the proliferation of primary human mammary epithelial cells and breast tumor progression. Cancer Res. 2004;64:6732–9. doi: 10.1158/0008-5472.CAN-04-0489. [DOI] [PubMed] [Google Scholar]
- 39.Hernandez-Alcoceba R, Saniger L, Campos J, Nunez MC, Khaless F, Gallo MA, Espinosa A, Lacal JC. Choline kinase inhibitors as a novel approach for antiproliferative drug design. Oncogene. 1997;15:2289–301. doi: 10.1038/sj.onc.1201414. [DOI] [PubMed] [Google Scholar]
- 40.Krishnamachary B, Glunde K, Wildes F, Mori N, Takagi T, Raman V, Bhujwalla ZM. Noninvasive detection of lentiviral-mediated choline kinase targeting in a human breast cancer xenograft. Cancer Res. 2009;69:3464–71. doi: 10.1158/0008-5472.CAN-08-4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yalcin A, Clem B, Makoni S, Clem A, Nelson K, Thornburg J, Siow D, Lane AN, Brock SE, Goswami U, Eaton JW, Telang S, Chesney J. Selective inhibition of choline kinase simultaneously attenuates MAPK and PI3K/AKT signaling. Oncogene. 2010;29:139–49. doi: 10.1038/onc.2009.317. [DOI] [PubMed] [Google Scholar]
- 42.Grinde MT, Skrbo N, Moestue SA, Rodland EA, Borgan E, Kristian A, Sitter B, Bathen TF, Borresen-Dale AL, Maelandsmo GM, Engebraaten O, Sorlie T, Marangoni E, Gribbestad IS. Interplay of choline metabolites and genes in patient-derived breast cancer xenografts. Breast cancer research : BCR. 2014;16:R5. doi: 10.1186/bcr3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao MD, Sitter B, Bathen TF, Bofin A, Lonning PE, Lundgren S, Gribbestad IS. Predicting long-term survival and treatment response in breast cancer patients receiving neoadjuvant chemotherapy by MR metabolic profiling. NMR Biomed. 2012;25:369–78. doi: 10.1002/nbm.1762. [DOI] [PubMed] [Google Scholar]
- 44.Cao MD, Giskeodegard GF, Bathen TF, Sitter B, Bofin A, Lonning PE, Lundgren S, Gribbestad IS. Prognostic value of metabolic response in breast cancer patients receiving neoadjuvant chemotherapy. BMC cancer. 2012;12:39. doi: 10.1186/1471-2407-12-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nature reviews Cancer. 2003;3:362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 46.Thompson EW, Reich R, Shima TB, Albini A, Graf J, Martin GR, Dickson RB, Lippman ME. Differential regulation of growth and invasiveness of MCF-7 breast cancer cells by antiestrogens. Cancer Res. 1988;48:6764–8. [PubMed] [Google Scholar]
- 47.Begley JK, Redpath TW, Bolan PJ, Gilbert FJ. In vivo proton magnetic resonance spectroscopy of breast cancer: a review of the literature. Breast cancer research : BCR. 2012;14:207. doi: 10.1186/bcr3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baltzer PA, Dietzel M, Kaiser WA. MR-spectroscopy at 1.5 tesla and 3 tesla. Useful? A systematic review and meta-analysis. Eur J Radiol. 2012;81(Suppl 1):S6–9. doi: 10.1016/S0720-048X(12)70003-7. [DOI] [PubMed] [Google Scholar]
- 49.Moestue SA, Borgan E, Huuse EM, Lindholm EM, Sitter B, Borresen-Dale AL, Engebraaten O, Maelandsmo GM, Gribbestad IS. Distinct choline metabolic profiles are associated with differences in gene expression for basal-like and luminal-like breast cancer xenograft models. BMC cancer. 2010;10:433. doi: 10.1186/1471-2407-10-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giskeodegard GF, Grinde MT, Sitter B, Axelson DE, Lundgren S, Fjosne HE, Dahl S, Gribbestad IS, Bathen TF. Multivariate modeling and prediction of breast cancer prognostic factors using MR metabolomics. Journal of proteome research. 2010;9:972–9. doi: 10.1021/pr9008783. [DOI] [PubMed] [Google Scholar]
- 51.Gallazzini M, Ferraris JD, Burg MB. GDPD5 is a glycerophosphocholine phosphodiesterase that osmotically regulates the osmoprotective organic osmolyte GPC. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11026–31. doi: 10.1073/pnas.0805496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Topanurak S, Ferraris JD, Li J, Izumi Y, Williams CK, Gucek M, Wang G, Zhou X, Burg MB. High NaCl- and urea-induced posttranslational modifications that increase glycerophosphocholine by inhibiting GDPD5 phosphodiesterase. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7482–7. doi: 10.1073/pnas.1305220110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Devi GR. siRNA-based approaches in cancer therapy. Cancer Gene Ther. 2006;13:819–29. doi: 10.1038/sj.cgt.7700931. [DOI] [PubMed] [Google Scholar]
- 54.Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, Paz-Ares L, Cho DC, Infante JR, Alsina M, Gounder MM, Falzone R, Harrop J, White AC, Toudjarska I, Bumcrot D, Meyers RE, Hinkle G, Svrzikapa N, Hutabarat RM, Clausen VA, Cehelsky J, Nochur SV, Gamba-Vitalo C, Vaishnaw AK, Sah DW, Gollob JA, Burris HA., 3rd First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer discovery. 2013;3:406–17. doi: 10.1158/2159-8290.CD-12-0429. [DOI] [PubMed] [Google Scholar]