

Abstract
Thrombosis is caused by the activation of platelets at the site of ruptured atherosclerotic plaques. This activation involves engagement of G protein–coupled receptors (GPCR) on platelets that promote their aggregation. Although it is known that protein kinases and phosphatases modulate GPCR signaling, how serine/threonine phosphatases integrate with G protein signaling pathways is less understood. Because the subcellular localization and substrate specificity of the catalytic subunit of protein phosphatase 1 (PP1c) is dictated by PP1c-interacting proteins, here we sought to identify new PP1c interactors. GPCRs signal via the canonical heterotrimeric Gα and Gβγ subunits. Using a yeast two-hybrid screen, we discovered an interaction between PP1cα and the heterotrimeric G protein Gβ1 subunit. Co-immunoprecipitation studies with epitope-tagged PP1c and Gβ1 revealed that Gβ1 interacts with the PP1c α, β, and γ1 isoforms. Purified PP1c bound to recombinant Gβ1-GST protein, and PP1c co-immunoprecipitated with Gβ1 in unstimulated platelets. Thrombin stimulation of platelets induced the dissociation of the PP1c-Gβ1 complex, which correlated with an association of PP1c with phospholipase C β3 (PLCβ3), along with a concomitant dephosphorylation of the inhibitory Ser1105 residue in PLCβ3. siRNA-mediated depletion of GNB1 (encoding Gβ1) in murine megakaryocytes reduced protease-activated receptor 4, activating peptide-induced soluble fibrinogen binding. Thrombin-induced aggregation was decreased in PP1cα−/− murine platelets and in human platelets treated with a small-molecule inhibitor of Gβγ. Finally, disruption of PP1c-Gβ1 complexes with myristoylated Gβ1 peptides containing the PP1c binding site moderately decreased thrombin-induced human platelet aggregation. These findings suggest that Gβ1 protein enlists PP1c to modulate GPCR signaling in platelets.
Keywords: ADP, G protein-coupled receptor (GPCR), phosphoprotein phosphatase 1 (PP1), protein phosphatase 1, platelet, thrombin, megakaryocytes
Introduction
The process of thrombosis entails activation of platelets at the site of ruptured atherosclerotic plaques. At these sites, platelets are likely exposed to several soluble agonists including thrombin, adenosine diphosphate (ADP), and thromboxane A2 (TXA2). These agonists engage distinct G protein–coupled receptors (GPCRs)4 on platelets and initiate inside-out signaling (1, 2). This signaling process activates integrin αIIbβ3, enables the binding of plasma fibrinogen, and facilitates platelet aggregation. Binding of fibrinogen to activated αIIbβ3 in turn generates outside-in signals that modulate the rearrangement of platelet cytoskeleton and stabilizes the growing platelet clot (3).
An essential mediator of the GPCR signaling is the G protein Gαβγ heterotrimers. Agonist binding to GPCR triggers the exchange of Gα subunit–associated guanosine 5′-diphosphate (GDP) with guanosine 5′-triphosphate (GTP). This event induces a conformational change in the Gα subunit, which may lead to the separation or dissociation of Gα and Gβγ subunits, and thereby enable the Gα and Gβγ subunits to engage with distinct downstream effectors (4). Propagation of signals involves the assembly and disassembly of multiprotein complexes that are regulated in part by protein phosphorylation in a spatial and temporal fashion. Although phosphorylation is regulated by both protein kinases and protein phosphatases, our understanding of how serine/threonine (Ser/Thr) protein phosphatases integrate with the GPCR signaling pathways has lagged behind that of the Ser/Thr protein kinases.
Protein phosphatase 1 (PP1) is a Ser/Thr phosphatase that regulates an entire range of cellular functions including cell division, cell metabolism, and cell death (5). The major isoforms of the catalytic subunit of PP1 (PP1cα, PP1cβ, and PP1cγ) share ∼90% amino acid similarity and do not exist freely in cells. Instead, they associate with a wide range of proteins referred to as phosphatase interacting proteins (PIP) in a spatial and temporal fashion. PP1 interacts with PIP via short peptide motifs, and many PIPs combine multiple docking motifs to form stable complex with PP1. The protein complexes of PP1c-PIP largely dictate where, when, and which substrates can be dephosphorylated by PP1c (6). We have noticed previously that PP1cγ−/− mice displayed moderately delayed time to occlusion in an in vivo thrombosis model and PP1cγ−/− platelets displayed decreased aggregation to low-dose thrombin but not collagen (7). The current study was undertaken to ascertain how PP1 couples with the GPCR-initiated signaling pathways by identifying unique PP1c interactors. To this end, we screened for the interaction partners of PP1cα by yeast two-hybrid analyses and identified the Gβ1 subunit, a component of the heterotrimeric G protein as a novel PP1c binding protein. Depletion of Gβ1 or PP1cα in murine megakaryocytes/platelets and disruption of PP1c-Gβ1 complex by the myristoylated Gβ1 peptide or blockade of Gβγ signaling in human platelets decreased GPCR signaling, suggesting a functional coupling of PP1c with Gβ1 in platelets.
Results
Gβ1 is a new PP1c interacting protein
To identify PP1c interacting partners in the GPCR signaling pathways, we utilized a yeast two-hybrid (Y2H) system with the full-length PPP1Ca (PP1cα) as a bait and screened the human bone marrow library. One of the interacting clones was identified as GNB1 (Gβ1 subunit). Gβ1 is a component of the heterotrimeric G proteins that associate with GPCRs. To validate the findings from yeast two-hybrid studies, we performed co-immunoprecipitation assays in epitope-tagged cells. Gβ1 co-immunoprecipitated with PP1cα, PP1cβ, and PP1cγ1 from human kidney embryonal 293 cells transfected with HA-tagged PP1cα, PP1cβ, PP1cγ1, or FLAG-tagged Gβ1 plasmids (Fig. 1A). To ascertain the nature of PP1c-Gβ1 interaction, we performed interaction studies with purified PP1c and Gβ1-GST proteins (Fig. 1B). In a glutathione-S-transferase (GST) pulldown assay, purified PP1c bound to Gβ1-GST fusion protein but not to GST protein (Fig. 1C), suggesting that Gβ1 can interact directly with PP1c. These studies identify Gβ1 as a novel PP1c interacting protein.
Figure 1.
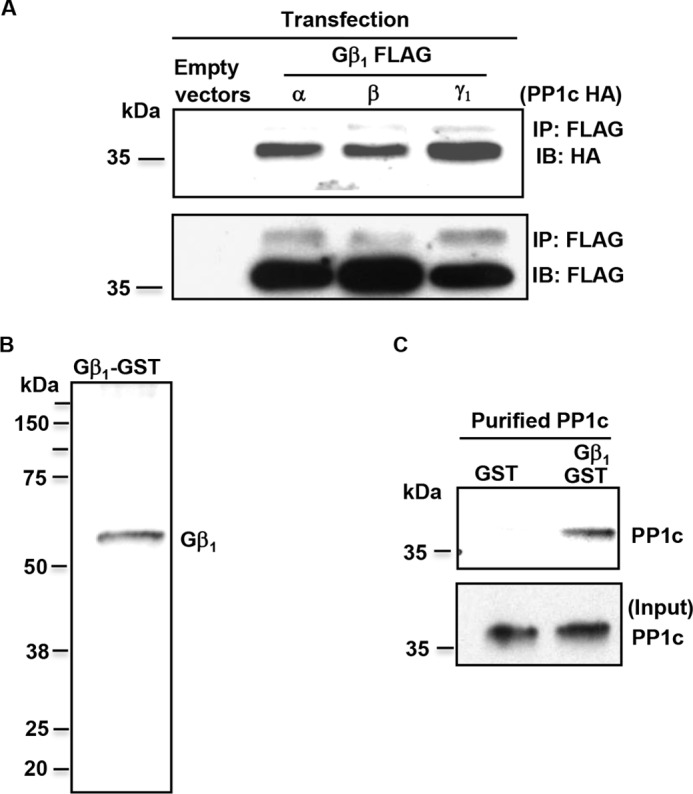
Gβ1 is a novel PP1c interacting protein. A, lysate from 293 cells transfected with HA-tagged PP1c α, β, and γ and FLAG-tagged Gβ1 or empty vector were immunoprecipitated (IP) and immunoblotted (IB) with anti-HA and FLAG antibodies. Blots are representative of three experiments. B, characterization of Gβ1-GST protein by immunoblotting with anti-Gβ1 antibody. C, Gβ1-GST or GST beads coupled to glutathione beads were used to pull down purified PP1c. Blots are representative of two experiments.
Presence of PP1c-Gβ1 protein complex in platelets and its modulation during platelet activation
To examine if PP1c-Gβ1 protein complex exists endogenously in platelets, we performed co-immunoprecipitation (Co-IP) assays with platelet lysate. Gβ1 was immunoprecipitated using anti-Gβ1 antibody from lysate obtained from unstimulated platelets and platelets treated with thrombin or ADP. Immunoprecipitated samples were immunoblotted with anti-PP1c antibody. Immunoblots of Gβ1 immunoprecipitates from the resting (Res) platelets detected PP1c. However, the interaction of Gβ1 with PP1c was decreased following platelet stimulation with GPCR agonist thrombin or ADP (Fig. 2, A and B). These studies suggest that PP1c-Gβ1 protein complex may dissociate following the engagement of GPCRs.
Figure 2.

Platelet activation disrupts endogenous PP1c-Gβ1 complex, facilitates PP1c-PLCβ3 interaction, and dephosphorylates PLCβ3 at Ser1105. A, C, and E, platelets were maintained in resting (Res) state or treated with thrombin (Thr) (0.5 unit/ml) or ADP (10 μm) for 2 mins. A, lysate was immunoprecipitated with control IgG or Gβ1 antibodies. Samples separated on SDS-PAGE were immunoblotted with anti-PP1c or Gβ1 antibodies. B, densitometry of the relative intensity of PP1c in arbitrary units (AU) from four experiments. C, lysate was immunoprecipitated with control IgG or PP1c antibodies. Samples were loaded in alternate lanes and separated on SDS-PAGE and immunoblotted with anti-PLCβ3 and PP1c antibodies. **, nonspecific bands. D, densitometry of the relative intensity of PLCβ3 in arbitrary units (AU) from four experiments. E, lysate was loaded in alternate lanes and separated on SDS-PAGE and immunoblotted with anti-Ser 1105 PLCβ3 or PLCβ3 antibodies. F, densitometric quantification of PLCβ3 Ser1105/total PLCβ3 from three to four experiments. Error bars, mean ± S. E.
One of the early consequences of GPCR signaling is the activation of the family of phospholipase C beta (PLCβ). Platelets deficient in both PLCβ2 and PLCβ3 showed decreased aggregation to low doses of thrombin and ADP (8). Among the PLCβ isoforms, we noticed that only PLCβ3 contained one of the consensus PP1c binding motifs ((K/R)X(V/I)X(F/W)) 27KFIKW31. Because the subcellular localization of PP1c is regulated by its ability to partner with distinct proteins, some of which may bear the PP1c binding motifs, we evaluated whether PP1c could interact with PLCβ3 in platelets using co-immunoprecipitation assays. Compared with the resting platelets, we observed an increased interaction of PLCβ3 with PP1c from thrombin- or ADP-stimulated platelets (Fig. 2, C and D). These studies suggest that the engagement of GPCR could lead to an association of PP1c with PLCβ3.
PLCβ3 is phosphorylated, and in particular Ser1105 phosphorylation of PLCβ3 by protein kinase A (PKA) inhibits PLCβ3 activity (9). Therefore, we tested if agonist-induced association of PLCβ3 with PP1c effected Ser1105 PLCβ3 phosphorylation. Basal phosphorylation of Ser1105 PLCβ3 was evident in unstimulated platelets, and agonist stimulation led to a decrease in the phosphorylation of Ser1105 PLCβ3 (Fig. 2, E and F). Taken together, these studies suggest that thrombin or ADP stimulation of platelets induces the dissociation of PP1c-Gβ1complex, which correlates with the association of PP1c with PLCβ3 and the subsequent dephosphorylation of the inhibitory Ser1105 in PLCβ3.
Genetic and pharmacological inhibition of Gβ1-reduced GPCR signaling
Compared with the Gα subunits, the contribution of Gβ and Gγ subunits in platelet GPCR signaling-dependent functions is unclear. Studies with Gβ1−/− platelets were not pursued because Gβ1−/− mice die within 2 days of birth (10). Megakaryocytes, from which platelets are derived, have emerged as an alternate system to study GPCR-induced signaling. Specifically, this model system is amenable to genetic manipulation and is most responsive to thrombin receptor protease-activated receptor 4 (PAR4)–dependent signaling (11, 12). Megakaryocytes cultured from the bone marrow of the wild-type mice were treated with murine GNB1 siRNA. Depletion of Gβ1 was evident in megakaryocytes (Fig. 3A). Compared with the control siRNA-treated megakaryocytes, Gβ1-depleted megakaryocytes displayed decreased PAR4 activating peptide (PAR4-AP)–stimulated soluble fibrinogen binding (Fig. 3B). Fibrinogen binding was specific to integrin αIIbβ3 because addition of EDTA, a cation chelator, decreased PAR4-AP–dependent fibrinogen binding to base line (unstimulated). For the rest of the manuscript, all functional studies were performed with GPCR agonist thrombin. Next, we tested the effect of gallein (13), a small-molecule inhibitor of Gβγ, on human platelet aggregation. Thrombin-induced platelet aggregation was moderately reduced by gallein treatment (Fig. 3, C and D). Collectively, these studies suggest that genetic depletion of Gβ1 in megakaryocytes and pharmacological blockade of Gβγ signaling in platelets can decrease PAR4/thrombin-induced inside-out integrin αIIbβ3 signaling.
Figure 3.

Genetic depletion of Gβ1 and pharmacological inhibition of Gβγ signaling reduce thrombin receptor–induced fibrinogen binding and platelet aggregation. A, Gβ1 expression in control and Gβ1 siRNA-treated murine megakaryocytes. Blots were reprobed for actin (loading control). Blots are representative of two to three experiments. B, megakaryocytes were mixed with 1 mm PAR4-AP, nonblocking αIIb antibody, 7-AAD dye, and Alexa 488 conjugated fibrinogen. Specific fibrinogen binding was evaluated as mean fluorescence intensity in a gated population of large megakaryocytes that are viable and expressed αIIb. n = 4. C, aggregation profile of washed platelets preincubated with DMSO and 10 μm or 20 μm gallein and then stimulated with thrombin (0.05 unit/ml). The time to achieve 50% aggregation was increased by gallein treatment as seen by the rightward shift in the tracings. D, final platelet aggregation from four subjects is shown. Error bars, mean ± S. E.
Decreased thrombin-induced aggregation in PP1cα−/− platelets
To further study the contribution of PP1cα in thrombin signaling, we generated mice that lacked PP1cα in platelets. PP1cα-loxp (fl) mice (14) were crossed with megakaryocyte/platelet lineage–specific platelet factor 4 (PF4) Cre recombinase knock-in mice (15). The resulting PP1ca−/− mice displayed loss of PP1cα only in platelets but not in lymphocytes (Fig. 4A). Consistent with the previous study, wherein PP1cβ isoform expression was enhanced in PP1cα−/− cardiac tissue (14), we also noticed ∼2-fold up-regulation of PP1cβ isoform in PP1cα−/− platelets (Fig. 4A). Next, we assessed the role of PP1cα in thrombin-induced murine platelet aggregation. Compared with the PP1cα-loxp fl/fl Cre negative (wild type), PP1cα-loxp fl/fl Cre positive (PP1cα−/−) platelets displayed decreased aggregation to low-dose (0.02 unit/ml) but not to high-dose (1 unit/ml) thrombin (Fig. 4, B and C). Moreover, 0.02 unit/ml thrombin-induced soluble fibrinogen binding was also reduced in PP1cα−/− platelets (Fig. 4D). Thus, loss of PP1cα decreased low-dose thrombin–induced soluble fibrinogen binding and platelet aggregation.
Figure 4.

Knock-out of PP1cα in platelets decreases thrombin-induced platelet aggregation and soluble fibrinogen binding. A, characterization of platelet-specific PP1cα knock-out mice. Lysate from platelets and non-megakaryocytic tissue (lymphocytes) from Pf4Cre− and PP1cαfl/fl (WT+/+) and Pf4Cre+ and PP1cαfl/fl (PP1cα−/−) were immunoblotted with anti-PP1cα, PP1cβ, PP1cγ, and actin antibodies. B, aggregation profile of washed WT and PP1cα−/− platelets stimulated with 0.02 unit/ml and 1 unit/ml thrombin. C, final platelet aggregation from six experiments is shown. D, platelets (basal, resting) stimulated with thrombin (0.02 unit/ml) in the presence of Alexa 488 conjugated fibrinogen were analyzed by flow cytometry, and soluble fibrinogen binding is shown as mean fluorescence intensity from six experiments. Error bars, mean ± S. E.
Disruption of PP1c-Gβ1 complexes reduced thrombin-induced platelet aggregation
We noticed a variant of the RVXF PP1c binding motif in Gβ1 at amino acids 78 through 82, 78KLIIW82. To investigate if this region in Gβ1 can support the interaction of PP1c, we constructed full-length Gβ1 plasmids with alanine substitutions at amino acids Lys, Ile, and Trp corresponding to positions 78, 80, and 82, respectively. In addition, a Gβ1 plasmid with all three amino acids (KIW) mutated to alanine (triple mutant) K27A/I29A/W31A was also generated. FLAG-tagged alanine mutants of Gβ1 were co-transfected with HA-tagged PP1c α, β, and γ1 in HEK 293 cells and lysate subjected to co-immunoprecipitation assays. Cells expressing the wild-type Gβ1 retained PP1c in the co-immunoprecipitation assay. Unexpectedly, PP1c co-immunoprecipitated with Gβ1 containing the alanine substitutions at amino acids Lys, Ile, and Trp. However, Gβ1 with the triple alanine mutations failed to support PP1c interaction (Fig. 5A). PP1c was adequately expressed under all transfection conditions (Fig. 5A, lowest panel). These studies reveal that simultaneous disruption of amino acids Lys, Ile, and Trp within the variant PP1c binding region on Gβ1 can diminish PP1c interaction. Thus, this region in Gβ1 may support an interaction with PP1c.
Figure 5.

Identification of PP1c binding site on Gβ1 and use of myristoylated Gβ1 peptide to disrupt PP1c-Gβ1 complex in platelets. A, 293 cells were transfected with either empty vector (EV), FLAG-tagged wild-type Gβ1 (WT), or FLAG-tagged Gβ1 with single alanine point mutations at residue Lys27, Ile29, Trp31, or triple alanine mutations at these locations along with HA-tagged PP1c α, β, and γ. Lysate was immunoprecipitated (IP) with anti-FLAG antibody and immunoblotted (IB) with anti-FLAG and HA antibodies. Input panel shows PP1c HA expression in cells transfected with Gβ1 full-length and mutant constructs. Similar results were obtained from two independent experiments. B, platelets were treated with vehicle DMSO, 200 μΜ Myr Gβ1 peptide, or Myr control scrambled (Scr) peptide, and lysate was immunoprecipitated with control (IgG) and anti-PP1c antibodies and blotted with anti-Gβ1 and PP1c antibodies. Blots are representative of three independent experiments.
Based on the interaction studies in HEK 293 cells, we generated a synthetic cell permeable myristoylated (Myr) Gβ1 peptide (75QDGKLIIWDSY85) and a scrambled control peptide. Thus, the Gβ1 peptide contained the variant PP1c binding motif and encompassed amino acids 75–85 of the intact protein. We investigated whether the cell-permeable Gβ1 peptide was capable of disrupting the endogenous PP1c-Gβ1 complex by co-immunoprecipitation studies. PP1c was detected in Gβ1 immunoprecipitates obtained from DMSO and the scrambled peptide-treated platelets. Treatment of platelets with the Gβ1 peptide reduced the interaction of PP1c with Gβ1 (Fig. 5B). These studies indicate that the myristoylated cell-permeable Gβ1 peptide can decrease the interaction of PP1c with Gβ1 complex in platelets.
To evaluate the consequence of perturbing PP1c-Gβ1 complex on platelet GPCR signaling, we analyzed platelet aggregation in response to low-dose (0.02 unit/ml) thrombin. Pretreatment of human platelets with cell-permeable Gβ1 peptide but not the scrambled peptide moderately decreased platelet aggregation in response to thrombin (Fig. 6, A and B). Gβ1 peptide treatment delayed the time to achieve 50% thrombin-induced aggregation. These studies indicate that the complex of PP1c and Gβ1 participates in platelet function.
Figure 6.

Myristoylated Gβ1 peptide reduces thrombin-induced platelet aggregation. A, aggregation profile of washed platelets preincubated with DMSO, Myr control scrambled (Scr) peptide, and Myr Gβ1 peptide and then stimulated with thrombin (0.02 unit/ml). B, final platelet aggregation from six subjects is shown. Error bars, mean ± SEM.
Discussion
Platelet activation, in part, via GPCR signaling is critical for hemostasis and thrombosis. Heterotrimeric G proteins Gα and Gβγ subunits that associate with GPCRs transduce signals by engaging multiple effectors, including the protein kinases. Here, we show that Gβ1 subunit interacts with the catalytic subunit of protein phosphatase 1 and modulates GPCR signaling in platelets. Specifically, GPCR signaling–dependent functions such as soluble fibrinogen binding and/or platelet aggregation was reduced if (a) Gβ1 subunit was depleted by siRNA in murine megakaryocytes, (b) PP1cα was deleted by Cre-lox approach in murine platelets, (c) Gβγ signaling was blocked by a small-molecule inhibitor in human platelets, and (d) PP1c-Gβ1 protein complex was disrupted by a cell-permeable Gβ1 peptide bearing the PP1c binding motif in human platelets. These studies suggest the importance of coupling G protein–induced inside-out αIIbβ3 signaling with phosphatase-driven signal transduction during platelet activation.
Gα and Gβγ subunits engage in multiprotein complexes in a spatial and temporal fashion to facilitate transmission of signals from the GPCR. For example, after GPCR activation, the Gβγ subunits engage proteins including arrestin, G protein receptor kinase (GRK), PLCβ, PI3Kγ, etc. (16). In a yeast two-hybrid screen, we noticed a novel interaction of PP1cα with the Gβ1 subunit. Co-immunoprecipitation assays in epitope-tagged cells revealed that all isoforms of PP1c could interact with Gβ1, whereas in vitro studies using purified proteins suggested that the interaction of Gβ1 with PP1c was direct and independent of the Gα and Gγ subunits (Fig. 1). Similar to the ability of Gβ1 to associate with PP1, Gβ-like (GβL) protein can interact with other subtypes of Ser/Thr phosphatases such as PP4, PP2A, and PP6 in transfected 293 cells (17). Such interactions likely facilitate the recruitment of these phosphatases to the IκB kinase (IKK) complex and regulate NF-κB signaling. Besides the Gβ subunit, Gα subunits (Gα12 and Gα13) can also interact with PP2A (18) and PP5 (19) and stimulate phosphatase activity in COS cells.
What is the significance of Ser/Thr phosphatase containing multiprotein complexes? Unlike Ser/Thr kinases that are diversified by gene duplication, the diversity of PP1 holoenzyme is achieved by the ability of PP1c to engage with a variety of proteins in a dynamic fashion to form separate PP1c-protein complexes. Indeed, over 200 distinct preassembled or signal-induced phosphatase complexes have been identified, wherein, phosphatase-interacting proteins generally enable targeting of the catalytic subunit of the phosphatase to distinct subcellular locations and thereby provide specificity to the holoenzyme (6, 20). Our studies identify the existence of two phosphatase complexes (PP1c-Gβ1 and PP1c-PLCβ3) in platelets, not previously described in any cell types (Fig. 2). PP1-Gβ1 complex, predominately noticed as a preformed complex in nonstimulated platelets, may facilitate the targeting of PP1 to the vicinity of GPCR and contribute to GPCR signaling. Indeed, we noticed that genetic depletion of Gβ1 in murine megakaryocytes reduced GPCR signaling (Fig. 3). However, depletion of Gβ1 is unlikely to ensure an unregulated PP1c that can indiscriminately dephosphorylate substrates. Rather, it is widely believed that PP1c does not exist freely in cell and is regulated by a multiprotein complex with PIP. Therefore, under conditions of Gβ1 depletion, PP1c might be interacting with other isoforms of Gβ or other PIP and respond to thrombin treatment. Pharmacological inhibition of Gβγ or disruption of PP1c-Gβ1 complex also reduced GPCR signaling in human platelets (Figs. 3 and 6). Loss of PP1cα or PP1cγ (an isoform that also interacts with Gβ1) reduced low-dose thrombin-induced platelet aggregation (Fig. 4) (7). Although we have not investigated the molecular target(s) of PP1c-Gβ1 complex in platelets, previous studies in other cell types have noticed PP1cα- (21), PP1cβ- (22), and PP1cγ-mediated (23) dephosphorylation of GPCRs as one of the features for the GPCR resensitization process. It is important to note that besides PP1, PP2A (24), PP2B (25), and PP2C (26) are also reported to regulate GPCR phosphorylation.
PP1c-PLCβ3 complex observed in agonist-stimulated platelets might target PP1 to regulate PLCβ3 phosphorylation. In line with this argument, we noticed that Ser1105 PLCβ3 phosphorylation was reduced in agonist-stimulated platelets (Fig. 2). Because previous studies suggest that Ser1105 phosphorylation of PLCβ3 dampens its activity (9, 27), the dephosphorylation of Ser1105 PLCβ3 upon agonist stimulation in platelets may represent one mechanism of PLCβ3 activation to transmit GPCR signals. It is important to note that the expression of PLCβ2 is higher than PLCβ3 in human platelets (28). Moreover, deficiency of PLCβ2 in human patients was associated with reduced GPCR-induced calcium mobilization, impaired aggregation, and bleeding diathesis (28–30). Although PLCβ2 isoform is well recognized for relaying GPCR signals, it is possible that PLCβ3 may have a redundant role in signal propagation downstream of GPCR. Indeed, platelets deficient in both PLCβ2 and PLCβ3 showed decreased aggregation to low doses of thrombin and ADP and reduced calcium mobilization and were unable to form stable clots in vivo (8).
Our studies bear some resemblance to previous observations in platelets and heterologous cells wherein PP1cα and PP1cγ formed a complex with the PP1 regulatory protein spinophilin (31), and loss of PPP1R9B (spinophilin) reduced platelet aggregation to thrombin and thromboxane A2 (32). In another study, PP1c associated with a protein complex containing at least 14–3-3γ and the regulator of G protein signaling 18 (RGS18) in platelets when the cyclic nucleotide levels were elevated (33). PP1c likely facilitated the dephosphorylation of RGS18 Ser9 and Ser218 and the subsequent dissociation of RGS18 from 14–3-3γ complex that enabled RGS18 to dampen platelet activation. The detailed understanding of PP1c multiprotein complexes is not limited to academic interest because this knowledge may enable the development of peptides or small molecules that can bind to PP1 and thus disrupt specific protein complexes for functional studies and/or therapeutic modalities. Indeed, a cell-permeable PP1 disrupting peptide (PDP3) containing the PP1c binding motifs of nuclear inhibitor of PP1 (NIPP1) dephosphorylated mitotic substrates of PP1 and sensitized tumors to chemotherapy, which was induced by protein kinase inhibitors (34). In a similar approach, our studies revealed that Gβ1 cell-permeable peptide disrupted PP1c-Gβ1 interaction in platelets and reduced thrombin-induced platelet aggregation (Figs. 5 and 6).
There are some intrinsic limitations with this study. Gβ has five isoforms (16, 35), and one or more of the Gβ isoforms may have facilitated PAR4-dependent signaling in Gβ1-depleted megakaryocytes. In the case of Gβ1 peptide study, potential coupling of additional phosphatase subtypes PP2A or PP2B with Gβ1 and/or other Gβ isoforms or Gα subunits may have facilitated signaling in human platelets. Multiple redundant signaling pathways triggered by the high dose of thrombin could have activated PP1cβ and PP1cγ isoforms in the absence of PP1cα and sustained platelet activation. These caveats may have contributed to the moderate functional phenotypes we noticed with the different approaches. In addition, the use of peptides that contain PP1c binding motifs can disrupt additional PP1c-PIP interactions. Nevertheless, our observations from several lines of investigation suggest that PP1c-Gβ1 complex along with PP1c-PLCβ3 complex may facilitate GPCR signaling as shown in a proposed model (Fig. 7).
Figure 7.
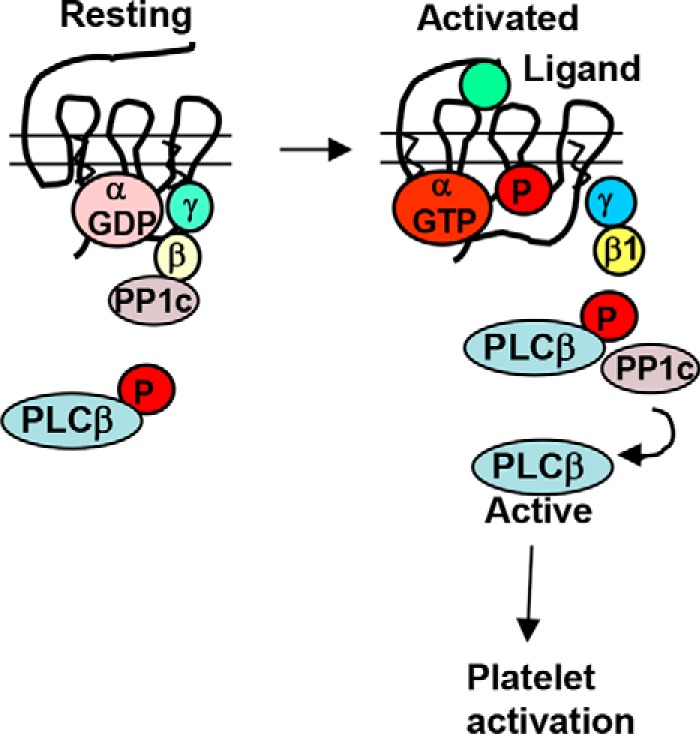
Proposed model depicting the role of PP1c-Gβ1 and PP1c-PLCβ3 complexes in GPCR-induced platelet activation. A multiprotein complex of Gβ1 and PP1c exists presumably in the vicinity of GPCR. Engagement of GPCR with thrombin or ADP leads to the disruption of this complex. A new signal-induced complex of PP1c-PLCβ3 with a concomitant dephosphorylation of PLCβ3 Ser1105 occurs. Based on previous findings, dephosphorylation of PLCβ3 may aid in signal transmission. Forced disruption of PP1c-Gβ1 complex prior to thrombin stimulation reduced platelet aggregation. Collectively, these studies suggest that PP1 facilitates thrombin signaling by coupling with Gβ1 and PLCβ3 in the GPCR pathway.
In conclusion, using platelets as a model system, our studies provide a molecular basis for the functional coupling of PP1c with GPCR function/signaling and thus identify a novel phosphatase complex (PP1c-Gβ1) as potential target for therapeutic intervention in thrombotic and inflammatory conditions. Because GPCR, PP1c, and Gβ1 are also ubiquitously expressed proteins that are involved in signal transduction in a variety of cell types, our findings will likely have relevance beyond platelets.
Experimental procedures
Yeast two-hybrid screening
Yeast two-hybrid screening was performed using the Mate & Plate human bone marrow library from Clontech (Palo Alto, CA) (catalog no. 630477). Full-length PP1cα (amino acids 1–473) was generated by PCR using primers 5′-GCCGAATTCATGGCGGATTTAGATAAAC-3′ (forward) and 3′-CGGGGATCCCTATTTCTTTGCTTGCTTTG-5′ (reverse) and ligated between EcoR1 and BamH1 sites of the bait vector pGBKT7. The pGBKT7-PP1cα vector was transformed into the yeast strain AH109 using lithium acetate. To study the interaction, AH109 yeasts containing the bait vector were mated with Y187 yeasts containing the cDNA libraries cloned in a prey vector pGADT7. The mated yeasts were plated on selection media lacking leucine (L) and tryptophan (W) and incubated at 30 °C until the colonies appear. Potential false positive colonies that grew on dropout medium (−LW) were ruled out by also qualitatively analyzing the expression of β-galactosidase (β-gal) marker gene activity by nitrocellulose filter lift assays. Briefly, colonies were transferred to Whatman No. 3 MM filter paper, permeated by brief immersion in liquid nitrogen, and incubated on filter paper saturated with Z-buffer containing 1 mg/ml X-gal at 30 °C for 0.5–8 h. β-gal–positive transformants were re-plated on synthetic medium lacking adenine, leucine, and tryptophan supplemented with histidine (-ALWH) to screen for Ade2 reporter gene activity assay. Plasmids from the positive colonies were recovered and amplified in Escherichia coli DH5α cells and selected for ampicillin and kanamycin resistance. Colonies which showed resistance for both the markers were analyzed by DNA sequencing using the T7 primers. Approximately 5 × 106 transformants were screened. Plasmids recovered from yeast were re-transformed into native AH109 yeast with the bait construct or nonspecific control selected on high stringency plates with SD/−Ade/−His/−Leu/−Trp/X-β-galactosidase to confirm the interaction.
Human platelet isolation
Blood from healthy donors was drawn in an acid citrate dextrose anticoagulant or 3.8% citrate after all the donors signed the informed consent, which was approved by the Institutional Review Board of Baylor College of Medicine. Blood was centrifuged at 189 g for 15 min at room temperature to obtain platelet-rich plasma (PRP). In some experiments, platelet-rich plasma supplemented with 75 nm prostaglandin E1 was recentrifuged at 524 g for 10 min. The resultant platelet pellet was resuspended in Tyrode's buffer and centrifuged. Washed platelets were then suspended in Tyrode's buffer and counts adjusted to 2.5 × 108 platelets/ml.
GST pulldown assays
GST or Gβ1GST (Abnova) (4 μg) pre-coupled with glutathione beads was mixed with 1 μg of recombinant polyhistidine-tagged PP1c (Santa Cruz Biotechnology) for 3 h at 4 °C. Beads were washed three times and the interacting proteins separated by SDS-PAGE and immunoblotted with anti-PP1c antibody.
Generation of DNA constructs and transfection assays
FLAG-tagged cDNA encoding full-length GNB1 and cloned in pCDNA3.1 was purchased from the cDNA Resource Center, Bloomsburg University. Generation of Gβ1 constructs with single alanine mutations (K27A, I29A, W31A) and triple alanine mutations (K27A/I29A/W31A) was performed using site-directed mutagenesis kit (QuikChange II XL) (Agilent Technologies). All constructs were sequenced and verified to contain the desired mutations. Full-length cDNAs for PP1c α, β, and γ cDNA were subcloned into pCMV vector between EcoR1 and Not1 sites to generate HA-tagged PP1c. HEK 293 cells were co-transfected with HA-tagged PP1c α, β, γ and FLAG-tagged full-length Gβ1 using Lipofectamine supplemented with PLUS reagent (Life Technologies). In some experiments, FLAG-tagged Gβ1 alanine mutants were used. Cells transfected with empty FLAG and HA vector (EV) served as control. After 48 h, cells were washed with phosphate-buffered saline (PBS) and lysed with radioimmune precipitation assay buffer supplemented with 1 mm Na3VO4 and 1 μg/ml leupeptin.
Generation of platelet-specific PP1cα−/− mice
All experiments with mice were performed following approval by the Baylor College of Medicine Institutional Animal Care and Use Committee. PP1cα-loxP flox (fl) mice were generated in collaboration with Lexicon Genetics as described previously (14). Briefly, exon 3 of PP1cα was flanked with loxP sequence that can be recognized by Cre recombinase. PP1cα flox/flox homozygote mice were crossed with a megakaryocyte/platelet-specific Pf4 Cre knock-in mice (The Jackson Laboratory) (15) to obtain platelet-specific PP1cα knock-out mice (PP1cα−/−). All studies were performed using male and female littermates (PP1cα fl/fl and Cre positive, referred to as PP1cα−/−) and PP1cα fl/fl and Cre negative, referred to as wild type (WT+/+).
Co-immunoprecipitation assays and immunoblotting
Lysate from transfected 293 cells were immunoprecipitated with anti-FLAG antibody. Samples were separated on a SDS-PAGE and immunoblotted with anti-HA and anti-FLAG antibodies. Platelets (resting) or following stimulation with thrombin (0.5 unit/ml) or ADP (10 μm) for 2 min under non-stirring conditions were lysed with radioimmune precipitation assay buffer and subjected to immunoprecipitation assays. In some experiments, platelets were treated with 0.2% vehicle DMSO, 200 μm myristoylated Gβ1 peptide (75QDGKLIIWDSY85-NH2) (Selleck Chemicals) or Myr-control scrambled peptide (DQILYDSKGWI) for 30 min at 37 °C before lysis. Platelet lysates were immunoprecipitated with control IgG, anti-Gβ1 (Santa Cruz) or anti-PP1c (Santa Cruz) antibodies and immunoblotted with anti-PP1c or anti-PLCβ3 antibodies, respectively. In some experiments, lysate of platelets simulated with agonist was separated on a SDS-PAGE gel and immunoblotted with anti-phospho PLCβ3 (Ser1105) (Cell Signaling) and anti-PLCβ3 (Cell Signaling) antibodies, followed by HRP-conjugated secondary antibodies, and blots were developed using SuperSignal chemiluminescent substrate (Thermo Scientific). Densitometry was performed using ImageJ software (National Institutes of Health).
Murine megakaryocytes, knockdown, and flow cytometry
Megakaryocytes were obtained from the bone marrow culture of the C57BL/6J black mice as we have described previously (12). Megakaryocytes on day 5 were transfected with 100 nm control siRNA or GNB1 siRNA using Mirus transfecting agent (Mirus Bio) for 48 h at 37 °C. On day 7, megakaryocyte suspension was lysed and immunoblotted with anti-Gβ1 and actin antibodies. An aliquot (50 μl) of megakaryocyte suspension was mixed with 1 mm PAR4-AP (AYPGKF) (protein sequencing Advanced Technology Core Laboratories, Baylor College of Medicine) along with nonblocking anti-αIIb antibody, 7-amino-actinomycin D (7-AAD) (BD Biosciences), and Alexa 488 conjugated fibrinogen (20 μg/ml final concentration) (Thermo Scientific) in the presence and absence of 10 mm EDTA for 20 min. Using the Coulter EPICS XL Flow Cytometry System (Beckman Coulter), Alexa fibrinogen binding was measured as mean fluorescence intensity (MFI) in FL1 channel only from a gated population of large megakaryocytes (size) that expressed αIIbβ3 (FL2) and were viable (negative for 7-AAD in the FL3).
Aggregation assays and soluble fibrinogen binding
Platelet aggregation was analyzed under stirring conditions using eight-channel PAP-8C aggregometer (Bio/Data Corp.) following one of the treatments described below. Washed human platelets were preincubated with DMSO (0.2%), 10 μm and 20 μm gallein (Tocris Bioscience) for 30 min, and then challenged with thrombin (0.05 unit/ml). Washed murine platelets (WT and PP1cα−/−) were stimulated with low (0.02 unit/ml) and high (1 unit/ml) thrombin. In other studies, washed human platelets were treated with DMSO, 200 μm Gβ1, or scrambled peptide for 30 min and then challenged with 0.02 unit/ml thrombin. To study the binding of soluble fibrinogen, washed resting (basal)- and thrombin (0.02 unit/ml)-stimulated murine platelets diluted to 2.5 × 107/ml were incubated with Alexa 488 conjugated fibrinogen (Thermo Scientific) and analyzed by flow cytometry as we have described previously (7).
Statistics
Data are expressed as mean ± S.E. and the statistical significance of the data analyzed using GraphPad Prism. For analysis of two groups, paired Student's t test was used, and one-way analysis of variance (ANOVA) followed by Tukey's post hoc test were used for comparison of differences among multiple groups. p < 0.05 was considered statistically significant.
Author contributions
S. P. performed all studies related to human platelets and murine megakaryocytes and analyzed all data. T. K. performed the yeast two-hybrid studies, generated the platelet-specific PP1c knock-out mice, conducted murine platelet studies, and analyzed data. A. C. N. provided the PP1cα flox mice and edited the paper. K. V. V. conceived, designed, and interpreted the study and wrote the paper.
Acknowledgment
This material is the result of work supported with resources and the use of facilities of the Michael E. DeBakey Veterans Affairs Medical Center.
This work was supported by National Institutes of Health Grants HL081613 and GM112806. K. V. V. is supported by the Mary R. Gibson Foundation. The authors declare that they have no conflicts of interest with the contents of this article. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs, the United States government, or the National Institutes of Health.
- GPCR
- G protein–coupled receptor
- PP1c
- catalytic subunit of protein phosphatase 1
- PAR4
- protease-activated receptor 4
- PAR4-AP
- PAR4 activating peptide
- Myr
- myristoylated
- PIP
- phosphatase interacting proteins
- RGS18
- regulator of G protein signaling 18.
References
- 1. Shankar H., Kahner B., and Kunapuli S. P. (2006) G-protein dependent platelet signaling–perspectives for therapy. Curr. Drug Targets 7, 1253–1263 [DOI] [PubMed] [Google Scholar]
- 2. Stalker T. J., Welsh J. D., and Brass L. F. (2014) Shaping the platelet response to vascular injury. Curr. Opin. Hematol. 21, 410–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Estevez B., Shen B., and Du X. (2015) Targeting integrin and integrin signaling in treating thrombosis. Arterioscler. Thromb. Vasc. Biol. 35, 24–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ford C. E., Skiba N. P., Bae H., Daaka Y., Reuveny E., Shekter L. R., Rosal R., Weng G., Yang C. S., Iyengar R., Miller R. J., Jan L. Y., Lefkowitz R. J., and Hamm H. E. (1998) Molecular basis for interactions of G protein βγ subunits with effectors. Science 280, 1271–1274 [DOI] [PubMed] [Google Scholar]
- 5. Ceulemans H., and Bollen M. (2004) Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol. Rev. 84, 1–39 [DOI] [PubMed] [Google Scholar]
- 6. Heroes E., Lesage B., Görnemann J., Beullens M., Van Meervelt L., and Bollen M. (2013) The PP1 binding code: A molecular-Lego strategy that governs specificity. FEBS J. 280, 584–595 [DOI] [PubMed] [Google Scholar]
- 7. Gushiken F. C., Hyojeong H., Pradhan S., Langlois K. W., Alrehani N., Cruz M. A., Rumbaut R. E., and Vijayan K. V. (2009) The catalytic subunit of protein phosphatase 1 gamma regulates thrombin-induced murine platelet αIIbβ3 function. PLoS One 4, e8304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lian L., Wang Y., Draznin J., Eslin D., Bennett J. S., Poncz M., Wu D., and Abrams C. S. (2005) The relative role of PLCβ and PI3Kγ in platelet activation. Blood 106, 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yue C., Dodge K. L., Weber G., and Sanborn B. M. (1998) Phosphorylation of serine 1105 by protein kinase A inhibits phospholipase Cβ3 stimulation by Gαq. J. Biol. Chem. 273, 18023–18027 [DOI] [PubMed] [Google Scholar]
- 10. Okae H., and Iwakura Y. (2010) Neural tube defects and impaired neural progenitor cell proliferation in Gβ1-deficient mice. Dev. Dyn. 239, 1089–1101 [DOI] [PubMed] [Google Scholar]
- 11. Yuan W., Leisner T. M., McFadden A. W., Wang Z., Larson M. K., Clark S., Boudignon-Proudhon C., Lam S. C., and Parise L. V. (2006) CIB1 is an endogenous inhibitor of agonist-induced integrin αIIbβ3 activation. J. Cell Biol. 172, 169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gushiken F. C., Patel V., Liu Y., Pradhan S., Bergeron A. L., Peng Y., and Vijayan K. V. (2008) Protein phosphatase 2A negatively regulates integrin αIIbβ3 signaling. J. Biol. Chem. 283, 12862–12869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lehmann D. M., Seneviratne A. M., and Smrcka A. V. (2008) Small molecule disruption of G protein βγ subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol. Pharmacol. 73, 410–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu R., Correll R. N., Davis J., Vagnozzi R. J., York A. J., Sargent M. A., Nairn A. C., and Molkentin J. D. (2015) Cardiac-specific deletion of protein phosphatase 1β promotes increased myofilament protein phosphorylation and contractile alterations. J. Mol. Cell Cardiol. 87, 204–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tiedt R., Schomber T., Hao-Shen H., and Skoda R. C. (2007) Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood 109, 1503–1506 [DOI] [PubMed] [Google Scholar]
- 16. Smrcka A. V. (2008) G protein βγ subunits: Central mediators of G protein-coupled receptor signaling. Cell. Mol. Life Sci. 65, 2191–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. You D. J., Kim Y. L., Park C. R., Kim D. K., Yeom J., Lee C., Ahn C., Seong J. Y., and Hwang J. I. (2010) Regulation of IκB kinase by GβL through recruitment of the protein phosphatases. Mol. Cells 30, 527–532 [DOI] [PubMed] [Google Scholar]
- 18. Zhu D., Kosik K. S., Meigs T. E., Yanamadala V., and Denker B. M. (2004) Gα12 directly interacts with PP2A: Evidence for Gα12-stimulated PP2A phosphatase activity and dephosphorylation of microtubule-associated protein, tau. J. Biol. Chem. 279, 54983–54986 [DOI] [PubMed] [Google Scholar]
- 19. Yamaguchi Y., Katoh H., Mori K., and Negishi M. (2002) Gα12 and Gα13 interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity. Curr. Biol. 12, 1353–1358 [DOI] [PubMed] [Google Scholar]
- 20. Cohen P. T. (2002) Protein phosphatase 1—targeted in many directions. J. Cell Sci. 115, 241–256 [DOI] [PubMed] [Google Scholar]
- 21. Gehret A. U., and Hinkle P. M. (2013) siRNA screen identifies the phosphatase acting on the G protein-coupled thyrotropin-releasing hormone receptor. ACS Chem. Biol. 8, 588–598 [DOI] [PubMed] [Google Scholar]
- 22. Pöll F., Doll C., and Schulz S. (2011) Rapid dephosphorylation of G protein-coupled receptors by protein phosphatase 1β is required for termination of β-arrestin-dependent signaling. J. Biol. Chem. 286, 32931–32936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Croci C., Sticht H., Brandstätter J. H., and Enz R. (2003) Group I metabotropic glutamate receptors bind to protein phosphatase 1C. Mapping and modeling of interacting sequences. J. Biol. Chem. 278, 50682–50690 [DOI] [PubMed] [Google Scholar]
- 24. Vasudevan N. T., Mohan M. L., Gupta M. K., Hussain A. K., and Naga Prasad S. V. (2011) Inhibition of protein phosphatase 2A activity by PI3Kγ regulates β-adrenergic receptor function. Mol. Cell 41, 636–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shih M., Lin F., Scott J. D., Wang H. Y., and Malbon C. C. (1999) Dynamic complexes of β2-adrenergic receptors with protein kinases and phosphatases and the role of gravin. J. Biol. Chem. 274, 1588–1595 [DOI] [PubMed] [Google Scholar]
- 26. Flajolet M., Rakhilin S., Wang H., Starkova N., Nuangchamnong N., Nairn A. C., and Greengard P. (2003) Protein phosphatase 2C binds selectively to and dephosphorylates metabotropic glutamate receptor 3. Proc. Natl. Acad. Sci. U.S.A. 100, 16006–16011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yue C., Ku C. Y., Liu M., Simon M. I., and Sanborn B. M. (2000) Molecular mechanism of the inhibition of phospholipase C β3 by protein kinase C. J. Biol. Chem. 275, 30220–30225 [DOI] [PubMed] [Google Scholar]
- 28. Lee S. B., Rao A. K., Lee K. H., Yang X., Bae Y. S., and Rhee S. G. (1996) Decreased expression of phospholipase C-β 2 isozyme in human platelets with impaired function. Blood 88, 1684–1691 [PubMed] [Google Scholar]
- 29. Rao A. K., Kowalska M. A., and Disa J. (1989) Impaired cytoplasmic ionized calcium mobilization in inherited platelet secretion defects. Blood 74, 664–672 [PubMed] [Google Scholar]
- 30. Yang X., Sun L., Ghosh S., and Rao A. K. (1996) Human platelet signaling defect characterized by impaired production of inositol-1,4,5-triphosphate and phosphatidic acid and diminished Pleckstrin phosphorylation: Evidence for defective phospholipase C activation. Blood 88, 1676–1683 [PubMed] [Google Scholar]
- 31. Ma P., Foote D. C., Sinnamon A. J., and Brass L. F. (2015) Dissociation of SHP-1 from spinophilin during platelet activation exposes an inhibitory binding site for protein phosphatase-1 (PP1). PLoS One 10, e0119496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma P., Cierniewska A., Signarvic R., Cieslak M., Kong H., Sinnamon A. J., Neubig R. R., Newman D. K., Stalker T. J., and Brass L. F. (2012) A newly identified complex of spinophilin and the tyrosine phosphatase, SHP-1, modulates platelet activation by regulating G protein-dependent signaling. Blood 119, 1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gegenbauer K., Nagy Z., and Smolenski A. (2013) Cyclic nucleotide dependent dephosphorylation of regulator of G-protein signaling 18 in human platelets. PLoS One 8, e80251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chatterjee J., Beullens M., Sukackaite R., Qian J., Lesage B., Hart D. J., Bollen M., and Köhn M. (2012) Development of a peptide that selectively activates protein phosphatase-1 in living cells. Angew. Chem. Int. Ed. Engl. 51, 10054–10059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clapham D. E., and Neer E. J. (1997) G protein β γ subunits. Annu. Rev. Pharmacol. Toxicol. 37, 167–203 [DOI] [PubMed] [Google Scholar]