

Abstract
The Mycobacterium tuberculosis rv2466c gene encodes an oxidoreductase enzyme annotated as DsbA. It has a CPWC active-site motif embedded within its thioredoxin fold domain and mediates the activation of the prodrug TP053, a thienopyrimidine derivative that kills both replicating and nonreplicating bacilli. However, its mode of action and actual enzymatic function in M. tuberculosis have remained enigmatic. In this study, we report that Rv2466c is essential for bacterial survival under H2O2 stress. Further, we discovered that Rv2466c lacks oxidase activity; rather, it receives electrons through the mycothiol/mycothione reductase/NADPH pathway to activate TP053, preferentially via a dithiol–disulfide mechanism. We also found that Rv2466c uses a monothiol–disulfide exchange mechanism to reduce S-mycothiolated mixed disulfides and intramolecular disulfides. Genetic, phylogenetic, bioinformatics, structural, and biochemical analyses revealed that Rv2466c is a novel mycothiol-dependent reductase, which represents a mycoredoxin cluster of enzymes within the DsbA family different from the glutaredoxin cluster to which mycoredoxin-1 (Mrx1 or Rv3198A) belongs. To validate this DsbA–mycoredoxin cluster, we also characterized a homologous enzyme of Corynebacterium glutamicum (NCgl2339) and observed that it demycothiolates and reduces a mycothiol arsenate adduct with kinetic properties different from those of Mrx1. In conclusion, our work has uncovered a DsbA-like mycoredoxin that promotes mycobacterial resistance to oxidative stress and reacts with free mycothiol and mycothiolated targets. The characterization of the DsbA-like mycoredoxin cluster reported here now paves the way for correctly classifying similar enzymes from other organisms.
Keywords: enzyme catalysis, Mycobacterium tuberculosis, oxidation-reduction (redox), oxidative stress, phylogenetics, oxidoreductase, thiol–disulfide exchange
Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB),4 is the second deadliest infectious agent in the world after HIV (1–3). In 2014, TB caused nearly 1.5 million deaths, with an estimated 9.6 million new cases of infection. One of the major features making M. tuberculosis a successful human pathogen is its exquisite ability to survive under anaerobic and aerobic conditions. M. tuberculosis effectively adapts to persist under hypoxic conditions, such as those found in granulomas, by promoting an overall down-regulation of its metabolism while up-regulating specific genes involved in respiration and central metabolism (4, 5). In contrast, to survive and multiply within the oxidizing and acidic environment of macrophages, M. tuberculosis has acquired a variety of mechanisms to maintain its redox equilibrium (6, 7). One of these mechanisms that is clearly different from the human host is the use of a different low-molecular weight (LMW) defense mechanism. In the human host, the tripeptide GSH (γ-l-glutamyl-l-cysteinylglycine) is the main LMW thiol, whereas in M. tuberculosis, mycothiol (MSH; 1-d-myo-inosityl 2-(N-acetylcysteinyl)amido-2-deoxy-α-d-glucopyranoside) is the main LMW thiol involved in detoxification and in maintaining redox homeostasis (8, 9). Protein S-mycothiolation, which is a post-translational modification that protects sensitive cysteines from overoxidation, is probably the link between stress resistance and mycothiol (10). To date, the only enzyme known to reverse the post-translational mycothiolation, using a monothiolic or a dithiolic mechanism, is mycoredoxin-1 (Mrx1) (11, 12). Mrx1 is exclusively linked to the mycothiol electron transfer pathway, and its main function is to reduce protein–MSH mixed disulfides (11). Structures of reduced and oxidized Mrx1 revealed a thioredoxin fold with a CGYC catalytic site motif (11). Mrx1 activity was demonstrated with S-mycothiolated mycothiol peroxidase (Mpx) and thiol peroxidase (Tpx) from Corynebacterium glutamicum, methionine sulfoxide reductase A (MsrA) from C. glutamicum and Corynebacterium diphtheriae, and alkylhydroperoxide reductase E (AhpE) from M. tuberculosis (10, 13–16).
For anti-TB drugs, efficacy against both replicating and non-replicating bacilli is an important feature; however, only a limited number of compounds fulfill these criteria (3). Recently, a new series of thienopyrimidine (TP) compounds able to effectively kill replicating and non-replicating M. tuberculosis were discovered, and the overexpression of Rv2466c in M. tuberculosis H37Rv increased the sensitivity to one of these TP compounds (TP053), supporting a potential role of TP053 as prodrug (17).
Rv2466c is a soluble protein, annotated as a DsbA, and its expression is transcriptionally regulated during oxidative stress response (18, 19). Kinetic data indicated that Rv2466c catalyzes the reduction of TP053 through the action of DTT and an unknown heat-stable cofactor present in a boiled Mycobacterium smegmatis extract. However, the real mode of action of Rv2466c and the nature of the electron donor pathway as well as the biological function of Rv2466c in Mycobacterium spp. remain largely unknown.
Here we report on Rv2466c, a newly uncovered mycoredoxin, renamed as Mrx2. We show that this enzyme belongs to a DsbA-like mycoredoxin cluster, which is evolutionarily different from the glutaredoxin cluster to which Mrx1 belongs. Rv2466c is directly involved in the H2O2 response of M. tuberculosis and is essential to mediate the TP053 prodrug activation in a M. tuberculosis H37Rv cell culture model. Compared with Mrx1, Rv2466c is less specific in receiving electrons, because both the mycothiol/mycothione reductase/NADPH (MSH/Mtr/NADPH) and the thioredoxin/thioredoxin reductase/NADPH (TrxC/TrxR/NADPH) pathways regenerate oxidized Rv2466c. Although more promiscuous compared with Mrx1, kinetics show that the MSH/Mtr/NADPH pathway is the preferred one.
Results
M. tuberculosis rv2466c null mutant (TB461) is sensitive to oxidative stress
Because rv2466c is one of the main targets of SigH (18, 19), which is strongly linked to the oxidative stress response in M. tuberculosis, we constructed an rv2466c null mutant in M. tuberculosis H37Rv (TB461b). We tested the sensitivity of the Tb461b mutant to three thiol-specific oxidative agents, diamide, hydrogen peroxide, and sodium hypochlorite, using an agar-based disc diffusion assay (Table 1 and supplemental Fig. S1). Tb461b mutant was more sensitive to diamide and hydrogen peroxide, when compared with the WT parental strain. In addition, phenotypes were fully restored by the reintroduction of a WT copy of rv2466c at an ectopic locus of the chromosome. Similarly, we analyzed the impact of the three oxidative agents in the sigH null mutant ST49 (18). Interestingly, the inhibition patterns were not significantly different from those obtained with the Tb461b mutant, suggesting that ST49 sensitivity to oxidative stress is mainly due to its inability to induce the expression of rv2466c (Table 1). Although Rv2466c is viewed as non-essential in M. tuberculosis under normal growth conditions (20), under oxidative stress conditions, both SigH and Rv2466c are essential for survival. These findings strongly support the hypothesis that Mrx1 and Rv2466c have similar roles in the maintenance of M. tuberculosis redox homeostasis under oxidative stress; both are individually essential. However, care must be taken because Mrx1 oxidative stress sensitivity was evaluated in the model organism M. smegmatis (11).
Table 1.
Inhibition growth parameters of M. tuberculosis H37Rv variants exposed to oxidative stress
| Strain | Diamide | H2O2 | NaClO |
|---|---|---|---|
| mm | |||
| H37Rv | NDa | 4.25 ± 0.25 | 1.35 ± 0.07 |
| Tb461b (Δ-rv2466c) | >8 | 7.0 ± 0.06b | 1.4 ± 0.2 |
| Tb462 (complemented) | ND | 4.1 ± 0.1 | 1.2 ± 0.01 |
| ST49 (Δ-sigH) | >8 | >8c | 1.25 ± 0.07 |
a ND, no inhibition zone was detected.
b Statistically significant difference compared with values of H37Rv and complemented strain; p < 0.01.
c Statistically significant difference compared with values of H37Rv and complemented strain; p < 0.005.
Rv2466c is essential to mediate TP053 activation in M. tuberculosis
TP053 is the most promising molecule from a series of TP derivatives targeting replicating and non-replicating M. tuberculosis. It has been shown that TP053 is a prodrug activated by Rv2466c (17). To further understand the molecular mechanism by which Rv2466c activates the prodrug TP053, the sensitivity of the Tb461 mutant to TP053 was compared with that of its WT parental strain H37Rv using a resazurin microtiter assay (REMA). This assay measures cell respiration through conversion of resazurin to resorufin and is commonly used to determine mycobacterial MICs to drugs (21). We found that the Tb461b mutant was more resistant than WT H37Rv, with an MIC > 4 μg/ml versus an MIC of 0.25 μg/ml (Fig. 1). Reintroduction of a WT copy of rv2466c in an ectopic locus of the chromosome (Tb462) fully restored the sensitivity to TP053, confirming Rv2466c as essential for TP053 activation (Fig. 1).
Figure 1.
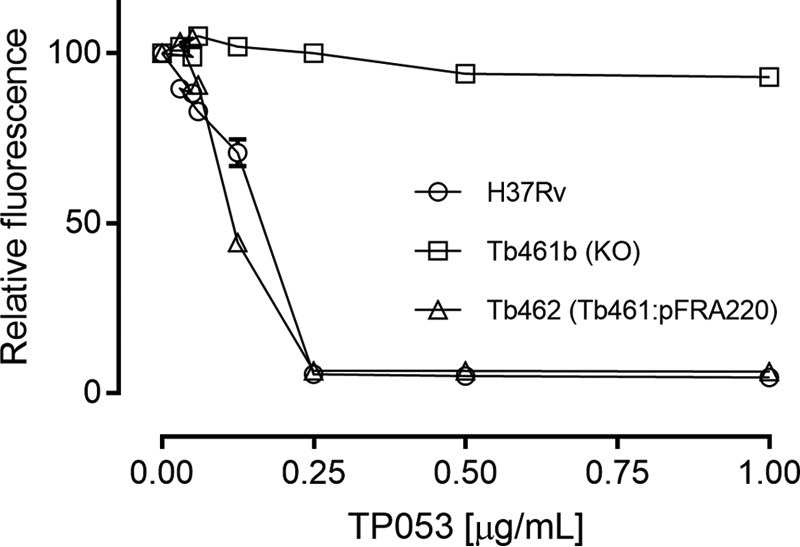
Rv2466c is essential to mediate TP053 activation in M. tuberculosis. Determination of MIC to TP053 by REMA. Cells were exposed to increasing concentrations of TP053. An MIC of 0.25 μg/ml was found for WT strain. No apparent growth inhibition was observed in the null variant (Tb461b). WT copy of rv2466c reintroduction fully restored the sensitivity (Tb462). Concentrations from 2 to 4 μg/ml are not shown for the sake of clarity because there was no growth variation. The experiment was performed in triplicate. Error bars, S.D.
The MSH/Mtr/NADPH pathway as an electron source for Rv2466c
Actinomycetes, including Streptomyces and Mycobacterium spp., produce LMW thiols to maintain a reducing cellular environment under oxidative stress (11, 22, 23). Specifically, MSH is the major LMW thiol produced by mycobacteria (9). The intracellular levels of MSH are tightly regulated during exponential and stationary phases of growth and synchronized in the course of various stresses, playing a critical role in the survival and pathogenicity of mycobacteria (23).
To identify possible electron donor pathways coupled to Rv2466c reduction, two of the most important electron transfer pathways responsible for keeping the redox potential in balance were reconstructed in vitro, that of the MSH/Mtr/NADPH and TrxC/TrxR/NADPH (23). First, we mycothiolated Rv2466c with the intent to observe Rv2466c–MSH mixed disulfide complex formation using an anti-MSH dot blot visualization (supplemental Fig. S2). The Rv2466c–MSH complex was observed from 0.5 to 10 min of room temperature exposure to a 1:1 molar excess of hydrogen peroxide, indicating a stable mixed-disulfide complex. In the absence of H2O2, a weaker anti-MSH signal was also observed, indicating that untreated Rv2466c is already partially in its oxidized state (disulfide or/and sulfenic acid).
Following steady-state kinetics with Rv2466c oxidized with a 4-fold molar excess of diamide (Rv2466cox) as substrate, initial velocities of 11 ± 2 nm·min−1·mg−1 and 1.25 ± 0.5 nmol·min−1·mg−1 were measured for the MSH/Mtr/NADPH and the TrxC/TrxR/NADPH electron donor pathways, respectively (Fig. 2A). It is worth noting that reduction of the oxidized form of Mrx1 (Mrx1ox) through the MSH/Mtr/NADPH pathway resulted in an initial velocity of 88 ± 6 nmol·min−1·mg−1 (Fig. 2A), and Mrx1 has been shown previously not to use the TrxC/TrxR/NADPH electron donor pathway (11). Here, we clearly show that, although Rv2466cox can be reduced by both electron-shuttling pathways, the enzyme is reduced 8.8-fold faster with MSH/Mtr/NADPH compared with the TrxC/TrxR/NADPH pathway. Interestingly, Mrx1ox catalyzed an 8-fold faster reaction coupled to MSH/Mtr/NADPH when compared with Rv2466cox, indicating a different specificity for both enzymes (Fig. 2A).
Figure 2.

Rv2466c is preferably reduced by the MSH pathway, is not an oxidase, and demycothiolates mixed disulfides. A, steady-state Rv2466c reduction utilizing the TrxC/TrxR/NADPH and MSH/Mtr/NADPH pathway and Mrx1 reduction utilizing the MSH/Mtr/NADPH pathway. The TrxC/TrxR/NADPH pathway with Mrx1 has no activity (data not shown). Trx, TrxC/TrxR/NADPH; MSH, MSH/Mtr/NADPH. B, Rv2466c is not a DsbA-oxidoreductase. The methylene blue RNA intercalating assay was utilized to quantify the activity of RNase I (0.5 μm) (29). E. coli RNase I was reduced, and the recovering of activity was monitored in the presence of several enzymes (5 μm DsbA, 5 μm DsbC, 5 μm DsbA or 5 μm DsbC, 5 μm Rv2466c WT, and 5 μm Mrx1). Rv2466c was unable to catalyze the disulfide bond formation in a previously reduced RNase I, indicating that Rv2466c is not a DsbA-oxidoreductase as predicted by sequence similarity. Reactions were performed in duplicate. C, Rv2466c demycothiolates HED–MSH mixed disulfide. An increase in HED–MSH conversion of 15% was observed when the resolving Cys22 was substituted by a serine. No activity was found in the presence of C19S as a catalyst. D, Rv2466c reduces the HED–GSH mixed disulfide. In a reaction mixture containing HED–GSH, a very low activity was observed in the presence of Rv2466c. No activity was measurable for Mrx1 in the presence of HED–GSH. Demycothiolation of HED–MSH (C) and deglutathionylation of HED–GSH (D) were continuously monitored at 340 nm and are represented by NADP+ formation per min in μm. Control measurements were performed in the absence of Rv2466c (WT, C19S, or C22S) or Mrx1. Reactions were performed in duplicate. Error bars, S.D.
The pKa values of the cysteines within the active-site motif of Rv2466c are more acidic
Accessibility and pKa values of cysteine residues are the most important features in determining the reactivity of the thiolate group toward disulfide bonds (24). To understand the reactivity of Cys19 and Cys22 within the active-site motif, we measured the cysteine pKa of WT and the C19S and C22S variants by monitoring the difference in absorption at 240 nm between the thiol and thiolate groups (R–S−) across a pH range from 5.0 to 9.4 (supplemental Fig. S3). For the resolving Cys22, pKa values of 8.17 ± 0.07 and 8.75 ± 0.04 were calculated for the WT and Rv2466c C19S, respectively. A reliable measurement of the pKa of Cys19 was not possible, because the C22S mutant precipitated at low pH. However, based on the pH profile, we estimated a value lower than 6 (supplemental Fig. S3). For Mrx1, the pKa values of both respective cysteines were more basic; the nucleophilic Cys14 was measured to be 6.8, whereas the resolving Cys17 has a pKa value of 11.1 (11).
Rv2466c reduces mycothiolated mixed disulfides and intramolecular disulfide bonds via a monothiol mechanism
Mycothiolation and glutathionylation are crucial post-translational cysteine modifications that protect cysteine residues from overoxidation during oxidative stress (9, 25). To test the ability of Rv2466c to reduce mixed disulfides, we compared its reactivity with Mrx1 using 2-hydroxyethyl disulfide (HED)–MSH as a source of mixed disulfide (Fig. 2, C and D). We followed the NADPH consumption coupled to MSH and Mtr at 340 nm. In a reaction mixture containing HED-MSH, initial velocities of 8.3 ± 0.9 nmol·min−1·mg−1 and 2.8 ± 0.2 nmol·min−1·mg−1 were measured for Mrx1 and Rv2466c, respectively (Fig. 2C). The resolving cysteine mutant (Rv2466c C22S) also displayed a slightly higher reducing activity of 3.2 ± 0.6 nmol·min−1·mg−1 toward mixed disulfides, whereas the C19S was inactive. This cysteine substitution experiment indicated that Rv2466c is functioning under a monothiol mode of action where only the nucleophilic Cys19 is required to catalyze mixed disulfide bond reduction. Interestingly, the resolving cysteine mutant C22S is 15% faster than the WT enzyme. For comparison, we also checked the reactivity with HED–GSH as a source of mixed disulfide coupled to glutathione reductase and NADPH (Fig. 2D). We measured a low reactivity of 0.77 ± 0.03 nmol·min−1·mg−1, whereas Mrx1 has been shown to be unable to reduce the HED–GSH mixed disulfide (11).
We further checked the ability of Mrx1 and Rv2466c to reduce intramolecular disulfide bonds in an insulin assay (26) (Table 2 and supplemental Fig. S4). Using DTT as an electron donor, a similar reactivity toward insulin disulfide bond reduction was observed. However, after introducing the MSH/Mtr/NADPH pathway as an electron donor, the activity of Mrx1 became almost 2-fold faster in reducing insulin compared with Rv2466c. In the absence of the resolving cysteine (Rv2466c C22S), a rate increase of 35% was observed, which is in line with a rate increase of 15% for an HED–MSH mixed disulfide reduction with this mutant.
Table 2.
Insulin reduction parameters
| DTT |
MSH/Mtr/NADPH |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Controla | TrxB | Mrx1 | Rv2466c | Controla | Mrx1 | Rv2466c | C22S | C19S | |
| Rate of precipitation (A600 × 10−5 s−1) | 14.09 ± 0.08 | 16.42 ± 0.03 | 21.56 ± 0.03 | 20.24 ± 0.03 | 2.8 ± 0.1 | 22.67 ± 0.02 | 13.03 ± 0.02 | 17.56 ± 0.01 | 2.9 ± 0.1 |
| Starting point (s) | 2,025 | 1,535 | 1,785 | 1,725 | 2,640 | 1,300 | 1,667 | 1,422 | 2,575 |
a Control, reaction without catalyst.
Rv2466c activates TP053 coupled to the MSH/Mtr/NADPH pathway preferably via a dithiol mechanism
Because Rv2466c is preferentially reduced by the MSH pathway, we decided to assay the two compounds with the lowest MIC values of the TP series (TP053 = 0.125 μg/ml and TP092 = 0.2 μg/ml) and a structurally similar compound ineffective against M. tuberculosis (TP055 > 100 μg/ml) as substrates for Rv2466c in a reaction mixture containing MSH, Mtr, and NADPH. For TP053 and TP092, we measured initial velocities of 0.0034 ± 0.0001 and 0.0052 ± 0.0001 s−1, respectively. No electron transfer with TP055 as substrate was found (Fig. 3A). Activation of TP092 is in the same range as TP053 activation, which is in line with the previously published MIC values (17).
Figure 3.

Rv2466c coupled to the MSH/Mtr/NADPH pathway activates TP053, preferably via a dithiol mechanism. The steady-state kinetic parameters for the activation of the TP compounds are shown and are in good agreement with the values previously published (17). A, TP derivative activation follows the Rv2466c/MSH/Mtr/NADPH pathway. Steady-state activation of TP053, TP055, and TP092 compounds by the Rv2466c/MSH/Mtr/NADPH pathway was evaluated by following the consumption of NADPH as a function of time. TP055 has no activity in this pathway (data not shown). Control measurements were performed in the absence of Rv2466c or Mrx1. B, steady-state kinetic parameters were determined at varying concentrations of TP053 (1–15 μm) and initial velocities calculated from the linear portion of the reaction curve. C, relative activity of C22S indicating a decrease of 75%. No activity was detected in the reaction mixture containing the C19S variant as catalyst. D, docked configuration of TP053 in the active-site cleft of Rv2466c. TP053 is colored by atoms. The Rv2466c α-helical bundle is shown in yellow, and the thioredoxin fold is shown in orange. Reactions were performed in duplicate. Error bars, S.D.
Because TP053 has been demonstrated to have a slightly lower MIC value than TP092, this compound was chosen for further biochemical characterization. Steady-state kinetic experiments carried out with the MSH/Mtr/NADPH pathway and Rv2466c yielded a Km value of 12 ± 2 μm and kcat of 0.016 ± 0.002 s−1 for TP053 (Fig. 3B). These kcat and Km values for TP053 measured through the MSH/Mtr/NADPH pathway are in the same range as those measured by DTT and an M. smegmatis methanol extract (Km = 6.4 μm and kcat = 0.048 s−1). Activation of TP053 decreased with 75% in the absence of the resolving cysteine (Cys22), and for the C19S mutant, no activation was measured (Fig. 3C). This led us to conclude that activation of TP053 preferably occurs via a dithiol mechanism. Interestingly, Mrx1 was not able to catalyze TP053 activation under the same experimental conditions utilized for Rv2466c (data not shown), which shows that Rv2466c is specific for TP053 activation. A schematic representation of TP053 activation through the MSH/Mtr/NADPH pathway is shown in Fig. 4.
Figure 4.

Schematic representation of the MSH/Mtr/NADPH/Rv2466c/TP053 catalytic cycle. Rv2466c in its reduced form reacts with TP053, leading to the formation of uncharacterized compounds. Subsequently, disulfide bond Rv2466c is recycled by the MSH/Mtr/NADPH pathways. The kcat value in the presence of MSH/Mtr/NADPH is in a good agreement with the values previously published in the presence of DTT and M. smegmatis methanol extract. However, the kobs for DTT and methanol extract is 80-fold faster than the value with MSH/Mtr/NADPH (17).
TP053 localizes close to the nucleophilic cysteine
The crystal structure of the reduced form of Rv2466c has been solved recently, revealing a canonical thioredoxin fold with a Cys19-Pro20-Trp21-Cys22 active-site motif, a typical feature of the thioredoxin superfamily of oxidoreductases (27). Rv2466c is a homodimer in which a β-strand is swapped between the thioredoxin fold domains of each subunit (28). As a consequence, the exposed face of the extended β-sheets forms a large, mostly hydrophobic groove flanked by an array of five α-helices on each side, where the active site is located (supplemental Fig. S5A). To observe the structure of complex formation within the binding cleft, we performed a computational docking of TP053 at the catalytic site of Rv2466c (Protein Data Bank entry 4NXI) (Fig. 3D). The binding signature indicates that TP053 is localized in the vicinity of the nucleophilic Cys19, suggesting a crucial role for Cys19 during TP053 activation.
Formation of Cys19–MSH mixed disulfide and a Cys19–Cys22 disulfide
To gain further insight into the reaction mechanism used by Rv2466c, we decided to identify possible enzyme adducts from the reaction of TP053 with Rv2466c and its cysteine mutants. We analyzed the mass of WT and both Cys variants after the reaction with TP053 and the MSH/Mtr/NADPH pathway with LC–MS (supplemental Fig. S6). Before the reaction, we checked the molecular mass of all three enzymes. For Rv2466c WT, a mass of 23,086 Da (supplemental Fig. S6A); for Rv2466c C22S a mass of 23,086 Da (supplemental Fig. S6C; +16 Da from a sulfenic acid); and for Rv2466c C19S, a mass of 23,073 Da (supplemental Fig. S6E) was obtained. This result shows that by removing the resolving cysteine, Cys19 becomes more sensitive to oxidation, forming a sulfenic acid.
After the reaction with Rv2466c WT, two species were found, the enzyme with a mass of 23,084 Da (supplemental Fig. S6B) showing the formation of a disulfide and the mycothiolated form with a mass of 23,571 Da (supplemental Fig. S6B). After the reaction with Rv2466c C22S, a mycothiolated form was detected (23,554 Da) (supplemental Fig. S6D), and for Rv2466c C19S, no mycothiolation was found.
All in all, the molecular mass for Rv2466c and its variants shows that at the end of the reaction and in the absence of Cys22, MSH directly interacts with Cys19. Further, we showed with Rv2466c WT that Cys22 resolves the Cys19–MSH mixed disulfide, leading to the formation of a Cys19–Cys22 disulfide.
Rv2466c has no oxidase properties like DsbA
Amino acid sequence similarity searches between Rv2466c and the non-redundant sequence database using the BLAST web server retrieve only DsbA oxidoreductases (CPWC motif) in the top 100 matches, which means that Rv2466c was annotated as a DsbA oxidoreductase. To test its putative DsbA-oxidoreductase activity, we used Escherichia coli RNase I as a substrate. RNase I is active with its four disulfide bonds correctly formed, making it an ideal model enzyme for oxidative protein folding evaluation (29). We used methylene blue intercalated RNA as a substrate to check the RNase activity at 659 nm after the incubation of reduced unfolded RNase I with DsbA, DsbC, Rv2466c, and Mrx1 (30). Unlike E. coli DsbA, Rv2466c did not catalyze the oxidative refolding of RNase I (Fig. 2B). Reduced RNase I (unfolded) demonstrated 14.9% of activity relative to folded RNase I (100%). In contrast, in the presence of DsbA, 64.7% of activity was recovered. Interestingly, combining DsbC with DsbA triggered the recovery of the refolding up to 79.9% of the folded RNase I. Thus, despite its high sequence similarity to DsbA, Rv2466c clearly did not act as an oxidase.
Rv2466c is a novel mycothiol-dependent reductase
To support the existence of a new class of enzymes currently wrongly classified as DsbA-oxidoreductases, a phylogenetic reconstruction of mycoredoxins, oxidoreductases, and glutaredoxins was performed. To this end, 63 sequences were chosen from the NCBI database, including a diverse range of biochemically/structurally characterized and uncharacterized enzymes. According to the catalytic CXXC motif, sequences were classified into seven different groups: CPHC, CPWC, CPFC, CPYC, CGYC, CGFS, and 3×CGFS (Fig. 5 and supplemental Fig. S7). Strikingly, Rv2466c forms a new cluster with other putative DsbA-like redox enzymes, preserving the CPWC active-site sequence motif (Fig. 5). In addition, this cluster contains two enzymes from C. glutamicum exhibiting the catalytic motif CPFC. Pro-154 was the only amino acid strictly conserved over the 63 sequences analyzed (according to Rv2466c numbering and thereafter), an essential residue for maintaining the Trx fold architecture (31). Residues Asp16 and His99 were conserved over the Rv2466c cluster (shown in salmon and deep purple in Fig. 5) in the phylogenetic analysis, although they are not present in the M. tuberculosis and C. glutamicum Mrx1 sequences (Fig. 6A). It has been demonstrated previously that the substitution of His99 by serine leads to a remarkable decrease in activity (28). His99 is proposed to stabilize the interface between the thioredoxin domain and the α-helical subdomain of Rv2466c and, as such, is directly involved in protein stability. This prediction comes from the observation that the H99S mutant undergoes a large structural change to adopt a conformation similar to that of the oxidized form of Rv2466c, indicative of a crucial structural role for His99 (28).
Figure 5.

A phylogenetic reconstruction (cladogram) of mycoredoxins, glutaredoxins, and oxidoreductases (DsbA) points to a distinct group of mycoredoxins. The new group of mycoredoxins is shown in salmon and deep purple. To give experimental support to the phylogenetic reconstruction, biochemically characterized enzymes were utilized, such as E. coli DsbA (45), S. cerevisiae Grx (64), and H. sapiens Grx (65), among others (32).
Figure 6.

Mrx2 cluster alignment and active-site interaction. A, alignment of the amino acid sequences of the Mrx2 cluster (salmon and deep purple) proposed by phylogenetic analysis. The figure was prepared with Boxshade, a web-based program hosted by Expasy (www.expasy.org). B, the hydrogen bonding environment of the Cys19 sulfur is depicted by black dashed lines, and the respective distances between heavy atoms are given in Å. C, by rotating the side chain of Asp16 (the stereochemistry of this residue was also optimized using the “regularize zone” function in COOT) the carboxylate oxygen forms a close interaction with the sulfur of Cys22. The Rv2466c α-helical bundle is shown in yellow, and the thioredoxin fold is shown in orange.
Additionally, this phylogenetic reconstruction positions Mrx1 in the cluster comprising the classical E. coli Grx1 and its homologs from Homo sapiens and Saccharomyces cerevisiae (32, 33) (Fig. 5), indicating that Mrx1 is a Grx-like mycoredoxin displaying a Grx classical mode of action but utilizing MSH instead of GSH. Altogether, we propose Rv2466c as a novel mycothiol-dependent reductase, mycoredoxin-2 (Mrx2), embedded in a biochemically related cluster of DsbA-like mycoredoxins (shown in salmon and deep purple in Fig. 5).
Difference between the DsbA-like and Grx-like mycoredoxin cluster validated
To further validate the discovery of a new class of DsbA-like mycoredoxins, the enzymatic properties of one of its members were studied. We decided to focus on the gene product of NCgl2339 from C. glutamicum and wanted to show that this enzyme is able to demycothiolate in vitro and in vivo. We recombinantly expressed NCgl2339 and its C13S and C16S variants and purified the proteins to homogeneity. Because Mrx1 from C. glutamicum (CgMrx1) and NCgl2339 are enzymes representing both mycoredoxin clusters, we decided to compare their demycothiolating activity toward an As(V)-MSH thiol–arseno adduct, as described (12). Following steady-state kinetic data analysis, we found an initial velocity of 2.8 ± 0.01 s−1 for NCgl2339 and 0.006 ± 0.001 s−1 for CgMrx1 (Fig. 7, A and B), clearly indicating a different rate (NCgl2339 is 466-fold faster than CgMrx1) for both enzymes belonging to different mycoredoxin clusters. As expected, the nucleophilic cysteine mutant was not active, and the resolving cysteine NCgl2339 C16S mutant has a 10-fold lower initial velocity of 0.29 ± 0.02 s−1 (Fig. 7, A and B).
Figure 7.
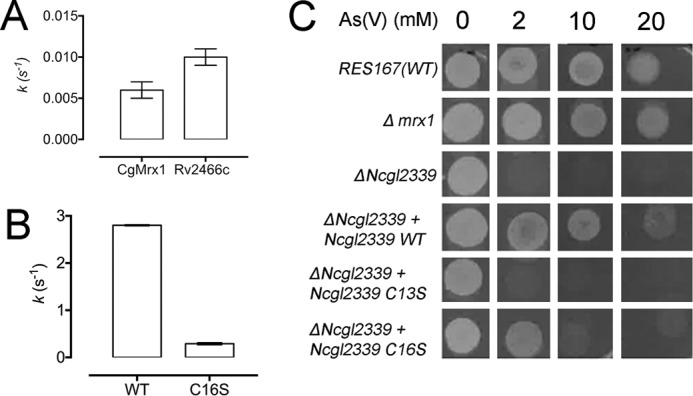
NCgl2339 from C. glutamicum is a Mrx2-like enzyme involved in As(V) resistance. Steady-state As(V) reduction via As(V)-MSH thiol–arseno complex formation utilizing CgMrx1 (A), Rv2466c (A), NCgl2339 WT (B), and NCgl2339 C16S (B) as catalysts. NCgl2339 is a mycoredoxin under a monothiol mode of action showing remarkably higher activity than CgMrx1. Rv2466c reduces the thiol–arseno complex as fast as CgMrx1. C, As(V) resistance-containing CgMrx1 and NCgl2339 variants. Genetic experiments show a pronounced susceptibility to oxidative stress when the gene NCgl2339 is not present. Complementation was performed by introducing the plasmids pECMx-WT, pECMC13S, and pECMC16S. The absence of CgMrx1 did not affect bacterial growth, corroborating the low steady-state activity observed. Reactions were performed in triplicate. To ensure that the reduction of As(V)–MSH thiol–arseno complex is the rate-limiting step, we used enzyme concentrations varying from 0.5 to 500 nm. A linear initial velocity increase was observed. Error bars, S.D.
To further understand the role of NCgl2339 and CgMrx1 in As(V) resistance at a cellular level, we disrupted (i) the NCgl2339 gene and (ii) the gene encoding CgMrx1 (NCgl0808). The NCgl2339 disruption mutant strain was found to be more sensitive to As(V) than the WT strain (RES167) (Fig. 7C). The resistant phenotype was fully restored by complementation with the WT NCgl2339 enzyme. Interestingly, complementation with the C16S mutant partially restored the phenotype, whereas complementation with C13S mutant did not alter the killing phenotype to As(V). It has been shown that CgMrx1 is directly involved in As(V) resistance (12); however, disruption of the gene encoding for CgMrx1 did not result in lowered resistance to As(V) (Fig. 7C). This cell culture experiment shows different properties for both mycoredoxins representing the DsbA and the Grx cluster.
Discussion
It is well known that M. tuberculosis produces and uses MSH as its main LMW thiol involved in detoxification and signaling. However, even today, important pieces in this redox puzzle are missing, like enzymes receiving electrons via MSH (34, 35). In this work, we uncovered a DsbA-like mycoredoxin (Rv2466c) that reacts with free mycothiol and mycothiolated targets, using a similar mode of action as the previously characterized Grx-like Mrx1 (Rv3198A) from M. tuberculosis (11). Rv2466c activates the prodrug TP053 (17), but no activation could be observed in the presence for Mrx1. Despite sharing common features, Rv2466c and Mrx1 show different enzymatic rates and substrate preferences.
The phylogenetic reconstruction indicates that Mrx1 forms a cluster closer to glutaredoxins (Fig. 5, blue clusters) instead of clustering with the DsbA-like Mrx2 cluster (Fig. 6, salmon and deep purple clusters). Grx1 enzymes are described as classical glutaredoxins displaying the dithiol CPYC motif (32). This class of enzymes can reduce disulfides containing targets like ribonucleotide reductase, a key player in the cell metabolism maintaining the necessary balance of deoxynucleotides for DNA repair and synthesis (36, 37). Interestingly, the well-characterized E. coli Grx1 is the closest sequence to Mrx1 in the present phylogenetic reconstruction and corroborates with the fact that Mrx1 has a similar mode of action as E. coli Grx1, but utilizing MSH instead of GSH (11, 38, 39). The closest biochemically characterized enzyme to Rv2466c is Grx5 from S. cerevisiae, an enzyme involved in oxidative and osmotic stress (40). Grx5 knock-out showed a possible role in iron metabolism due to the increased level of intracellular iron (41). Whether Rv2466c will have a similar role in iron–sulfur cluster formation within M. tuberculosis still needs to be determined.
The pKa value of the nucleophilic Cys19 of Rv2466c is at least one pH unit lower than the pKa observed for the corresponding nucleophilic Cys14 of Mrx1 (supplemental Fig. S5B). Interestingly, the respective conserved cysteines of DsbA enzymes have a pKa of ∼3.5 (42, 43), so the pKa value of nucleophilic Cys of Rv2466c lies between the pKa values of the respective cysteines of Mrx1 and DsbA. As a consequence of the lower pKa value of the nucleophilic cysteine of Rv2466c compared with Mrx1, Rv2466c is 8-fold less reactive compared with Mrx1 when coupled to the MSH/Mtr/NADPH pathway (Fig. 2A). A close inspection of the active site of Rv2466c suggests that the low pKa observed for the nucleophilic Cys19 directly depends on the hydrogen-bonding network of the sulfur (44). The crystal structure of Rv2466cRED (Protein Data Bank entry 4NXI) revealed that rotation of the Asp16 side chain certainly promotes a close interaction of its carboxylate moiety with both the Cys19 (3.8 Å) and Cys22 (2.9 Å) sulfur groups (Fig. 6, B and C). This carboxylate moiety of Asp23 in E. coli Trx displays an unusual high pKa of 7.58, allowing it to perform a protonation (acid) or deprotonation (base) of Cys35, thereby regulating the sulfur pKa reactivity (45–47). According to the active-site configuration, Asp16 acts as a proton shuttle, deprotonating and polarizing Cys19 or Cys22 through its carboxylate group (Fig. 8, step 3). Therefore, the presence of a conserved aspartate residue in the vicinity of both Cys residues in Rv2466c and E. coli Trx points to a common catalytic activation mechanism. An alternative mechanism would rely on a water cluster, which can be observed in the vicinity of Cys19 and Cys22 in the crystal structure of Rv2466cRED, providing the potential for polar interactions and proton transfer (48, 49). Another possible mechanism is the Rv2466c Cys19 hydrogen-bonding with backbone atoms like the nitrogens belonging to Trp21 and Cys22 that are 3.4 Å from Cys19 sulfur or the hydroxyl portion of Thr153 that is 3.1 Å from the sulfur atom, similar to the mechanism proposed for Bacillus subtilis (48). Similarly, the pKa of Cys22 could be regulated by the presence of Asp16 in its vicinity, increasing sulfur reactivity and leading to a nucleophilic attack toward Cys19 sulfur, generating a disulfide bond (Fig. 8, step 4).
Figure 8.

Proposed mechanistic model for Rv2466c reduction by mycothiol and disulfide formation. The proposed mechanism includes four steps. As previously demonstrated, Cys19 and Cys22 form a disulfide bond when under oxidative stress (17, 28). 1, in the first chemical step, one molecule of mycothiol reacts with the nucleophilic Cys19, forming a mixed disulfide (R-S-SM). Subsequently, two pathways can take place. 2, in the presence of a second molecule of mycothiol, a nucleophilic attack takes place at the mixed disulfide, releasing mycothione and the reduced form of Cys19. 3, a general base performs the deprotonation at Cys19, increasing the reactivity of the sulfur atom, leading to its thiolate form. 4, in a parallel reaction, the resolving Cys22 pKa is lowered via deprotonation and performs a nucleophilic attack on the mixed disulfide, releasing one molecule of mycothiol and forming an intramolecular disulfide, leading back to the start of the reaction scheme. This chemical step slows down the overall reaction.
The biochemical data presented herein led to the general hypothesis that resolving and nucleophilic cysteine residues in a CXXC domain are in competition, where in this case the resolving Cys22 slows down Rv2466c reduction by MSH via intramolecular disulfide bond formation. The reductive pathway (Fig. 8) describes the chemical steps used to reduce the intramolecular disulfide formed by Cys19 and Cys22 under oxidative stress, giving a reduced form of Rv2466c and mycothione (MSSM). MSSM is converted back to MSH by Mtr, which is an NADPH-dependent flavoenzyme (Fig. 8). An alternative pathway can take place, where mycothiolated Cys19 (R-S-SM) is under nucleophilic attack of Cys22. In the present mechanism, a general base deprotonates Cys22 to produce the thiolate form activated for nucleophilic attack (Fig. 8, step 4) (48). This alternative pathway would then be in competition with the reducing pathway (Fig. 8, steps 2 and 3). Interestingly, in the absence of Cys22, a rate increase of 15% was observed in HED–MSH reduction, and a 35% increase was seen in the rate of intramolecular disulfide bond reduction (Table 2 and Figs. 2 and 4).
Based on the LC–MS data (supplemental Fig. S5A), in the presence of the resolving Cys22, disulfide bond formation depicted in step 4 (Fig. 8) takes place concomitantly with the reductive pathway in steps 2 and 3. In the absence of the resolving Cys22, only the reductive pathway is functional. These data are in good agreement with a mechanism where the nucleophilic attack of Cys22 slows down the reduction of Rv2466c by MSH. It has been demonstrated that the Mpx from C. glutamicum is under the same mode of action as proposed here (14). Mpx contains three cysteine residues, Cys36, Cys64, and Cys79. Under oxidative stress, Cys36 is oxidized to a sulfenic acid and can be rescued from overoxidation by nucleophilic attack from MSH or the resolving Cys79. In this scenario, reduction of Mpx is in competition with the formation of a Cys36–Cys79 disulfide bond in a similar mode of action proposed for Rv2466c. Moreover, a C79S mutation increased the MSH peroxidase activity of Mpx, corroborating the competitive mechanism. A similar mechanism was also shown for MsrA from C. diphtheriae, which displays an increase of 15% in catalytic efficiency (kcat/Km) when the resolving Cys215 is substituted by serine (16). On the other hand, TP053 reduction by Rv2466c is 75% faster than its resolving cysteine mutant (Fig. 3C), and in As(V) reduction, NCgl2339 demonstrated a decrease in activity of 90% upon mutation of the resolving Cys16 to serine (Fig. 7B). The decrease in activity observed in the absence of Cys22 might be due to a higher reactivity of its resolving cysteine sulfur compared with the reactivity of the MSH sulfur. The pKa of the Cys22 sulfur (8.17) is already lower than the pKa of the MSH sulfur (8.76) (50), and it will most likely even be lowered by hydrogen bonding with the carboxylate of Asp16 (Fig. 6, B and C). Moreover, Cys22 is always present in the correct orientation within the structural fold for a nucleophilic attack, whereas MSH first needs to form a complex and reach the correct orientation before a nucleophilic attack. This competitive system is controlled by the rates of disulfide formation and MSH reduction and is a common feature for every protein that can form an intramolecular disulfide and a mixed disulfide with an LMW thiol.
Experimental procedures
Rv2466c bacterial strains and growth conditions
M. tuberculosis H37Rv and derivatives were grown at 37 °C in Middlebrook 7H9 broth (Difco) or on Middlebrook 7H10, both supplemented with 0.2% glycerol, 0.05% Tween 80, and 10% albumin-dextrose-NaCl. Hygromycin (100 μg/ml) or kanamycin (50 μg/ml) was added when necessary. For cloning purposes, DH5α E. coli was grown in shaking flasks at 37 °C in LB broth or on LB agar with kanamycin (50 μg/ml).
Construction of an M. tuberculosis rv2466c null mutant
An rv2466c null mutant was obtained by introducing a 519-bp in-frame deletion by two-step recombination as described previously (51). Briefly, two DNA fragments flanking and overlapping the distal parts of rv2466c were amplified from genomic DNA of M. tuberculosis H37Rv using Q5® high-fidelity DNA polymerase (New England Biolabs). Primers used for the amplification were as follows: upstream fragment, RP1696 (5′-tttaaatcagccacagatcgtcccac-3′/RP1697 (5′-agatctatcggcgacagacttctggg-3′) (amplification product 1,032 bp); downstream fragment, RP1698 (5′-ttcgcttcctacccgcacttt-3′/RP1699 (5′-ctgcaggcaaccacgtccgcaacctc-3′) (amplification product 1,098 bp). After amplification, the two fragments were cloned in the suicide vector p1NIL. The cassette from pGOAL19 containing lacZ sacB and a gene conferring hygromycin resistance was then inserted into the unique PacI site of the resulting vector to give the final construct pFRA218, which was introduced in M. tuberculosis H37Rv by electroporation as described previously (52) and plated on Middlebrook 7H10 containing hygromycin (Invitrogen) and X-gal 50 μg/ml (Sigma). Blue hygR transformants were isolated and controlled by PCR to verify the first crossover event. Three colonies with the correct integration were grown in no-drug Middlebrook 7H9 until stationary phase and then plated on no-drug Middlebrook 7H10 containing X-gal and sucrose (2%) to select the second crossover. White colonies able to grow on sucrose were collected, and the structure of their rv2466c locus was verified by PCR to confirm the gene deletion (not shown). A colony with the right structure was chosen for further work and called Tb461c.
To obtain a complemented strain, rv2466c was amplified with its upstream region (603 bp) using the primers RP1766 (5′-gatatccgccgttggagatcaccttc-3′/RP1767 (5′-gatatcccgacattgtgcccgaca-3′) and cloned into in an integrative plasmid based on pMV306 (53). The resulting plasmid was then electroporated into Tb461c and selected on Middlebrook 7H9 plates added with kanamycin to obtain the complemented strain, which was called Tb462.
Construction of the NCgl2339 gene-disrupted mutant C. glutamicum ΔNCgl2339
For disruption of the NCgl2339 gene, a 198-bp internal fragment of the NCgl2339 gene was PCR-amplified using the primer pair Mrx-intUp (5′-taaggatcctcgctgtcctaaacgaag-3′)/Mrx-intDown (5′-taaaagcttcgatgacatcattgaagg-3′). The amplified fragment was BamHI/HindIII-digested and cloned into the plasmid pK18mob (equivalently digested) for further ligation and E. coli transformation assays, obtaining the recombinant plasmid pKNCgl2339. This plasmid was used to transform the E. coli strain S17-1 (donor strain), and the strain was further used for plasmid mobilization to the recipient strain C. glutamicum RES167. The transferred plasmids behave as suicide in corynebacteria; therefore, only transconjugant clones with the chromosomally integrated plasmid should be selected under the pressure of kanamycin (12.5 μg/ml). PCR amplification analysis and further fragment sequencing validated the correct chromosomal plasmid integration.
Plasmidic constructions for in vivo complementation analysis of NCgl2339 (WT) and its variants
The recombinant pET28a derivative plasmids (pETMrx-WT, pETMrx-C13S, and pETMrx-C16S) were first used for site-directed mutagenesis, to remove the BglII target present inside the original NCgl2339 gene (and also within its mutagenic variants C13S and C16S), using the mismatched primer pair NCgl2339/176Fw (5′-gaggaagcaggagagatattcgacgcttccttcaag)/NCgl2339/176Rv (5′-cttgaaggaagcgtcgaatatctctcctgcttcctc); the nucleotide mutagenesis (underlined) does not modify the encoded amino acid. The clones lacking the BglII target were selected by BglII digestion of the isolated plasmids and further gene sequencing. Then the NCgl2339 and its Cys variants (lacking in all the cases the BglII sites) were PCR-amplified using the primer pair NCgl2339-up/NCgl2339-down (indicated above) and digested with NdeI/XhoI, and the isolated fragments were used for subcloning with the equally digested E. coli vector pXHisNpro (12). The obtained recombinant plasmids, named pXHMx-wt, pXHMC13S, and pXHMC16S, were BglII-digested, and the individual 2-kbp cassettes (containing the NCgl2339 gene variants under the control of the constitutive kanamycin promoter (Pkan-NCgl2339)) were used for ligation to the bifunctional mobilizable vector pECM2 (BamHI-digested), obtaining plasmids pECMx-WT, pECMC13S, and pECMC16S. These plasmids were adequately transferred by conjugation to the C. glutamicum strains WT and ΔNCgl2339, and complementation of the corresponding activity was evaluated.
Mycothiol and protein expression and purification
Recombinant DsbA, DsbC, Grx, and RNase I from E. coli and TrxB, TrxC, TrxR, Mtr, Mrx1, Rv2466c, and Rv2466c C19S and C22S mutants from M. tuberculosis were produced in E. coli and purified to homogeneity as described previously (11, 14, 17, 54). After Rv2466c-His6 purification, the enzyme was dialyzed against 20 mm Tris, 150 mm NaCl, pH 7.6, to remove residual imidazole and incubated with 1 mg of tobacco etch virus nuclear inclusion-a endopeptidase per 10 mg of enzyme. Cleaved Rv2466c was injected to a HisTrap chelating column (GE HealthCare) and separated from the cleaved tag. Rv2466c fractions were pooled and stored at −20 °C. MSH was overexpressed and purified as described previously (12).
Cloning, expression, and purification of NCgl2339
NCgl2339 was cloned into the expression vector pET28a (+) (Novagen) using the primer pair NCgl2339-Fw (5′tatcatatggctcaaaaagtaaccttctgg-3′)/NCgl2339-Rv (5′-tattctcgagttagtcgaactgtgggttctcag-3′) and the NdeI/XhoI restriction sites. Recombinant expression was done in E. coli BL21 (DE3) in LB. Upon reaching 0.7 A600, 1 mm isopropyl β-d-thiogalactopyranoside (Sigma-Aldrich) was added to induce protein expression. Cells were harvested after overnight incubation at 30 °C. The NCgl2339 purification procedure was the same as for Rv2466c (17). For cloning, expression, and purification of C13S and C16S variants, the same procedures as for the WT were followed.
Detection of Rv2466c mycothiolation on dot blot
Rv2466c was incubated with a 1:1 molar excess of hydrogen peroxide at room temperature, and aliquots were withdrawn at different time points (0.5, 1, 1.5, 2, 4, 6, and 10 min). Subsequently, a 10-fold molar excess of MSH was added to the samples. The PVDF membrane was activated by soaking in methanol for 1 min and equilibrated in 15 mm Tris-HCl, pH 7.6, 192 mm glycine, 20% methanol, and 0.03% SDS for 3 min. 4 μg of Rv2466c was transferred to the PVDF membrane, and 1% milk powder in PBS buffer was added to block the membrane. Primary antibody anti-MSH (14) (1:20,000 dilution) was added and incubated for 1 h at room temperature. The PVDF membrane was washed three times with PBS before adding the anti-rabbit alkaline phosphatase (1:10,000), followed by a 1-h incubation at room temperature. The membrane was washed three times and developed in 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, 5 mm MgCl2 buffer solution containing nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate p-toluidine for 30 min in the dark.
Computational docking of TP053 in Rv2466c
To assess the feasibility of the binding of TP053 at the catalytic site of Rv2466c, computational docking of TP053 in the active-site cleft of Rv2466c was performed using AutoDock Vina (55). AutoDock Vina is a computational docking program that applies an iterated local search global optimizer algorithm to perform rapid gradient optimization searches of ligand docking conformations. The aim of using this program was to confirm that TP053 is able to bind in the vicinity of the nucleophilic cysteine of Rv2466c with reasonable affinity. Of the 20 calculated conformations of docked TP053, the seventh highest affinity binding mode (−6.4 kcal/mol) gave a docked configuration in which C19 of Rv2466c was within a reasonable distance for a potential nucleophilic attack on TP053 (Fig. 3D). The overall reaction for TP053 activation catalyzed by Rv2466c through the MSH/Mtr/NADPH pathway is schematically represented in Fig. 8.
Agar-based disk diffusion assay for M. tuberculosis
Bacterial strains were grown to early exponential phase and 100 μl of culture containing about 3 × 106 cfu was spread onto 20-ml Middlebrook 7H10 plates. Paper disks soaked with 10 μl of a stock solution of the inhibitory reagent were placed on top of the agar. Stock solutions were diamide (0.5 m), H2O2 (3.6%), sodium hypochlorite (5%) (bleach). The diameter of the inhibition zones was measured after 15 days of incubation at 37 °C. Experiments were performed in triplicate.
Resazurin microtiter assay
TP053 susceptibility of M. tuberculosis was determined by using REMA, as described previously (21), Briefly, a log-phase bacterial culture was diluted up to a theoretical A540 nm = 0.0005 and dispensed in a Nunclon 96-well flat bottom black plate (Thermo Scientific) in the presence of serial compound dilution. A growth control containing no compound and a sterile control without inoculum were also included. After a 1-week incubation at 37 °C, 10% (v/v) Alamar Blue (10 μl) (Invitrogen) was added to each well. After a further 24 h of incubation at 37 °C, fluorescence values were measured using an Infinite 200Pro microplate reader (Tecan Group Ltd.; excitation = 535 nm, emission = 590 nm). The lowest drug concentration that resulted in at least 90% inhibition of fluorescence development was considered as the MIC. Experiments were performed in triplicate.
In vivo arsenate resistance analysis of WT, mutants, and complemented C. glutamicum strains
C. glutamicum strains RES167 (ATCC 13032 derivative strain used as a control), the mutant ΔNCgl2339, and the complemented ΔNCgl2339 derivatives were precultured in complex medium (tryptone soy broth) at 30 °C overnight; flasks containing minimal medium for corynebacteria (56) were inoculated with the precultures to an A600 of 0.05 and cultured up to reach an A600 of 1.0. Cultures were then washed, and cells were resuspended in an equal volume of water with cells ready to be assayed. Samples of 10 μl were disposed on dishes containing minimal medium for corynebacteria supplemented with different concentrations of arsenate (0, 2, 10, and 20 μg/ml) and further incubated at 30 °C for 24–36 h.
Identification of Rv2466c species via LC–MS
The samples were analyzed by electrospray ionization mass spectroscopy on a Micromass Q-Tof micro system coupled to a Waters Breeze analytical HPLC system equipped with a Waters 2489 UV/visible detector (at a wavelength of 215 nm). The runs were performed on a Grace Vydac C18 column (15 cm × 2.1 mm, 3 μm) at a flow rate of 0.3 ml/min. The solvent system consists of water and acetonitrile (containing 0.1% of formic acid) with a linear gradient from 3 to 100% acetonitrile over 20 min. Electrospray data were acquired on electrospray positive ionization mode scanning over the mass-to-charge ratio (m/z) scale from 100 to 2,000 at a scan time of 1 s and a cone voltage of 38 V. Data collection was done with Masslynx software. The molecular mass was determined using the maximum entropy approach (57, 58).
Rv2466c pKa determination
The extinction coefficient of thiol groups (R-SH) at 240 nm is the main readout utilized to measure pKa values of cysteine residues due to the lack of absorption of its un-ionized counterpart (R-S−) in the same wavelength (44). To cover a broad pH range, a reaction mixture containing a poly-buffer solution composed of 10 mm sodium acetate, 10 mm sodium phosphate, 10 mm sodium borate, and 10 mm sodium citrate, pH 9.4, was used. Rv2466c was first reduced by incubation with 100 mm DTT for 30 min and oxidized by incubation with 40 mm diamide for 30 min, both at room temperature. For the oxidation of cysteine mutants, a 10-fold excess of H2O2 was used. Excess of DTT, diamide, or H2O2 was removed by gel filtration on a Superdex200 10/300 GL column equilibrated with the poly-buffer. A final reaction mixture of 20 μm Rv2466c (reduced or oxidized) was titrated with 100 mm HCl. The pKa of the Rv2466c C22S and C19S mutants was determined in the same conditions as described for Rv2466c wild type. All of the measurements were carried out in a Carry UV spectrophotometer (Agilent Technologies) precooled at 10 °C.
The sigmoidal pH-dependent saturation curve was fitted to the Henderson–Hasselbalch equation (54) (see Equation 2), where Aexp is the experimental value A240/A280, ASH is the A240/A280 value for the protonated form, and AS− is the A240/A280 for the deprotonated form. The data were fitted to Equation 1 using GraphPad Prism version 7.0.
| (Eq. 1) |
RNase I activity assay
Oxidase activity was measured as described previously (54). DsbC was reduced with 20 mm DTT for 30 min at room temperature, and DsbA, Rv2466c, and Mrx1 were oxidized with a 4-fold molar excess of diamide for 30 min at room temperature. Excess DTT or diamide was removed with pre-equilibrated Biospin6 columns (Bio-Rad) in 50 mm Hepes, pH 7.5, 150 mm NaCl. Reduced RNase I was preincubated with the thiol/disulfide oxidoreductases in 50 mm Hepes, pH 7.5, 150 mm NaCl for 3 min at room temperature at a final concentration of 0.5 μm reduced RNase I before RNase activity measurements. The RNA hydrolysis activity was measured in a methylene blue RNase assay using a buffer solution containing 1 mg of methylene blue in 200 ml of MOPS buffer solution (0.1 m MOPS, pH 7.5, 2 mm EDTA). The absorbance was followed as a function of time at 659 nm, the wavelength with a maximum difference between methylene blue intercalated with RNA and without RNA. The measured RNA activities are the initial velocities determined on the first 5% of the progress curves.
TrxC/TrxR/NADPH electron transfer assay
Rv2466c and Mrx1 were oxidized using a 4-fold molar excess of diamide for 30 min at room temperature, and the excess of oxidizing agent was removed on a Superdex200 10/300 GL column equilibrated with 20 mm Tris, 150 mm NaCl, pH 7.6. Rv2466c-dependent oxidation of NADPH in the TrxC/TrxR/NADPH pathway was continuously monitored at 340 nm (ϵ = 6.22 × 103 m−1·cm−1) in a 96-well plate reader at 37 °C in a reaction mixture containing 500 μm NADPH, 5 μm TrxR, 5 μm TrxC, and 50 μm Rv2466c in 50 mm Hepes, pH 8.0, and 50 mm NaCl. All reactions were carried out at 37 °C and started by the addition of oxidized Rv2466c or Mrx1 in a reaction mixture previously incubated for 3 min at 37 °C. Control measurements were performed in the absence of TrxC. Reactions were performed in duplicate.
MSH/Mtr/NADPH electron transfer assay
Rv2466c and Mrx1 were oxidized using a 4-fold molar excess of diamide for 30 min at room temperature, and the excess of oxidizing agent was removed on a Superdex200 10/300 GL column equilibrated with 20 mm Tris, 150 mm NaCl, pH 7.6. Rv2466c- or Mrx1-dependent oxidation of NADPH utilizing the MSH/Mtr/NADPH pathway was continuously monitored at 340 nm (NADPH ϵ = 6.22 × 103 m−1·cm−1) in a 96-well plate reader at 37 °C in a reaction mixture containing 500 μm NADPH, 200 μm MSH, 5 μm Mtr, and 50 μm Rv2466c or Mrx1 in 50 mm Hepes, pH 8.0, and 50 mm NaCl. Initial velocities were measured from the linear portion of the reaction curve. All reactions were carried out at 37 °C, incubated for 3 min, and started by the addition of oxidized Rv2466c or Mrx1. Control measurements were performed in the absence of Rv2466c or Mrx1. Reactions were performed in duplicate.
Arsenate electron transfer assay
Assays were performed as described previously, with some modifications (12). Rv2466c, C. glutamicum Mrx1, and C. glutamicum NCgl2339 (WT, C13S, or C16S) was previously reduced by incubation with 20 mm DTT for 30 min at room temperature, and the excess of DTT was removed with a Biospin6 column (Bio-Rad). The final assay mixture contained 1 μm C. glutamicum ArsC1, 5 μm C. glutamicum Mtr, 200 μm MSH, and 250 μm NADPH and included 0.5 nm WT NCgl2339, 2.5 nm C16S NCgl2339, or 0.5 μm CgMrx1, Rv2466c, or C13S NCgl2339 (the concentrations were optimized to find the rate-limiting condition). Reactions were started with the addition of 100 mm arsenate (Na2HAsO4-7H2O; Sigma). The reaction was performed at 37 °C in 50 mm Hepes, pH 8.0, and monitored at 340 nm (NADPH ϵ = 6.22 × 103 m−1·cm−1).
HED assay
The HED assay for GSH (26) was modified for MSH, as described by Van Laer et al. (11). Briefly, the mixed disulfide between MSH or GSH and 2-HED was formed by incubating 700 μm HED and 1 mm MSH or GSH, respectively, at 30 °C for 3 min. A final concentration of 250 μm MSH–HED or GSH–HED substrate was added to the reaction mixture containing 500 μm NADPH, 200 μm MSH, 5 μm Mtr or GR, and 50 μm Rv2466c or Mrx1. Control measurements were performed in the absence of Rv2466c (WT, C19S, or C22S) or Mrx1. The assay was performed at 25 °C in 50 mm Hepes, 50 mm NaCl, pH 8.0, and the absorption was monitored at 340 nm. All reactions were started by the addition of the catalyst. Reactions were performed in duplicate.
NADPH-dependent reduction of insulin
Rv2466c, TrxB, and Mrx1 was previously reduced by incubation with 20 mm DTT for 30 min at room temperature. Excess of DTT was removed with a Biospin6 column (Bio-Rad). Enzyme-dependent reduction of insulin was monitored continuously at 600 nm in a 96-well plate reader at 37 °C in a reaction mixture containing PBS, pH 7.4, 1 mm EDTA, 500 μm NADPH, 5 μm Mtr, 250 μm MSH, 100 μm insulin, and 5 μm reduced Rv2466c, Mrx1, or TrxB. Insulin reduction containing DTT as reductant was performed in the same conditions as described for the MSH/Mtr/NADPH pathway, containing 100 μm DTT instead of MSH/Mtr/NADPH. The reaction was started by the addition of insulin, and control measurements were performed in the absence of Rv2466c, Mrx1, or TrxB. The precipitation starting point was defined as an increase of 0.02 absorbance units at A600 after a stable baseline recording, and the rate of precipitation was calculated using a linear regression composed by A600 ranging from 2,000 to 2,500 s (26).
TP055, TP092, and TP053 steady-state kinetics
Steady-state kinetic parameters of MSH/Mtr/NADPH/Rv2466c-dependent TP053 activation were determined at varying concentrations of TP053 (1–15 μm) in a reaction mixture containing 500 μm NADPH, 250 μm MSH, 0.5 μm Mtr, and 5 μm Rv2466c reduced (WT, C19S, or C22S) or Mrx1 in 50 mm Hepes, 50 mm NaCl, pH 8.0. TP053 activation was continuously monitored at 412 nm (TP053 ϵ = 17.962 × 103 m−1·cm−1), and initial velocities were calculated from the linear portion of the reaction curve. All reactions were carried out at 37 °C and started by the addition of Rv2466c. Control measurements were performed in the absence of Rv2466c, and the final amount of 10% DMSO used to solubilize the compounds was taken into account (59). TP055 and TP092 activation were performed in the same conditions as TP053 activation. Non-linear regression was used to fit the hyperbolic saturation curve to the Michaelis–Menten equation (Equation 1) where v is the steady-state velocity, S is the substrate concentration, Vmax is the maximum rate, and Km is the Michaelis–Menten constant. The data were analyzed using GraphPad Prism version 6.0 software.
| (Eq. 2) |
Phylogenetic reconstruction
To create an enzyme classification based on functionality, 63 amino acid sequences were selected to include mycoredoxins, oxidoreducatases, and glutaredoxins. The sequences were aligned using Muscle (Mega 6 software) (60). The resulting alignment was utilized to generate a neighbor-joining phylogenetic three (61), and the Jones–Taylor–Thornton matrix method (62) was utilized to generate the distance matrix. A pairwise deletion was utilized, and the level of confidence was measured based on 2,000 bootstrap replicates. The web-based program iTOL was utilized to generate the final figure (63).
Author contributions
L. A. R., K. W., G. D., B. P., D. Y., V. M., W. V., G. V., D. A-J., M. E. G., L. M. M., R. M., and J. M. designed the research. L. A. R., K. W., G. D., B. P., D. Y., A. G. R., F. B., E. M., L.-M. P., E. S.-V., R. P., and C. M. performed the research. L. A. R., K. W., G. D., B. P., D. Y., F. B., E. S.-V., R. P., C. M., G. V., L. M. M., R. M., and J. M. analyzed the data. L. A. R., K. W., G. D., B. P., D. Y., C. M., W. V., G. V., M. E. G., L. M. M., R. M., and J. M. wrote the paper.
Supplementary Material
Acknowledgments
J. M. thanks Rudi Glockshuber for bringing him in contact with M. E. G., Mamta Rawat, and Inge Van Molle for fruitful discussions on this project.
This work was supported by grants from the European Commission (More Medicines for Tuberculosis, Contract HEALTH-F3–2011-260872) (to M. E. G. and R. M), the Spanish Ministry of Economy and Competitiveness (Contract BIO2013-49022-C2-2-R), the Basque Government (to M. E. G.), Agentschap voor Innovatie door Wetenschap en Technologie (IWT) (to B. P.), Vlaams Instituut voor Biotechnologie (VIB) (to J. M.), the SRP34 project of the Vrije Universiteit Brussel (VUB) (to J. M. and W. V.), Research Foundation Flanders (FWO) (to J. M.), Flanders Hercules Foundation (Grant HERC16) for the purification platform (to J. M.), and Junta Castilla y León Grants LE326U14 and ULE-UXXI-2016/00127 (to L. M. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S7.
- TB
- tuberculosis
- LMW
- low-molecular weight
- MSH
- mycothiol
- Mrx
- mycoredoxin
- Mpx
- mycothiol peroxidase
- Tpx
- thiol peroxidase
- TP
- thienopyrimidine
- Mtr
- mycothione reductase
- TrxC
- thioredoxin
- TrxR
- thioredoxin reductase
- REMA
- resazurin microtiter assay
- MIC
- minimal inhibitory concentration
- HED
- 2-hydroxyethyl disulfide
- MSSM
- mycothione
- MsrA
- methionine sulfoxide reductase A.
References
- 1. Dheda K., Barry C. E. 3rd, and Maartens G. (2016) Tuberculosis. Lancet 387, 1211–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Russell D. G., Barry C. E. 3rd, and Flynn J. L. (2010) Tuberculosis: What we don't know can, and does, hurt us. Science 328, 852–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zumla A., Nahid P., and Cole S. T. (2013) Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 12, 388–404 [DOI] [PubMed] [Google Scholar]
- 4. Cook G. M., Berney M., Gebhard S., Heinemann M., Cox R. A., Danilchanka O., and Niederweis M. (2009) Physiology of mycobacteria. Adv. Microb. Physiol. 55, 81–182, 318–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sacchettini J. C., Rubin E. J., and Freundlich J. S. (2008) Drugs versus bugs: in pursuit of the persistent predator Mycobacterium tuberculosis. Nat. Rev. Microbiol. 6, 41–52 [DOI] [PubMed] [Google Scholar]
- 6. Schnappinger D., Ehrt S., Voskuil M. I., Liu Y., Mangan J. A., Monahan I. M., Dolganov G., Efron B., Butcher P. D., Nathan C., and Schoolnik G. K. (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trivedi A., Singh N., Bhat S. A., Gupta P., and Kumar A. (2012) Redox biology of tuberculosis pathogenesis. Adv. Microb. Physiol. 60, 263–324 [DOI] [PubMed] [Google Scholar]
- 8. Newton G. L., Arnold K., Price M. S., Sherrill C., Delcardayre S. B., Aharonowitz Y., Cohen G., Davies J., Fahey R. C., and Davis C. (1996) Distribution of thiols in microorganisms: mycothiol is a major thiol in most Actinomycetes. J. Bacteriol. 178, 1990–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reyes A. M., Pedre Perez B., De Armas M. I., Tossounian M. A., Radi R., Messens J., and Trujillo M. (2017) Chemistry and redox biology of mycothiol. Antioxid. Redox. Signal. 10.1089/ars.2017.7074 [DOI] [PubMed] [Google Scholar]
- 10. Chi B. K., Busche T., Van Laer K., Bäsell K., Becher D., Clermont L., Seibold G. M., Persicke M., Kalinowski J., Messens J., and Antelmann H. (2014) Protein S-mycothiolation functions as redox-switch and thiol protection mechanism in Corynebacterium glutamicum under hypochlorite stress. Antioxid. Redox. Signal. 20, 589–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van Laer K., Buts L., Foloppe N., Vertommen D., Van Belle K., Wahni K., Roos G., Nilsson L., Mateos L. M., Rawat M., van Nuland N. A., and Messens J. (2012) Mycoredoxin-1 is one of the missing links in the oxidative stress defence mechanism of Mycobacteria. Mol. Microbiol. 86, 787–804 [DOI] [PubMed] [Google Scholar]
- 12. Ordóñez E., Van Belle K., Roos G., De Galan S., Letek M., Gil J. A., Wyns L., Mateos L. M., and Messens J. (2009) Arsenate reductase, mycothiol, and mycoredoxin concert thiol/disulfide exchange. J. Biol. Chem. 284, 15107–15116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hugo M., Van Laer K., Reyes A. M., Vertommen D., Messens J., Radi R., and Trujillo M. (2014) Mycothiol/mycoredoxin 1-dependent reduction of the peroxiredoxin AhpE from Mycobacterium tuberculosis. J. Biol. Chem. 289, 5228–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pedre B., Van Molle I., Villadangos A. F., Wahni K., Vertommen D., Turell L., Erdogan H., Mateos L. M., and Messens J. (2015) The Corynebacterium glutamicum mycothiol peroxidase is a reactive oxygen species-scavenging enzyme that shows promiscuity in thiol redox control. Mol. Microbiol. 96, 1176–1191 [DOI] [PubMed] [Google Scholar]
- 15. Si M., Zhang L., Chaudhry M. T., Ding W., Xu Y., Chen C., Akbar A., Shen X., and Liu S. J. (2015) Corynebacterium glutamicum methionine sulfoxide reductase A uses both mycoredoxin and thioredoxin for regeneration and oxidative stress resistance. Appl. Environ. Microbiol. 81, 2781–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tossounian M. A., Pedre B., Wahni K., Erdogan H., Vertommen D., Van Molle I., and Messens J. (2015) Corynebacterium diphtheriae methionine sulfoxide reductase a exploits a unique mycothiol redox relay mechanism. J. Biol. Chem. 290, 11365–11375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Albesa-Jové D., Chiarelli L. R., Makarov V., Pasca M. R., Urresti S., Mori G., Salina E., Vocat A., Comino N., Mohorko E., Ryabova S., Pfieiffer B., Lopes Ribeiro A. L., Rodrigo-Unzueta A., Tersa M., et al. (2014) Rv2466c mediates the activation of TP053 to kill replicating and non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 9, 1567–1575 [DOI] [PubMed] [Google Scholar]
- 18. Manganelli R., Voskuil M. I., Schoolnik G. K., Dubnau E., Gomez M., and Smith I. (2002) Role of the extracytoplasmic-function σ factor σ(H) in Mycobacterium tuberculosis global gene expression. Mol. Microbiol. 45, 365–374 [DOI] [PubMed] [Google Scholar]
- 19. Raman S., Song T., Puyang X., Bardarov S., Jacobs W. R. Jr., and Husson R. N. (2001) The alternative σ factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J. Bacteriol. 183, 6119–6125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Griffin J. E., Gawronski J. D., Dejesus M. A., Ioerger T. R., Akerley B. J., and Sassetti C. M. (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palomino J. C., Martin A., Camacho M., Guerra H., Swings J., and Portaels F. (2002) Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 46, 2720–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fahey R. C. (2001) Novel thiols of prokaryotes. Annu. Rev. Microbiol. 55, 333–356 [DOI] [PubMed] [Google Scholar]
- 23. Kumar A., Farhana A., Guidry L., Saini V., Hondalus M., and Steyn A. J. (2011) Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev. Mol. Med. 13, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jensen K. S., Pedersen J. T., Winther J. R., and Teilum K. (2014) The pKa value and accessibility of cysteine residues are key determinants for protein substrate discrimination by glutaredoxin. Biochemistry 53, 2533–2540 [DOI] [PubMed] [Google Scholar]
- 25. Roos G., and Messens J. (2011) Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic. Biol. Med. 51, 314–326 [DOI] [PubMed] [Google Scholar]
- 26. Holmgren A. (1979) Glutathione-dependent synthesis of deoxyribonucleotides: purification and characterization of glutaredoxin from Escherichia coli. J. Biol. Chem. 254, 3664–3671 [PubMed] [Google Scholar]
- 27. Collet J. F., and Messens J. (2010) Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox Signal. 13, 1205–1216 [DOI] [PubMed] [Google Scholar]
- 28. Albesa-Jové D., Comino N., Tersa M., Mohorko E., Urresti S., Dainese E., Chiarelli L. R., Pasca M. R., Manganelli R., Makarov V., Riccardi G., Svergun D. I., Glockshuber R., and Guerin M. E. (2015) The redox state regulates the conformation of Rv2466c to activate the antitubercular prodrug TP053. J. Biol. Chem. 290, 31077–31089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Messens J., Collet J. F., Van Belle K., Brosens E., Loris R., and Wyns L. (2007) The oxidase DsbA folds a protein with a nonconsecutive disulfide. J. Biol. Chem. 282, 31302–31307 [DOI] [PubMed] [Google Scholar]
- 30. Greiner-Stoeffele T., Grunow M., and Hahn U. (1996) A general ribonuclease assay using methylene blue. Anal. Biochem. 240, 24–28 [DOI] [PubMed] [Google Scholar]
- 31. Sinha S., Langford P. R., and Kroll J. S. (2004) Functional diversity of three different DsbA proteins from Neisseria meningitidis. Microbiology 150, 2993–3000 [DOI] [PubMed] [Google Scholar]
- 32. Lillig C. H., Berndt C., and Holmgren A. (2008) Glutaredoxin systems. Biochim. Biophys. Acta 1780, 1304–1317 [DOI] [PubMed] [Google Scholar]
- 33. Vlamis-Gardikas A., and Holmgren A. (2002) Thioredoxin and glutaredoxin isoforms. Methods Enzymol. 347, 286–296 [DOI] [PubMed] [Google Scholar]
- 34. Newton G. L., and Fahey R. C. (2008) Regulation of mycothiol metabolism by sigma(R) and the thiol redox sensor anti-σ factor RsrA. Mol. Microbiol. 68, 805–809 [DOI] [PubMed] [Google Scholar]
- 35. Antelmann H., and Hamilton C. J. (2012) Bacterial mechanisms of reversible protein S-thiolation: structural and mechanistic insights into mycoredoxins. Mol. Microbiol. 86, 759–764 [DOI] [PubMed] [Google Scholar]
- 36. Wei Y., Funk M. A., Rosado L. A., Baek J., Drennan C. L., and Stubbe J. (2014) The class III ribonucleotide reductase from Neisseria bacilliformis can utilize thioredoxin as a reductant. Proc. Natl. Acad. Sci. U.S.A. 111, E3756–E3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gon S., Faulkner M. J., and Beckwith J. (2006) In vivo requirement for glutaredoxins and thioredoxins in the reduction of the ribonucleotide reductases of Escherichia coli. Antioxid. Redox Signal. 8, 735–742 [DOI] [PubMed] [Google Scholar]
- 38. Prinz W. A., Aslund F., Holmgren A., and Beckwith J. (1997) The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 272, 15661–15667 [DOI] [PubMed] [Google Scholar]
- 39. Porat A., Lillig C. H., Johansson C., Fernandes A. P., Nilsson L., Holmgren A., and Beckwith J. (2007) The reducing activity of glutaredoxin 3 toward cytoplasmic substrate proteins is restricted by methionine 43. Biochemistry 46, 3366–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rodríguez-Manzaneque M. T., Ros J., Cabiscol E., Sorribas A., and Herrero E. (1999) Grx5 glutaredoxin plays a central role in protection against protein oxidative damage in Saccharomyces cerevisiae. Mol. Cell. Biol. 19, 8180–8190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rodríguez-Manzaneque M. T., Tamarit J., Bellí G., Ros J., and Herrero E. (2002) Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Mol. Biol. Cell 13, 1109–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jacobi A., Huber-Wunderlich M., Hennecke J., and Glockshuber R. (1997) Elimination of all charged residues in the vicinity of the active-site helix of the disulfide oxidoreductase DsbA: influence of electrostatic interactions on stability and redox properties. J. Biol. Chem. 272, 21692–21699 [DOI] [PubMed] [Google Scholar]
- 43. Messens J., and Collet J. F. (2006) Pathways of disulfide bond formation in Escherichia coli. Int. J. Biochem. Cell Biol. 38, 1050–1062 [DOI] [PubMed] [Google Scholar]
- 44. Roos G., Foloppe N., and Messens J. (2013) Understanding the pKa of redox cysteines: the key role of hydrogen bonding. Antioxid. Redox Signal. 18, 94–127 [DOI] [PubMed] [Google Scholar]
- 45. Chivers P. T., and Raines R. T. (1997) General acid/base catalysis in the active site of Escherichia coli thioredoxin. Biochemistry 36, 15810–15816 [DOI] [PubMed] [Google Scholar]
- 46. LeMaster D. M. (1996) Structural determinants of the catalytic reactivity of the buried cysteine of Escherichia coli thioredoxin. Biochemistry 35, 14876–14881 [DOI] [PubMed] [Google Scholar]
- 47. Dyson H. J., Jeng M. F., Tennant L. L., Slaby I., Lindell M., Cui D. S., Kuprin S., and Holmgren A. (1997) Effects of buried charged groups on cysteine thiol ionization and reactivity in Escherichia coli thioredoxin: structural and functional characterization of mutants of Asp 26 and Lys 57. Biochemistry 36, 2622–2636 [DOI] [PubMed] [Google Scholar]
- 48. Roos G., Foloppe N., Van Laer K., Wyns L., Nilsson L., Geerlings P., and Messens J. (2009) How thioredoxin dissociates its mixed disulfide. PLoS Comput. Biol. 5, e1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Røhr Å. K., Hammerstad M., and Andersson K. K. (2013) Tuning of thioredoxin redox properties by intramolecular hydrogen bonds. PLoS One 8, e69411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sharma S. V., Van Laer K., Messens J., and Hamilton C. J. (2016) Thiol redox and pKa properties of mycothiol, the predominant low-molecular-weight thiol cofactor in the Actinomycetes. Chembiochem 17, 1689–1692 [DOI] [PubMed] [Google Scholar]
- 51. Parish T., and Stoker N. G. (2000) Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146, 1969–1975 [DOI] [PubMed] [Google Scholar]
- 52. Maciag A., Dainese E., Rodriguez G. M., Milano A., Prowedi R., Pasca M. R., Smith I., Palu G., Riccardi G., and Manganelli R. (2007) Global analysis of the Mycobacterium tuberculosis zur (FurB) regulon. J. Bacteriol. 189, 4974–4974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stover C. K., de la Cruz V. F., Fuerst T. R., Burlein J. E., Benson L. A., Bennett L. T., Bansal G. P., Young J. F., Lee M. H., Hatfull G. F., Snapper S. B., Barletta R. G., Jacobs W. R., and Bloom B. R. (1991) New use of Bcg for recombinant vaccines. Nature 351, 456–460 [DOI] [PubMed] [Google Scholar]
- 54. Roos G., Garcia-Pino A., Van Belle K., Brosens E., Wahni K., Vandenbussche G., Wyns L., Loris R., and Messens J. (2007) The conserved active site proline determines the reducing power of Staphylococcus aureus thioredoxin. J. Mol. Biol. 368, 800–811 [DOI] [PubMed] [Google Scholar]
- 55. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Letek M., Valbuena N., Ramos A., Ordóñez E., Gil J. A., and Mateos L. M. (2006) Characterization and use of catabolite-repressed promoters from gluconate genes in Corynebacterium glutamicum. J. Bacteriol. 188, 409–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ferrige A. G., Seddon M. J., Green B. N., Jarvis S. A., Skilling J., and Staunton J. (1992) Disentangling electrospray spectra with maximum entropy. Rapid Commun. Mass Spectrom. 6, 707–711 [Google Scholar]
- 58. Ferrige A. G., Seddon M. J., Jarvis S., Skilling J., and Aplin R. (1991) Maximum entropy deconvolution in electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 5, 374–377 [Google Scholar]
- 59. Tjernberg A., Markova N., Griffiths W. J., and Hallén D. (2006) DMSO-related effects in protein characterization. J. Biomol. Screen. 11, 131–137 [DOI] [PubMed] [Google Scholar]
- 60. Tamura K., Stecher G., Peterson D., Filipski A., and Kumar S. (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Saitou N., and Nei M. (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 [DOI] [PubMed] [Google Scholar]
- 62. Jones D. T., Taylor W. R., and Thornton J. M. (1992) The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 [DOI] [PubMed] [Google Scholar]
- 63. Letunic I., and Bork P. (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mesecke N., Spang A., Deponte M., and Herrmann J. M. (2008) A novel group of glutaredoxins in the cis-Golgi critical for oxidative stress resistance. Mol. Biol. Cell 19, 2673–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Johansson C., Lillig C. H., and Holmgren A. (2004) Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J. Biol. Chem. 279, 7537–7543 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.