

Abstract
The prodiginine family of bacterial alkaloids is a diverse set of heterocyclic natural products that have likely been known to man since antiquity. In more recent times, these alkaloids have been discovered to span a wide range of chemical structures that possess a number of interesting biological activities. This review provides a comprehensive overview of research undertaken toward the isolation and structural elucidation of the prodiginine family of natural products. Additionally, research toward chemical synthesis of the prodiginine alkaloids over the last several decades is extensively reviewed. Finally, the current, evidence-based understanding of the various biosynthetic pathways employed by bacteria to produce prodiginine alkaloids is summarized.
Graphical Abstract
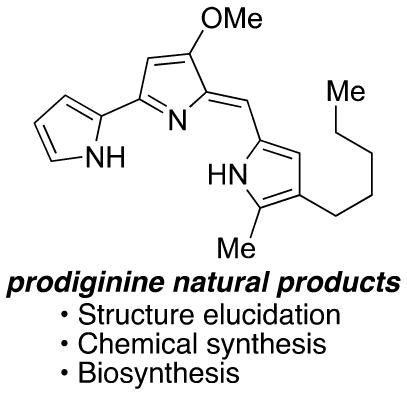
1. INTRODUCTION
In the summer of 1819, the apparently spontaneous, brilliant reddening of a farmer’s polenta (boiled cornmeal) created a stir in Padua, Italy.1 Local peasants called the occurrence “bloody polenta”, believing it to be of diabolical origin, and implored priests to banish the evil spirits behind the event. The hype over “bloody polenta” achieved such a state that the Paduan police department appointed a committee, composed mostly of professors from the University of Padua, to investigate the reports. Among the committee members was the pharmacist Bartolomeo Bizio, who demonstrated through experiment that the development of red color on polenta was dependent on humidity, that the presence of “foul air” accelerated the development of red color, and that the cause of red color was transmissible both by air and by contact.
Bizio therefore concluded that the discoloration was not supernatural but caused by the growth of a microorganism, erroneously classifying the bacterium as a fungus due to its ability to reproduce even after exposure to camphor, turpentine, or tobacco vapors and high temperature. Bizio named the organism Serratia marcescens. The descriptor marcescens, derived from the Latin word meaning “decaying,” was chosen due to his observation that the organism dissolved into a red fluid and mucus-like matter upon reaching maturity. Bizio’s deductions were remarkable as they were made before the birth of Pasteur. The brilliant red pigment that Bizio attempted to isolate from Serratia would be identified much later and named prodigiosin (Figure 1).
Figure 1.
Prodigiosin (1), the prototypical prodiginine.
Today, it is speculated that because of its ability to grow on cereal grains, its dripping fluid-like appearance, and its ability to produce the bright red pigment prodigiosin, S. marcescens is responsible for the numerous accounts of “bleeding bread” phenomena in history, akin to the aforementioned 19th-century event in Padua.2 Recorded events date as far back as 322 B.C., when soldiers under Alexander the Great believed they saw blood inside a piece of bread, an omen interpreted as foreshadowing victory in a coming battle. Perhaps the best known example of such events is the Eucharistic miracle of Bolsena. In a 1263 celebration of Mass, a Roman Catholic priest skeptical of the Catholic doctrine of transubstantiation is believed to have found blood smattered over the bread he broke as the host for Communion. It was only after the investigation of this incident that Pope Urban IV issued a papal bull in 1264 establishing Corpus Christi as the first universal feast throughout the Latin Rite. The event is immortalized in the Vatican by one of Raphael’s Stanze, “The Mass at Bolsena” (1512). The role played by prodigiosin in such “prodigious” events is aptly reflected in its name.
Interest in the prodiginines is not limited to their role in human culture. As early as 1823, Bizio thought to apply the red alcohol extract of Serratia in the dyeing of silk and wool.1 Though prodigiosin itself was found to be too light-sensitive for practical application as a dye, in more recent times, the red color of prodigiosin has made its biosynthetic pathway a useful model system for bacterial quorum sensing research3 as well as a useful educational tool for university-level biology students.4
The prodiginines have been continuously investigated for medically relevant properties including antimalarial activity,5–9 a distinct mechanism of immunosuppression from cyclosporine A, FK506, and rapamycin,10–19 and their ability to selectively induce apoptosis in many human cancer cell lines.8,20–27 A synthetic analogue based on the prodiginine family, obatoclax, was used in multiple phase I and II combination cancer chemotherapy studies.28–33 The quorum sensing control of prodigiosin biosynthesis and medicinal properties of the prodiginines have been previously reviewed.3,8,24,34,35
In this review, efforts in the isolation, structural elucidation, total syntheses, and biosyntheses of the prodiginine family of natural products will be discussed, placing in context the relationships between discoveries in the aforementioned fields. In particular, the role of total synthesis in the structural elucidation and structural revision of the prodiginines will be highlighted, updating and elaborating on a prior review from 2003 by Fürstner.36
2. ISOLATION AND STRUCTURAL ELUCIDATION
The structures of the prodiginines have consistently elicited curiosity in the chemical and biological communities, and new structural questions about these natural products have continued to arise even since the complete identification of prodigiosin in 1962. The following section recounts efforts to isolate and elucidate the structures of prodiginines between 1920 and 2015.
2.1. Prodigiosin
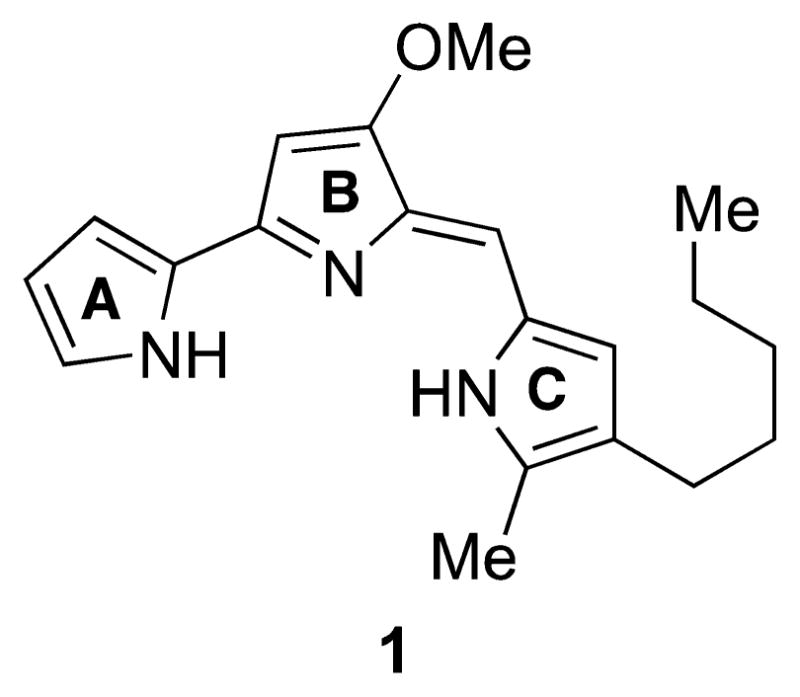
Prodigiosin was first isolated in pure form from Serratia marcescens (then called Bacillus prodigiosus) in 1929 by Wrede and Hettche37 at the University of Greifswald. On the basis of degradation studies, Wrede determined that prodigiosin contained three pyrrole groups linked in an unknown fashion: pyrrole, 3-methoxypyrrole, and 2-methyl-3-amylpyrrole.38 In 1933, Wrede and Rothhaas39,40 suggested structures 1, 2, and 3 for prodigiosin, arbitrarily favoring the tripyrrylmethene structure 3 (Figure 2).
Figure 2.

Structures originally proposed by Wrede and Rothhaas39,40 for prodigiosin. Stucture 1 is now known to be correct.
For the next 20 years, synthetic efforts focused on preparing tripyrrylmethene structures related to 2 and 3, but comparisons of synthetic model compounds (e.g., 4, Figure 3) with isolated prodigiosin could neither definitively confirm nor refute Wrede’s favored structure 3, since the synthetic tripyrrylmethenes differed considerably in substitution from prodigiosin.
Figure 3.

Early structural studies attempted to match the UV spectrum of prodigiosin to synthetic model compounds such as 4.
It was not for another three decades after Wrede’s proposal that definitive evidence against structures 2 and 3 became available. In 1956, Santer and Vogel41 isolated a compound with the formula C10H10O2N2 from an S. marcescens mutant blocked in prodigiosin biosynthesis and demonstrated that this compound could be converted to prodigiosin by a second mutant. Wasserman et al. realized that prodigiosin (C20H25ON3) was the formal condensation product of Vogel’s compound and Wrede’s degradation product 2-methyl-3-amylpyrrole (C10H17N, 7), and in 196042 they reported the partial synthesis of prodigiosin through treatment of Vogel’s compound with 7 under acidic conditions. Further degradation studies on Vogel’s compound by the same group suggested that it contained two α,α′-linked pyrroles and an aldehyde group, implying that tripyrrylmethene structures 2 and 3 for prodigiosin were highly unlikely.
In 1962, Rapoport and Willson43 reported the syntheses of bipyrrole aldehydes 5 and 6, as well as the results of condensation of both materials with 2-methyl-3-amylpyrrole 7 and isomers of 7. They found that only the condensation of 4-methoxy-2,2′-bipyrrole-5-carbaldehyde (MBC, 6) with pyrrole 7 provided material that was analytically identical to naturally occurring prodigiosin, firmly establishing the identity of Vogel’s C10H10O2N2 compound as 6 and of prodigiosin as 1 (Figure 4).
Figure 4.

Rapoport and Willson’s43 condensation of aldehyde 5 with pyrrole 7 yielded a compound, iso-1, whose UV–vis spectrum differed significantly from natural prodigiosin. Condensation of aldehyde 6 with pyrrole 7 yielded a compound identical to natural prodigiosin (1).
2.2. Close Analogues of Prodigiosin
Since Rapoport and Willson’s43 1962 structural elucidation of prodigiosin, several closely related natural products have been discovered (Figure 5). Among them are alkyl-chain homologues, such as propylprodigiosin (8),44 butylprodigiosin (9),44 hexylprodigiosin (11),44,45 and heptylprodigiosin (12).46 Such metabolites likely arise through incorporation of a different number of malonyl-CoA extender units during fatty acid biosynthesis or the utilization of propionyl-CoA instead of acetyl-CoA as a starter unit by the fatty acid synthase (see section 4.4).47
Figure 5.

Structures of alkyl-chain homologues of prodigiosin (8–12) and norprodigiosin (13), a demethylated prodigiosin analogue.
To date, only mass spectrometric data have been reported in support of the structures of the alkyl-chain homologues other than heptylprodigiosin.48 The O-desmethyl analogue of prodigiosin, norprodigiosin, was isolated from S. marcescens mutants and chemically characterized in 1964 by Hearn et al.49 Norprodigiosin (13) likely arises through the condensation of 4-hydroxy-2,2′-bipyrrole-5-carbaldehyde, MBC, with 2-methyl-3-amylpyrrole, MAP (see section 4.5).
In 2008, Hemscheidt and co-workers50 reported the isolation of the first prodigiosin analogue with a substituent at C-2 of ring A, 2-(p-hydroxybenzyl)prodigiosin (14), from Pseudoalteromonas rubra (Figure 6). Taken from the surface of a nudibranch obtained from the waters off Oahu, Hawaii, this compound was found to have broad-spectrum activity against Escherichia coli, Staphylococcus aureus, methicillin-resistant S. aureus, Candida albicans, and human ovarian adenocarcinoma cells. This represents the first member of a new class of acyclic 2-substituted prodiginines. On the basis of the currently proposed biosynthetic pathway for S. marcescens (see section 4.2), it is not clear how such a compound would arise. Hemscheidt and co-workers have speculated that 14 may be the result of nonribosomal peptide synthase incorporation of p-hydroxybenzyl-substituted proline into pyrrole ring A in place of proline (see section 4.2).
Figure 6.
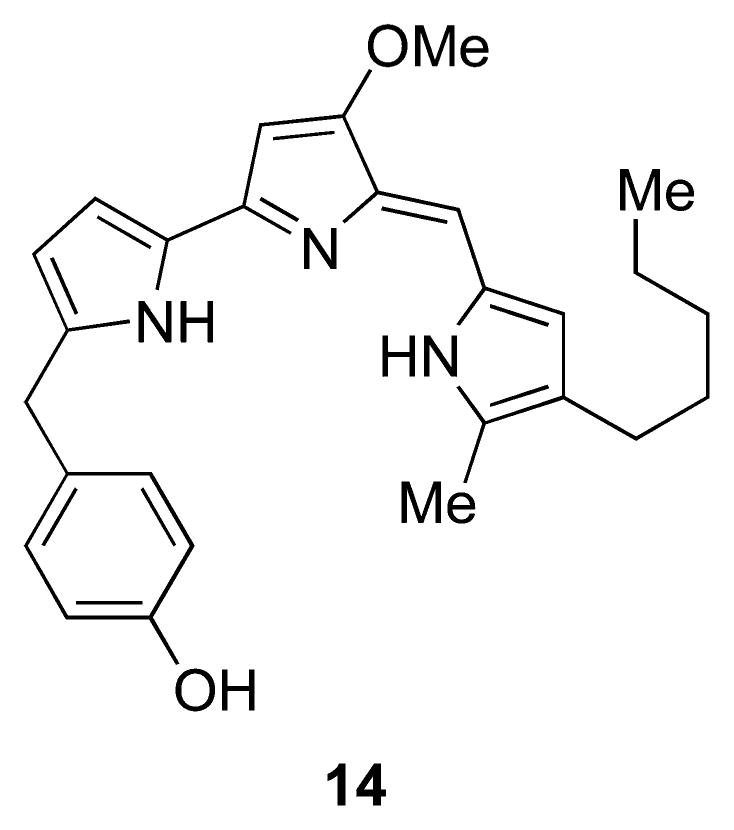
Structure of a unique prodigiosin analogue (14) from P. rubra with a substituent at C-2 of ring A.
2.3. Cycloprodigiosin
In 1979, nearly 20 years after the structural elucidation of prodigiosin, Gerber and Gauthier51 isolated a unique cyclic analogue of prodigiosin from Alteromonas rubra, a marine bacterium from Mediterranean coastal waters. Notably, this new prodigiosin was not produced by strains previously found to produce prodigiosin, including S. marcescens. On the basis of mass spectrometric and 1H NMR data, Gerber assigned the cyclopentane structure 15 to the new compound (Figure 7).
Figure 7.

Originally proposed (15) and subsequently revised (16) structures for cycloprodigiosin.
Four years later, Lattasch and Thomson52 reisolated the cyclic prodigiosin analogue and came to the conclusion that this compound, which they called cycloprodigiosin, should be assigned the structure 16 instead of 15. Lattasch and Thomson attributed Gerber’s misassignment to the presence of an aliphatic impurity in Gerber’s original sample, which caused a triplet to appear at 0.95 ppm in the 1H NMR spectrum, leading Gerber to incorrectly conclude that a methylene group was present between the methyl group and the aliphatic ring. Simultaneously, Gerber53 discovered a salt-water marsh bacterium, Beneckea gazogenes, which provided much higher yields of cycloprodigiosin than A. rubra. With greater quantities of cycloprodigiosin in hand, Gerber was able to procure 13C NMR data that allowed her to reassign its structure to 16. To resolve lingering confusion regarding the structure of cycloprodigiosin, Wasserman and Fukuyuma54 completed a short total synthesis of the racemate of 16. Wasserman’s synthetic material proved to be “identical with samples of the natural product derived from both B. gazogenes and A. rubra”, confirming the structure of cycloprodigiosin as 16.
It was a further three decades after the gross structural assignment of cycloprodigiosin (16) was confirmed that information regarding its stereochemical configuration came to light. In 2015, Sarpong and co-workers55 reported the synthesis of both enantiomers of cycloprodigiosin (16) through an efficient synthetic route (see section 3.2). Isolation of natural cycloprodigiosin (16) from Pseudoalteromonas rubra (Gauthier) (ATCC 29570) enabled subsequent determination that the natural material was produced by the microorganism as a scalemic mixture of (R)-16 and (S)-16 in a ratio of 83:17 (Figure 8).
Figure 8.

Sarpong and co-workers55 determined that naturally occurring cycloprodigiosin (16) is a 83:17 mixture of (R) and (S) enantiomers.
2.4. Undecylprodigiosin
In 1961, shortly after Wasserman reported a partial synthesis of prodigiosin, Perry56 reported the identification of a red pigment from an actinobacterium belonging to the Streptomyces genus. Perry claimed that the isolated pigment was identical to that isolated from S. marcescens, based on UV–vis spectra recorded in acid and alkaline solutions and on thin-layer chromatographic (TLC) analysis. In 1966, however, Wasserman et al.58 at Yale and Nagatsu and co-workers57 in Tokyo independently disclosed the isolation of the same pigment from two strains of Streptomyces and determined that it possessed the chemical formula C25H35N3O. Nagatsu and co-workers assigned structure 17 to this compound and named it “prodigiosin-25 C” on the basis of NMR, mass spectrometric, and degradation studies (Figure 9).
Figure 9.
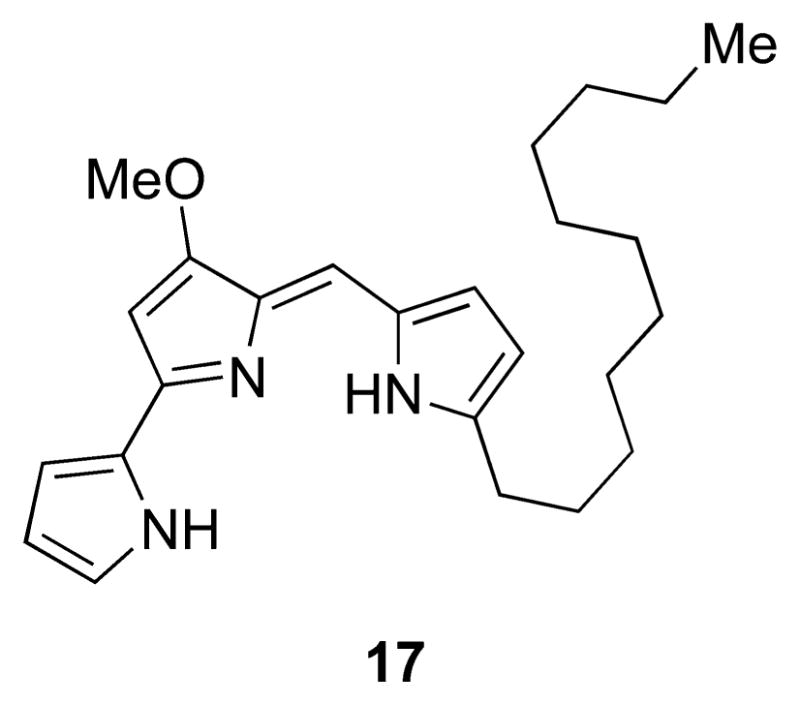
Structure of undecylprodigiosin (17), the 25-carbon prodiginine identified independently by Wasserman et al.58 and Nagatsu and co-workers.57
The Wasserman group found that the isolated pigment could be synthesized by condensation of 2-undecylpyrrole with previously synthesized aldehyde 6, and thus named the compound undecylprodigiosin. Since 1966, undecylprodigiosin has been isolated from numerous other actinobacterial genera, including Streptoverticillium, Actinomadura, and Saccharopolyspora, but it has not been isolated from proteobacteria.3 Similarly, prodigiosin has been isolated from several different genera of proteobacteria (Serratia, Hahella, Pseudomonas, Vibrio), but it has not been reported to be produced by an actinobacterium. An evolutionary explanation for these observations has yet to be proposed.
2.5. Metacycloprodigiosin
When Wasserman et al.58 first elucidated the structure of undecylprodigiosin from Streptomyces longisporus ruber, they noted that a second, more complex, 25-carbon pigment with an additional element of unsaturation (C25H33N3O, vs C25H35N3O for undecylprodigiosin) was also produced. Several years earlier, Wasserman et al.59 had suggested that this compound might be a prodigiosin analogue derived from 2-methyl-3-heptyl-4-propylpyrrole (Figure 10). Shortly thereafter, they showed this hypothesis was false by synthesizing 2-methyl-3-heptyl-4-propylpyrrole (18) and condensing it with MBC 6 (Figure 10). This reaction yielded a prodiginine (19) with properties that differed from those of the natural product in many respects.60
Figure 10.

Wasserman’s original structural proposal for metacycloprodigiosin (19), based on the incorrect assumption that Streptomyces species produce prodigiosin (1). The properties of synthetic 19 did not match those of isolated metacycloprodigiosin.
Three years after they reported the structural elucidation of undecylprodigioisn, Wasserman et al.61 deduced the correct structure for the desaturated undecylprodigiosin derivative produced by S. longisporus ruber. The structure was confirmed by total synthesis of a racemic sample and the compound was named metacycloprodigiosin (20, Figure 11).62 With the benefit of hindsight, this was a confusing choice of nomenclature, because other meta-disubstituted prodiginines were subsequently discovered (e.g., see section 2.6).
Figure 11.
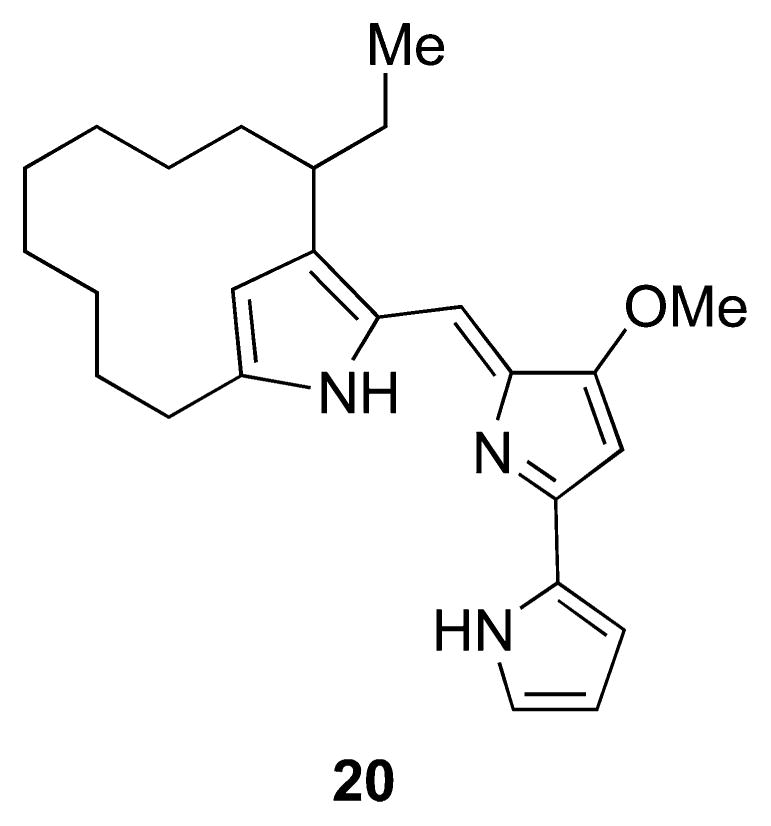
Structure of metacycloprodigiosin (20), deduced by Wasserman and co-workers61 and confirmed by total synthesis.62 The absolute stereochemistry was not defined.
Wasserman et al.62 also noted that metacycloprodigiosin is optically active. This was significant because it indicated that carbocyclic prodiginine derivatives, such as metacycloprodigiosin and cycloprodigiosin, are enzymatically biosynthesized and do not simply result from adventitious oxidation of the corresponding precursors (1 and 17, respectively). Indeed, in 2001, Challis and co-workers63 postulated that an orthologue of the Streptomyces coelicolor Rieske oxygenase-like enzyme RedG is likely responsible for the oxidative cyclization of undecylprodigiosin (17) to metacycloprodigiosin (20). A decade later, Challis and co-workers64 reported that RedG catalyzes the conversion of undecylprodigiosin (17) to streptorubin B (22) and that McpG, a RedG orthologue from S. longisporus ruber, catalyzes the conversion of undecylprodigiosin (17) to metacycloprodigiosin (20) (see section 4.6).
In 2009, 40 years after metacycloprodigiosin’s discovery, Clift and Thomson65 completed the first enantioselective total synthesis of metacycloprodigiosin in an effort to determine the absolute configuration of the natural product (see section 3.3). Although a natural sample of metacycloprodigiosin could not be obtained at the time of the report, subsequent circular dichroic (CD) spectroscopic comparison of synthetic (R)-metacycloprodigiosin with natural metacycloprodigiosin isolated from S. longisporus ruber showed that the absolute configuration of natural metacycloprodigiosin is R (as shown in Figure 12).66
Figure 12.
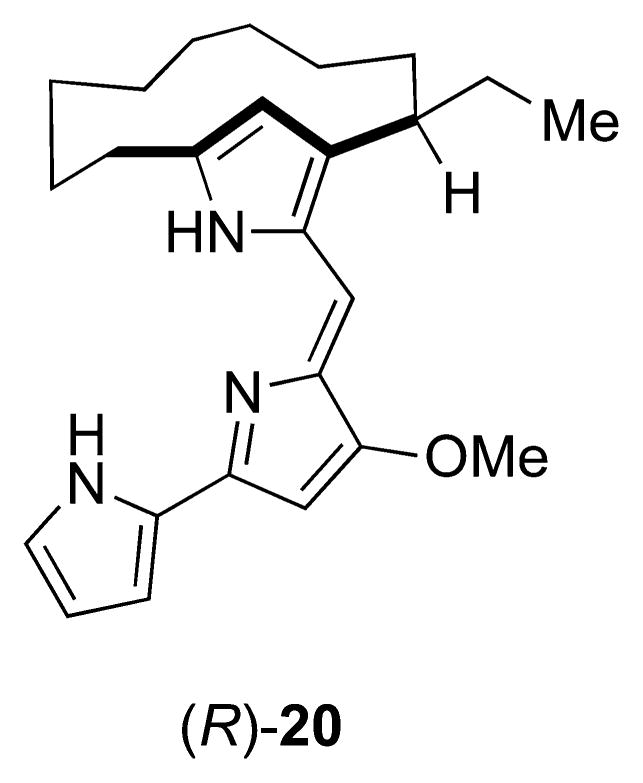
Absolute configuration of metacycloprodigiosin was assigned as R through total synthesis.
2.6. Streptorubin B
In 1975, Gerber67 reported the isolation and structural elucidation of two pink pigments from Streptomyces sp. Y-42, isolated from leaf and grass compost. One pigment was easily identified as undecylprodigiosin (17) on the basis that its chromic acid oxidation product was undecanoic acid. The physical properties of the second pigment strongly resembled those of metacycloprodigiosin (20), but the two compounds had different TLC Rf values and produced significantly different fragment ions in mass spectrometric analyses. On the basis of chemical degradation of the second pigment and the observation that it possessed an additional element of unsaturation compared to undecylprodigiosin (m/z = 391 for the unidentified pigment versus 393 for undecylprodigiosin-H+), Gerber narrowed down the structure of the mysterious pigment to ortho- and meta-bridged isomers of butylcycloheptylprodigiosin, 21 and 22, respectively (Figure 13).
Figure 13.

Two structures initially suggested by Gerber67 for the desaturated derivative of undecylprodigiosin produced by Streptomyces sp. Y-42. In early work, structure 21 was favored.
On the basis of comparisons of the NMR spectra for other ortho- and meta-substituted pyrroles with those for the desaturated undecylprodigiosin derivative, Gerber assigned structure 21 to the isolate. A few months later, Gerber67 reported the isolation of two pink pigments from Streptoverticillium rubrireticuli, a bacterium known to cause problematic pink staining of PVC plastics, and assigned them the same structures (17 and 21) as the Y-42 pigments. Interestingly, in 1964, Thirumalachar et al.68 isolated a pink pigment from a Streptomyces species that they called “streptorubrin B”. Detailed structural characterization of this pigment was not, however, reported. In 1976, Gerber and Lechevalier69 found that butylcycloheptylprodigiosin was identical to streptorubrin B, which has been arbitrarily renamed as streptorubin B in recent literature. In an effort to remain consistent, we will henceforth refer to streptorubrin B as streptorubin B.
Curiously, in the course of subsequent biosynthetic studies, Gerber et al.70 reassigned the structure of streptorubin B from the ortho-bridged isomer 21 to the meta-bridged isomer 22, without commenting on the reasons for the reassignment, which appeared to go unnoticed. In 1985, Floss and co-workers71 reported the isolation of two pigments from S. coelicolor A3(2): undecylprodigiosin (17), and a pink pigment of molecular mass 391 Da with spectral data “closely match[ing] those reported [by Gerber] for butylcycloheptylprodigiosin”. Thus, Floss assigned structure 21 to his pigment, apparently unaware that Gerber had reassigned the structure of streptorubin B to 22.
In 1991, Weyland and co-workers72 isolated an actinomycete strain (B 4358) that was found also to produce a prodiginine alkaloid with the molecular formula C25H33N3O. On the basis of extensive NMR spectroscopic analysis, structure 22 was assigned to this compound, which had strikingly similar NMR data to that reported by Gerber for streptorubin B. Thus, it was proposed that streptorubin B should also be assigned structure 22, in agreement with Gerber’s 1978 reassignment. Moreover, an additional signal with a chemical shift of −1.55 ppm was noted in the 1H NMR spectrum of streptorubin B that had not been previously reported. This provided further evidence that streptorubin B is the meta isomer of butylcycloheptylprodigiosin, because force field calculations predicted that one of the hydrogen atoms attached to C-4′ would lie within the anisotropy cone of the ansa-bridged pyrrole (Figure 14).
Figure 14.

Calculations predict that the meta-bridged isomer 21 has a proton located directly within the anisotropy cone of the ansa-bridged pyrrole. Consistent with this hypothesis, one of the C-4′ protons of streptorubin B is observed to be significantly shielded in the 1H NMR spectrum. This is confirmed by the X-ray structure of 22·HCl shown.66
In 2005, the structure of butylcycloheptylprodigiosin isolated by Gerber and Floss was called into question by Fürstner et al.,73 who noted that neither had reported a signal at −1.55 ppm in the 1H NMR spectra of the pigments they isolated (note, however, that Floss and co-workers71 did not collect data below −0.5 ppm). Because Gerber, Floss, and Weyland had isolated their pigments from different strains of bacteria, Fürstner argued that there was a possibility that o-butylcycloprodigiosin 21 could, in fact, be a natural product. Thus, Fürstner et al.73 undertook a total synthesis of 21 (see section 3.5), reporting that the 1H NMR spectrum of this material was an excellent match with the corresponding spectrum of the natural product from S. coelicolor recorded by Floss. On this basis, Fürstner concluded that o-butylcycloheptylprodigiosin (21) is a distinct natural product from streptorubin B (22), as originally suggested by Gerber in 1975. In 2007, Reeves74 also published a synthesis of racemic 21 (see section 3.5) and came to the same conclusion, following correspondence with Floss and Fürstner.
In 2008, during the course of prodiginine biosynthetic studies in S. coelicolor A3(2) (the same species from which Floss isolated undecylprodigiosin and butylcycloheptylprodigiosin in 1985), Challis and co-workers75 isolated the carbocyclic undecylprodigiosin derivative produced by S. coelicolor and showed, by an extensive array of 1D and 2D NMR experiments, that this possesses the meta-bridged structure 22. Moreover, there was no evidence for production of a metabolite with structure 21. In 2011, Thomson and co-workers66 completed the first enantioselective total synthesis of streptorubin B (22) (see section 3.4) and compared its 1H NMR spectrum with that provided by Floss for the carbocyclic derivative of undecylprodigiosin he had originally isolated from S. coelicolor. Due to significant impurities in the material isolated by Floss, Thomson was unable to draw any firm conclusions from this comparison. However, a short total synthesis of 21 was devised (see section 3.5), and comparison of the fragment ions observed in electron ionization (EI) mass spectra of synthetic 21 and 22 showed significant, reproducible differences.76 Further comparison of the fragment ions observed for the synthetic compounds with both Gerber’s and Floss’s original fragmentation data revealed an excellent match between the natural isolates and synthetic 22 but not synthetic 21. Thus, Thomson and co-workers confirmed that o-butylcycloheptylprodigiosin (21) is a structural misassignment, as originally proposed by Gerber and Weyland.
Thomson and co-workers76 speculated that structural changes between o-butylcycloheptylprodigiosin 21 and streptorubin B (25) might alter the energetic differences between conformational isomers resulting from potential rotation of the bis(pyrrole) side arm relative to the carbocyclic core (Figure 15). The cis isomer (23 or 25b) is believed to possess an optimal juxtaposition of functional groups for activities such as anion transport and cation binding.77,78 An evolutionary advantage may therefore be conferred on bacteria that have evolved oxidative carbocyclization enzymes (e.g., RedG and McpG) that allow the C ring of prodigiosin and undecylprodigiosin to be functionalized at the carbon atom directly adjacent to the azafulvene core. Such functionalization generates significant 1,3-allylic strain in the trans isomer. This is lacking in the cis isomer (see 23 vs 24, Figure 15), and thus it is energetically favored. Conversion of undecylprodigiosin 17 to o-butylcycloheptylprodigiosin 21 would be unlikely to significantly affect the position of equilibrium between the cis isomer 23 and the trans isomer 24. In contrast, conversion of undecylprodigiosin to the meta-bridged product 22 would greatly increase the concentration of the cis isomer 25 relative to the trans isomer 26. In this context, it is striking that numerous carbocyclic derivatives of prodigiosin and undecylprodigiosin in which the cis conformation is enforced (e.g., cycloprodigiosin, cyclomethyldecylprodigiosin, cyclononylprodigiosin, streptorubin B, metacycloprodigiosin, and roseophilin) have been isolated, whereas derivatives that do not enforce a cis conformation have yet to be identified.
Figure 15.

Bacteria appear to have evolved oxidative cyclization enzymes to enforce the cis configuration, in which counteranion and cation binding is favored by the protonated and neutral forms of the molecules, respectively.
The resolution of the ambiguity surrounding the structure of streptorubin B set the stage for elucidating the stereochemistry of this fascinating metabolite. In 2011, Thomson and co-workers66 and Challis and co-workers79 independently reported that streptorubin B exists as a pair of slowly interconverting atropisomers (Figure 16). The major atropisomer at equilibrium possesses an anti relationship between the 4-methoxypyrrolyldipyrromethene core and the n-butyl side chain. The barrier for interconversion of the atropisomers was estimated to be ~20.5 kcal·mol−1 (see section 3.4).
Figure 16.

Structures of the atropisomers of streptorubin B discovered in the course of stereochemical studies by Challis and co-workers79 and synthetic studies by Thomson and co-workers.66 The natural product has the 7′S absolute configuration as shown, and the n-butyl side chain is oriented anti to the exocyclic methine group in the thermodynamically more stable atropisomer.
Thomson and co-workers66 utilized X-ray crystallographic analysis to determine the absolute configuration of streptorubin B prepared by enantioselective total synthesis (see section 3.4) and then compared the CD spectrum of the synthetic material with natural streptorubin B isolated by Challis and co-workers from S. coelicolor. In a complementary approach (Figure 17), Challis and co-workers79 stereoselectively synthesized both enantiomers of [4′-2H]2-undecylpyrrole (27) and fed them separately to a mutant of S. coelicolor unable to produce 2-undecylpyrrole (a key intermediate in streptorubin B biosynthesis). 1H and 2H NMR spectroscopic analysis of the resulting stereoselectively deuterium-labeled streptorubin B hydrochloride salt led to assignment of the 7′S configuration for the natural product,79 in complete agreement with the absolute stereochemical assignment made on the basis of the total synthesis studies.66
Figure 17.

Feeding experiment by Challis and co-workers79 to elucidate the absolute configuration of streptorubin B.
HPLC comparisons on a homochiral stationary phase of racemic synthetic streptorubin B with the natural product isolated from S. coelicolor showed that the latter is in fact a 95:5 mixture of 7′S and 7′R enantiomers.79 Remarkably, CD spectroscopic comparisons of metacycloprodigiosin prepared by Clift and Thomson65 via enantioselective total synthesis with metacycloprodigiosin isolated by the Challis group from S. longisporus ruber revealed that the natural product has the 9′R absolute configuration;66 that is, it is the antipode of streptorubin B (Figure 18).
Figure 18.

Naturally occurring streptorubin B and metacycloprodigiosin are pseudoenantiomeric. It is not clear whether metacycloprodigiosin possesses a great enough torsional barrier about its 12-membered ring to allow observation of atropisomers as in the case of streptorubin B.
2.7. Nonylprodigiosin, Cyclononylprodigiosin, and Analogues
In 1969, three years after the structural elucidation of undecylprodigiosin, Gerber80 isolated 13 strains of Actinomadura (Nocardia) pelletieri and three strains of Actinomadura madurae that produced prodiginines (it was at this time that the use of the generic name “prodiginine” to describe prodigiosin-like compounds was first suggested). From a structural and biosynthetic standpoint, Gerber’s 1969 isolation experiments were significant for the identification of a new 23-carbon prodiginine, nonylprodigiosin (30), from A. madurae (Figure 19). Nonylprodigiosin is closely related to undecylprodigiosin; it differs only in the number of methylene groups in the hydrocarbon chain. This difference presumably arises because the RedJ homologue in A. madurae preferentially hydrolyzes decanoyl-ACP rather than dodecanoyl-ACP and/or the adenylation domain at the N-terminus of the A. madurae RedL homologue is selective toward decanoic acid (see section 4.4).
Figure 19.
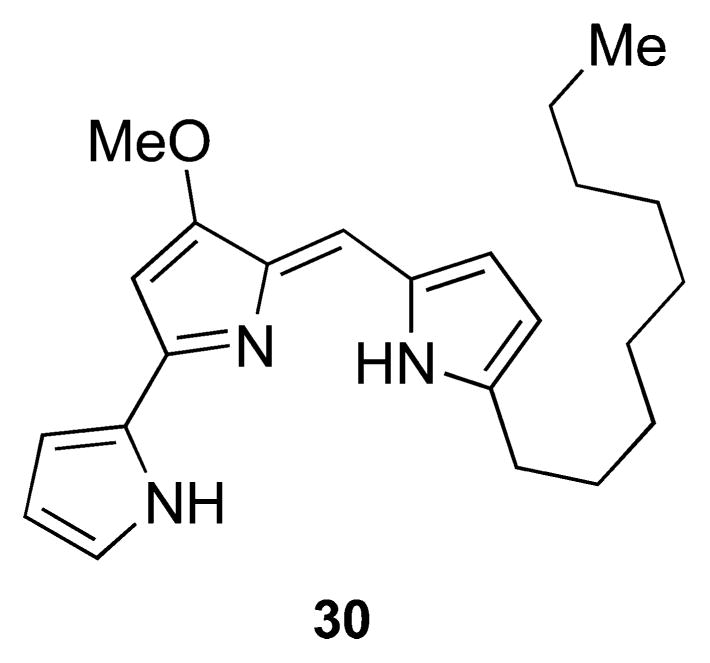
Structure of nonylprodigiosin isolated from A. madurae.
In her 1969 report, Gerber also noted that A. pelleteri produces a 25-carbon pigment (“pelletrin”) with an additional element of unsaturation compared to undecylprodigiosin, indicating that it may contain a carbocycle. The spectroscopic data for this compound differed from that reported for metacycloprodigiosin and its identity could thus not be unambiguously established. A year later, Gerber81 reported that A. madurae also makes a carbocyclic derivative of nonylprodigiosin (30) under different growth conditions. Degradation and spectroscopic studies led to the identification of this new carbocyclic pigment as cyclononylprodigiosin (31, Figure 20), the first member of the prodiginine family with additional functionalization of the A ring. Having elucidated the structure of cyclononylprodigiosin, Gerber returned to the “pelletrin” compound reported in 1969 and, through spectroscopic and degradation studies, assigned structure 32 to the carbocyclic undecylprodigiosin derivative, which she named methylcyclodecylprodigiosin. No signals were observed in the CD spectrum of cyclononylprodigiosin (31), whereas methylcyclodecylprodigiosin (32) exhibited a clear Cotton effect, indicating that it is enantiomerically enriched.82 However, the absolute configuration of the stereogenic center in methylcyclodecylprodigiosin remains undefined.
Figure 20.

Structures of carbocyclic derivatives 31 and 32 of nonylprodigiosin and undecylprodigiosin isolated from A. madurae and A. pelletieri, respectively.
While preparing larger quantities of carbocyclic prodiginines 31 and 32 for antimalarial testing, Gerber83 discovered that A. madurae and A. pelletieri produce the minor congeners methylcyclooctylprodigiosin (33) and ethylcyclononylprodigiosin (34), respectively (Figure 21).
Figure 21.

Structures of the minor prodiginine congeners 33 and 34, produced by A. madurae and A. pelletieri, respectively.
In 1999, 30 years after the report of its isolation, Fürstner et al.84 completed the first total synthesis of cyclononylprodigiosin (31) using ring-closing metathesis methodology (see section 3.6), and were able to obtain an X-ray structure of a key intermediate (35) in its synthesis. From this it was concluded that the major tautomer of cyclononylprodigiosin (as well as other prodiginines) has a central rather than a peripheral azafulvene (i.e., 35 as opposed to 36 in Figure 22).
Figure 22.

Two possible tautomers of a key intermediate in cyclononylprodigiosin synthesis, which were discriminated by use of X-ray crystallographic analysis.
2.8. Roseophilin, Dechlororoseophilin, and Prodigiosin R1
In 1992, Seto and co-workers85 disclosed the structure of a new antibiotic, roseophilin (37), from Streptomyces griseoviridis (Figure 23). Roseophilin shows significant resemblance to the carbocyclic prodiginines yet possesses numerous distinct features. First, it contains two (rather than one) C–C bonds between the hydrocarbon chain and the conjugated heterocyclic ring system, resulting in a tricyclic cyclopentylpyrrolophane. Second, the characteristic central azafulvene of the prodiginines is replaced by a furan. Consequently, the pyrrole linked to the hydrocarbon chain becomes an azafulvene. Finally, roseophilin possesses a chlorine substituent on the A-ring pyrrole.
Figure 23.
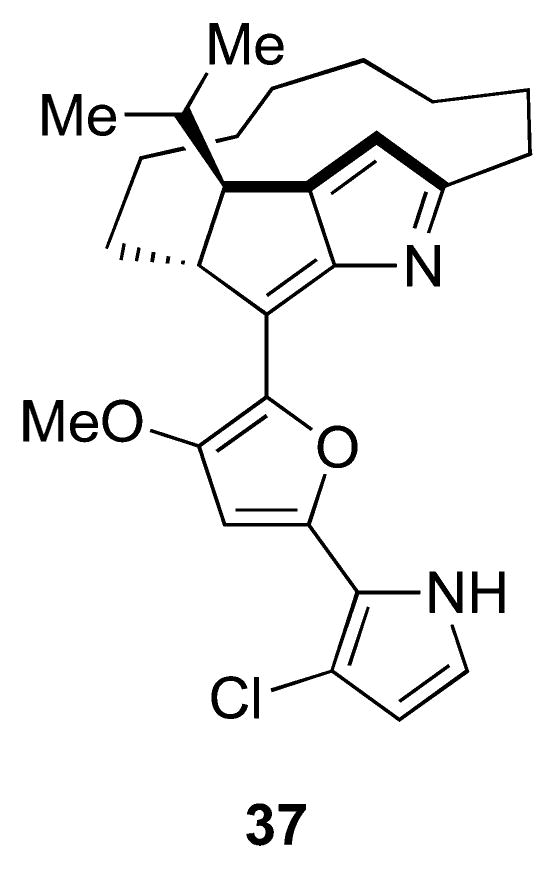
Structure of roseophilin (only the relative stereochemistry was reported at the time of isolation).
The planar structure and relative stereochemistry of roseophilin were confirmed in 1998 through total synthesis of the racemate by Fürstner and Weintritt86 (see section 3.7). Although these authors were able to separate the two enantiomers of synthetic roseophilin by HPLC, they were unable to determine the absolute configuration of the natural product. In 2001, Boger and Hong87 and Harrington and Tius88 disclosed independent enantioselective total syntheses of (22S,23S)-37 and (22R,23R)-37, respectively (see section 3.7). Comparison of CD spectroscopic data for Tius’s synthetic (22R,23R)-roseophilin and the natural product established that they had the same absolute stereochemistry.
Despite several clear differences in the chromophore of roseophilin and the carbocyclic prodiginines discussed previously, roseophilin appears to share a common biosynthetic origin, likely originating from oxidative modifications to a hitherto unidentified dimethylated undecylprodigiosin analogue (39, Figure 24). Circumstantial evidence for the existence of 11′-dimethylundecylprodigiosin (39) and its role as a roseophilin precursor is provided by the recently reported isolation of prodigiosin R1 (40)89 and dechlororoseophilin (38)90 from the roseophilin producer S. griseoviridis (see section 4.8).
Figure 24.

Structures of 11′-dimethylundecylprodigiosin (39), dechlororoseophilin (38), and prodigiosin R1 (40) and their proposed biosynthetic relationship.
2.9. Marineosins A and B
Marineosins A and B were isolated from a marine-derived Streptomyces-related actinobacterium (strain CNQ-617) by Fenical and co-workers in 200891 (Figure 25). The structure and relative stereochemistry of marineosins A (41) and B (42) were assigned on the basis of an extensive series of NMR experiments. Although the marineosins are structurally distinct from other prodiginines, they appear to derive from undecylprodigiosin via a series of redox transformations, including one that is similar to the oxidative carbocyclization reactions involved in streptorubin B and metacycloprodigiosin biosynthesis (see section 4.7). A total synthesis of the two compounds remains elusive, but recent studies by Reynolds and co-workers92 have established the absolute stereochemistry to be as shown in Figure 25.
Figure 25.

Structures of marineosins A and B.
3. TOTAL SYNTHESES
Due to the diverse and fascinating structures of the prodiginines, as well as their intriguing biological activities, numerous total and formal syntheses have been reported over the last 60 years. Here, we provide a comprehensive overview of the major chemical syntheses to date.
3.1. Prodigiosin and Undecylprodigiosin
In 1962, Rapoport and Willson43 reported the syntheses of several isomers of prodigiosin in order to determine its complete structure (Scheme 1).
Scheme 1.

Synthesis of Prodigiosin Isomer iso-1 by Rapoport and Willson43
Synthesis of iso-1 initiated with a tandem conjugate addition/Dieckmann cyclization with diethyl fumarate 43 and sodium glycinate 44 to yield 3-oxoprolinate 45.43,93 Decarboxylation of 45 under acidic conditions, followed by dimethylsulfite-mediated ketalization, provided pyrrolidine 46. Treatment of 46 with catalytic palladium on carbon in a dehydrogenation vessel at high temperature led to a pyrrole ethyl ester, which was converted to the corresponding methyl ester (47) under standard conditions. Aromatic substitution of 47 with dehydropyrrolidine 48 at elevated temperature provided amine 49, which was again aromatized with catalytic palladium at high temperature. The resulting bis(pyrrole) ester 50 was subjected to a three-step McFayden–Stevens reduction to provide aldehyde 5, which was summarily condensed with pyrrole 7 to provide prodigiosin iso-1 after alumina chromatography.
Unfortunately, spectral data of iso-1 did not match that of the prodigiosin from Serratia. Rapoport and Willson43 proceeded to synthesize (Scheme 2) the next most likely isomer, based on previous studies by Wasserman et al.42 (i.e., 1).
Scheme 2.

Synthesis of Prodigiosin (1) by Rapoport and Willson43
Synthesis of 1 began with a similar tandem conjugate addition/Dieckmann condensation sequence to that employed in the synthesis of iso-1.43 Sodium glycinate 44 was combined with vinylogous ester 51 to afford hydroxypyrrole 52. O-Methylation of 52, followed by a two-step decarboxylation sequence, provided methoxypyrrole 54. Aromatic substitution of 54 with cyclic imine 48 provided amine 55, which was dehydrogenated at high temperature under palladium catalysis. A three-step McFayden–Stevens reduction revealed aldehyde 6, which could be condensed with amylpyrrole 7 under acidic conditions to provide prodigiosin (1). Prodigiosin (1) was found to be identical in all aspects to naturally isolated prodigiosin, fully confirming the natural product’s elusive structure more than three decades after its isolation.
Since this first synthesis of prodigiosin (1), numerous other syntheses of the key bis(pyrrole) aldehyde 6 have been reported, with applications toward the syntheses of many prodiginine natural products. In 1988, Boger and Patel94 disclosed the total synthesis of prodigiosin and several prodigiosin analogues for structure–activity relationship (SAR) studies via azadiene Diels–Alder methodology (Scheme 3).
Scheme 3.

Synthesis of Prodigiosin (1) by Boger and Patel (1988)94
The synthesis of Boger and Patel94 began with the tandem [4 + 2]/retro-[4 + 2] cycloaddition reaction of azadiene 57 and vinyl ether 58 to generate pyrazine 59 in excellent yield. Reductive pyrrole formation followed by selective hydrolysis of a methyl ester provided carboxylic acid 60, which could be decarboxylatively iodinated to form diiodide 61. Hydrogenolysis of the carbon–halogen bonds, followed by N-acylation of resulting pyrrole 62 with acyl chloride 63, provided carbonyldipyrrole 64, which could be cleanly converted to fused compound 65 by action of stoichiometric, polymer-supported palladium(II). Methanolysis of 65 followed by a McFayden–Stevens reduction of ester 66 generated the key prodigiosin intermediate, bis(pyrrole) aldehyde 6, which could be converted to the natural product as previously reported.
In 1989, Wasserman and Lombardo95 disclosed a new total synthesis of prodigiosin (1) based on newly developed vicinal tricarbonyl methodology (Scheme 4). The first key transformation in Wasserman and Lombardo’s synthesis was a twostep condensation between pyrrole aldehyde 67 and acetoacetate dianion 68 to form unsaturated ketone 69.95 Oxidation of the β-keto ester in 69 by action of nitrosoaniline 70 provided tricarbonyl 71, which provided bis(pyrrole) ester 73 upon condensation with benzylamine 72. Methylation and deprotection of 73 revealed ester 56, which was subjected to a McFayden–Stevens reduction to provide the key bis(pyrrole) aldehyde (6). Aldehyde 6 was finally condensed with amylpyrrole 7 to provide prodigiosin (1).
Scheme 4.

Synthesis of Prodigiosin (1) by Wasserman and Lombardo (1989)95
In 1999, Wasserman et al.96 reported a new, shorter synthesis of the bis(pyrrole) aldehyde based on singlet-oxygen methodology developed in their group (Scheme 5). Wasserman et al. found that pyrrole 74 could be converted to bis(pyrrole) ester 75, on a small scale, by sequential exposure of 74 to singlet oxygen at low temperature in the presence of excess pyrrole. Compound 75 could then be converted to bis(pyrrole) aldehyde 6 by a McFayden–Stevens reduction, as used in numerous previous syntheses. Aldehyde 6 was identical in all aspects to the naturally occurring aldehyde and was used by Wasserman et al.96 to synthesize prodiginines bearing simple substitutions on the Aring pyrrole.
Scheme 5.

Synthesis of Key Bis(pyrrole) Aldehyde 6 by Wasserman et al. (1999)96
In 1996, driven by interest in undecylprodigiosin as an immunosuppressive agent, D’Alessio and Rossi97 at Pharmacia & Upjohn in Italy disclosed a synthesis of undecylprodigiosin which did not rely on the condensation of a pyrrole with bis(pyrrole) aldehyde 6, as many of the previously reported synthetic routes to 6 were not suitable for scaling up in case of possible lead development. Their synthesis began with the conversion of undecylpyrrole 76 to pyrrole aldehyde 77 under Vilsmeier– Haack conditions (Scheme 6). Aldehyde 77 was then subject to a base-mediated condensation with lactam 78 to provide conjugated lactam 79. Treatment of 79 with triflic anhydride provided the corresponding triflate 80, which could be cross-coupled to pyrroleboronic acid 81 with concomitant t-butyloxycarbonyl (Boc) deprotection under Suzuki–Miyaura reaction conditions to provide the natural product (16). D’Alessio et al.11 later applied this method to the synthesis of several undecylprodigiosin derivatives for SAR studies for immunosuppressive activity. Similar reaction sequences were later employed by Fürstner et al.73,98 in their synthesis of butylcycloheptylprodigiosin (see section 3.5) and by Clift and Thomson65 in their synthesis of metacycloprodigiosin (see section 3.3).
Scheme 6.

Synthesis of Undecylprodigiosin (17) by D’Alessio and Rossi97
Most recently, in 2006, Lavallée and co-workers99 at Gemin X found that the method developed by D’Alessio and Rossi could be applied to synthesis of the commonly used bis(pyrrole) aldehyde 6 (Scheme 7). They found that lactam 78 could be converted to bromoenamine 82 by action of a Vilsmeier–Haack reagent. Bromoenamine 82 could then be employed in a Suzuki– Miyaura cross-coupling with boronic acid 81 under basic, aqueous conditions to provide bis(pyrrole) aldehyde 6. This process has been used by Gemin X to prepare multikilogram quantities of a synthetic prodigiosin analogue, obatoclax, for clinical trials.33
Scheme 7.

Synthesis of Bis(pyrrole) Aldehyde 6 by Lavallée and Co-workers99
3.2. Cycloprodigiosin
In 1984, to confirm the structure of cycloprodigiosin after revisions by Gerber and Laatsch, Wasserman and Fukuyama54 at Yale undertook its total synthesis. Their synthesis began with a condensation of methylcyclohexanone (83) with sulfonylhydrazine 84 to afford hydrazone 85 (Scheme 8). Treatment of 85 with sec-butyllithium generated a vinyl anion, which was trapped with N,N-dimethylformamide (DMF) to form aldehyde 86. Reaction of 86 with thioacetal anion 87 at low temperature generated 88, the deprotection of which revealed 1,4-dicarbonyl 89. Treatment of 89 with ammonium carbonate at high temperature allowed the formation of pyrrole 90, which was condensed with bis(pyrrole) aldehyde 6 to afford a compound that was identical with samples of the natural product derived from both B. gazogenes and A. rubra, establishing the structure of cycloprodigiosin as 16.
Scheme 8.

Synthesis of Racemic Cycloprodigiosin (16) by Wasserman and Fukuyama (1984)54
The first enantioselective synthesis of cycloprodigiosin was reported in 2013 by Schultz and Sarpong100 (Scheme 9). Their approach centered upon an application of a novel method they had developed for the construction of fused pyrroles. The requisite methyl stereogenic center was set at the beginning of the synthesis by way of a Myers auxiliary-controlled enolate alkylation to afford amide 93. Functional group interconversions then allowed for synthesis of terminal alkyne 95, which was further processed to provide propargylic alcohol 96 over five steps. Generation of the 1,2-disubstituted allene 97 was achieved by a Myers allene synthesis, which set the stage for the key rhodium-catalyzed cycloaddition cascade to fused pyrrole 98. Removal of the N-tosyl group, followed by condensation with bis(pyrrole) 99 and in situ Boc group cleavage, provided cycloprodigiosin in 71% yield over three steps.
Scheme 9.

Enantioselective Synthesis of Cycloprodigiosin (16) by Schultz and Sarpong (2013)100
In a subsequent publication in 2015, Sarpong and coworkers55 reported an efficient second-generation route to cycloprodigiosin (15) that enabled access to both enantiomers of the natural product (Scheme 10).
Scheme 10.

Enantioselective Synthesis of Both Enantiomers of Cycloprodigiosin (16) by Sarpong and Co-workers (2015)55
Application of the Schöllkopf–Barton–Zard pyrrole synthesis using enantiopure menthol-derived isonitrile 101 in conjunction with racemic 100 allowed direct access to both diasteromeric pyrroles 102 and 103 in a combined yield of 62%. Separation of the diastereomers allowed further processing to either of the methyl-substituted pyrroles, 104 or 105, through a three-step procedure of bromination, pyrrole protection, and palladium-catalyzed methylation. Cleavage of the chiral auxiliary within 104 or 105 was achieved with concomitant decarboxylation under basic conditions, allowing for an acid-promoted condensation with pyrrole 6 to deliver either enantiomeric form of the natural product in good yield.
3.3. Metacycloprodigiosin
The first total synthesis of metacycloprodigiosin (20) was conducted by Wasserman and co-workers in 196961 as part of a sustained effort to elucidate the structures of the prodigiosin family.101 Their synthesis (Scheme 11) began with cyclododecanone (106), which was alkylated and protected as ketal 107. Regioselective bromination of ketal 107 under mildly acidic conditions provided bromide 108 in quantitative yield. Elimination at high temperature followed by acid-catalyzed deprotection revealed cyclododecenone 109, which was epoxidized to provide keto-epoxide 110 as a mixture of diastereomers. Wharton fragmentation of epoxide 110 with hydrazine afforded allylic alcohol 111, the oxidation and cyanation of which provided nitrile 112. Nitrile 112 was then converted to the corresponding 1,4-dicarbonyl 113 through trivial functional-group manipulations. Condensation of 113 with an ammonia equivalent produced pyrrole 114, which was further condensed with bis(pyrrole) aldehyde 6 to provide the natural product as the hydrochloride salt. The final condensation step in Wasserman’s synthesis of metacycloprodigiosin, cycloprodigiosin, and other prodiginines is often referred to as “biomimetic”,102 but in the true biosynthesis of cyclic prodiginines such as metacycloprodigiosin, the condensation first occurs with 2-methyl-3-amylpyrrole (MAP) or 2-undecylpyrrole (2-UP) before an enzyme-catalyzed oxidative radical cyclization forms the ring64 (e.g., 114 and related cyclic pyrroles are not true biosynthetic precursors of 20 and corresponding prodiginines—see section 4.5).
Scheme 11.

Total Synthesis of Metacycloprodigiosin by Wasserman et al. (1969)61
Three decades after Wasserman’s first synthesis of metacycloprodigiosin, two elegant nonenantioselective formal total syntheses based on the success of Wasserman’s condensation were reported by Fürstner et al. in 1998102 and 1999.103 Fürstner’s 1998 formal synthesis of metacycloprodigiosin (Scheme 12A) was based on the development of an enynemetathesis reaction. To begin the synthesis, cyclodecene 115 was treated with an in situ-generated diiminoselenium reagent to afford aminated cyclodecene 116.102 N-Alkylation of 116 with propargyl bromide provided alkyne 117, which could be acylated via the intermediacy of an alkynylzinc reagent. Treatment of the resulting ynone 118 with either catalytic platinum(II) or stoichiometric boron trifluoride resulted in a rearrangement to form bridged pyrrolidine 119. Reduction of the alkene in 119 provided ketone 120, which could be reduced and thionylated to provide thionocarbonate 121. Barton–McCombie dehydroxylation of 121 then generated pyrrolidine 122. The choice of a radical dehydroxylation process was critical, as any pathway involving the generation of a carbocation at the hydroxyl position led to significant rearrangement in the molecule. Final elimination of the tosyl group from 122 was achieved with excess KAPA (potassium anion of 1,3-diaminopropane), providing Wasserman’s pyrrole 114 in 5% overall yield from cyclodecene.
Scheme 12.

Formal Syntheses of Metacycloprodigiosin by Fürstner et al. (1998, 1999)102,103
Fürstner and Krause’s 1999 formal synthesis of metacycloprodigiosin103 (Scheme 12B) employed methodology similar to that used in Fürstner and Weintritt’s 1998 total synthesis of roseophilin86 (see section 3.7). The synthesis was initiated with a Corey–Chaykovsky reaction between sulfonium 124 (available in five steps from allylic chloride 123) and 8-bromooctanal.103 The formed epoxide 125 was then used to alkylate potassium enolate 126, generating 127. Subjection of 127 to palladium(0) catalysis under high dilution conditions resulted in an intramolecular Tsuji–Trost allylation to form cyclododecanene 128 as an inconsequential mixture of diastereomers. Protecting group removal and oxidation with Dess–Martin periodinane (DMP) provided enone 130, which rearranged with loss of phenylsulfinate under basic conditions to form pyrone 131. Methanolysis of 131 provided α,β-unsaturated aldehyde 132, which was condensed with benzylamine under acidic conditions to generate pyrrole 133. Removal of the acetyl group followed by ruthenium-mediated oxidation provided β-keto ester 134, which in turn revealed ketone 135 after subjection to Krapcho decarboxylation conditions. Finally, Wittig olefination of 135 and hydrogenation of the resulting alkene with Crabtree’s catalyst produced benzylpyrrole 136. Removal of the benzyl group would afford pyrrole 114 and thence 20, but the actual transformation was not reported.
In 2009, 40 years after metacycloprodigiosin’s discovery, Clift and Thomson65 at Northwestern University completed the first enantioselective total synthesis of metacycloprodigiosin in an effort to finally identify the absolute configuration of the natural product (Scheme 13). Their synthesis employed an asymmetric Cu-catalyzed conjugate addition to trans-dienone 137 as the enantioselective step. The nascent magnesium enolate was then trapped with an equivalent of chloroenolsilane 138 to afford silylbis(enol) ether 139. Treatment of unpurified 139 with ceric ammonium nitrate (CAN) provided the 1,4-dicarbonyl 140, which underwent ring-closing metathesis to provide substituted dodecanone 141. Upon hydrogenation and condensation with ammonium acetate, 141 was converted to 2-methylpyrrole 142. In the final steps of the synthesis, an alteration of the prodigiosin core synthesis of D’Alessio and Rossi97 was employed (see section 3.1). After oxidation of 142, aldehyde 143 was treated with trimethylsilyl trifluoromethanesulfonate (TMSOTf) and lactam 78 to afford the vinylogous aldol product 144. Acidic fragmentation of 144 provided lactam 145, which was converted to the corresponding triflate and cross-coupled with Bocprotected pyrroleboronic acid 81 under Suzuki–Miyaura reaction conditions. Deprotection under basic conditions furnished enantiopure metacycloprodigiosin (93:7 enantiomeric ratio, er). Although a natural sample could not be obtained at the time of the report, two years later, in 2011,66 comparisons of CD spectra of synthetic (R)-metacycloprodigiosin with natural metacycloprodigiosin isolated from S. longisporus ruber were used to assign the absolute configuration of natural metacycloprodigiosin as R (as it appears in Scheme 13).
Scheme 13.

Enantioselective Synthesis of Metacycloprodigiosin (20) by Clift and Thomson (2009)65
In 2015, Thomson and co-workers reported a second enantioselective total synthesis of metacycloprodigisoin (20) that cut the number of operations required to complete the synthesis from 12 to nine steps (Scheme 14). Their new route took advantage of the late-stage union of pyrrole 114 with bispyrrole aldehyde 99, in a fashion analogous to that described by Wasserman and co-workers in their inaugural synthesis of 20. Thomson’s enantioselective synthesis of Wasserman’s pyrrole (114) began with the synthesis of aldehyde 149 in four steps from diol 146. As in their previous synthesis, enantioenrichment was achieved through the use of an asymmetric Feringa conjugate addition. Oxidative coupling of aldehyde 149 with enol silane 150 was then carried out via enamine catalysis to generate 1,4-dicarbonyl 152 in 22% yield. Ring-closing metathesis once again forged the requisite 12-membered ring. Hydrogenation and Paal–Knorr pyrrole condensation delivered Wasserman’s pyrrole 114, which was then converted in one step to the natural product upon acid-catalyzed condensation with 99. The brevity of this route, which is currently the shortest synthesis of metacycloprodigiosin (20), goes some way toward making up for the disappointingly low yield obtained during the key oxidative coupling event (149 → 152).
Scheme 14.

Enantioselective Synthesis of Metacycloprodigiosin (20) by Thomson and Co-workers (2015)
3.4. Streptorubin B
Although it was first isolated in 1964, the first synthetic studies toward streptorubin B were not reported until 1998, when Fürstner et al.102 disclosed a synthesis of streptorubin’s pyrrole core (Scheme 15A).104 Fürstner’s work was based on their development of an enyne-metathesis reaction and paralleled their synthetic studies toward metacycloprodigiosin. To begin the synthesis, cyclooctene 154 was treated with an intermediate generated in situ from chloramine T and elemental selenium to afford aminated cyclooctene 155. N-Alkylation of 155 with propargyl bromide provided alkyne 156, which could be acylated via the intermediacy of an alkynylzinc reagent. Treatment of the resulting ynone 157 with catalytic platinum(II) resulted in a highly efficient rearrangement to form bridged pyrrolidine 158. Radical reduction of the alkene in 158 provided ketone 159, which could be reduced and thionylated to provide thionocarbonate 160. Radical dehydroxylation of 160 then generated pyrrolidine 161. Final elimination of the tosyl group from 161 was effected with excess KAPA, affording synthetic pyrrole 162 for the first time. The conversion of 162 into streptorubin B (22) was not reported, though related condensations had been previously reported for other prodigionines.
Scheme 15.

Synthetic Efforts toward Total Synthesis of Streptorubin B (22)
Seven years after Fürstner’s publication, Chang et al.105 at the National University of Kaohsiung in Taiwan reported an approach to streptorubin B based on Fürstner’s pyrrolidine intermediate 161 (Scheme 15B). Chang’s synthesis centered on a ring-closing metathesis reaction to create the cyclodecane backbone. Pyrrolidine 163 (available in four steps from trans-4-hydroxyproline) was oxidized and homologated with ylide 164 to provide ester 165. A two-step reduction of both the alkene and ester functionalities provided alcohol 166, which was oxidized and converted to terminal alkene 167 by Wittig methodology. Deprotection and oxidation of ether 167 in one pot revealed ketone 168, which was converted to requisite diene 170 upon treatment with Grignard reagent 169. Exposure of 170 to Grubbs’ first-generation catalyst under high dilution conditions provided cycloalkene 171 as an inconsequential mixture of diastereomers and geometric isomers. Hydrogenation of the double bond followed by dehydration provided pyrrolidine 161 again as a mixture of diastereomers, which may be converted to 162 according to the procedure of Fürstner et al.102
In 2011, Thomson and co-workers66 disclosed the first enantioselective total synthesis of streptorubin B (Scheme 15C). To begin the synthesis, heptanedial 172 was treated with catalytic (S)-proline according to a procedure developed by List to induce an enantioselective exo-enol-6-exo-trig aldol reaction. The transient aldehyde 173 was trapped in one pot with Wittig reagent 159 to afford cyclohexanol 174. Oxidation of 174 under Swern conditions, followed by the 1,2-addition of organolithium reagent 175, generated dienol 176. Treatment of the dienol with potassium hexamethyldisilazide, in the presence of stoichiometric 18-crown-6 ether, induced an anionic oxy-Cope rearrangement to cyclodecanone 178 with excellent transfer of stereochemistry through a chair-like transition state (177). Tandem hydrogenation and benzyl group hydrogenolysis afforded alcohol 179, which was oxidized with Dess–Martin periodinane and condensed with ammonium acetate to afford the pyrrole core 162. The synthesis was completed with an acidpromoted condensation between pyrrole 162 and Boc-protected bis(pyrrole) aldehyde 99, followed by protecting group removal under basic conditions in one pot. Analysis of the bright-red material thus obtained revealed an approximately 10:1 mixture of two compounds in which the major compound did not match the natural product. Reexamination of the NMR sample after 10 days, however, revealed that the mixture had transformed almost completely to streptorubin B (22). The synthetic HCl salt was identical to the natural product as determined by 1H and 13C NMR spectroscopy and mass spectrometry.
Intrigued by the initial formation of an apparent isomer of the natural product, Thomson and co-workers66 conducted a series of nuclear Overhauser effect spectroscopic (NOESY) NMR experiments to determine its identity. They realized that the pyrrole precursor 162 had been formed atropdiastereoselectively from alcohol 179 as the syn atropisomer 181 (Figure 26), which was favored in equilibrium with the anti atropisomer 180. Condensation of the isomeric mixture led to a mixture of streptorubin B atropisomers 22·HCl and 182·HCl, initially favoring 182·HCl. Relaxation over a period of several days at room temperature then led to a mixture strongly favoring isomer 22·HCl. Kinetic measurements of the equilibrium allowed the torsional barrier to be approximated as ca. 20.5 kcal·mol−1.66
Figure 26.

Atropisomerism phenomenon in streptorubin B and its synthetic pyrrole intermediate, discovered in the course of total synthesis by Thomson and co-workers.66
3.5. Butylcycloheptylprodigiosin
In 2005, Fürstner et al.73 conducted a total synthesis of butylcycloheptylprodigiosin (21) in an effort to determine whether or not it had in fact been previously isolated in nature (Scheme 16).
Scheme 16.

Synthesis of Butylcycloheptylprodigiosin (21) by Fürstner et al. (2005)73
Cyclononadienone 184 (available in six steps from cyclooctanone) was reduced and acetylated under standard conditions to provide acetate 185. Exposure of allyl acetate 185 to sodium methyl acetoacetate with catalytic Pd(0), followed by thermal decarboxylation, led to diene 186 as the major product. Condensation of 186 with hydroxylamine, followed by acylation with pentafluorobenzoyl chloride, afforded oxime ester 187. A unique Narasaka–Heck cyclization was then conducted on a multigram scale with Pd(0) and P(o-tolyl)3 to provide bicyclic imine 188, which further rearranged under strongly basic conditions to a conjugated pyrrole (Boc-protected as 189). The protected pyrrole 189 was then subjected to a hydroboration– oxidation sequence, providing alcohol 190, which could be further oxidized with Dess–Martin periodinane to reveal ketone 191. Wittig olefination of 191 at elevated temperature provided olefin 192 in good yield. Catalytic hydrogenation of the double bond, along with oxidation of the pyrrole methyl substituent, proceeded smoothly to generate aldehyde 193, which could be condensed under basic conditions with lactam 78 to afford lactam 194.
Formation of the corresponding triflate and Suzuki–Miyaura cross-coupling with boronic acid 81 then furnished the desired o-butylcycloheptylprodigiosin 21. On the basis of comparisons of synthetic 21 to unpublished spectral data obtained from Floss, Fürstner and co-workers concluded that butylcycloheptylprodigiosin was in fact a natural product (see section 2.6).
In 2007, Reeves74 at Boehringer Ingelheim Pharmaceuticals reported a short synthesis of butylcycloheptylprodigiosin based on a method for the synthesis of 2-formyl-4,5-disubstituted pyrroles that he had recently disclosed (Scheme 17). The synthesis began with cyclononenone (available in three steps from cyclooctanone). Treatment of cyclononenone 195 with n-butylmagnesium chloride under copper catalysis at low temperature generated a magnesium enolate, which could be diastereoselectively trapped with formyloxazole 196 to provide keto alcohol 197. Elimination of the hydroxyl group in 197 by action of mesyl chloride under basic conditions provided 2-formylpyrrole 198, which could be extended to lactam 194 by condensation with 78. As per Fürstner’s synthesis, lactam 194 could be converted to the appropriate triflate and coupled to boronic acid 81 under Suzuki–Miyaura conditions to provide butylcycloheptylprodigiosin 21.
Scheme 17.

Synthesis of Butylcycloheptylprodigiosin (21) by Reeves (2007)74
In 2013, Thomson and co-workers76 developed a short synthesis of butylcycloheptylprodigiosin using a tandem catalytic approach in order to provide material for comparisons with their synthetic streptorubin B.66 In the synthesis (Scheme 18), a copper-catalyzed conjugate addition of butylmagnesium bromide to cyclononenone 195 generated a transient magnesium enolate, which could be efficiently allylated by allyl bromide in the presence of catalytic Pd(0). Alkene 199 was then subjected to Lemieux–Johnson oxidation conditions to afford 1,4-dicarbonyl 200, which could be smoothly condensed with ammonium acetate to afford pyrrole 201. Condensation with protected bis(pyrrole) aldehyde 99 under dry acidic conditions, followed by basic deprotection in situ, afforded the desired prodigiosin 21. Thomson and co-workers76 were able to use the same route to synthesize alkyl-chain analogues of butylcyclohepylprodigiosin for mass spectrometric studies that invalidated the identity of 21 as a natural product (see section 2.6).
Scheme 18.

Synthesis of Butylcycloheptylprodigiosin (21) by Thomson and Co-workers (2013)76
3.6. Cyclononylprodigiosin
In 1999, 30 years after cyclononylprodigiosin’s isolation, Fürstner’s research group at the Max Planck Institute completed its first total synthesis using ring-closing metathesis (RCM) methodology (Scheme 19).84
Scheme 19.

Synthesis of Cyclononylprodigiosin (31) by Fürstner et al. (1999),84 Part I
Fürstner’s synthesis began with the assembly of formylpyrrole 203 in four trivial steps from 5-hexenoic acid (202). Condensation of 203 with lactam 78 in basic dimethyl sulfoxide (DMSO) according the method of D’Alessio resulted in the smooth formation of oxodipyrrylmethene 204, which could be easily converted to corresponding triflate 205. Suzuki coupling of 205 with pyrroleboronic acid 206 with concomitant deprotection generated the tripyrrylmethene 207. At this point, the Fürstner group faced a synthetic dilemma. Compound 207 was not a suitable substrate for ring-closing metathesis, as free amines were known to shut down the metal catalysts. While one solution would be to protect the amine as the hydrochloride salt, it was known that the protonated forms of prodiginines such as undecylprodigiosin and PNU-156804 (208) favored the trans-rotomeric configuration (Figure 27).106
Figure 27.

Rotomer interconversion phenomenon.
At high pH, 208 had been shown to exist exclusively in the cis configuration as opposed to the trans configuration (i.e., as 208, not 209)84 due to the existence of a N–H–N hydrogen bond in the dipyrrylmethene moiety. Upon protonation with strong acid, however, the equilibrium shifted in the opposite direction (i.e., 211 was favored over 210) due to an O–H–N hydrogen bond (which had been previously observed in the crystal structure of a prodigiosin analogue).107 Fortunately, however, Fürstner’s group was able to take advantage of conformational dynamics to close the macrocyclic ring (Scheme 20). Since 212·HCl would not react intramolecularly, only 213·HCl would be converted to the macrocycle. Evolution of ethylene gas would thus slowly drive the intramolecular reaction to completion. By performing the RCM with catalyst 214 at high dilution (1.4 mM), they were able to recover macrocycle 215 as the HCl salt in good yield. Finally, catalytic hydrogenation of 215 with Wilkinson’s catalyst furnished the natural product as its HCl salt (31·HCl).
Scheme 20.

Synthesis of Cyclononylprodigiosin (31) by Fürstner et al.,84 Part II
3.7. Roseophilin
Due to its unique structure and corresponding biological activity, roseophilin (37) was a popular synthetic target from the moment of its reported isolation.85 Beginning in 1995 with the report of Terashima and co-workers108 on synthesis of the pyrrolylfuran moiety of roseophilin (216, Figure 28), numerous creative synthetic studies have been reported in the last two decades. Major total synthesis contributions have been made by groups including Fuchs and co-workers (1997, formal),109,110 Terashima and co-workers (1998, formal),108,111 Fürstner and Weintritt (1998),86 Robertson et al. (1999, formal),112,113 Hiemstra and Speckamp and co-workers (2000, enantioselective formal),114,115 Trost and Doherty (2000, enantioselective formal),116 Boger and Hong (2001, enantioselective),87 Harrington and Tius (2001, enantioselective),88,117 Bitar and Frontier (2009, formal),118 and Frederich and Harran (2013).119 As our review is primarily concerned with the structure of the prodiginines, we will focus our coverage on the works of Fürstner, Boger, and Tius and their co-workers, whose syntheses first shed new light on the structure and properties of roseophilin. We then conclude with a summary of the most recent synthesis by the Harran lab, due to its unique synthetic strategy, which does not employ ketone 217 as an intermediate (unlike every other synthesis to date).
Figure 28.

Commonly targeted synthetic roseophilin fragments.
The first total synthesis of roseophilin was completed in 1998 by Fürstner and Weintritt,86 who developed a convergent approach involving the coupling of a pyrrylfuran fragment (216) and a macrotricyclic core (217) (Scheme 21). The first key step in Fürstner and Weintritt’s synthesis of roseophilin was a Corey– Chaykovsky epoxidation of 9-bromononanal (219) with sulfonium 124 (available in three steps from alcohol 218) to afford the allylic epoxide 220. Displacement of the bromide in 220 with potassium sulfonate 221 provided α-sulfonyl ester 222. Treatment of 222 with a palladium(0) catalyst caused an intramolecular Tsuji–Trost-type macrocyclization to afford allylic alcohol 223. Fluoridic deprotection of 223 with concomitant esterification under basic conditions resulted in the formation of lactone 224, which could be oxidized with DMP and treated with benzylamine and catalytic palladium(0) to afford pyrrole 225. Activation of the carboxylic acid of 225 with Vilsmeier–Haack reagent 226, followed by exposure to stannous chloride, allowed an intramolecular Friedel–Crafts reaction to take place, forming α-sulfonyl ketone 227. Elimination of the sulfinate group followed by a zincate-conjugate addition reaction provided the benzyl-protected roseophilin core structure, 228. Reductive debenzylation and reprotection of the pyrrole nitrogen with 2-(trimethylsilyl)ethoxymethyl chloride (SEMCl) resulted in tricyclic species 229, which was treated with a carefully prepared furanyl cerate, 230, to afford tertiary alcohol 231. While Terashima and co-workers108 had reported the acid condensation of pyrrylfuran 216 with simple ketones, Fürstner and Weintritt86 found that such a condensation was ineffective on more complex substrates. Finally, desilylation and dehydration of 231 afforded the natural product (37) in racemic form (1% yield after 18 linear steps from alcohol 218). A comparison of the synthetic product to an authentic sample of natural roseophilin confirmed the structure and relative stereochemistry originally postulated in 1992 (see section 2.8).
Scheme 21.

Synthesis of Roseophilin (37) by Fürstner and Weintritt (1998)86
Boger and Hong’s synthesis87 involved oxidation of alcohol 233 (available in three steps from oxazolidinone 232) followed by Wittig homologation of the corresponding aldehyde to enol ether 234 (Scheme 22A). A [4 + 2]/retro-[4 + 2] cycloaddition sequence with tetrazine 235, with concomitant elimination of methanol, afforded diazine 236. Reductive ring contraction of 236 with zinc under acidic conditions, followed by benzyl group hydrogenolysis and acid-catalyzed lactonization, provided 237. A series of trivial functional-group interconversions from 237 provided lactone 238, which could be subjected to oxidation, Wittig olefination, and hydrogenation to furnish alcohol 239. Oxidation and olefination of 239 led to alkene 40, which could be hydrolyzed and similarly olefinated to afford triene 241. Ring closing metathesis with Grubbs’ first-generation catalyst provided a macrocyclic diene, which was further elaborated to acyl selenide 242. Treatment of 242 with azobis- (isobutyronitrile) (AIBN) and tributyltin hydride led to the formation of an acyl radical, which cyclized in an intramolecular fashion to afford the tricyclic ketone 229 after catalytic hydrogenation over a platinum catalyst. The enantioenriched ketone 229 could then be converted to (22S,23S)-37 according to Fürstner’s cerate-addition procedure.
Scheme 22. (A) Synthesis of (22S,23S)-Roseophilin by Boger and Hong87 and (B) Enantioselective Synthesis of (22R,23R)-Roseophilin (37) by Tius and Harrington88,117 a.

aHFIP = 1,1,1,3,3,3-hexafluoro-2-propanol; TFE = 2,2,2-trifluoroethanol.
Tius and Harrington’s synthesis88,117 targeted the opposite enantiomer to that of Boger and Hong, and it used a method they had developed to introduce asymmetry at a later stage. Their synthesis initiated with an amination of 5-hexenal with tert-butylamine to afford imine 244 (Scheme 22B). C-Silylation of 244, followed by a Peterson-type olefination and imine hydrolysis, afforded enal 245. Pinnick oxidation of 245, followed by amidation of the resulting carboxylic acid, led to morpholine amide 246. Monoaddition of lithiated allene 247 to 246, followed by transfer of the solution to a cold acidic bath, led to the enantioselective formation of cyclopentenone 248 through a Nazarov cyclization process. Benzoylation of 248 under standard conditions provided benzoate 249, which was subject to Stetter reaction conditions with 6-heptenal to generate 1,4-dicarbonyl 252. Ring-closing metathesis with Grubbs’ first-generation catalyst provided macrocycle 253 as a mixture of E/Z isomers, which were simultaneously hydrogenated to diketone 254. Paal– Knorr condensation and deprotection of 254, followed by SEM protection of the resulting pyrrole, afforded the roseophilin core structure 229, which was converted to enantiopure (22R,23R)-roseophilin according to the method of Fürstner.
A comparison of the CD spectrum of enantiopure, synthetic (22S,23S)-roseophilin from Boger and Hong87 to that of an authentic sample provided by Seto and co-workers85 revealed that natural roseophilin was the enantiomer of Boger and Hong’s synthetic roseophilin (Figure 29). Indeed, a direct comparison of Tius’s synthetic (22R,23R)-roseophilin to the natural product revealed a perfect spectroscopic match, setting the absolute configuration of roseophilin as (22R, 23R).88
Figure 29.

Absolute configuration of natural roseophilin, (+)-37, and suspected configuration of natural prodigiosin R1, 40.
Based on the structure of roseophilin and the fact that S. griseoviridis produces both natural products, the absolute configuration of prodigiosin R1 might be expected to be as shown in Figure 29. However, as mentioned in the section discussing the structure of streptorubin B (see section 2.6), streptorubin B and its very similar congener metacycloprodigiosin are pseudoenantiomeric in nature,66 and the situation may in fact be the same for prodigiosin R1 and roseophilin. Clift and Thomson65 completed an enantioselective synthesis of both enantiomers of prodigiosin R1 using the same strategy as that used to prepare metacycloprodigiosin (see section 3.3). However, without natural material for comparison, the absolute configuration of prodigiosin R1 remains unknown.
Most recently, Frederich and Harran119 reported a concise synthesis of (+)-roseophilin (37) through a unique strategy (Scheme 23). Unlike all previous syntheses of 37, which used Fürstner’s end-game, Frederich and Harran utilized a completely different series of end-game bond constructions. Their synthesis involved initial generation of alkene 262 in a short number of steps from furan 260 (for which they developed an improved synthesis). Phosphorylation of the pyrrole nitrogen within 262, and subsequent formation of ketone 263 by cross-metathesis and conjugate reduction, set the stage for the first critical cyclization to generate pyrrolophane 267. In this cleverly designed step, enolization of ketone 263 with potassium bis(trimethylsilyl)-amide (KHMDS) allows for reversible formation of the thermodynamically disfavored macrocyclic aldol adduct 265, which is kinetically trapped by a subsequent N-to-O migration of the phosphoryl group to generate 266. In situ elimination affords the pyrrolophane 267 in a remarkable 66% yield. Enantioselective hydrogenation of the thus-formed tetrasubstituted alkene with JosiPhos ligand 268 afforded ketone 269 in 92% yield and 67% enantiomeric excess (ee), with a diastereomeric ratio (dr) > 25:1. Completion of the synthesis was achieved through an intramolecular Friedel–Crafts condensation that initially generates azafulvene 270. In situ treatment with acid cleaves the SEM protecting group and facilitates isomerization of the azafulvene subunit to the fully conjugated isomer, which in this case is the desired natural product, (+)-roseophilin (37).
Scheme 23.

Enantioselective Synthesis of (+)-Roseophilin (37) by Frederich and Harran119
4. BIOSYNTHESIS
Due to their fascinating structures and wide range of biological activities, numerous studies of prodiginine alkaloid biosynthesis have been reported over the last century. Here, we give a historical perspective of these studies and summarize modern understanding of the natural mechanisms for prodiginine assembly.
4.1. Precursor Incorporation Experiments and Early Genetic Studies
Due to the original tripyrrylmethene structure proposed for prodigiosin (see section 2.1) and the proposed involvement of a tripyrrylmethene intermediate in porphyrin biosynthesis, prior to 1960 it was hypothesized that prodigiosin (1) and porphyrin (274) are biosynthesized via similar pathways (Figure 30).120 Supporting this notion, in 1950 Hubbard and Rimington121 found that, as is the case for porphyrins, radiolabeled glycine and acetate were incorporated into prodigiosin (1). In 1958, Gibson et al.122 found that eukaryotic cells converted glycine (271) and succinyl-CoA to 5-aminolevulinic acid (5-ALA, 272), a key intermediate in porphobilinogen (274) biosynthesis.
Figure 30.

Before 1960, scientists speculated that a biosynthetic relationship might exist between prodiginines and porphyrins.
In 1960, however, Marks and Bogorad123 fed radiolabeled 5-aminolevulinic acid (5-ALA) to S. marcescens and found that it was not incorporated into prodigiosin (1), indicating that the 4-methoxypyrrolyldipyrromethene moiety of 1 does not derive from porphobilinogen, despite the fact that glycine is a common precursor of both 1 and porphyrins. While investigations into a possible biosynthetic relationship between prodiginines and porphyrins were being carried out, evidence that prodigiosin derives from a bifurcated pathway that is distinct from porphyrin biosynthesis began to emerge. In 1956, Santer and Vogel41 isolated 4-methoxy-2,2′-bipyrrole-5-carboxaldehyde (MBC, 6) from an S. marcescens mutant (9-3-3) blocked in prodigiosin biosynthesis and demonstrated that it could be converted to prodigiosin by a second strain also blocked in prodigiosin production. In 1960, Wasserman et al.124 found that exposure of S. marcescens strain 9-3-3 to vapors of synthetic methylamylpyrrole (MAP, 7) resulted in the formation of prodigiosin within a few minutes. Evidence that the condensation of 6 and 7 (Figure 31) is enzyme-catalyzed was reported by Williams et al. in 1965,125 who found that the condensation of MBC and MAP by a cell lysate of strain 9-3-3 is significantly less efficient at slightly elevated temperatures (>35 °C).
Figure 31.

Structures of MBC (6) and MAP (7), key intermediates in prodigiosin (1) biosynthesis.
In 1966, Morrison126 was able to further delineate the prodigiosin biosynthetic pathway through the isolation of several hundred S. marcescens mutants blocked at different points in prodigiosin biosynthesis. These mutants were labeled B or M according to whether they are blocked in MBC or MAP biosynthesis, respectively. He was able to determine the point at which the biosynthesis was blocked in most of the mutants by cosynthesis experiments; mutants blocked at late stages in the biosynthetic pathway could restore prodigiosin production in those blocked at earlier stages (e.g., B2 could restore prodigiosin production in B1; Figure 32). Through such cosynthesis studies, Morrison was also able to confirm the bifurcated nature of the prodigiosin biosynthetic pathway, because different classes of mutants existed that could restore prodigiosin production in other classes regardless of order (e.g., M2 and M1 could restore prodigiosin biosynthesis in both B2 and B1). Morrison126 also noted the appearance of single mutants that appeared to be simultaneously blocked in both MAP and MBC biosynthesis. On the basis of this observation, he speculated that there might be a step common to both MAP and MBC biosynthesis, though current knowledge suggests such mutations were in regulatory genes (see sections 4.2 and 4.3).
Figure 32.

Morrison’s map of prodigiosin biosynthesis.126 Mutants blocked at different stages of the pathway are indicated by the vertical lines through the arrows.
In the early 1970s, Wasserman et al.127,128 conducted a series of incorporation experiments utilizing 13C and 14C labeled precursors to determine the metabolic origins of prodigiosin (1) and undecylprodigiosin (17) (Figure 33). Both A-rings were found to derive from L-proline and both B-rings were found to originate from L-serine, a unit of acetate, and the S-methyl group of methionine. The early observation that glycine is incorporated into prodigiosin can be explained by the well-known metabolic interconversion of glycine and serine. The precursor incorporation pattern was found to diverge for the C-ring, which was found to derive from acetate and L-alanine in prodigiosin (1), but from acetate and glycine in undecylprodigiosin (17).
Figure 33.

Precursor incorporation pattern determined by Wasserman et al.127,128 for prodigiosin (1) and undecylprodigiosin (17). Colored dots indicate 13C or 14C labels.
These studies led to the hypothesis that undecylprodigiosin is biosynthesized in Streptomyces species via a pathway analogous to that for prodigiosin biosynthesis in Serratia species, in which the common intermediate MBC (6) is condensed with 2-undecylpyrrole (2-UP, 76) instead of MAP (7). In 1980, Rudd and Hopwood129 used genetic analysis of nonpigmented mutants to identify and map the prodiginine biosynthetic gene cluster (the red cluster) in a mutant of Streptomyces coelicolor A3(2) deficient in actinorhodin biosynthesis. A decade later, Hopwood and co-workers130 reported cloning of the complete red cluster from S. coelicolor and expression in a heterologous host (Streptomyces lividans). In 2001, the sequence of the entire red cluster became available through the S. coelicolor genome sequencing project (Figure 34),63 facilitating elucidation of the biosynthetic pathway to undecylprodigiosin (17) and streptorubin B (22) via molecular genetics and biochemical studies (vida infra).
Figure 34.

Organization of the red gene cluster in S. coelicolor A3(2), responsible for undecylprodigiosin (17)/streptorubin B (22) biosynthesis, and the pig gene cluster in Serratia species, responsible for prodigiosin (1) biosynthesis.
In 2000, while screening a Serratia sp. ATCC 39006 chromosomal cosmid library in Erwinia carotovora mutants deficient in carbapenem biosynthesis for genes that could restore carbapenem production, Salmond and co-workers131 inadvertently created red-pigmented E. carotovora transformants that were found to produce prodigiosin (1). Four years later, Salmond and co-workers132 reported sequencing and bioinformatics analysis of the cosmid responsible for inducing red pigment production upon introduction into E. carotovora, leading to identification of the cluster of genes (the pig cluster) that directs prodigiosin biosynthesis in Serratia species (Figure 34). Subsequent molecular genetic studies established the role played by each of the genes within the pig cluster in prodigiosin biosynthesis.133
4.2. 4-Methoxy-2,2′-bipyrrole-5-carboxaldehyde (MBC) Biosynthesis
The biosynthesis of MBC (6) in both Serratia and Streptomyces begins with the activation of L-proline as a thioester and subsequent conversion to the corresponding pyrrole. In 2001, Challis and co-workers63 hypothesized that the redM, redO, and redW genes, respectively encoding a standalone nonribosomal peptide synthetase (NRPS) adenylation (A) domain, a peptidyl carrier protein (PCP), and a flavin adenine dinucleotide (FAD)-dependent dehydrogenase, are responsible for these transformations, in analogy to the initial steps of pyoluteorin biosynthesis proposed by Gould and co-workers.134 The following year, Walsh and co-workers135 experimentally confirmed that oxidation of the pyrrolidine of a L-prolyl thioester to the corresponding pyrrole is common to pyoluteorin and undecylprodigiosin biosynthesis and is catayzed by the PltF/RedM, PltL/RedO, and PltE/RedW enzymes. The pigI, pigG, and pigA genes in Serratia species were subsequently assigned similar roles in prodigiosin biosynthesis,133 and Walsh, Kelleher, and co-workers136 later verified the functions of the corresponding proteins. Independent deletion of the redM and redW genes in S. coelicolor abolished the biosynthesis of undecylprodigiosin (17) and streptorubin B (22).137 Production of these metabolites could be restored by feeding synthetic MBC to the mutants, confirming that RedM and RedW are involved in MBC assembly.137 Thus, the initial steps of MBC biosynthesis in S. coelicolor and Serratia species consist of L-proline activation by RedM/PigI, loading onto the peptidyl carrier protein RedO/PigG, and four-electron oxidation of the resulting prolyl thioester 277 by the FAD-dependent oxidase RedW/PigA to generate the pyrrole-2-carboxyl thioester 278 (Scheme 24).
Scheme 24. Proposed Pathway for Assembly of MBC, a Common Intermediate in Biosynthesis of Prodigiosin in Serratia Species and Undecylprodigiosin in S. coelicolora.

aAbbreviations: A, adenylation; ACP, acyl carrier protein; KS, ketosynthase; OAS, α-oxamine synthase; OMT, O-methyltransferase; PCP, peptidyl carrier protein; PPTase, phosphopantetheinyl transferase.
A plausible mechanism for oxidation of thioester 277 to pyrrolyl thioester 278 can be proposed, based on mechanisms of other FAD-dependent dehydrogenases (Scheme 25).138 Removal of the acidic α-proton of thioester 277, followed by net elimination of the β-hydride (presumably via single-electron transfer from enolate 283 to the FAD cofactor 284, followed by abstraction of the β-hydride by the resulting flavin radical) would result in Δ2-pyrrolinyl-2-carboxyl-PCP 285. Conversion of the FADH2 formed in this process back to FAD by molecular oxygen would allow subsequent oxidation of Δ2-pyrrolinyl-2-carboxyl-PCP 285 to pyrrole-2-carboxyl-PCP 278 via removal of the acidic γ-proton, followed by net δ-hydride elimination from 286.
Scheme 25.

Possible Mechanism for Oxidation of L-Prolyl-PCP to Pyrrole-2-carboxyl-PCP by Flavin-dependent Dehydrogenases RedW, PigA, and PltE
PCPs such as RedO and PigG must undergo post-translational phosphopantetheinylation, catalyzed by a phosphopantetheinyl transferase (PPTase), to convert them to their active holo forms. RedU and PigL show significant sequence similarity to known PPTases. Challis and co-workers137 reported that deletion of redU in S. coelicolor abolishes the biosynthesis of undecylprodigiosin (17) and streptorubin B (22). Production of these metabolites could be restored by feeding synthetic MBC (6) to the mutant, indicating that redU is involved in the phosphopantetheinylation of carrier proteins involved in MBC biosynthesis. Feeding pyrrole-2-carboxylic acid N-acetylcysteamine (NAC) thioester 288, which mimics the pyrrole-2-carboxyl-PCP intermediate in MBC biosynthesis (Figure 35), to the redU mutant also restored production of undecylprodigiosin (17) and streptorubin B (22), indicating that RedU is required only for the phosphopantetheinylation of the RedO PCP and not the Red N acyl carrier protein (ACP) domains (Scheme 24).
Figure 35.

Phosphopanteteine is a prosthetic group essential for the function of PCPs and ACPs in metabolic pathways. N-Acetylcysteamine (NAC) 288 mimics the phosphopantetheine prosthetic group, and NAC thioesters are thus able to replace acyl-ACPs and PCPs in vitro and in vivo. Condensation of pyrrole 2-carboxylic acid with NAC generates thioester 289, which mimics the pyrrole 2-carboxyl-PCP intermediate 279 in MBC biosynthesis.
The pyrrole-2-carboxyl-PCP intermediate (279) in MBC biosynthesis is proposed to be elongated by decarboxylative condensation with a malonyl-CoA-derived thioester to yield β-keto thioester 280 (Scheme 24). RedX and RedN in S. coelicolor, and their homologues PigJ and PigH in Serratia species, are hypothesized to be involved in this step. RedN/PigH possesses two ACP domains, but in-frame deletion experiments have shown that either one is sufficient to support MBC biosynthesis. RedX/PigJ contains two ketosynthase-like domains, though only the C-terminal domain is predicted to possess ketosynthase activity because the active-site cysteine residue in the N-terminal domain is replaced by aspartate. The transfer of the pyrrole-2-carboxyl intermediate from RedO/PigG to the ketosynthase domain of RedX/PigJ has been inferred to occur by direct transthioesterificaton rather than via hydrolysis, reactivation, and acylation. This is because production of undecylprodigiosin (17) and streptorubin B (22) in redM and redW mutants of S. coelicolor was restored when the NAC thioester of pyrrole-2-carboxylic acid 289 (Figure 35) was added to the culture, but not when pyrrole-2-carboxylic acid was fed.137
RedN/PigH possesses a putative α-oxoamine synthase (OAS) domain at its C-terminus, which is predicted to catalyze chain release. OASs are pyridoxal 5′-phosphate (PLP)-dependent enzymes that catalyze the decarboxylative condensation of amino acids with acyl-CoA thioesters.139 The OAS domain of RedN/PigH is therefore proposed to catalyze decarboxylative condensation of L-serine with RedN/PigH-bound β-keto thioester intermediate 280 to form α,γ-dioxoamine 281, which likely undergoes spontaneous cyclization, dehydration, and tautomerization to yield 4-hydroxyl-2,2′-bipyrrole-5-methanol (HBM) 282 (Scheme 24).
A plausible mechanism for the reaction catalyzed by the RedN/PigH OAS domain can be proposed, based on the well-studied catalytic mechanism of 7-oxo-8-aminononanoate synthase (Scheme 26).140 In the resting state, the PLP cofactor is bound to the OAS domain via an iminium linkage to an activesite lysine residue. Transimination with L-serine forms the external aldimine 290, which is deprotonated by the liberated lysine amino group to form 291. β-Keto thioester 280 acylates 291 to form 292. Decarboxylation of 292 and subsequent protonation yields the product aldimine 293, which undergoes transimination with the active-site lysine residue to release α,γ-dioxoamine 281. Subsequent cyclization, dehydration, and tautomerization of 281 to yield HBM 282 can likely occur spontaneously, but catalysis of these transformations by RedN/PigH cannot be excluded.
Scheme 26.

Proposed Mechanism for Release of β-Keto Thioester Intermediate 280 from RedN/PigH, Catalyzed by OAS Domain, and Subsequent Spontaneous Conversion of the Resulting α,γ-Dioxoamine 281 to HBM 282
Mutants of Serratia lacking pigM were shown to produce alcohol 282 instead of MBC (6), implicating PigM/RedV in the oxidation of 282 to aldehyde 275. PigM and RedV show modest sequence similarity to flavin mononucleotide (FMN)-dependent bacterial nitroreductases,133 suggesting that the oxidation of alcohol 282 to aldehyde 275 utilizes a flavin cofactor.
The final step in MBC (6) biosynthesis is methylation of the hydroxyl group of HBC 275 (Scheme 24). PigF and PigN have both been implicated in this transformation in Serratia species, but RedI is the only enzyme known to be involved in this reaction in S. coelicolor (note, however, that RedF is a homologue of PigN). Sequence comparisons suggest that the homologous RedI and PigF proteins function as S-adenosylmethionine (SAM)-dependent O-methyltransferases (OMTs). Deletion of the genes encoding these proteins in S. coelicolor and Serratia species results in the formation of O-demethylated analogues of undecylprodigiosin and prodigiosin, respectively.64,133 Although PigN appears to be necessary for the efficient conversion of aldehyde 275 to MBC 6, its precise function remains unknown.133
4.3. Methylamylpyrrole (MAP) Biosynthesis in Serratia Species
The same researchers133 have reported that only three genes are required for MAP (7) biosynthesis in Serratia: pigD, pigE, and pigB (Figure 34). Sequence comparisons of the proteins encoded by these genes with proteins of known function led initially to a proposed pathway for MAP biosynthesis from 2-octenal and pyruvate. However, this proposal was subsequently revised in light of further experimental data.141,142 (Scheme 27).
Scheme 27.

Proposed Pathway for MAP (7) Biosynthesis in Serratia Species
The first step of MAP biosynthesis was originally hypothesized to involve decarboxylative condensation of pyruvate with 2-octenal catalyzed by PigD, a thiamine diphosphate (TDP)-dependent enzyme.133 This hypothesis explains the pattern of acetate incorporation into the MAP-derived moiety of prodigiosin 1 observed in earlier feeding experiments (Figure 33)141 and is consistent with derivation of the n-pentyl chain and C-3/C-4/C-5 of the pyrrole from an eight-carbon fatty acid or polyketide precursor.142 However, investigations of the substrate tolerance and product profile of purified recombinant PigD suggest that it catalyzes the condensation of pyruvate with a 2-octenoyl-ACP or CoA thioester (294) rather than 2-octenal.143
The catalytic mechanism of PigD likely involves addition of the heterocyclic carbene resulting from deprotonation of TDP to the keto group of pyruvate, followed by decarboxylation to form a C-1 acetaldehyde anion equivalent that adds to C-3 of 2-octenoyl thioester 294 (Scheme 28). Protonation of the resulting enolate, followed by deprotonation of the tertiary alcohol and elimination of the TDP carbene, would yield 3-acetyloctanoyl thioester 295. Such a mechanism explains the 1968 observation of Goldschmidt and Williams144 that thiamin promotes the formation of MAP in Serratia. The incorporation of C-3 of L-alanine into the methyl group attached to C-2 of the MAP pyrrole (Figure 33) is also consistent with this catalytic mechanism, because L-alanine is known to act as an amino donor in vivo in numerous PLP-mediated transamination reactions that yield pyruvate as a byproduct.
Scheme 28.

Proposed Catalytic Mechanism of TDP-dependent PigD Enzyme
The second step in MAP biosynthesis was originally hypothesized to involve reductive amination of the aldehyde derived from PigD-catalyzed condensation of octenal with pyruvate by PigE, a putative PLP-dependent transaminase, followed by spontaneous cyclization of amino ketone 296 to generate dihydro-MAP 297.133 Consistent with this hypothesis, deletion of pigB (which encodes the putative final enzyme in MAP biosynthesis) resulted in accumulation of a metabolite that gave rise to ions in liquid chromatographic–mass spectrometric (LC-MS) analyses with m/z = 152.1, corresponding to the protonated form of 297.133 Treatment of mycelial extracts of this mutant with sodium cyanoborohydride (NaBH3CN), an imine-specific reducing agent, converted the accumulated metabolite to a new species, which gave rise to ions with m/z = 154.1 in LC-MS analyses, corresponding to the protonated form of the pyrrolidine resulting from reduction of 297.133 More recently, however, it has been proposed that PigE is a bifunctional thioester reductase/PLP-dependent transaminase that catalyzes reduction of γ-keto thioester 295 to the corresponding aldehyde, followed by transamination to yield aminoketone 296 (Scheme 27).145
It has been proposed that the enamine tautomer of 297 is oxidized to MAP (7) by PigB, which shows sequence similarity to FAD-dependent amine oxidases. Interestingly, PigB possesses three putative transmembrane helices at its N-terminus and is thus likely to be membrane-associated. This is presumably due to the hydrophobic nature of the PigB substrate, which causes it to partition preferentially into the membrane bilayer.133
4.4. 2-Undecylpyrrole (2-UP) Biosynthesis in Streptomyces Species
Various genetic and biochemical experiments have implicated redP, redQ, redR, redJ, redK, and redL in the biosynthesis of 2-UP 76 (Scheme 29).75,146,147 RedP, a homologue of the fatty acid biosynthetic enzyme FabH, was proposed to initiate 2-UP biosynthesis by catalyzing the decarboxylative condensation of a malonyl group, attached via a thioester linkage to the RedQ ACP, with acetyl-CoA (Scheme 29).63 Consistent with this, deletion of redP in S. coelicolor strongly impaired prodiginine biosynthesis and led to the production of several undecylprodigiosin (17) analogues with altered alkyl chains.146 These analogues are believed to result from FabH-mediated condensation of alternative starter units, such as isobutyryl-CoA and isovaleryl-CoA, with malonyl-RedQ. The keto group in acetoacetyl-RedQ resulting from RedP-catalyzed condensation is reduced to a methylene group, presumably by ketoreductase (KR), dehydratase (DH) and enoylreductase (ER) components of the core metabolic fatty acid synthase (FAS), because no genes encoding such enzymes are present in the red gene cluster (Figure 34; Scheme 29).63 The butanoyl group generated by this process is then proposed to translocate onto the active-site cysteine residue of the FabF homologue RedR, where it undergoes chain elongation via a further round of decarboxylative condensation with malonyl-RedQ and subsequent ketoreduction, dehydration, and enoylreduction, again presumably catalyzed by the requisite components of the core metabolic FAS (Scheme 29).63 Three further iterations of this cycle result in the formation of dodecanoyl-RedQ,63 which is hydrolytically cleaved by the RedJ thioesterase to generate dodecanoic acid 298.147 Deletion of redR in S. coelicolor strongly impaired undecylprodigiosin production, which could be restored to wild-type levels by feeding of dodecanoic acid.75 These results are consistent with the proposed role of RedR and RedJ in 2-UP biosynthesis. Purified recombinant RedJ has a preference for decanoyl-and dodecanoyl-ACP thioester substrates but is also able to catalyze the hydrolysis of acetyl-ACPs. The X-ray crystal structure of RedJ reveals a large hydrophobic pocket adjacent to its Ser-His-Asp catalytic triad that is capable of accommodating the hydrophobic alkyl chains of decanoyl- and dodecanoyl-ACPs,147 explaining the substrate preference of RedJ. However, levels of prodiginine production are approximately 4-fold lower in a redJ mutant of S. coelicolor than in the wild type.147 Feeding of dodecanoic acid to the mutant does not increase the amount of undecylprodigiosin 17/streptorubin B 22 produced, but in-trans expression of redJ in the mutant restores wild-type production levels.147 These data indicate that RedJ may also play a role in deacetylation of acetylated prodiginine biosynthetic ACPs/PCPs, which arise from utilization of acetyl-CoA in place of coenzyme A by the PPTase(s) responsible for converting them from apo to holo forms. Such partial acetylation of the phosphopantetheine thiols of these ACPs/PCPs would be expected to reduce the overall efficiency of prodiginine biosynthesis.
Scheme 29. Proposed Pathway for 2-UP Biosynthesis in S. coelicolora.

aAbbreviations: A, adenylation; ACP, acyl carrier protein; AT, acyltransferase; DH, dehydratase; ER, enoyl reductase; KS, ketosynthase; KR, ketoreductase; OAS, α-oxoamine synthase; OR, oxidoreductase; TE, thioesterase. Enzymes proposed to be “borrowed” from the core metabolic fatty acid synthase are shown in green.
Deletion of redL in S. coelicolor abrogates the biosynthesis of undecylprodigiosin 17 and streptorubin B 22.75 Production of both metabolites is restored by feeding chemically synthesized 2-UP 76 to the redL mutant, demonstrating that RedL plays a role in the biosynthesis of 2-UP 76, but not MBC 6, and that streptorubin B 22 arises from functionalization of the alkyl chain of either 2-UP 76 or undecylprodigiosin 17 (see section 4.6).75 Sequence analysis of RedL indicates that it contains an adenylation (A) domain, two ACP domains, a KS domain, an acyltransferase (AT) domain, and an OAS domain (Scheme 29). The A domain is proposed to catalyze adenylation of dodecanoic acid, prior to transfer of the dodecanoyl chain to the phosphopantetheine thiol of the adjacent ACP domain. Similarly, the AT domain is proposed to catalyze malonylation of the ACP domain appended to its C-terminus. Decarboxylative condensation of the malonyl thioester with the dodecanoyl thioester, catalyzed by the KS domain, would yield the corresponding β-keto tetradecanoyl thioester. Condensation of this thioester with glycine and subsequent decarboxylation is proposed to be catalyzed by the PLP-dependent OAS domain, via a mechanism analogous to that employed by the RedN OAS domain in MBC biosynthesis (Schemes 26 and 29). Cyclodehydration of the resulting α-oxoamine yields 5-undecylpyrrolin-3-one 299, which is proposed to undergo reduction to 300 catalyzed by RedK, a putative NAD(P)H-dependent oxidoreductase, and subsequent dehydration to yield 2-UP 76. Consistent with this hypothesis, the production of both undecylprodigiosin 17 and streptorubin B 22 was abolished in a redK mutant of S. coelicolor, and feeding of synthetic 2-UP 76 to the mutant restored the production of both metabolites.75 LC-MS analyses showed that the redK mutant accumulated two new metabolites with m/z = 238.0 and 410.3, corresponding to the [M + H]+ ions for 5-undecylpyrrolin-3-one 299 and a hydroxylated derivative of undecylprodigiosin, respectively.75 The instability of these shunt metabolites precluded their isolation and structural elucidation by NMR spectroscopy. However, the hydroxylated undecylprodigiosin derivative was assigned structure 301 on the basis of UV–vis and high-resolution and tandem mass spectrometric (HRMS and MS/MS) data, biosynthetic considerations, and the result of the feeding experiment described above.75 Compound 301 presumably arises from condensation of MBC 6 with 5-undecylpyrrolin-3-one 299 (Figure 36).
Figure 36.

Proposed origin of shunt metabolite 301 accumulated in a redK mutant of S. coelicolor.
4.5. Condensation of 4-Methoxy-2,2′-bipyrrole-5-carboxaldehyde (MBC) with Methylamylpyrrole (MAP) or 2-Undecylpyrrole (2-UP)
The final step in biosynthesis of prodigiosin (1) and undecylprodigiosin (17) is the condensation of MAP (7) and 2-UP (76), respectively, with MBC (6, Scheme 24). These reactions were shown to be catalyzed by PigC in Serratia species and RedH in S. coelicolor.148,149 Mutants in which redH and pigC have been deleted are unable to produce prodiginines, even when fed with MBC, MAP, or 2-UP.148,149 Moreover, feeding of 2-UP and MBC to Streptomyces venezuelae expressing redH resulted in the production of undecylprodigiosin 17.148 Similarly, when MAP and MBC were fed to E. coli expressing pigC, prodigiosin 1 was produced.149 Both RedH and PigC are able to utilize a variety of MBC analogues with alterations to the monosubstituted pyrrole as substrates. This results in production of the corresponding undecylprodigiosin and prodigiosin analogues. 148,149 However, RedH cannot tolerate an MBC analogue in which the trisubstituted pyrrole is replaced with a furan, indicating that the NH group of the trisubstituted pyrrole may play an important role in the condensation reaction.148
Sequence analyses indicated that RedH contains three functional domains (Scheme 30).63 Two of these domains show significant sequence similarity to the ATP-binding and phosphotransfer domains of phosphoenolpyruvate synthase (PEPS) and pyruvate phosphate dikinase (PPDK), suggesting that RedH is an ATP-dependent enzyme that transfers a phosphate group to one of its substrates.63 PigC has an identical domain architecture to RedH.133 The third domain of RedH/PigC is not similar to any proteins of known function and has been proposed to be an MBC-binding domain.133,148 More recently it has been shown that PigC partitions to the membrane bilayer when it is overproduced in E. coli, and it has been confirmed that ATP is required for the condensation of MBC and MAP by PigC-containing membrane preparations.145
Scheme 30. Proposed Mechanism for RedH-catalyzed Condensation of MBC 6 with 2-UP 76a and for PigC-catalyzed Condensation of MBC 6 with MAP 7b,c.

aR1 = H, R2 = C11H23. bR1 = C5H11, R2 = Me. cOrganization of catalytic domains within RedH/PigC is shown in the inset. PT, phosphotransferase domain.
A plausible mechanism for the RedH-catalyzed condensation reaction has been proposed, based on the previously studied catalytic mechanisms of PEPS and PPDK (Scheme 30).148,150 Thus, a conserved histidine residue in the phosphotransfer domain reacts with the γ-phosphate group of ATP. The resulting phosphoryl-histidine intermediate then phosphorylates the carbonyl group of MBC 6 to form doubly vinylogous iminium ion 302. Nucleophilic attack of 302 by C-5 of 2-undecylpyrrole 76, followed by elimination of phosphate and concomitant deprotonation to rearomatize the pyrrole, yields undecylprodigiosin 17 (Scheme 30). A similar mechanism for PigC-catalyzed condensation of MBC 6 and MAP 7 to form prodigioisin 1 has also been proposed, and site-directed mutagenesis of the conserved histidine residue in the phosphotransfer domain has shown it plays an essential role in catalysis (Scheme 30).133,145
4.6. Oxidative Carbocyclizations of Undecylprodigiosin
Carbocyclic derivatives of undecylprodigiosin 17, such as streptorubin B 22 and metacycloprodigiosin 20, have been shown to result from oxidative C–C bond-forming reactions catalyzed by enzymes belonging to the Rieske non-heme iron-dependent oxygenase family.64,151 RedG, which shares conserved [2Fe-2S] cluster and non-heme iron-binding residues with the well-studied Rieske oxygenase naphthalene dioxygenase (NDO), is responsible for the conversion of undecylprodigiosin 17 to streptorubin B 22.64 RedG was first implicated in this oxidative carbocyclization reaction via deletion of its encoding gene, which abrogated the production of streptorubin B 22, but not undecylprodigiosin 17.64 Furthermore, a redO mutant of S. coelicolor accumulated desmethylundecylprodigiosin 303 (Figure 37) but not desmethylstreptorubin B, implying that undecylprodigiosin 17, rather than 2-undecylpyrrole 76, is the substrate of RedG.64
Figure 37.
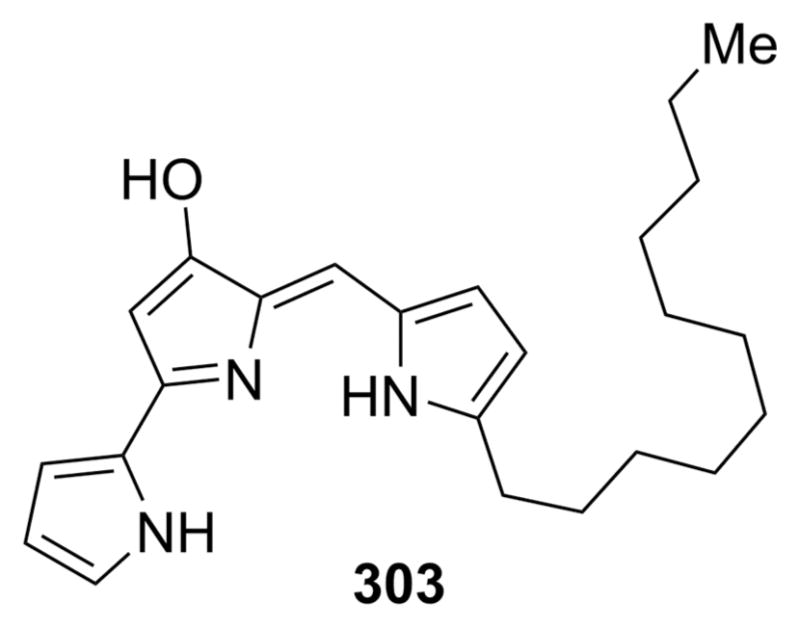
A redO mutant of S. coelicolor accumulates desmethylundecylprodigiosin (303) but not desmethylstreptorubin B, suggesting that undecylprodigiosin is the substrate of RedG.
Feeding of undecylprodigiosin 17 to S. venezuelae in which redG or redHG have been constitutively expressed resulted in the production of streptorubin B 22, demonstrating that RedG alone is responsible for the oxidative carbocyclization reaction. Streptorubin B 22 production levels were higher in the strain expressing both redG and redH than in the strain expressing redG alone, suggesting that RedG may form a complex with RedH that results in enhanced catalytic efficiency. No transformation to a carbocyclic derivative was observed when 2-undecylpyrrole 76 was fed to the strains.64
Orthologues of redH and redG, named mcpH and mcpG, respectively, were identified in the genome of the metacycloprodigiosin 20 producer S. longisporus ruber and sequenced. Expression of mcpG in the redG mutant of S. coelicolor led to the exclusive production of metacycloprodigiosin 20, demonstrating that RedG and McpG catalyze the regio- and stereodivergent oxidative carbocyclizations of undecylprodigiosin 17 to form streptorubin B 22 and metacycloprodigiosin 20, respectively (Scheme 31).64
Scheme 31.

RedG and McpG Catalyze Regio- and Stereodivergent Oxidative Carbocyclizations of Undecylprodigiosin (17) To Form Streptorubin B (22) and Metacycloprodigiosin (20), Respectively
A mechanism for the RedG-catalyzed oxidative carbocyclization reaction has recently been proposed, based on what is known about the catalytic mechanism of the well-studied Rieske oxygenase NDO (Figure 38).151 Binding of undecylprodigiosin 17 to the active site is hypothesized to trigger loss of a water ligand from the non-heme iron center, allowing dioxygen to bind. Transfer of an electron from the [2Fe-2S] cluster to the nonheme iron center, coupled with loss of a second water ligand and proton transfer, leads to formation of Fe(III)OOH complex 304. Abstraction of a hydrogen atom (or hydride ion) from C-7′ of enzyme-bound undecylprodigiosin 17 by either the Fe(III)OOH complex, or an Fe(V)O(OH) complex resulting from rearrangement of the Fe(III)OOH complex, would result in formation of a carbon-centered radical 305 (or the corresponding cation in the case of hydride abstraction) and an Fe(IV)O(OH2) complex 306. Addition of the C-7′ radical (or cation) to C-4 would result in formation of the C-5 radical 307 (or the corresponding cation), either of which can be stabilized by delocalization into the adjacent π-system. Abstraction of a hydrogen atom from C-4 of 307 by the Fe(IV)O(OH2) complex 306 (or a proton in the case of the cationic intermediate) would yield enzyme-bound streptorubin B 22, the release of which is coupled to transfer of a second electron from the [2Fe-2S] cluster to the non-heme iron center and protonation of the hydroxide ligand.
Figure 38.

Mechanism proposed by Challis and co-workers151 for the conversion of undecylprodigiosin 17 to streptorubin B 22, catalyzed by the Rieske oxygenase-like RedG enzyme. An Fe(V)O(OH) species and C-7′/C-5 cations are plausible alternatives to intermediates 304 and 305/307, respectively.
Very recently, the use of 2-(5-pentoxypentyl)-pyrrole 308 to probe the mechanism of RedG-catalyzed conversion of undecylprodigiosin (17) to streptorubin B (22) has been reported.152 Feeding of 2-(5-pentoxypentyl)pyrrole 308, along with MBC 6, to Streptomyces albus expressing redH and redG resulted in production of the expected 6′-oxa undecylprodigiosin analogue 309. However, none of the corresponding 6′-oxa streptorubin B analogue could be detected. Instead, a small quantity of 5-hydroxypentylprodigioisn 310 was produced (Scheme 32).
Scheme 32.

Fate of the 6′-Oxa Analog (308) of Undecylprodigiosin When Fed Along With MBC (6) to S. Albus Expressing redHG
5-Hydroxypentylprodigiosin 310 presumably arises via abstraction of a hydrogen atom from C-7′ of 309 by Fe(III)OOH complex 304. The resulting radical/cation is stabilized by the adjacent oxygen lone pair, preventing it from adding to C-4. Instead it reacts with the Fe(IV)O(OH2) complex 306 to form the hemiketal complex 311, which collapses with loss of n-pentanal 312 to yield 5-hydroxypentylprodigiosin 310 (Scheme 33).
Scheme 33.

Proposed Mechanism for RedG-catalyzed Formation of 5-Hydroxypentylprodigiosin 310 from the 6′-Oxa Analogue 309 of Undecylprodigiosin
The same study152 also investigated the stereochemical course of RedG-catalyzed conversion of undecylprodigiosin 17 to streptorubin B 22. Feeding of [7′-2H](7′R)-2-undecylpyrrole 314, along with MBC 6, to S. albus expressing redH and redG resulted in substantial loss of the deuterium label from the resulting streptorubin B 22 (Scheme 34). In contrast, the deuterium label was largely retained when [7′-2H](7′S)-2-undecylpyrrole 315 and MBC 6 were fed to S. albus expressing redH and redG (Scheme 34).152
Scheme 34. Loss and Retention of Deuterium Label in Biosynthesis of Streptorubin B (22)a.

aBiosynthesis of streptorubin B (22) via feeding of [7′-2H]-(7′R)-2-undecylpyrrole 314 and MBC 6 to S. albus expressing redHG results in substantial loss of deuterium label, whereas the label is retained in streptorubin B produced by feeding of [7′-2H]-(7′S)-2-undecylpyrrole 315 and MBC 6 to S. albus expressing redHG.
These results show that RedG abstracts predominantly the pro-R hydrogen atom from C-7′ of undecylprodigiosin (Scheme 34). Streptorubin B 22 has predominantly the 7′S configuration (see section 2.6). Thus, the conversion of undecylprodigiosin 17 to streptorubin B 22 proceeds with inversion of configuration at C-7′. This contrasts with the functionalization of unactivated carbon centers via oxidative heterocyclization reactions catalyzed by non-heme iron-dependent oxygenases such as isopenicillin N synthase and clavaminate synthase. In such cases the reactions proceed with retention of configuration at the carbon atom undergoing functionalization.153,154
4.7. Marineosin Biosynthesis
In their paper reporting the isolation and structural elucidation of marineosins A and B (41 and 42), Fenical and co-workers91 noted that both structures contain a retron for an inverse electron-demand Diels–Alder reaction. This led them to propose that the marineosins may be biosynthesized via an intramolecular [4 + 2] cycloaddition reaction of 316 (Scheme 35).
Scheme 35.

Pathway for Marineosin Biosynthesis Proposed by Fenical and Co-workers in 200891
In early 2010, Lindsley and co-workers155 reported the synthesis of 316 and attempts to complete a biomimetic synthesis of the marineosins inspired by Fenical’s biosynthetic proposal. Despite trying numerous different reaction conditions, they were unable to observe the conversion of 316 to either marineosin A (41) or marineosin B (42). Although this did not disprove Fenical’s biosynthetic proposal, because the Diels– Alder reaction may require enzymatic catalysis, these findings called its feasibility into question.
Later the same year, Snider and co-workers156 reported a different route toward the synthesis of marineosins and put forward an alternative biosynthetic proposal, involving abstraction of a hydrogen atom from C-8′ of undecylprodigiosin 17 to create a radical that undergoes cyclization to generate intermediate 317 (Scheme 36). Intramolecular hydrogen transfer via a six-membered transition state would convert intermediate 317 to the C-10′ radical 318, which could be trapped by enzyme-mediated hydroxylation to give 319. Tautomerization of 319 followed by nonselective spirocyclization would yield marineosins A and B (41 and 42; Scheme 36).
Scheme 36.

Alternative Proposal for Marineosin Biosynthesis Put Forward by Snider and Co-workers156
In 2014, Reynolds and co-workers157 reported the cloning, sequencing, and heterologous expression of the mar gene cluster responsible for marineosin biosynthesis in Streptomyces CNQ-617. The gene content and organization of the mar cluster are identical to those of the red cluster responsible for streptrorubin B biosynthesis in S. coelicolor. The only exception is that the mar cluster contains an additional gene, marA, which was shown to be involved in the last step of marineosin biosynthesis, the reduction of premarineosins 321 to 41 and 42 (Scheme 37). Deletion of marG, which encodes a RedG homologue (see section 4.6), from the heterologous expression construct abrogated marineosin production and resulted in the accumulation of 10′-hydroxyundecylprodigiosin. Feeding of (10′S)-10′-hydroxyundecylprodigiosin 320 deuterated at C-10′ to S. venezuelae expressing marG resulted in the production of premarineosins 321 in which the deuterium label had been retained, whereas an analogous experiment employing (10′R)-10′-hydroxyundecylprodigiosin deuterated at C-10′ resulted in loss of the deuterium label from premarineosins 321.92 Taken together, these data suggest that the marineosins are biosynthesized via the pathway shown in Scheme 37.
Scheme 37.

Pathway for Marineosin Biosynthesis Elucidated by Reynolds and Co-workers157
On the basis of their recent mechanistic studies of RedG, Challis and co-workers152 have proposed a MarG catalytic mechanism involving abstraction of a hydrogen atom from C-8′ of (10′S)-10′-hydroxyundecylprodigiosin 320 by the Fe(III)-OOH complex 304 to yield a methylene radical that cyclizes onto C-1″. The resulting delocalized radical 322 can then react with Fe(IV)O(OH2) complex 306 to form the hydroxylated product 323 (Figure 39).
Figure 39.

Proposed mechanism for MarG-catalyzed oxidative cyclization and hydroxylation of (10′S)-10′-hydroxyundecylprodigiosin 320. As for the RedG catalytic mechanism, alternative Fe(V)O(OH) and carbocation intermediates are also possible.
The product 323 of the MarG-catalyzed reaction is hypothesized by Challis and co-workers152 to undergo elimination of water to form 324, followed by addition of the C-10 hydroxyl group to afford the premarineosins 321 (Scheme 38).
Scheme 38.

Mechanism for Conversion of Product 312 of the MarG-catalyzed Reaction to Premarineosins 311
4.8. Roseophilin Biosynthesis
Although roseophilin has been the target of numerous synthetic studies over the last two decades, relatively little has been known about its biosynthesis until recently. It seems likely that chlorination is the final step in roseophilin biosynthesis, because dechlororoseophilin 38 was isolated by Kawasaki and coworkers90 from the roseophilin producer S. griseoviridus in 2009. Prodigiosin R1 40 has also been isolated from this strain and it seem likely that both 38 and 40 are derived from the common precursor 11′-dimethylundecylprodigiosin 39.89
In 2009, Kawasaki et al.158 reported identification of the probable dechlororoseophilin 38 and prodigiosin R1 40 biosynthetic gene cluster in S. griseoviridis. This gene cluster is very similar to the red cluster in S. coelicolor, but it contains four genes (rphG, rphG2, rphG3, and rphG4) encoding RedG orthologues. The protein encoded by the rphG3 gene is likely nonfunctional because its N-terminal Rieske domain lacks the conserved Cys and His residues that ligate the [2Fe-2S] cluster. Two of the other RedG orthologues have recently been proposed by Challis and co-workers152 to be involved in the assembly of dechlororoseophilin 38, via a RedG-like oxidative carbocyclization between C-9′ and C-4 of 11′-dimethylundecylprodigiosin 39 to yield 325, followed by a MarG-like oxidative carbocylization– hydroxylation to yield 326 (Scheme 39). Ring opening of 326, followed by tautomerization, cyclization, and elimination of ammonia would yield dechlororoseophilin (38). The remaining RedG orthologue is hypothesized to catalyze the conversion of 11′-dimethylundecylprodigiosin 39 to prodigiosin R1 (Scheme 39).152
Scheme 39.

Pathways for Biosynthesis of Dechlororoseophilin 38 and Prodigiosin R1 40 from 11′-Dimethylundecylprodigiosin 39, Proposed by Challis and Co-workers152
5. CONCLUDING REMARKS
The prodiginine family of natural products has stimulated research in diverse fields of science for well over a century due to their wide range of fascinating properties, from their color to their chirality. In writing this review, we aimed to provide a comprehensive overview of developments in the chemistry and biology of the prodiginine alkaloids over the past century, while providing the historical context in which major discoveries were made. We have also taken care to note the last century’s rich interplay between natural product structural elucidation and chemical synthesis, between chemical synthesis and biosynthetic theory, and between biosynthetic theory and molecular genetics. Nearly 200 years after the identification of Serratia, research into prodiginine chemistry and biology continues to provide leads in fascinating fields including aliphatic C–H functionalization, cellular ion transport, and modular natural product biosynthesis. We hope that our organization of recent research and newly proposed classification of prodiginine natural products will assist modern researchers in natural products chemistry, organic synthesis, and microbial natural product biosynthesis alike.
Acknowledgments
We are grateful for support from the National Institutes of Health (1R01GM085322 to R.J.T.), Northwestern University (summer undergraduate research grant to D.X.H.), the BBSRC for a doctoral training grant, and the Royal Society of Chemistry for the Briggs scholarship (both to D.M.W.).
Biographies
Dennis X. Hu graduated from Northwestern University in 2011 with B.A./M.S. degrees in chemistry carried out under the mentorship of Professor Regan J. Thomson. After a year of graduate study at the University of Cambridge with Professor Steven Ley as a Churchill fellow, he moved to Stanford University as an NSF graduate fellow in the lab of Professor Noah Z. Burns. Following the completion of his Ph.D. in 2015, Dennis accepted a position as a medicinal scientist at FLX Biosciences, Inc., South San Francisco, CA.
David M. Withall graduated with an M. Chem. in chemistry with industrial training from the University of Warwick in 2010 and completed Ph.D. studies on the biosynthesis of streptorubin B at the same institution under the supervision of Professor Greg Challis in 2014. He is currently a postdoctoral fellow at Rothamsted Research with Dr. Mike Birkett and Professor John Pickett. He was the recipient of a Briggs scholarship from the Royal Society of Chemistry (2010–2013).
Gregory L. Challis was born in the United Kingdom in 1973. He graduated with a B.Sc. in chemistry from Imperial College London in 1994 and was awarded a Ph.D. by the University of Oxford in 1998, for research carried out under the supervision of Professor Sir Jack Baldwin. After postdoctoral studies as a Wellcome Trust International Prize travelling research fellow with Professor Craig Townsend at Johns Hopkins University and with Professor Keith Chater at the John Innes Center, he joined the faculty of the University of Warwick in 2001. Greg’s research interests encompass the discovery, biosynthesis, bioengineering, and mechanism of action of bioactive natural products. He is the recipient of the Royal Society of Chemistry’s Meldola Medal (2002) and Hickinbottom Award (2009), the Fleming Prize of the Microbiology Society (2007), and the Gabor Medal (2009) and a Wolfson Research Merit Award (2013–2018) from the Royal Society.
Regan J. Thomson was born in New Zealand in 1976 and received his Ph.D. in 2003 at The Australian National University, working with Professor Lewis N. Mander. Following postdoctoral studies with Professor David A. Evans at Harvard University, he joined the faculty at Northwestern University in 2006. Regan’s research interests include reaction development, total synthesis, natural product discovery and biosynthesis, and atmospheric chemistry. He is the recipient of an NSF Career Award (2009), an Amgen Young Investigator Award (2010), an Illinois Division American Cancer Society Research Scholar Award (2012), and a Novartis Chemistry Lectureship (2015–2016).
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Merlino CP. Bartolomeo Bizio’s Letter to the Most Eminent Priest, Angelo Bellani, Concerning the Phenomenon of the Red-Colored Polenta [Translated from the Italian] J Bacteriol. 1924;9(6):527–543. doi: 10.1128/jb.9.6.527-543.1924. ( http://jb.asm.org/content/9/6/527.full.pdf+html) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett JW, Bentley R. Seeing Red: The Story of Prodigiosin. Adv Appl Microbiol. 2000;47:1–31. doi: 10.1016/s0065-2164(00)47000-0. [DOI] [PubMed] [Google Scholar]
- 3.Williamson NR, Fineran PC, Leeper FJ, Salmond GPC. The Biosynthesis and Regulation of Bacterial Prodiginines. Nat Rev Microbiol. 2006;4:887–899. doi: 10.1038/nrmicro1531. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt EV. Genetic Control of Cell Chemistry Using Serratia marcescens. In: Goldman CA, editor. Tested Studies for Laboratory Teaching; Proceedings of the 14th Workshop/Conference of the Association for Biology Laboratory Education (ABLE); 1993. pp. 21–34. http://www.ableweb.org/volumes/vol-14/2-schmidt.pdf. [Google Scholar]
- 5.Castro AJ. Antimalarial Activity of Prodigiosin. Nature. 1967;213:903–904. doi: 10.1038/213903a0. [DOI] [PubMed] [Google Scholar]
- 6.Kim H, Hayashi M, Shibata Y, Wataya Y, Mitamura T, Horii T, Kawauchi K, Hirata H, Tsuboi S, Moriyama Y. Cycloprodigiosin Hydrochloride Obtained from Pseudoalteromonas denitrificans Is a Potent Antimalarial Agent. Biol Pharm Bull. 1999;22(5):532–534. doi: 10.1248/bpb.22.532. [DOI] [PubMed] [Google Scholar]
- 7.Isaka M, Jaturapat A, Kramyu J, Tanticharoen M, Thebtaranonth Y. Potent in Vitro Antimalarial Activity of Metacycloprodigiosin Isolated from Streptomyces spectabilis BCC 4785. Antimicrob Agents Chemother. 2002;46(4):1112–1113. doi: 10.1128/AAC.46.4.1112-1113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laurent D, Pietra F. Antiplasmodial Marine Natural Products in the Perspective of Current Chemotherapy and Prevention of Malaria. A Review. Mar Biotechnol. 2006;8:433–447. doi: 10.1007/s10126-006-6100-y. [DOI] [PubMed] [Google Scholar]
- 9.Papireddy K, Smilkstein M, Kelly JX, Shweta, Salem SM, Alhamadsheh M, Haynes SW, Challis GL, Reynolds KA. Antimalarial Activity of Natural and Synthetic Prodiginines. J Med Chem. 2011;54:5296–5306. doi: 10.1021/jm200543y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stepkowski SM, Erwin-Cohen RA, Behbod F, Wang ME, Qu X, Tejpal N, Nagy ZS, Kahan BD, Kirken RA. Selective Inhibitor of Janus Tyrosine Kinase 3, PNU156804, Prolongs Allograft Survival and Acts Synergistically with Cyclosporine but Additively with Rapamycin. Blood. 2002;99:680–689. doi: 10.1182/blood.v99.2.680. [DOI] [PubMed] [Google Scholar]
- 11.D’Alessio R, Bargiotti A, Carlini O, Colotta F, Ferrari M, Gnocchi P, Isetta A, Mongelli N, Motta P, Rossi A, et al. Synthesis and Immunosuppressive Activity of Novel Prodigiosin Derivatives. J Med Chem. 2000;43(13):2557–2565. doi: 10.1021/jm001003p. [DOI] [PubMed] [Google Scholar]
- 12.Mortellaro A, Songia S, Gnocchi P, Ferrari M, Fornasiero C, D’Alessio R, Isetta A, Colotta F, Golay J. New Immunosuppressive Drug PNU156804 Blocks IL-2-Dependent Proliferation and NF-KB and AP-1 Activation. J Immunol. 1999;162:7102–7109. ( http://www.jimmunol.org/content/162/12/7102) [PubMed] [Google Scholar]
- 13.Songia S, Mortellaro A, Taverna S, Fornasiero C, Scheiber EA, Erba E, Colotta F, Mantovani A, Isetta A, Golay J. Characterization of the New Immunosuppressive Drug Undecylprodigiosin in Human Lymphocytes: Retinoblastoma Protein, Cyclin-Dependent Kinase-2, and Cyclin-Dependent Kinase-4 as Molecular Targets. J Immunol. 1997;158(8):3987–3995. [PubMed] [Google Scholar]
- 14.Kawauchi K, Shibutani K, Yagisawa H, Kamata H, Nakatsuji S, Anzai H, Yokoyama Y, Ikegami Y, Moriyama Y, Hirata H. A Possible Immunosuppresant, Cycloprodigiosin Hydrochloride, Obtained from Pseudoalteromonas denitrificans. Biochem Biophys Res Commun. 1997;237:543–547. doi: 10.1006/bbrc.1997.7186. [DOI] [PubMed] [Google Scholar]
- 15.Magae J, Miller MW, Nagai K, Shearer G. Effect of Metacycloprodigiosin, an Inhibitor of Killer T Cells, on Murine Skin and Heart Transplants. J Antibiot. 1996;49(1):86–90. doi: 10.7164/antibiotics.49.86. [DOI] [PubMed] [Google Scholar]
- 16.Tsuji RF, Magae J, Yamashita M, Nagai K, Yamasaki M. Immunomodulating Properties of Prodigiosin 25-C, an Antibiotic Which Preferentially Suppresses Induction of Cytotoxic T Cells. J Antibiot. 1992;45(8):1295–1302. doi: 10.7164/antibiotics.45.1295. [DOI] [PubMed] [Google Scholar]
- 17.Tsuji RF, Yamamoto M, Nakamura A, Kataoka T, Magae J, Nagai K, Yamasaki M. Selective Immunosuppresion of Prodigiosin 25-C and FK506 in the Murine Immune System. J Antibiot. 1990;43(10):1293–1301. doi: 10.7164/antibiotics.43.1293. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura A, Magae J, Tsuji RF, Yamasaki M, Nagai K. Suppression of Cytotoxic T Cell Induction in Vivo by Prodigiosin 25-C. Transplantation. 1989;47(6):1013–1016. doi: 10.1097/00007890-198906000-00019. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura A, Nagai K, Ando K, Tamura G. Selective Suppression by Prodigiosin of the Mitogenic Response of Murine Splenocytes. J Antibiot. 1986;39(8):1155–1159. doi: 10.7164/antibiotics.39.1155. [DOI] [PubMed] [Google Scholar]
- 20.Bojo ZP, Deano CD, Jacinto SD, Concepcion GP. Synergistic in Vitro Cytotoxicity of Adociaquinone B and Heptyl Prodigiosin against MCF-7 Breast Cancer Cell Line. Philippine Sci Lett. 2010;3(2):48–58. ( http://www.philsciletters.org/pdf/2010n2.8a.pdf) [Google Scholar]
- 21.Ho TF, Ma CJ, Lu CH, Tsai YT, Wei YH, Chang JS, Lai JK, Cheuh PJ, Yeh CT, Tang PC, et al. Undecylprodigiosin Selectively Induces Apoptosis in Human Breast Carcinoma Cells Independent of P53. Toxicol Appl Pharmacol. 2007;225:318–328. doi: 10.1016/j.taap.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Francisco R, Perez-Tomas R, Gimenez-Bonafe P, Soto-Cerrato V, Gimenez-Xavier P, Ambrosio S. Mechanisms of Prodigiosin Cytotoxicity in Human Neuroblastoma Cell Lines. Eur J Pharmacol. 2007;572:111–119. doi: 10.1016/j.ejphar.2007.06.054. [DOI] [PubMed] [Google Scholar]
- 23.Fürstner A, Reinecke K, Prinz H, Waldmann H. The Core Structures of Roseophilin and the Prodigiosin Alkaloids Define a New Class of Protein Tyrosine Phosphatase Inhibitors. Chem Bio Chem. 2004;5:1575–1579. doi: 10.1002/cbic.200400135. [DOI] [PubMed] [Google Scholar]
- 24.Perez-Tomas R, Montaner B, Llagostera E, Soto-Cerrato V. The Prodigiosins, Proapoptotic Drugs with Anticancer Properties. Biochem Pharmacol. 2003;66:1447–1452. doi: 10.1016/s0006-2952(03)00496-9. [DOI] [PubMed] [Google Scholar]
- 25.Fürstner A, Grabowski J. Studies on DNA Cleavage by Cytotoxic Pyrrole Alkaloids Reveal the Distinctly Different Behavior of Roseophilin and Prodigiosin Derivatives. Chem Bio Chem. 2001;2(9):706–709. doi: 10.1002/1439-7633(20010903)2:9<706::AID-CBIC706>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto D, Kiyozuka Y, Uemura Y, Yamamoto C, Takemoto H, Hirata H, Tanaka K, Hioki K, Tsubura A. Cycloprodigiosin Hydrochloride, a H+/Cl− Symporter, Induces Apoptosis in Human Breast Cancer Cell Lines. J Cancer Res Clin Oncol. 2000;126:191–197. doi: 10.1007/s004320050032. [DOI] [PubMed] [Google Scholar]
- 27.Montaner B, Navarro S, Pique M, Vilaseca M, Martinell M, Giralt E, Gil J, Perez-Tomas R. Prodigiosin from the Supernatant of Serratia marcescens Induces Apoptosis in Haematopoietic Cancer Cell Lines. Br J Pharmacol. 2000;131:585–593. doi: 10.1038/sj.bjp.0703614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smoot RL, Blechacz BRA, Werneburg NW, Bronk SF, Sinicrope FA, Sirica AE, Gores GJ. A Bax-Mediated Mechanism for Obatoclax-Induced Apoptosis of Cholangiocarcinoma Cells. Cancer Res. 2010;70:1960–1969. doi: 10.1158/0008-5472.CAN-09-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paik PK, Rudin CM, Brown A, Rizvi N, Takebe N, Travis W, James L, Ginsberg MS, Juergens R, Markus S, et al. A Phase I Study of Obatoclax Mesylate, a Bcl-2 Antagonist, Plus Topotecan in Solid Tumor Malignancies. Cancer Chemother Pharmacol. 2010;66:1079–1085. doi: 10.1007/s00280-010-1265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Brien SM, Claxton DF, Crump M, Faderl S, Kipps T, Keating MJ, Viallet J, Cheson BD. Phase I Study of Obatoclax Mesylate (GX15-070), a Small Molecule Pan Bcl-2 Family Antagonist, in Patients with Advanced Chronic Lymphocytic Leukemia. Blood. 2009;113:299–305. doi: 10.1182/blood-2008-02-137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, Bornmann W, Kantarjian H, Viallet J, Samudio I, et al. Mechanisms of Antileukemic Activity of the Novel Bcl-2 Homology Domain-3 Mimetic GX15-070 (Obatoclax) Cancer Res. 2008;68(9):3413–3420. doi: 10.1158/0008-5472.CAN-07-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Madiraju SRM, Goulet D, Viallet J, Belec L, Billot X, et al. Small Molecule Obatoclax (GX15-070) Antagonizes MCL-1 and Overcomes MCL-1-Mediated Resistance to Apoptosis. Proc Natl Acad Sci US A. 2007;104(49):19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dairi K, Yao Y, Faley M, Tripathy S, Rioux E, Billot X, Rabouin D, Gonzalez G, Lavallée J, Attardo G. A Scalable Process for the Synthesis of the Bcl Inhibitor Obatoclax. Org Process Res Dev. 2007;11(6):1051–1054. [Google Scholar]
- 34.Perez-Tomas R, Vinas M. New Insights on the Antitumoral Properties of Prodiginines. Curr Med Chem. 2010;17(21):2222– 2231. doi: 10.2174/092986710791331103. [DOI] [PubMed] [Google Scholar]
- 35.Williamson NR, Fineran PC, Gristwood T, Chawrai SR, Leeper FJ, Salmond GPC. Anticancer and Immunosuppresive Properties of Bacterial Prodiginines. Future Microbiol. 2007;2(6):605– 618. doi: 10.2217/17460913.2.6.605. [DOI] [PubMed] [Google Scholar]
- 36.Fürstner A. Chemistry and Biology of Roseophilin and the Prodigiosin Alkaloids: A Survey of the Last 2500 Years. Angew Chem, Int Ed. 2003;42(31):3582–3603. doi: 10.1002/anie.200300582. [DOI] [PubMed] [Google Scholar]
- 37.Wrede F, Hettche O. Uber Das Prodigiosin, Den Roten Farbstoff Des Bacillus prodigiosus, I. Ber Dtsch Chem Ges B. 1929;62:2678. [Google Scholar]
- 38.Wrede F, Rothhaas A. Uber Das Prodigiosin, Den Roten Farbstoff Des Bacillus prodigiosus, IV. Hoppe-Seyler’s Z Physiol Chem. 1933;219:267. [Google Scholar]
- 39.Wrede F, Rothhaas A. Uber Das Prodigiosin, Den Roten Farbstoff Des Bacillus prodigiosus, VI. Hoppe-Seyler’s Z Physiol Chem. 1934;226:95. [Google Scholar]
- 40.Wrede F, Rothhaas A. Uber Das Prodigiosin, Den Roten Farbstoff Des Bacillus prodigiosus, V. Hoppe-Seyler’s Z Physiol Chem. 1933;222:203. [Google Scholar]
- 41.Santer UV, Vogel HJ. Prodigiosin Synthesis in Serratia marcescens: Isolation of a Pyrrole-Containing Precursor. Biochim Biophys Acta. 1956;19:578–579. doi: 10.1016/0006-3002(56)90500-5. [DOI] [PubMed] [Google Scholar]
- 42.Wasserman HH, McKeon JE, Smith L, Forgione P. Prodigiosin. Structure and Partial Synthesis. J Am Chem Soc. 1960;82:506–507. [Google Scholar]
- 43.Rapoport H, Willson CD. The Preparation and Properties of Some Methoxypyrroles. J Am Chem Soc. 1962;84:630–635. [Google Scholar]
- 44.Kim D, Lee JS, Park YK, Kim JF, Jeong H, Oh TK, Kim BS, Lee CH. Biosynthesis of Antibiotic Prodiginines in the Marine Bacterium Hahella chejuensis KCTC 2396. J Appl Microbiol. 2007;102:937–944. doi: 10.1111/j.1365-2672.2006.03172.x. [DOI] [PubMed] [Google Scholar]
- 45.Gandhi NM, Patell JR, Gandhi J, De Souza NJ, Kohl H. Prodigiosin Metabolites of a Marine Pseudomonas Species. Mar Biol. 1976;34:223–227. [Google Scholar]
- 46.D’Aoust JY, Gerber NN. Isolation and Purification of Prodigiosin from Vibrio psychroerythrus. J Bacteriol. 1974;118(2):756– 757. doi: 10.1128/jb.118.2.756-757.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Hazzard C, Eustaquio AS, Reynolds KA, Moore BS. Biosynthesis of Salinosporamides from Alpha-Beta-Unsaturated Fatty Acids: Implications for Extending Polyketide Synthase Diversity. J Am Chem Soc. 2009;131(30):10376–10377. doi: 10.1021/ja9042824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sertan-de Guzman AA, Predicala RZ, Bernardo EB, Neilan BA, Elardo SP, Mangalindan GC, Tasdemir D, Ireland CM, Barraquio WL, Concepcion GP. Pseudovibrio denitrificans Strain Z143-1, a Heptylprodigiosin-Producing Bacterium Isolated from a Philippine Tunicate. FEMS Microbiol Lett. 2007;277(2):188–196. doi: 10.1111/j.1574-6968.2007.00950.x. [DOI] [PubMed] [Google Scholar]
- 49.Hearn WR, Worthington RE, Burgus RC, Williams RP. Norprodigiosin: Occurrence in a Mutant of Serratia marcescens. Biochem Biophys Res Commun. 1964;17(5):517–520. [Google Scholar]
- 50.Feher D, Barlow RS, Lorenzo PS, Hemscheidt TK. A 2-Substituted Prodiginine, 2-(p-Hydroxybenzyl)Prodigiosin, from Pseudoalteromonas rubra. J Nat Prod. 2008;71(11):1970–1972. doi: 10.1021/np800493p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerber NN, Gauthier MJ. New Prodigiosin-Like Pigment from Alteromonas rubra. Appl Environ Microbiol. 1979;37(6):1176– 1179. doi: 10.1128/aem.37.6.1176-1179.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lattasch H, Thomson RH. A Revised Structure for Cycloprodigiosin. Tetrahedron Lett. 1983;24(26):2701–2704. [Google Scholar]
- 53.Gerber NN. Cycloprodigiosin from Beneckea gazogenes. Tetrahedron Lett. 1983;24(27):2797–2798. [Google Scholar]
- 54.Wasserman HH, Fukuyama JM. The Synthesis of (±) Cycloprodigiosin. Tetrahedron Lett. 1984;25(13):1387–1388. [Google Scholar]
- 55.Johnson RE, de Rond T, Lindsay VNG, Keasling JD, Sarpong R. Synthesis of Cycloprodigiosin Identifies the Natural Isolate as a Scalemic Mixture. Org Lett. 2015;17(14):3474–3477. doi: 10.1021/acs.orglett.5b01527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perry J. Prodigiosin in an Actinomycete. Nature. 1961;191:77– 78. doi: 10.1038/191077a0. [DOI] [PubMed] [Google Scholar]
- 57.Harashima K, Tsuchida N, Tanaka T, Nagatsu J. Prodigiosin-25 C, Isolation and the Chemical Structure. Agric Biol Chem. 1967;31(4):481–489. [Google Scholar]
- 58.Wasserman HH, Rodgers GC, Keith DD. The Structure and Synthesis of Undecylprodigiosin. A Prodigiosin Analogue from Streptomyces. Chem Commun. 1966;22:825–826. [Google Scholar]
- 59.Wasserman HH, Keggi J, Bohlmann F, Luders W. Struktur Eines Prodigiosin-Ahnlichen Pilzfarbstoffs Aus Streptomyces longisporus ruber. Angew Chem. 1960;72(21):779. [Google Scholar]
- 60.Wasserman HH, Williams LL, Keggi JJ. Der Prodigiosin-Ahnliche Pilzfarbstoff Aus Streptomyces longisporus ruber. Angew Chem. 1961;73:467. [Google Scholar]
- 61.Wasserman HH, Rodgers GC, Keith DD. Metacycloprodigiosin, a Tripyrrole Pigment from Streptomyces longisporus ruber. J Am Chem Soc. 1969;91(5):1263–1264. doi: 10.1021/ja01033a065. [DOI] [PubMed] [Google Scholar]
- 62.Wasserman HH, Keith DD, Rodgers GC. The Structure of Metacycloprodigiosin. Tetrahedron. 1976;32:1855–1861. [Google Scholar]
- 63.Cerdeno AM, Bibb MJ, Challis GL. Analysis of the Prodiginine Biosynthesis Gene Cluster of Streptomyces coelicolor A3(2): New Mechanisms for Chain Initiation and Termination in Modular Multienzymes. Chem Biol. 2001;8:817–829. doi: 10.1016/s1074-5521(01)00054-0. [DOI] [PubMed] [Google Scholar]
- 64.Sydor PK, Barry SM, Odulate OM, Barona-Gomez F, Haynes SW, Corre C, Song L, Challis GL. Regio- and Stereodivergent Antibiotic Oxidative Carbocyclizations Catalysed by Rieske Oxygenase-like Enzymes. Nat Chem. 2011;3:388–392. doi: 10.1038/nchem.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clift MD, Thomson RJ. Development of a Merged Conjugate Addition/Oxidative Coupling Sequence. Application to the Enantioselective Total Synthesis of Metacycloprodigiosin and Prodigiosin R1. J Am Chem Soc. 2009;131(40):14579–14593. doi: 10.1021/ja906122g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu DH, Clift MD, Lazarski KE, Thomson RJ. Enantioselective Total Synthesis and Confirmation of the Absolute and Relative Stereochemistry of Streptorubin B. J Am Chem Soc. 2011;133(6):1799–1804. doi: 10.1021/ja109165f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerber NN. A New Prodiginine (Prodigiosin-Like) Pigment from Streptomyces. Antimalarial Activity of Several Prodiginines. J Antibiot. 1975;28(3):194–199. doi: 10.7164/antibiotics.28.194. [DOI] [PubMed] [Google Scholar]
- 68.Thirumalachar MJ, Bringi NV, Deshmukh PV, Rahalkar PW, Indira R, Gopalkrishnan KS. Streptorubrin A and B. New Antibiotics with Cytostatic Properties. Hindustan Antibiot Bull. 1964;7:18–24. [Google Scholar]
- 69.Gerber NN, Lechevalier MP. Prodiginine (Prodigiosin-Like) Pigments from Streptomyces and Other Aerobic Actinomycetes. Can J Microbiol. 1976;22:658–667. doi: 10.1139/m76-097. [DOI] [PubMed] [Google Scholar]
- 70.Gerber NN, McInnes AG, Smith DG, Walter JA, Wright JLC, Vining LC. Biosynthesis of Prodiginines. 13C Resonance Assignments and Enrichment Patterns in Nonyl-, Cyclononyl-, Methylcyclodecyl-, and Butylcycloheptylprodiginine Produced by Actinomycete Cultures Supplemented with 13C-Labeled Acetate and 15N-Labeled Nitrate. Can J Chem. 1978;56(9):1155–1163. [Google Scholar]
- 71.Tsao S, Rudd B, He X, Chang C, Floss HG. Identification of a Red Pigment from Streptomyces coelicolor A3(2) as a Mixture of Prodigiosin Derivatives. J Antibiot. 1985;38(1):128–131. doi: 10.7164/antibiotics.38.128. [DOI] [PubMed] [Google Scholar]
- 72.Laatsch H, Kellner M, Weyland H. Butyl-meta-Cycloheptylprodiginine - a Revision of the Structure of the Former Ortho-Isomer. J Antibiot. 1991;44(2):187–191. doi: 10.7164/antibiotics.44.187. [DOI] [PubMed] [Google Scholar]
- 73.Fürstner A, Radkowski K, Peters H. Chasing a Phantom by Total Synthesis: The Butylcycloheptylprodigiosin Case. Angew Chem, Int Ed. 2005;44:2777–2781. doi: 10.1002/anie.200462215. [DOI] [PubMed] [Google Scholar]
- 74.Reeves JT. A Concise Synthesis of Butylcycloheptylprodigiosin. Org Lett. 2007;9(10):1879–1881. doi: 10.1021/ol070341i. [DOI] [PubMed] [Google Scholar]
- 75.Mo S, Sydor PK, Corre C, Alhamadsheh MM, Stanley AE, Haynes SW, Song L, Reynolds KA, Challis GL. Elucidation of the Streptomyces coelicolor Pathway to 2-Undecylpyrrole, a Key Intermediate in Undecylprodiginine and Streptorubin B Biosynthesis. Chem Biol. 2008;15:137–148. doi: 10.1016/j.chembiol.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 76.Jones BT, Hu DX, Savoie BM, Thomson RJ. Elimination of Butylcycloheptylprodigiosin as a Known Natural Product Inspired by an Evolutionary Hypothesis for Cyclic Prodigiosin Biosynthesis. J Nat Prod. 2013;76(10):1937–1945. doi: 10.1021/np400531b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sessler JL, Eller LR, Cho WS, Nicolaou S, Aguilar A, Lee JT, Lynch VM, Magda DJ. Synthesis, Anion-Binding Properties, and in Vitro Anticancer Activity of Prodigiosin Analogues. Angew Chem, Int Ed. 2005;44(37):5989–5992. doi: 10.1002/anie.200501740. [DOI] [PubMed] [Google Scholar]
- 78.Park G, Tomlinson JT, Melvin MS, Wright MW, Day CS, Manderville RA. Zinc and Copper Complexes of Prodigiosin: Implications for Copper-Mediated Double-Strand DNA Cleavage. Org Lett. 2003;5(2):113–116. doi: 10.1021/ol027165s. [DOI] [PubMed] [Google Scholar]
- 79.Haynes SW, Sydor PK, Corre C, Song L, Challis GL. Stereochemical Elucidation of Streptorubin B. J Am Chem Soc. 2011;133:1793–1798. doi: 10.1021/ja109164t. [DOI] [PubMed] [Google Scholar]
- 80.Gerber NN. Prodigiosin-Like Pigments from Actinomadura (Nocardia) pelletieri and Actinomadura madurae. Appl Environ Microbiol. 1969;18(1):1–3. doi: 10.1128/am.18.1.1-3.1969. ( http://aem.asm.org/content/18/1/1.full.pdf+html) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gerber NN. A Novel, Cyclic, Tripyrrole Pigment from Actinomadura (Nocardia) madurae. Tetrahedron Lett. 1970;11:809– 812. doi: 10.1016/s0040-4039(01)97837-2. [DOI] [PubMed] [Google Scholar]
- 82.Gerber NN. Prodigiosin-Like Pigments from Actinomadura (Nocardia) pelletieri. J Antibiot. 1971;24(9):636–640. doi: 10.7164/antibiotics.24.636. [DOI] [PubMed] [Google Scholar]
- 83.Gerber NN. Minor Prodiginine Pigments from Actinomadura madurae and Actinomadura pelletieri. J Heterocycl Chem. 1973;10(6):925–929. [Google Scholar]
- 84.Fürstner A, Grabowski J, Lehmann CW. Total Synthesis and Structural Refinement of the Cyclic Tripyrrole Pigment Nonylprodigiosin. J Org Chem. 1999;64(22):8275–8280. doi: 10.1021/jo991021i. [DOI] [PubMed] [Google Scholar]
- 85.Hayakawa Y, Kawakami K, Seto H, Furihata K. Structure of a New Antibiotic, Roseophilin. Tetrahedron Lett. 1992;33(19):2701– 2704. [Google Scholar]
- 86.Fürstner A, Weintritt H. Total Synthesis of Roseophilin. J Am Chem Soc. 1998;120:2817–2825. [Google Scholar]
- 87.Boger DL, Hong J. Asymmetric Total Synthesis of ent- (−)-Roseophilin: Assignment of Absolute Configuration. J Am Chem Soc. 2001;123:8515–8519. doi: 10.1021/ja011271s. [DOI] [PubMed] [Google Scholar]
- 88.Harrington PE, Tius MA. Synthesis and Absolute Stereochemistry of Roseophilin. J Am Chem Soc. 2001;123(35):8509–8514. doi: 10.1021/ja011242h. [DOI] [PubMed] [Google Scholar]
- 89.Kawasaki T, Sakurai F, Hayakawa Y. A Prodigiosin from the Roseophilin Producer Streptomyces griseoviridis. J Nat Prod. 2008;71(7):1265–1267. doi: 10.1021/np7007494. [DOI] [PubMed] [Google Scholar]
- 90.Hayakawa Y, Nagatsuka S, Kawasaki T. Dechlororoseophilin: A New Cytotoxic Metabolite from Streptomyces griseoviridis. J Antibiot. 2009;62(9):531–532. doi: 10.1038/ja.2009.59. [DOI] [PubMed] [Google Scholar]
- 91.Boonlarppradab C, Kauffman C, Jensen P, Fenical W. Marineosins A and B, Cytotoxic Spiroaminals from a Marine-Derived Actinomycete. Org Lett. 2008;10(24):5505–5508. doi: 10.1021/ol8020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kancharla P, Lu W, Salem SM, Kelly JX, Reynolds KA. Stereospecific Synthesis of 23-Hydroxyundecylprodiginines and Analogues and Conversion to Antimalarial Premarineosins Via a Rieske Oxygenase Catalyzed Bicyclization. J Org Chem. 2014;79(23):11674– 11689. doi: 10.1021/jo5023553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kuhn R, Osswald G. Neue Synthese Von Beta-Pyrrolidonen; Darstellung Von DL-Gamma-Oxo-Prolin, DL-Allo-Hydroxy-Prolin Und 4-Athoxy-Pyrrol-Carbonsilure-(2) Chem Ber. 1956;89:1423– 1442. [Google Scholar]
- 94.Boger DL, Patel M. Total Synthesis of Prodigiosin, Prodigiosene, and Desmethoxyprodigiosin: Diels-Alder Reactions of Heterocyclic Azadienes and Development of an Effective Palladium(II)- Promoted 2,2′-Bispyrrole Coupling Procedure. J Org Chem. 1988;53(7):1405–1415. [Google Scholar]
- 95.Wasserman HH, Lombardo LJ. The Chemistry of Vicinal Tricarbonyls a Total Synthesis of Prodigiosin. Tetrahedron Lett. 1989;30(13):1725–1728. [Google Scholar]
- 96.Wasserman HH, Petersen AK, Xia M, Wang J. Pyrrole- Singlet Oxygen Reactions Leading to Alpha,Alpha′-Bipyrroles. Synthesis of Prodigiosin and Analogs. Tetrahedron Lett. 1999;40:7587– 7589. [Google Scholar]
- 97.D’Alessio R, Rossi A. Short Synthesis of Undecylprodigiosine. A New Route to 2,2′-Bipyrroyl-Pyrromethene Systems. Synlett. 1996;1996:513–514. [Google Scholar]
- 98.Fürstner A, Radkowski K, Peters H, Seidel G, Wirtz C, Mynott R, Lehmann C. Total Synthesis, Molecular Editing and Evaluation of a Tripyrrolic Natural Product: The Case of “Butylcycloheptylprodigiosin”. Chem - Eur J. 2007;13:1929–1945. doi: 10.1002/chem.200601639. [DOI] [PubMed] [Google Scholar]
- 99.Dairi K, Tripathy S, Attardo G, Lavallée J. Two-Step Synthesis of the Bipyrrole Precursor of Prodigiosins. Tetrahedron Lett. 2006;47:2605–2606. [Google Scholar]
- 100.Schultz EE, Sarpong R. Application of in Situ-Generated Rh- Bound Trimethylenemethane Variants to the Synthesis of 3,4-Fused Pyrroles. J Am Chem Soc. 2013;135(12):4696–4699. doi: 10.1021/ja401380d. [DOI] [PubMed] [Google Scholar]
- 101.Wasserman HH, Keith DD, Nadelson J. The Synthesis of Metacycloprodigiosin. Tetrahedron. 1976;32:1867–1871. doi: 10.1021/ja01033a066. [DOI] [PubMed] [Google Scholar]
- 102.Fürstner A, Szillat H, Gabor B, Mynott R. Platinum- and Acid-Catalyzed Enyne Metathesis Reactions: Mechanistic Studies and Applications to the Syntheses of Streptorubin B and Metacycloprodigiosin. J Am Chem Soc. 1998;120(33):8305–8314. [Google Scholar]
- 103.Fürstner A, Krause H. Flexible Synthesis of Metacycloprodigiosin and Functional Derivatives Thereof. J Org Chem. 1999;64(22):8281–8286. doi: 10.1021/jo991022a. [DOI] [PubMed] [Google Scholar]
- 104.Vega MM, Crain DM, Konkol LC, Thomson RJ. Enantioselective Synthesis of Metacycloprodigiosin via the Wasserman Pyrrole. Tetrahedron Lett. 2015;56(23):3228–3230. doi: 10.1016/j.tetlet.2014.12.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chang MY, Pai CL, Chen HP. Synthesis of Streptorubin B Core. Tetrahedron Lett. 2005;46:7705–7709. [Google Scholar]
- 106.Rizzo V, Morelli A, Pinciroli V, Sciangula D, D’Alessio R. Equilibrium and Kinetics of Rotamer Interconversion in Immunosuppressant Prodigiosin Derivatives in Solution. J Pharm Sci. 1999;88(1):73–78. doi: 10.1021/js980225w. [DOI] [PubMed] [Google Scholar]
- 107.Blake AJ, Hunter GA, McNab H. A Short Synthesis of Prodigiosin Analogues. J Chem Soc, Chem Commun. 1990:734–736. [Google Scholar]
- 108.Nakatani S, Kirihara M, Yamada K, Terashima S. Synthesis of Various Model Compounds for the Conjugated Heterocyclic Ring System of Antibiotic Roseophilin. Tetrahedron Lett. 1995;36(46):8461–8464. [Google Scholar]
- 109.Kim SH, Figueroa I, Fuchs PL. Application of the Grubbs Ring-Closing Metathesis for the Construction of a Macrocyclic Ansa- Bridge. Synthesis of the Tricyclic Core of Roseophilin. Tetrahedron Lett. 1997;38(15):2601–2604. [Google Scholar]
- 110.Kim SH, Fuchs PL. Conversion of Delta-(Sulfonyl)-Amino-Alpha-Epoxy Ketones to Bicyclic Ketopyrroles Via Intramolecular Conjugate-Addition to Azoene Intermediates. Synthesis of the Bicyclic Ketopyrrole Core of the 1-Azafulvene Roseophilin. Tetrahedron Lett. 1996;37(15):2545–2548. [Google Scholar]
- 111.Mochizuki T, Itoh E, Shibata N, Nakatani S, Katoh T, Terashima S. Studies toward the Total Synthesis of Antibiotic Roseophilin: A Novel Synthesis of the Macrotricyclic Part. Tetrahedron Lett. 1998;39:6911–6914. [Google Scholar]
- 112.Robertson J, Hatley RJD, Watkin DJ. Preparation of the Tricyclic Ketopyrrole Core of Roseophilin by Radical Macrocyclisation and Paal-Knorr Condensation. J Chem Soc, Perkin Trans 1. 2000:3389–3396. [Google Scholar]
- 113.Robertson J, Hatley RJD. Formal Synthesis of Roseophilin. Chem Commun. 1999:1455–1456. [Google Scholar]
- 114.Bamford SJ, Luker T, Speckamp WN, Hiemstra H. Enantioselective Formal Total Synthesis of Roseophilin. Org Lett. 2000;2(8):1157. doi: 10.1021/ol005750s. [DOI] [PubMed] [Google Scholar]
- 115.Luker T, Koot WJ, Hiemstra H, Speckamp WN. Michael Additions to (R)-1-Acetyl-5-Isopropoxy-3-Pyrrolin-2-One and Subsequent N-Acyliminium Ion Generation: Synthesis of Enantiopure 1-Azabicycles and Preparation of an Intermediate for a Projected Synthesis of Roseophilin. J Org Chem. 1998;63:220–221. [Google Scholar]
- 116.Trost BM, Doherty GA. An Asymmetric Synthesis of the Tricyclic Core and a Formal Total Synthesis of Roseophilin Via an Enyne Metathesis. J Am Chem Soc. 2000;122(16):3801–3810. [Google Scholar]
- 117.Harrington PE, Tius MA. A Formal Total Synthesis of Roseophilin: Cyclopentannelation Approach to the Macrocyclic Core. Org Lett. 1999;1(4):649–651. doi: 10.1021/ol990124k. [DOI] [PubMed] [Google Scholar]
- 118.Bitar AY, Frontier AJ. Formal Synthesis of (±) Roseophilin. Org Lett. 2009;11(1):49–52. doi: 10.1021/ol802329y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Frederich JH, Harran PG. Modular Access to Complex Prodiginines: Total Synthesis of (+)-Roseophilin Via Its 2-Azafulvene Prototropisomer. J Am Chem Soc. 2013;135(10):3788–3791. doi: 10.1021/ja400473v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Turner WJ. A Theory of Porphyrinogenesis. J Lab Clin Med. 1940;26(2):323–330. [Google Scholar]
- 121.Hubbard R, Rimington C. The Biosynthesis of Prodigiosin, the Tripyrrylmethene Pigment from Bacillus prodigiosus (Serratia marcescens) Biochem J. 1950;46(2):220–225. doi: 10.1042/bj0460220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gibson KD, Laver WG, Neuberger A. Initial Stages in the Biosynthesis of Porphyrins. The Formation of Delta-Aminolaevulic Acid from Glycine and Succinyl-CoA by Particles from Chicken Erythrocytes. Biochem J. 1958;70:71–81. doi: 10.1042/bj0700071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marks GS, Bogorad L. Studies on the Biosynthesis of Prodigiosin in Serratia marcescens. Proc Natl Acad Sci US A. 1960;46(1):25–28. doi: 10.1073/pnas.46.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wasserman HH, McKeon JE, Santer UV. Studies Related to the Biosynthesis of Prodigiosin in Serratia marcescens. Biochem Biophys Res Commun. 1960;3(2):146–149. doi: 10.1016/0006-291x(60)90211-4. [DOI] [PubMed] [Google Scholar]
- 125.Williams RP, Goldschmidt ME, Gott CL. Inhibition by Temperature of the Terminal Step in Biosynthesis of Prodigiosin. Biochem Biophys Res Commun. 1965;19(2):177–181. doi: 10.1016/0006-291x(65)90500-0. [DOI] [PubMed] [Google Scholar]
- 126.Morrison DA. Prodigiosin Synthesis in Mutants of Serratia marcesens. J Bacteriol. 1966;91(4):1599–1604. doi: 10.1128/jb.91.4.1599-1604.1966. http://jb.asm.org/content/91/4/1599.full.pdf+html() [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wasserman HH, Shaw CK, Sykes RJ, Cushley RJ. The Biosynthesis of Metacycloprodigiosin and Undecylprodigiosin. Tetrahedron Lett. 1974;15(33):2787–2790. [Google Scholar]
- 128.Wasserman HH, Sykes RJ, Peverada P, Shaw CK, Cushley RJ, Lipsky SR. Biosynthesis of Prodigiosin. Incorporation Patterns of 13C-Labeled Alanine, Proline, Glycine, and Serine Elucidated by Fourier Transform Nuclear Magnetic Resonance. J Am Chem Soc. 1973;95(20):6874–6875. doi: 10.1021/ja00801a080. [DOI] [PubMed] [Google Scholar]
- 129.Rudd BAM, Hopwood DA. A Pigmented Mycelial Antibiotic in Streptomyces coelicolor: Control by a Chromosomal Gene Cluster. Microbiology. 1980;119:333–340. doi: 10.1099/00221287-119-2-333. [DOI] [PubMed] [Google Scholar]
- 130.Malpartida F, Niemi J, Navarrete R, Hopwood DA. Cloning and Expression in a Heterologous Host of the Complete Set of Genes for Biosynthesis of the Streptomyces coelicolor Antibiotic Undecylprodigiosin. Gene. 1990;93(1):91–99. doi: 10.1016/0378-1119(90)90141-d. [DOI] [PubMed] [Google Scholar]
- 131.Thomson NR, Crow MA, McGowan SJ, Cox A, Salmond GPC. Biosynthesis of Carbapenem Antibiotic and Prodigiosin Pigment in Serratia Is under Quorum Sensing Control. Mol Microbiol. 2000;36(3):539–556. doi: 10.1046/j.1365-2958.2000.01872.x. [DOI] [PubMed] [Google Scholar]
- 132.Harris AKP, Williamson NR, Slater H, Cox A, Abbasi S, Foulds I, Simonsen HT, Leeper FJ, Salmond GPC. The Serratia Gene Cluster Encoding Biosynthesis of the Red Antibiotic, Prodigiosin, Shows Species- and Strain-Dependent Genome Context Variation. Microbiology. 2004;150:3547–3560. doi: 10.1099/mic.0.27222-0. [DOI] [PubMed] [Google Scholar]
- 133.Williamson NR, Simonsen HT, Ahmed RAA, Goldet G, Slater H, Woodley L, Leeper FJ, Salmond GPC. Biosynthesis of the Red Antibiotic, Prodigiosin, in Serratia: Identification of a Novel 2-Methyl-3-N-Amyl-Pyrrole (Map) Assembly Pathway, Definition of the Terminal Condensing Enzyme, and Implications for Undecylprodigiosin Biosynthesis. Mol Microbiol. 2005;56(4):971–989. doi: 10.1111/j.1365-2958.2005.04602.x. [DOI] [PubMed] [Google Scholar]
- 134.Nowak-Thompson B, Chaney N, Wing JS, Gould SJ, Loper JE. Characterization of the Pyoluteorin Biosynthetic Gene Cluster of Pseudomonas Fluorescens Pf-5. J Bacteriol. 1999;181(7):2166–2174. doi: 10.1128/jb.181.7.2166-2174.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Thomas MG, Burkart MD, Walsh CT. Conversion of LProline to Pyrrolyl-2-Carboxyl-S-PCP during Undecylprodigiosin and Pyoluteorin Biosynthesis. Chem Biol. 2002;9:171–184. doi: 10.1016/s1074-5521(02)00100-x. [DOI] [PubMed] [Google Scholar]
- 136.Garneau-Tsodikova S, Dorrestein PC, Kelleher NL, Walsh CT. Protein Assembly Line Components in Prodigiosin Biosynthesis: Characterization of PigA,G,H,I,J. J Am Chem Soc. 2006;128:12600– 12601. doi: 10.1021/ja063611l. [DOI] [PubMed] [Google Scholar]
- 137.Stanley AE, Walton LJ, Zerikly MK, Corre C, Challis GL. Elucidation of the Streptomyces coelicolor Pathway to 4-Methoxy-2,2′-Bipyrrole-5-Carboxaldehyde, an Intermediate in Prodiginine Biosynthesis. Chem Commun. 2006:3981–3983. doi: 10.1039/b609556a. [DOI] [PubMed] [Google Scholar]
- 138.Adachi O, Ano Y, Toyama H, Matsushita K. Modern Biooxidation: Enzymes, Reactions and Applications. Chapt. 1. Wiley–VCH Verlag; 2007. Biooxidation with PQQ- and FAD-Dependent Dehydrogenases; pp. 1–41. [DOI] [Google Scholar]
- 139.Eisenberg MA, Star C. Synthesis of 7-Oxo-8-Aminopelargonic Acid, a Biotin Vitamer, in Cell-Free Extracts of Escherichia coli Biotin Auxotrophs. J Bacteriol. 1968;96(4):1291–1297. doi: 10.1128/jb.96.4.1291-1297.1968. ( http://jb.asm.org/content/96/4/1291.full.pdf+html) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Webster SP, Alexeev D, Campopiano DJ, Watt RM, Alexeeva M, Sawyer L, Baxter RL. Mechanism of 8-Amino-7-Oxononanoate Synthase: Spectroscopic, Kinetic, and Crystallographic Studies. Biochemistry. 2000;39:516–528. doi: 10.1021/bi991620j. [DOI] [PubMed] [Google Scholar]
- 141.Cushley RJ, Anderson DR, Lipsky SR, Sykes RJ, Wasserman HH. Carbon-13 Fourier Transform Nuclear Magnetic Resonance Spectroscopy. II. The Pattern of Biosynthetic Incorporation of [1-13C]- and [2-13C]Acetate into Prodigiosin. J Am Chem Soc. 1971;93(23):6284–6286. [Google Scholar]
- 142.Rock CO, Cronan JE. Escherichia Coli as a Model for the Regulation of Dissociable (Type II) Fatty Acid Biosynthesis. Biochim Biophys Acta, Lipids Lipid Metab. 1996;1302:1–16. doi: 10.1016/0005-2760(96)00056-2. [DOI] [PubMed] [Google Scholar]
- 143.Dresen C, Richter M, Pohl M, Lüdeke S, Müller M. The enzymatic asymmetric conjugate umpolung reaction. Angew Chem, Int Ed. 2010;49:6600–6603. doi: 10.1002/anie.201000632. [DOI] [PubMed] [Google Scholar]
- 144.Goldschmidt ME, Williams RP. Thiamine-Induced Formation of the Monopyrrole Moiety of Prodigiosin. J Bacteriol. 1968;96(3):609–616. doi: 10.1128/jb.96.3.609-616.1968. ( http://jb.asm.org/content/96/3/609.full.pdf+html) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Williamson NR, Chawrai S, Leeper FJ, Salmond GPC. Chapter 15: Prodiginines and their potential utility as proapoptotic anticancer agents. In: Fialho A, Chakrabarty A, editors. Emerging Cancer Therapy: Microbial Approaches and Biotechnological Tools. John Wiley & Sons; 2010. [Google Scholar]
- 146.Mo S, Kim BS, Reynolds KA. Production of Branched-Chain Alkylprodiginines in S. coelicolor by Replacement of the 3-Ketoacyl ACP Synthase III Initiation Enzyme, RedP. Chem Biol. 2005;12:191–200. doi: 10.1016/j.chembiol.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 147.Whicher JR, Florova G, Sydor PK, Singh R, Alhamadsheh M, Challis GL, Reynolds KA, Smith JL. Structure and Function of the RedJ Protein, a Thioesterase from the Prodiginine Biosynthetic Pathway in Streptomyces coelicolor. J Biol Chem. 2011;286(25):22558– 22569. doi: 10.1074/jbc.M110.213512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Haynes SW, Sydor PK, Stanley AE, Song L, Challis GL. Chem Commun. 2008:1865–1867. doi: 10.1039/b801677a. [DOI] [PubMed] [Google Scholar]
- 149.Chawrai SR, Williamson NR, Salmond GPC, Leeper FJ. Chemoenzymatic Synthesis of Prodigiosin Analogues–Exploring Substrate Specificity of PigC. Chem Commun. 2008:1862–1864. doi: 10.1039/b719353j. [DOI] [PubMed] [Google Scholar]
- 150.Herzberg O, Chen CCH, Kapadia G, McGuire M, Carroll LJ, Noh SJ, Dunaway-Mariano D. Swiveling-Domain Mechanism for Enzymatic Phosphotransfer between Remote Reaction Sites. Proc Natl Acad Sci US A. 1996;93:2652–2657. doi: 10.1073/pnas.93.7.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Barry SM, Challis GL. Mechanism and Catalytic Diversity of Rieske Non-Heme Iron-Dependent Oxygenases. ACS Catal. 2013;3(10):2362–2370. doi: 10.1021/cs400087p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Withall DM, Haynes SW, Challis GL. Stereochemistry and Mechanism of Undecylprodigiosin Oxidative Carbocyclization to Streptorubin B by the Rieske Oxygenase RedG. J Am Chem Soc. 2015;137(24):7889–7897. doi: 10.1021/jacs.5b03994. [DOI] [PubMed] [Google Scholar]
- 153.Baldwin JE, Wan TS. Penicillin Biosynthesis. Tetrahedron. 1981;37(8):1589–1595. [Google Scholar]
- 154.Basak A, Salowe SP, Townsend CA. Stereochemical Course of the Key Ring-Forming Reactions in Clavulanic Acid Biosynthesis. J Am Chem Soc. 1990;112(4):1654–1656. [Google Scholar]
- 155.Aldrich LN, Dawson ES, Lindsley CW. Evaluation of the Biosynthetic Proposal for the Synthesis of Marineosins A and B. Org Lett. 2010;12(5):1048–1051. doi: 10.1021/ol100034p. [DOI] [PubMed] [Google Scholar]
- 156.Cai X, Wu X, Snider BB. Synthesis of the Spiroiminal Moiety of Marineosins A and B. Org Lett. 2010;12(7):1600–1603. doi: 10.1021/ol100333d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Salem SM, Kancharla P, Florova G, Gupta S, Lu W, Reynolds KA. Elucidation of Final Steps of the Marineosins Biosynthetic Pathway through Identification and Characterization of the Corresponding Gene Cluster. J Am Chem Soc. 2014;136(12):4565–4574. doi: 10.1021/ja411544w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kawasaki T, Sakurai F, Nagatsuka S, Hayakawa Y. Prodigiosin Biosynthesis Gene Cluster in the Roseophilin Producer S. griseoviridis. J Antibiot. 2009;62:271–276. doi: 10.1038/ja.2009.27. [DOI] [PubMed] [Google Scholar]