

Abstract
In many Gram-negative bacteria, including Rhodobacter capsulatus, cytochrome c maturation (Ccm) is carried out by a membrane-integral machinery composed of nine proteins (CcmA to I). During this process, the periplasmic thiol-disulfide oxidoreductase DsbA is thought to catalyze the formation of a disulfide bond between the Cys residues at the apocytochrome c heme-binding site (CXXCH). Subsequently, a Ccm-specific thioreductive pathway involving CcmG and CcmH reduces this disulfide bond to allow covalent heme ligation. Currently, the sequence of thioredox reactions occurring between these components and apocytochrome c and the identity of their active Cys residues are unknown. In this work, we first investigated protein–protein interactions among the apocytochrome c, CcmG, and the heme-ligation components CcmF, CcmH, and CcmI. We found that they all interact with each other, forming a CcmFGHI–apocytochrome c complex. Using purified wild-type CcmG, CcmH, and apocytochrome c, as well as their respective Cys mutant variants, we determined the rates of thiol-disulfide exchange reactions between selected pairs of Cys residues from these proteins. We established that CcmG can efficiently reduce the disulfide bond of apocytochrome c and also resolve a mixed disulfide bond formed between apocytochrome c and CcmH. We further show that Cys-45 of CcmH and Cys-34 of apocytochrome c are most likely to form this mixed disulfide bond, which is consistent with the stereo-specificity of the heme–apocytochrome c ligation reaction. We conclude that CcmG confers efficiency, and CcmH ensures stereo-specificity during Ccm and present a comprehensive model for thioreduction reactions that lead to heme–apocytochrome c ligation.
Keywords: cytochrome c, disulfide, heme, protein–protein interaction, thiol, Rhodobacter capsulatus, apocytochrome, cytochrome c maturation, thiol–disulfide exchange, thioreduction
Introduction
Cytochromes (cyt)2 c are electron transport proteins that play central roles in most energy transduction pathways (1). They differ from other heme-containing proteins in having their heme groups covalently ligated to the polypeptide at conserved CXXCH heme-binding sites (HBS) by a post-translational process designated cyt c maturation (Ccm).
In Rhodobacter capsulatus and many other bacteria, as well as in plant mitochondria, nine membrane-bound proteins (CcmABCDEFGHI) with different functions catalyze Ccm (Fig. 1A) (2–4). Usually, apocytochromes (apocyts) are translated in the cytoplasm and translocated to the periplasm via the general secretory system (5). In the highly oxidative environment of the periplasm, the Cys residues of the HBS of apocyts are oxidized forming a disulfide bond via the thiol-disulfide oxidoreductase DsbA (thio-oxidation) (6, 7), possibly minimizing their proteolytic degradation (Fig. 1A) (8). However, a prerequisite for heme-apocyt c ligation is the availability of reduced Cys residues at the HBS of apocyts c (9). The thiol-disulfide oxidoreductases CcmG and CcmH, together with CcdA, carry out this process (thioreduction).
Figure 1.
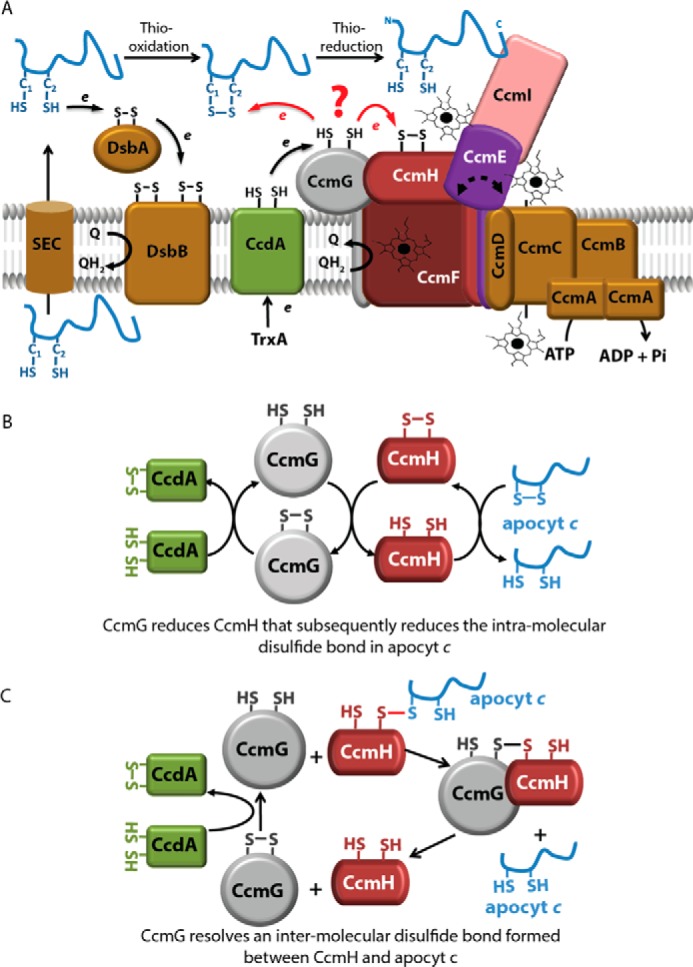
R. capsulatus Ccm system I and earlier proposed models for thioreduction of apocyt c HBS disulfide. A, nine membrane integral proteins (CcmABCDEFGHI) with different functions are responsible for covalent heme ligation to the apocyts to produce mature c-type cyts. Apocyts c are translocated via the SEC system to the periplasm, where the Cys residues at their HBS are oxidized by the DsbA–DsbB system (thio-oxidation). CcdA receives electrons from the cytoplasmic thioredoxin TrxA and reduces CcmG. The thiol-disulfide oxidoreductases CcmG and CcmH reduce the intramolecular disulfide bond at the apocyt HBS to allow heme ligation (thioreduction). CcmH together with CcmF and CcmI form the heme ligation core, whereas CcmABCD is an ATP-dependent ABC-type transporter that loads heme to CcmE to produce holo-CcmE. CcmI traps the C termini of the apocyt c substrates, whereas the heme delivered by holo-CcmE is covalently ligated to the apocyts c heme-binding sites by the CcmFHI core complex. B, CcmG–CcmH–apocyt c linear cascade of thiol-disulfide exchange. This proposal suggests that reduced CcmH (recycled by CcmG and CcdA) reduces directly apocyt c HBS disulfide bond (13, 24–27). C, CcmG reduces a mixed disulfide between CcmH and apocyt c. This proposal suggests that CcmG reduces a mixed disulfide formed between CcmH and apocyt c instead of oxidized CcmH or apocyt c HBS disulfide (12, 19, 25, 27).
CcmG is tethered to the membrane by a single N-terminal transmembrane (TM) helix and contains a thioredoxin motif (CXXC) facing the periplasm. The three-dimensional (3D) structures of the periplasmic domain of CcmG from Escherichia coli (10), Bradyrhizobium japonicum (11), and Pseudomonas aeruginosa (12) show a conserved thioredoxin-like fold with an acidic active site (11). The two catalytic Cys residues are essential for Ccm (13, 14), but CcmG might play an additional role (“holdase”) in chaperoning the apocyts (Fig. 1A) (14).
CcdA (DsbD in some species) is an integral membrane protein with six TM helices that conveys reducing equivalents from the cytoplasmic thioredoxins (e.g. TrxA) to CcmG (15, 16). In R. capsulatus, CcmG and CcdA interact directly, forming mixed disulfides in vivo (Fig. 1A) (17, 18).
CcmH has a single C-terminal TM helix and a periplasmic domain containing a conserved LRCXXCQ active site. The 3D structure of CcmH differs from the canonical thioredoxin fold showing a three-helical bundle structure and has its N-terminal Cys residue buried while its C-terminal Cys is solvent-exposed (Fig. 1A) (19–21).
Earlier genetic studies showed that in the absence of thio-oxidation by DsbA, thioreduction involving CcdA (22) or CcmG (14) is not required. In contrast, this thioredox compensation is not observed in mutants lacking both CcmH and DsbA (14), suggesting that CcmH plays another role in addition to thioreduction of the disulfide bond at the HBS of apocyts c. Indeed, CcmH together with CcmI and CcmF form the heme ligation complex CcmFHI (23).
Although the involvement of both CcmG and CcmH in thioreduction of the apocyts c is established, the sequence of the reactions between these components and their active Cys residues remain unknown. Early experiments using purified R. capsulatus CcmG and CcmH suggested a linear thiol-disulfide cascade based on the ability of CcmH to oxidize CcmG and to reduce a short peptide mimicking apocyt c HBS (Fig. 1B) (13, 21, 24–27). Consistent with this model were the observations that the Arabidopsis thaliana CcmH homologue is able to reduce a peptide mimicking the apocyt c HBS (13, 26), and P. aeruginosa CcmH interacts with a similar peptide at low (micromolar range) affinity (19). The unusual fold and biochemical properties of P. aeruginosa CcmH, together with the inability of CcmG to reduce the disulfide bond of CcmH, led to a different proposal in which CcmG is responsible for resolving a CcmH-apocyt c mixed disulfide formed during Ccm (Fig. 1C) (12, 19, 25, 27).
The establishment of the order of thioreduction reactions between CcmG, CcmH, and apocyt c is essential for elucidating the mechanism of heme ligation. In this work, we first investigated protein–protein interactions between CcmG, CcmH, and class I apocyts c (using apocyt c1 or c2 as model substrates) via co-purification assays, using native and Cys-less variants of purified proteins, as well as detergent-dispersed membrane fractions. We found that CcmG binds tightly to CcmH (and the other heme ligation components) and to apocyt c1, but with lower affinity (micromolar range), forming a CcmFGHI–apocyt c complex. Then, using purified mutant proteins we determined the rate constants of thiol-disulfide exchange reactions between selected Cys residues from CcmG, CcmH, and apocyt c1. Based on these rate constants and the interactions between these proteins, we propose a model for apocyt c thioreduction, suggesting that CcmG is required for efficient cyt c maturation and CcmH for stereo-specific heme-apocyt c ligation during Ccm.
Results
Purification of His6-CcmGWT, FLAG-CcmHWT, and Strep-apocyt c1WT and their derivatives, and protein–protein interactions between apocyt c1 and other Ccm components
CcmG and CcmH are Ccm-specific components that are responsible for thioreduction of the disulfide bond at the HBS of apocyts c. Currently, neither the order of the thiol-disulfide exchange reactions that occur during this process between the three components nor the identity of their active Cys residues that participate in these reactions are well defined. To address this issue, we overproduced in the cytoplasm of E. coli, and then purified by affinity chromatography (>95% purity) soluble derivatives of native (WT) (Fig. 2) as well as single and double Cys (*) mutant variants of His6-CcmG (CcmGWT, CcmGCys-75, CcmGCys-78, and CcmG*), FLAG-CcmH (CcmHWT, CcmHCys-42, CcmHCys-45, and CcmH*), and Strep-apocyt c1 (apocyt c1WT, apocyt c1Cys-34, apocyt c1Cys-37, and apocyt c1*). Fig. 2 shows as an example the purified wild-type derivatives of His6-CcmG, FLAG-CcmH, and Strep-apocyt c1. The Cys mutant derivatives of these proteins were also purified to the same degree of homogeneity (supplemental Fig. S1).
Figure 2.
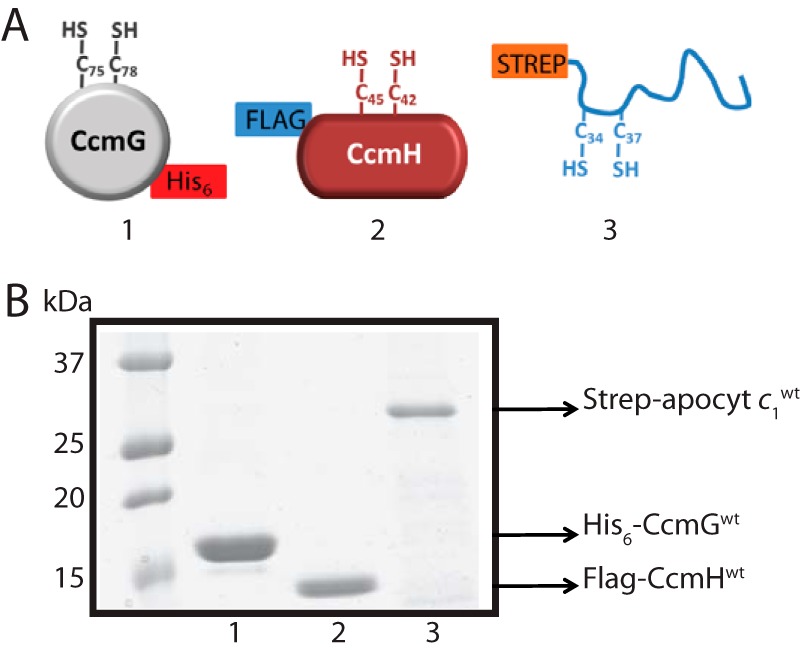
Purified CcmG, CcmH, and apocyt c1 proteins. A, schematic representations of (1) soluble CcmGWT lacking its TM anchor, with a C-terminal His6 tag and its Cys-75 and Cys-78 residues at its thioredoxin-like CXXC motif; (2) soluble CcmHWT lacking its signal sequence and C-terminal TM anchor, with an N-terminal FLAG-tag and its Cys-42 and Cys-45 residues at its unusual CXXC motif; (3) N-terminal Strep-tagged and soluble form of class I apocyt c1WT; this protein lacks its C-terminal TM anchor and has an N terminus located HBS with its Cys-34 and Cys-37 residues. B, Coomassie Blue-stained SDS-PAGE of ∼3 μg of nickel-Sepharose HP-purified His6-CcmGWT (lane 1), anti-FLAG (DYKDDDDK peptide) affinity gel-purified FLAG-CcmHWT (lane 2), and Strep-Tactin–Sepharose purified Strep-apocyt c1WT (lane 3). Mutant derivatives of purified CcmG, CcmH, and apocyt c1 are shown in supplemental Fig. S1.
In thioredoxins and thiol-disulfide oxidoreductases, substrate recognition relies mainly on non-covalent electrostatic and hydrophobic interactions as well as hydrogen bonding within the substrate–enzyme complex (28). Given that CcmG is a Ccm-specific thioredoxin, we investigated its interactions with apocyt c and other Ccm components, in particular CcmH, via co-purification assays using purified CcmG*, apocyt c1*, and CcmH* (Fig. 3). We chose the Cys-less variants of these proteins to avoid increased complexity that could emerge from inter-molecular disulfide bond formation during these assays. The data showed that FLAG-CcmH* co-purified with His6-CcmG* using a nickel-Sepharose HP resin (anti-His), indicating that they interact strongly with each other in vitro despite the absence of their Cys residues (Fig. 3A, right panel). As a control, we showed that in the absence of His6-CcmG*, FLAG-CcmH* was not retained by the anti-His resin (Fig. 3A, left panel). Next, anionic exchange chromatography (Q-Sepharose) was carried out using n-dodecyl β-d-maltoside (DDM)-dispersed membranes from R. capsulatus MTSRP1.r1 (a strain lacking CcmI but overproducing CcmF and CcmH) (Table 1). The fraction eluted at 150 mm NaCl contained many proteins (SDS-PAGE data not shown), but it was highly enriched in CcmF, CcmH, and CcmG, as evidenced using appropriate specific antibodies, as done before (Fig. 3B) (23). The presence of CcmG in this fraction was detected using the newly produced rabbit polyclonal antibodies (see under “Experimental procedures”). The specificity of anti-CcmG antibodies was confirmed using purified His6-CcmG* and appropriate R. capsulatus wild-type and mutant strains (supplemental Fig. S2). The fraction eluted at 150 mm NaCl was incubated with purified His10-CcmIWT and loaded into a nickel-Sepharose resin, as done previously (29). Analysis of the elution fraction using anti-CcmG antibodies revealed that CcmGWT was co-purified with CcmIWT (Fig. 3C). Thus, CcmG interacted with His10-CcmI either directly or indirectly via CcmH (Fig. 3A). CcmI is known to interact strongly with both CcmH and CcmF (23), and the data indicated that CcmG might also be associated with the heme ligation complex CcmFHI.
Figure 3.
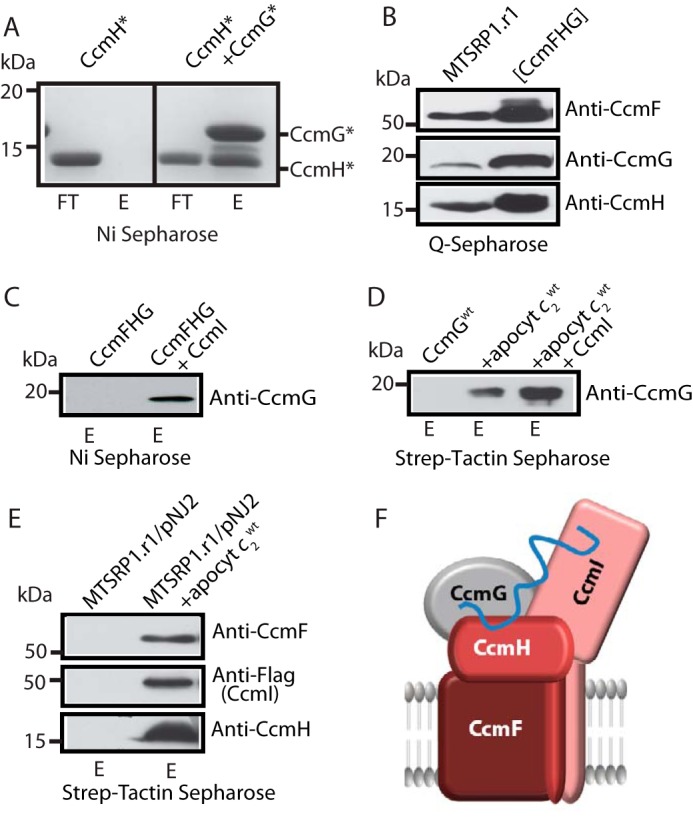
Protein–protein interactions between the apocyt c, thioredoxin CcmG, and heme ligation components CcmF, CcmH, and CcmI. A, co-purification of Cys-less FLAG-CcmH* with Cys-less His6-CcmG* using nickel-Sepharose resin is shown on the right panel. Note that FLAG-CcmH* does not bind to the resin in the absence of His6-CcmG* (left panel). In all panels, FT and E refer to flow-through and elution fractions, respectively. B, DDM-dispersed membrane proteins from R. capsulatus strain MTSRP1.r1 were separated using a Q-Sepharose column, and a fraction containing CcmF, CcmG, and CcmH co-eluting together was collected. 40 μg of total membrane proteins before fractionation (left lane) and 10 μg of the fraction containing CcmF, CcmG, and CcmH together (right lane) are shown. All proteins were detected by immunoblots using appropriate specific antibodies as indicated. C, co-purification of native CcmG from the CcmFGH-enriched fraction with purified CcmI. 10 μg of purified His10-CcmI was incubated with 100 μg of CcmFGH-containing fraction and re-purified in a nickel-Sepharose resin. Only in the presence of His10-CcmI is CcmG retained by the resin and found in the elution fraction (right lane). D, co-purification of equimolar concentration (∼1.5 μm) of His6-CcmGWT with Strep-apocyt c2WT using Strep-Tactin–Sepharose resin (middle lane). In the absence of apocyt c2, CcmG was not retained by the resin (left lane), and in the presence of His10-CcmI (∼0.5 μm) more His6-CcmG co-eluted with Strep-apocyt c2 (right lane). C and D, CcmG was detected by immunoblots using anti-CcmG-specific antibodies. E, 500 μg of DDM-dispersed membrane proteins from R. capsulatus strain MTSRP1.r1/pNJ2, containing CcmF, CcmH, and FLAG-CcmI were incubated with 10 μg of Strep-apocyt c2WT, which was then re-purified using Strep-Tactin-Sepharose resin. Binding to the Strep-Tactin column of the heme ligation components CcmF, CcmH, and FLAG-CcmI occurs only when apocyt c2WT is present (compare left and right lanes). Immunodetection was done with anti-CcmF, anti-CcmH, and anti-FLAG (for FLAG-CcmI detection) polyclonal antibodies, as appropriate. F, schematic representation of a hypothetical CcmFGHI-apocyt c complex that might occur during Ccm. Our data showing that apocyt c2 interacts with CcmG, CcmF, CcmH, and CcmI, together with CcmG interacting with the heme ligation core components CcmH, CcmI, and CcmF in the absence of apocyt c2, support the occurrence of such a multisubunit complex binding the apocyt substrates.
Table 1.
Strains and plasmids used in this work
| Strains/plasmids | Relevant propertiesa | Refs. |
|---|---|---|
| Bacteria | ||
| R. capsulatus | ||
| MT-SRP1.r1 | Δ(ccmI::kan) G488A up-promoter of ccmFH; Res+ Ps+ | 50 |
| MD11 | Δ(ccmG::kan); Res+ Ps− | 51 |
| MD14 | ccmH::spe; Res+ Ps− | 50 |
| E. coli | ||
| HB101 | F− Δ (gpt-proA)62 araC14 leuB6(Am) glnV44(AS) thi-1 galK2(Oc) lacY1 Δ(mcrC-mrr) rpsL20(Strr) xylA5 mtl-1 | Stratagene |
| XL1 Blue | endA1 gyrA96(NalR) thi-1 recA1 relA1 lac glnV44 F′[::Tn10 proAB+ lacIq Δ(lacZ)M15] hsdR17(rK−mK+) | Stratagene |
| BL21(DE3) | fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS λ DE3 = λ sBamHI oΔEcoRI-B int:: (lacI::PlacUV5::T7 gene1) i21 Δnin5 | New England Biolabs |
| Plasmids | ||
| pAV1 | pCS1302 derivative carrying truncated R. capsulatus cycA at the NdeI–BamHI sites with a Strep-tag II at its 5′-end and without a signal sequence, AmpR (Strep-apocyt c2WT)b | 29 |
| pAV10 | pFLAG-1 derivative carrying truncated R. capsulatus ccmH lacking its signal sequence and TM helix (C-terminal 46 amino acids) from pCS1604 at the NdeI–BamHI sites, AmpR (FLAG-CcmHWT) | This work |
| pAV10C42S | pAV10 derivative with C42S mutation in R. capsulatus ccmH, AmpR (FLAG-CcmHCys-45) | This work |
| AV10C45S | pAV10 derivative with C45S mutation in R. capsulatus ccmH, AmpR (FLAG-CcmHCys-42) | This work |
| pAV10C42SC45S | pAV10 derivative with C42S and C45S mutations in R. capsulatus ccmH, AmpR (FLAG-CcmH*) | This work |
| pBSK | AmpR | Stratagene |
| pCS1552 | pQE60-helX derivative with C75S mutation in R. capsulatus ccmG, AmpR | This work |
| pCS1553 | pQE60-helX derivative with C78S mutation in R. capsulatus ccmG, AmpR | This work |
| pCS1554 | pQE60-helX derivative with C75S and C78S mutations in R. capsulatus ccmG, AmpR | This work |
| pCS1555 | A truncated R. capsulatus ccmG from pQE60-helX cloned into the EcoRI–HindIII sites of pBSK, AmpR (His6–CcmGWT) | This work |
| pCS1556 | A truncated R. capsulatus ccmG with C75S mutation taken from pCS1552 and cloned into the EcoRI–HindIII sites of pBSK, AmpR (His6-CcmGCys-78) | This work |
| pCS1557 | A truncated R. capsulatus ccmG with C78S mutation taken from pCS1553 and cloned into the EcoRI–HindIII sites of pBSK, AmpR (His6-CcmGCys-75) | This work |
| pCS1558 | A truncated R. capsulatus ccmG with C75S and C78S mutations, taken from pCS1554 and cloned into the EcoRI–HindIII sites of pBSK, AmpR (His6-CcmG*) | This work |
| pCS1566 | pCHB500 derivative producing R. capsulatus ccmG with its signal peptide and His6-tagged at its 3′-end, cloned into the XbaI/KpnI sites, TetR (His6-CcmGWT) | 51 |
| pCS1604 | pACCyc3 derivative (52), producing a truncated R. capsulatus ccmH from pRGK291 (13), lacking both its signal sequence and TM helix (last 46 amino acids at its 3′-end), cloned into NdeI-BamHI sites. ClmR | This work |
| pFLAG-1 | pMADO5 derivative carrying 5′-FLAG-tag ccmI (instead of His10 tag), AmpR | 31 |
| pMADO5 | pCS1303 derivative, containing R. capsulatus ccmI carrying 5′-His10 tag, AmpR (His10-CcmIWT) | 29 |
| pMAM1 | pCS1302 derivative, containing truncated R. capsulatus petC* (derivative of petC with no signal sequence and lacking its last C-terminal 39 amino acids, and its non-heme ligating Cys-144 and Cys-167 residues) cloned into the NdeI–BamHI sites with a Strep-tag II fused at its 5′-end, AmpR (Strep-apocyt c1WT) | 30 |
| pMAM1C34S | pMAM1 derivative with C34S mutation of R. capsulatus petC*, AmpR (Strep-apocyt c1Cys-37) | This work |
| pMAM1C37S | pMAM1 derivative with C37S mutation of R. capsulatus petC*, AmpR (Strep-apocyt c1Cys-34) | This work |
| pMAM1C34SC37S | pMAM1 derivative with C34S and C37S mutations of R. capsulatus petC*, AmpR (Strep-apocyt c1*) | 30 |
| pNJ2 | pRK415 derivative producing R. capsulatus 5′-end FLAG-tagged ccmI with its own promoter, TetR (FLAG-CcmIWT) | 51 |
| pQE60 | Ampr | Qiagen |
| pQE60-helX | pQE60 derivative containing a truncated R. capsulatus ccmG without its signal sequence and with a C-terminal His6 tag cloned into the NcoI–BamHI sites | c |
| pST6 | pCHB500 derivative producing R. capsulatus ccmH, with a Strep-tag fused at its 3′-end, TetR (Strep-CcmHWT) | 14 |
a Res and Ps refer to respiratory and photosynthetic growth, respectively.
b Proteins produced by appropriate plasmids are indicated in bold fonts.
c F. Daldal, unpublished data.
Despite many attempts, co-purification of apocyt c1* with either CcmG* or CcmH* was not observed. Because the use of apocyt c1WT for these assays was not suitable due to its high tendency to dimerize in the presence of oxygen, we used biolayer interferometry to study these interactions. Real-time binding kinetics between purified CcmGWT and apocyt c1WT were determined, as done earlier (30). The association (kon of 9.97 ± 0.11 × 102 m−1 s−1) and dissociation (koff of 7.31 ± 1.16 × 10−3 s−1) rates thus determined yielded a KD value of 7.2 ± 1.8 μm using a 1:1 homogeneous kinetic model describing bimolecular interactions (30). Similarly, when native apocyt c2WT (another class I cyt c, known to interact with CcmI and CcmE (29–31)) was used instead of apocyt c1WT, its co-purification with CcmGWT was readily seen using specific antibodies (Fig. 3D). Furthermore, the amount of CcmGWT that co-purified with apocyt c2WT was higher upon addition of purified CcmI, a specific apocyt c chaperone, suggesting that the interactions between CcmG and apocyts c also involved additional Ccm partners (29). Finally, upon incubation with detergent-dispersed membrane fractions from R. capsulatus strain MTSRP1.r1 complemented with plasmid pNJ2 carrying FLAG-tagged CcmI (Table 1), native apocyt c2WT co-purified with not only FLAG-CcmI but also CcmF and CcmH (Fig. 3E), indicating that besides interacting with CcmG (above), apocyt c2 also interacts with CcmI (29), CcmF, and CcmH, forming a CcmFGHI-apocyt c complex (Fig. 3F). The need for the HBS Cys residues of apocyt c, similar to what was seen here with CcmG or CcmH and apocyt c1, has also been observed for the interactions of apocyt c2 and apoCcmE (31). Our earlier data, showing a ternary complex composed by apocyt c2, CcmI, and apoCcmE (31), together with the data presented here, are consistent with the existence of a multisubunit maturase supercomplex, composed of at least the CcmEFGHI-apocyt c components, as proposed previously (2).
Rate constants of thiol-disulfide exchange reactions between CcmG, CcmH, and apocyt c1
To define the sequence of thiol-disulfide exchange reactions occurring between CcmG, CcmH, and apocyts c during thioreduction and the Cys residues involved in this process, we used Ellman's reagent (5,5′-dithiobis(2-nitrobenzoic acid) (DTNB))-based assays carried out with purified single Cys mutant variants CcmGCys-75, CcmGCys-78, CcmHCys-42, CcmHCys-45, apocyt c1Cys-34, and apocyt c1Cys-37. DTNB is commonly used to quantitate the thiol groups in proteins, as it readily forms mixed disulfide bonds with accessible thiols, and the TNB2− ions released during this reaction can be easily monitored by visible spectroscopy at 412 nm (12, 32, 33). The protein–Cys–S–S–TNB (protein–TNB) adducts formed are good proxies for inter-molecular mixed disulfide bonds between a given protein and another Cys-containing partner protein (12, 32, 33). The faster a specific thiolate carries out a nucleophilic attack to the TNB-conjugated Cys residue (i.e. higher rate constant k), the higher is the likelihood of these two Cys residues to engage in thiol-disulfide exchange reactions (Fig. 4A). Purified single Cys mutant variants of CcmH and apocyt c1 were reacted with DTNB, and their protein–TNB adducts were isolated (see under “Experimental procedures”). Because CcmG is a bona fide thioredoxin (17, 22), we surmised that it would preferentially initiate a nucleophilic attack to an existing disulfide bond in oxidized CcmH or apocyt c1, or a mixed disulfide between them, hence its TNB adducts were not prepared. The rates of reduction of CcmH–TNB and apocyt c1–TNB adducts (CcmHCys-42–TNB, CcmHCys-45–TNB, apocyt c1Cys-34–TNB, and apocyt c1Cys-37–TNB) by the single Cys derivatives of CcmG (CcmGCys-75 and CcmGCys-78), CcmH (CcmHCys-42 and CcmHCys-45), and apocyt c1 (apocyt c1Cys-34 and apocyt c1Cys-37) were measured under pseudo-first order kinetics (i.e. excess of reducing partner versus the protein–TNB adduct), and their corresponding bimolecular rate constants were determined (Table 2). As an example, a set of data showing the release of TNB2− ions (increase in A412 nm) when 1 μm CcmHCys-45-TNB reacted with different amounts (up to 30 μm) of reduced CcmGCys-78 are shown in Fig. 4B. In a control experiment when CcmGCys-78 treated with iodoacetamide (IOA) was used, no signal increase at A412 nm was observed. For each concentration of reducing protein used (e.g. CcmGCys-78), the corresponding kobs value was determined (see under “Experimental procedures”). Plotting these kobs values against the concentrations of the reducing partner (e.g. CcmGCys-78) yielded the bimolecular rate constant (k, m−1 s−1) of the thiol-disulfide exchange reaction for this Cys pair (Fig. 4C, e.g. slope of the top line). In similar ways, the k values for all Cys pairs between CcmG, apocyt c1, and CcmH were determined (Table 2). We inferred from these k values the likelihood of occurrence of the corresponding thiol-disulfide exchange reactions as follows.
Figure 4.

Thiol–disulfide exchange reactions between CcmG, CcmH, and apocyt c1. A, schematic representation of the DTNB-based thiol–disulfide exchange assay. The thiolate of a fully reduced single Cys mutant derivative of CcmG (e.g. CcmGCys-78) initiates a nucleophilic attack on the mixed disulfide of purified CcmH–TNB adduct (e.g. CcmHCys-45–TNB), releasing TNB2− ions. B, TNB2− ions release kinetics during the reaction between CcmGCys-78 and CcmHCys-45–TNB. As an illustrative example, the data that are obtained using 1 μm CcmHCys-45–TNB and different concentrations of reduced CcmGCys-78 (1–30 μm) in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA buffer, are shown. During these reactions, the increase in A412 nm due to the release of the TNB2− ions was monitored in function of time. Under pseudo-first order kinetics conditions, the time-dependent absorbance changes follow single exponential curves. In each case, the initial rates were converted to kobs using 1 μm protein–TNB adduct (e.g. CcmHCys-45-TNB) and the absorption coefficient at 412 nm of TNB2− or the logarithmic linear plot to determine the rate. Reduced and IOA-alkylated CcmGCys-78, which is incapable of resolving the CcmHCys-45-TNB mixed disulfide, was used as a control. Similar assays were repeated with all selected Cys pairs between CcmG, CcmH, and apocyt c1, as appropriate. C, bimolecular rate constants for the thiol–disulfide exchange reactions. kobs values obtained above were plotted in function of the concentration of reduced protein (e.g. CcmGCys-78). Selected Cys pairs with high k values for TNB2− ion release are shown. CcmGCys-78 × CcmHCys-45-TNB (□), CcmGCys-75 × apocyt c1Cys-34-TNB (○), apocyt c1Cys-34 × CcmHCys-45-TNB (×), and CcmHCys-45× apocyt c1Cys-34-TNB (▿) are shown. In each case, the data points are average of at least two assays, and the linear curve is the best fit to the data points. The slope of this line represents the bimolecular rate constant (k) of the thiol–disulfide exchange reactions between the indicated Cys residues. Cumulative data obtained with all tested Cys pairs between CcmG, CcmH, and apocyt c1 are presented in Table 2.
Table 2.
Bimolecular rate constants (k) of thiol-disulfide exchange reactions between apocyt c1, CcmG, and CcmH
| apocyt c1Cys-34-TNB | apocyt c1Cys-37-TNB | CcmH Cys-42-TNB | CcmH Cys-45-TNB | |
|---|---|---|---|---|
| CcmGCys-75 | 6.7a | 2.7 | 0.15 | 18.0 |
| CcmGCys-78 | 4.4 | 1.7 | 0.22 | 23.0 |
| CcmHCys-42 | 2.2 | 1.1 | NDb | ND |
| CcmHCys-45 | 2.9 | 1.8 | ND | ND |
| apocyt c1Cys-34 | ND | ND | 0.50 | 4.3 |
| apocyt c1Cys-37 | ND | ND | 0.67 | 2.3 |
a The reduced single Cys mutant derivatives of CcmG, CcmH, or apocyt c1 were reacted with single Cys mutant derivatives of apocyt c1–TNB and CcmH–TNB adducts, and their bimolecular reaction rate constants k (102 m−1 s−1) were determined as described under “Experimental procedures.”
b ND means not determined.
First, both CcmGCys-75 and CcmGCys-78 could readily carry out a nucleophilic attack to either apocyt c1Cys-34-TNB (k of 6.7 × 102 and 4.4 × 102 m−1 s−1, respectively) or apocyt c1Cys-37-TNB (k of 2.7 × 102 and 1.7 × 102 m−1 s−1, respectively) (Table 2). Thus, reduced CcmG can clearly reduce a disulfide bond at the HBS of apocyt c1, and higher k values are seen with apocyt c1Cys-34–TNB adduct. Second, both CcmGCys-75 and CcmGCys-78 could attack either CcmHCys-42-TNB (k of 0.15 × 102 and 0.22 × 102 m−1 s−1, respectively) or CcmHCys-45-TNB (k of 18 × 102 and 23 × 102 m−1 s−1, respectively) (Table 2). However, much higher k values were observed with the latter TNB adduct, suggesting that Cys-42 of CcmH is less reactive and that a mixed disulfide involving CcmHCys-45 is more likely to be reduced by CcmGCys-75 or CcmGCys-78. Although the observed k values favor a nucleophilic attack initiated by the C-terminal Cys residue Cys-78, the canonical thioredoxin fold of CcmG suggests that its N-terminal Cys-75 might be the attacking residue (see under “Discussion,). Third, apocyt c1Cys-34 and apocyt c1Cys-37 could attack either CcmHCys-42-TNB (k of 0.50 × 102 and 0.67 × 102 m−1 s−1, respectively) or CcmHCys-45-TNB (k of 4.3 × 102 and 2.3 × 102 m−1 s−1, respectively) (Table 2). Again, higher k values were observed with the latter TNB adduct, consistent with Cys-42 of CcmH being less reactive (compared with Cys-45), and Cys-34 of apocyt c1 showing a higher reactivity (compared with Cys-37) toward Cys-45 of CcmH. Fourth, CcmHCys-42 or CcmHCys-45 could also reduce apocyt c1Cys-34-TNB (k of 2.2 × 102 and 2.9 × 102 m−1 s −1, respectively) or apocyt c1Cys-37-TNB (k of 1.1 × 102 and 1.8 × 102 m−1 s−1, respectively), indicating that Cys-45 of CcmH is more reactive than Cys-42 toward Cys-34 of apocyt c1 (Table 2). Finally, both Cys-34 and Cys-37 of apocyt c1 could be attacked by either CcmG or CcmH, but the k values were always higher for Cys-34 than Cys-37 of apocyt c1. We therefore concluded that among the tested pairs of Cys residues, the most favorable reactions would include Cys-75 or Cys-78 of CcmG reducing either a mixed disulfide involving Cys-45 of CcmH or a disulfide bond between Cys-34 and Cys-37 of apocyt c. Regarding CcmH and apocyt c1 interactions, Cys-45 (rather than Cys-42) of CcmH was more likely to react with Cys-34 (rather than Cys-37) of apocyt c1 to yield a mixed disulfide bond.
Formation of a mixed disulfide in vitro between CcmG and CcmH
To further substantiate the DTNB-based assays, we also measured the Cys reactivity of the single mutants by testing the formation in vitro of mixed disulfide bonds between the proteins examined. Based on the data presented in Table 2, we chose to react reduced CcmGCys-75 with CcmHCys-45-TNB (in a molar ratio of 2:1) to obtain a mixed disulfide in vitro under the DTNB assay conditions used (see under “Experimental procedures”). After 16 h of incubation at room temperature, the reaction mixture was analyzed using SDS-PAGE under non-reducing conditions. Fig. 5, lane 3, shows a faint band of ∼33 kDa, containing both CcmG and CcmH, as identified by nLC-MS/MS spectrometry (Fig. 5, right panel). Similar data confirmed that the band of ∼26 kDa corresponded to the CcmHCys-45 dimer and the lower bands to CcmG and CcmH monomers (Fig. 5 and supplemental Table S1). Overall data clearly showed the formation of a CcmGCys-75–Cys-45CcmH mixed disulfide upon incubation of these two proteins. However, compared with the amounts of reduced CcmGCys-75 (17 kDa, Fig. 5, lane 2) and CcmHCys-45-TNB (13.5 kDa, Fig. 5, lane 1) used, the yield of this reaction remained very low despite our many attempts using different buffers and incubation times. A possibility is that the pKa of CcmGCys-75 is unexpectedly high and that at pH 7.5 only a small amount of thiolate is produced to carry out efficiently a nucleophilic attack to the CcmHCys-45-TNB mixed disulfide. Another possibility is that the reactions are not being quenched at low pH, reverse reactions might have occurred under oxic conditions. Recent studies have indicated that the chemistry underlying DTNB-based reactions is complex (34). Other studies have also reported similar very low yields of mixed disulfides formed between other thiol-disulfide oxidoreductases (32, 35–37).
Figure 5.

Formation of a mixed disulfide between CcmGCys-75 and CcmHCys-45. 30 μm CcmGCys-75 and 15 μm CcmHCys-45 were incubated at room temperature for 16 h under the DTNB assay conditions (see “Experimental procedures”), and the samples were analyzed by SDS-PAGE. A small amount of CcmGCys-75-CcmHCys-45 species forming a mixed disulfide bond (∼ 33 kDa) together with CcmHCys-45 dimers (∼26 kDa) were detected (lane 3). Lanes 1 and 2 correspond to the same amounts of CcmHCys-45-TNB and CcmGCys-75 used as controls. NanoLC-MS/MS spectrometry was used to confirm the identity of the protein bands corresponding to CcmHCys-45 monomer (∼13.5 kDa), CcmGCys-75 monomer (∼17.5 kDa), and the mixed disulfide-containing band at ∼33 kDa, which contained both CcmGCys-75 and CcmHCys-45, as indicated by one related specific peptide on the right. The list of all peptides identified in all cases by MS analyses is presented in supplemental Table S1. The ∼26-kDa band was also identified as CcmHCys-45 dimer (supplemental Table S1). nd indicates not found, and * indicates oxidized methionine or carbamidomethylated cysteine residues.
Determination of the redox states of CcmG and CcmH in actively growing cells
Information about the steady-state redox states of CcmG and CcmH in actively growing wild-type cells is crucial for correctly attributing specific roles to the above-identified Cys residues during the thioreduction of apocyts c in vivo. For this purpose, we used the thiol-alkylating reagent 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) that reacts covalently with free thiolates in proteins and allows identification of their redox states. Proteins containing AMS-modified Cys residues exhibit migration shifts toward higher molecular weights during SDS-PAGE. Upon treatment of cell extracts with AMS, CcmG was shifted to a higher molecular weight (Fig. 6A, lane 2) compared with untreated samples (Fig. 6A, lane 1) when subjected to SDS-PAGE. This molecular weight shift was identical to that seen when cell cultures were reduced with DTT prior to AMS modification (Fig. 6A, lane 4). Thus, CcmG was mostly in a reduced state. On the contrary, CcmH showed no shift with or without AMS treatment, indicating that it was mainly in oxidized state (Fig. 6B, lanes 1 and 2). A molecular weight increase due to the modification of CcmH thiolates by AMS was seen only when cell cultures were reduced with DTT prior to AMS addition (Fig. 6B, lanes 3 and 4). We therefore concluded that, in actively growing R. capsulatus cells, CcmG and CcmH were mainly in the reduced and oxidized states, respectively, in agreement with earlier reports (13).
Figure 6.
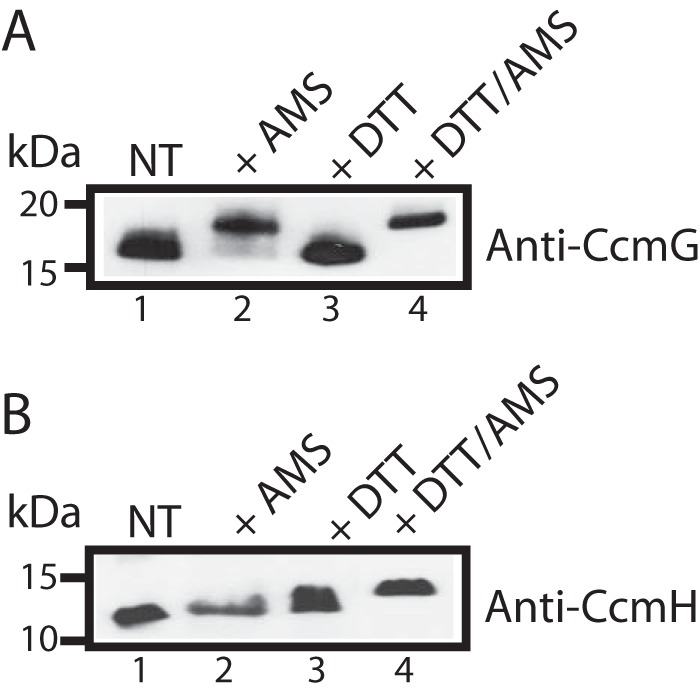
In vivo redox states of CcmG and CcmH in actively growing cells. Immunoblot analysis of cell extracts from appropriately complemented R. capsulatus mutants MD11/pCS1566 and MD14/pST6 (Table 1) using anti-CcmG- and anti-CcmH-specific polyclonal antibodies, respectively. Aliquots from cell cultures were TCA-precipitated with or without prior DTT reduction. Cell pellets were solubilized in buffer containing SDS with or without AMS. AMS alkylates free Cys thiolates, adding to the total protein molecular mass of ∼0.5 kDa per AMS-modified thiolate. Reduced and AMS-modified proteins show higher molecular weights in SDS-PAGE. The data show that in actively growing cells, CcmG is reduced (A) and CcmH is oxidized (B). NT refers to untreated cell cultures solubilized in buffer without AMS; + AMS indicates untreated cells solubilized in buffer containing AMS; + DTT indicates DTT-reduced cell cultures solubilized in buffer without AMS, and +DDT/AMS indicates cells first reduced with DTT and then reacted with AMS.
Discussion
Under oxic or anoxic growth conditions, and despite the presence of DsbA, which is an efficient periplasmic thiol oxidant, cells must keep the thiol groups of the apocyts c HBS Cys residues in the reduced state for heme ligation to occur (9). CcmG and CcmH are Ccm-dedicated thiol-disulfide oxidoreductases that carry out this process, but the mechanisms governing these reactions are not well defined (Fig. 1) (12, 13, 19, 25, 27). In this work, we addressed this issue systematically, using wild-type as well as single or double Cys mutant derivatives of CcmG (CcmGWT, CcmGCys-75, CcmGCys-78, and CcmG*), CcmH (CcmHWT, CcmHCys-42, CcmHCys-45, and CcmH*), and apocyt c1 (apocyt c1WT, apocyt c1Cys-34, apocyt c1Cys-37, and apocyt c1*). First, we examined the protein–protein interactions between these proteins, and then defined the catalytic abilities of their Cys residues to engage in thiol-disulfide exchange reactions.
Protein–protein interaction studies indicated that CcmG and CcmH interacted strongly with each other and that these interactions did not require their Cys residues. Moreover, CcmGWT also interacted, but weakly with the class I apocyts c (e.g. approximately micromolar KD for apocyt c1WT), and both the presence of the HBS Cys residues of apocyts c or the chaperone CcmI further enhanced these interactions. Finally, using dispersed membranes, we showed that apocyt c2WT interacted with the heme ligation components (CcmF–CcmH–CcmI), and altogether they yielded a CcmFGHI–apocyt c complex (Fig. 3F). Our earlier studies established that CcmI is an apocyt c chaperone that binds tightly the C terminus of class I apocyts (29, 30, 38) and that CcmG has oxidoreductase and chaperone (holdase) activities to assist the apocyts c and enhance cyt c maturation (14). Similar cooperation of CcmG with the heme ligation complex was reported with an engineered E. coli CcmFGH complex (equivalent of R. capsulatus CcmFGHI) that can carry out Ccm in the absence of CcmABCDE (39). Furthermore, genetic studies showed that the absence of CcmI can be bypassed by overexpression of CcmFH and CcmG and that CcmG is functionally related with CcmI (40). These data are consistent with the occurrence of a CcmFGHI–apocyt c complex as seen in this study (Fig. 3). They also support our recent proposal that the entire Ccm machinery might form an even larger supercomplex also including the CcmABCDE complex (2).
Next, to probe the catalytic abilities of the Cys residues of CcmG, CcmH, and apocyt c1, the bimolecular rate constants (k) of thiol-disulfide exchange reactions between a protein–TNB adduct and a thiolate of a partner protein were determined (Table 2). The k values obtained in this work are lower than those observed with the thiol-disulfide oxidoreductases of the thioredoxin family (106 to 107 m−1s−1) (28). However, these rates are higher than those reported for the reactions involving CcmGWT and single Cys mutant derivatives of P. aeruginosa CcmH–TNB adducts (23 and 4.1 m−1 s−1) (12). We note that P. aeruginosa CcmG and CcmH have similar (−215 and −213 mV, respectively) redox midpoint potential (Em) values, whereas a larger difference exists between those of R. capsulatus CcmG (−300 mV) and CcmH (−210 mV) (12, 19, 24).
Furthermore, by using the single Cys mutants of both CcmG and CcmH, we could visualize the occurrence of a mixed disulfide intermediate in vitro, and AMS labeling in vivo indicated that CcmG is mainly reduced, in agreement with its faster rate of reduction by CcdA (105 m−1 s−1) (41) than its oxidation during apocyt c thioreduction (Table 2). Similarly, CcmH was found mainly oxidized in actively growing R. capsulatus cells, which would be expected if it is indeed oxidized by DsbA, which exhibits high rates of oxidation for its substrates (∼ 105 m−1 s−1) (42).
The high k values observed with either CcmGCys-75 or CcmGCys-78 and apocyt c1Cys-34 (Table 2) suggested that CcmG can reduce directly and efficiently the disulfide bond at the HBS of apocyt c1 (its Cys-34 residue being the site of nucleophilic attack). During the thiol–disulfide exchange reactions mediated by thioredoxins, the target disulfide is first attacked by an active thiolate (N-terminal Cys), forming a mixed disulfide bond. Subsequent attack of this disulfide bond by the remaining thiolate (C-terminal Cys) of thioredoxin reduces the target protein, leaving thioredoxin oxidized. In members of this superfamily the N-terminal Cys residue is more solvent-exposed (i.e. acidic) than the C-terminal Cys residue. This residue is kept buried in a hydrophobic environment until the occurrence of conformational changes induced by the formation of the mixed disulfide bond (43, 44). Like a bona fide thioredoxin, it is likely that the Cys-75 (rather than Cys-78) of CcmG carries out the nucleophilic attack on Cys-34 of apocyt c1. Earlier studies attributed reduction of the apocyt c HBS disulfide bond to CcmH (13, 26, 27), and we also observed that CcmHCys-45 could carry out this task (k of 2.9 × 102 m−1 s−1) but slower than CcmG (k of 6.7 × 102 m−1 s−1) (Table 2). Hence, in wild-type cells (i.e. in the presence of DsbA), the N-terminal Cys-75 thiolate from reduced CcmG rapidly attacks oxidized apocyt c1WT HBS via Cys-34, followed by resolution of the mixed disulfide thus formed by its C-terminal Cys-78, yielding oxidized CcmG that is subsequently reduced by CcdA (Fig. 7, left side).
Figure 7.

Thioreduction of the disulfide bond at the HBS of apocyt c and stereo-specific heme ligation during Ccm. The comprehensive model depicted here uses apocyt c1 as an example, but the process is considered to be similar with other apocyts c as well. In wild-type cells, after translocation to the periplasm, apocyt c1 is oxidized by the thiol-disulfide oxidoreductase DsbA, resulting in the formation of a disulfide bond at its HBS (34CXXCH38) (step 1). Ccm-specific thioredoxin CcmG carries a nucleophilic attack via its N-terminal, solvent exposed Cys-75 to the N-terminal Cys-34 at the HBS of apocyt c1 (steps 2 and 3), forming CcmGCys-75–Cys-34apocyt c1 mixed disulfide bond between them (step 3). The C-terminal Cys-78 of CcmG resolves this mixed disulfide bond, resulting in reduced apocyt c1 and oxidized CcmG (step 4), which is then re-reduced by CcdA (via electrons coming from cytoplasmic thioredoxins, not shown) (step 5). Reduced apocyt c1 attacks oxidized CcmH with its N-terminal Cys-34 (step 6), forming an intermediate in which apocyt c1 Cys-34 forms a mixed disulfide with the C-terminal highly reactive Cys-45 of CcmH (step 7), defining the stereo-specificity of heme ligation. Once the CcmHCys-45–Cys-34apocyt c1 intermediate is formed, holoCcmE carrying heme attached via its vinyl-2 is assumed to interact with CcmH and apocyt c1 (step 8), leading to the formation of the first thioether bond between the available Cys-37 of apocyt c1 and vinyl-4 of heme. The CcmH–apocyt c1–heme–CcmE intermediate thus formed is resolved efficiently via Cys-75 of CcmG, attacking this mixed disulfide (step 9), to form a mixed disulfide with Cys-45 of CcmH and releasing apocyt c1 (step 10). The Cys-34 of apocyt c1 is then assumed to form the second thioether bond with vinyl-2 of heme and released from CcmE (possibly involving CcmF, not shown). The CcmGCys-75–Cys-45CcmH mixed disulfide bond is resolved via Cys-78 of CcmG, rendering CcmH reduced and CcmG oxidized (step 11). As before (steps 2–4), CcdA recycles oxidized CcmG (step 12), and CcmH is oxidized via DsbA or another periplasmic oxidant(s) (step 13, heavy dashed line). Conceivably, the CcmHCys-45–Cys-34apocyt c1 intermediate may also be formed by an alternative route via a nucleophilic attack of the HBS disulfide bond by Cys-45 of reduced CcmH (steps 6a–7a), and be resolved via Cys-42 of CcmH, yielding oxidized CcmH (steps 9a–10a). However these steps (indicated by lightly dotted arrows) are less favorable as CcmH is mainly oxidized in vivo, and Cys-42 of CcmH is not very reactive. The thiol–disulfide exchange rates k (102 m−1 s−1, small red arrows) determined in this work are used to develop this scheme, where the resolution of intra-molecular disulfide bonds is indicated in small brown or gray arrows.
Next, we observed that both Cys-75 and Cys-78 of CcmG could rapidly resolve a mixed disulfide between CcmHCys-45 and apocyt c1Cys-34 (Table 2), in agreement with another earlier proposal that a mixed disulfide bond involving CcmH might be the substrate of CcmG (12). The k values observed with CcmGCys-78 are slightly higher than those obtained with CcmGCys-75 in the attack of CcmH Cys-45 possibly due to conformation changes in CcmG inflicted by single Cys mutations. The 3D structure of CcmH shows that its Cys-42 residue is buried inside the protein and is less accessible, which is consistent with its lower reactivity seen here (12, 19). Similarly, the observed higher reactivity for the thiol-disulfide exchange reactions of apocyt c1Cys-34 compared with apocyt c1Cys-37 suggests that Cys-34 of apocyt c1 might be more solvent-exposed. However no structural information is available.
The overall findings of this study, combined with published data by us and other groups (12, 13, 19, 24, 25, 27), led us to a comprehensive model for apocyt c thioreduction and stereo-specific heme ligation during Ccm (Fig. 7, right side). Accordingly, in wild-type cells (i.e. in the presence of DsbA, reduced CcmG, and oxidized CcmH), the Cys-34 residue of apocyt c1 (previously reduced by CcmG (Fig. 7, steps 2–4)) would attack Cys-45 of oxidized CcmH yielding CcmHCys-45–Cys-34 apocyt c1 mixed disulfide bond (Fig. 7, steps 6 and 7, k of 4.3 × 102 m−1 s−1), leaving Cys-37 of apocyt c1 available to form a thioether bond with the vinyl-4 of heme, which is still attached to CcmE (Fig. 7, step 8) (2, 45). This mixed disulfide bond could then be resolved rapidly and efficiently by reduced CcmG (Fig. 7, steps 9 and 10, k of 18 × 102 m−1 s−1), releasing CcmE–heme–apocyt c1 (Fig. 7, step 10) and forming the CcmGCys-75–Cys-45CcmH mixed disulfide bond to be resolved intra-molecularly via Cys-78 of CcmG (Fig. 7, step 11), yielding oxidized CcmG and reduced CcmH. Afterward, these latter components would be recycled to their steady-state forms (i.e. reduced CcmG and oxidized CcmH) via CcdA (Fig. 7, step 12) and possibly DsbA (or an oxidant like O2) (Fig. 7, step 13), respectively. At an unknown stage, upon resolution of the CcmE–heme–apocyt c intermediate, whose existence has already been described (46), the second thioether bond between Cys-34 of apocyt c1 and heme vinyl-2 would be formed completing heme ligation. Indeed, efficient formation of CcmHCys-45–Cys-34apocyt c1 (instead of CcmHCys-45–Cys-37apocyt c1) mixed disulfide would ensure stereo-specific ligation of heme to apocyt c, as postulated earlier (2, 45), leaving the Cys-37 of apocyt c1 free to form a thioether linkage with the free vinyl-4 of heme held by CcmE (2, 45).
Conceivably, the mixed disulfide containing CcmHCys-45–Cys-34apocyt c1 intermediate described above (Fig. 7, step 7) could also be formed at a slower rate by an alternative route, via reduction of oxidized apocyt c with reduced CcmH (if available) (Fig. 7, steps 6a–7a, k of 2.9 × 102 m−1 s−1). However, if under steady-state growth conditions DsbA keeps both CcmH and apocyt c oxidized then in the absence of reduced CcmG, this alternative pathway would be unlikely to occur. Similarly, in the absence of reduced CcmG, the mixed disulfide containing CcmHCys-45–Cys-34apocyt c1 intermediate (if formed) could also be resolved intra-molecularly (Fig. 7, steps 9a–10a) via Cys-42 of CcmH. However, this alternative would occur at a much lower rate compared with CcmG, as Cys-42 thiolate is not very reactive (reduced CcmHCys-42 reacts with TNB-apocyt c1 at a k of 2.2 × 102 m−1 s−1).
Furthermore, the model depicted in Fig. 7 also accounts for low amounts of cyt c observed in several Ccm-related R. capsulatus mutants (14, 22). In mutants lacking DsbA, reduced apocyt c would be rapidly degraded, and moreover, CcmH would remain reduced until its oxidation by non-Ccm-dedicated components (e.g. O2), creating a rate-limiting step and reducing Ccm efficiency (∼ 50%) (14). Similarly, in the absence of both DsbA and CcdA or DsbA, CcdA, and CcmG, a severe decrease in cyt c amounts (to ∼10% of wild-type levels) would occur due to cumulative effects of the absence of DsbA and CcmG, which are required for stability of apocyt c and efficient resolution of CcmHCys-45–Cys-34apocyt c1 mixed disulfide, respectively.
In summary, this study provided a comprehensive description of thioreduction of apocyt c HBS disulfide bond, which is essential for Ccm to occur. Importantly, it identified the probable catalytic Cys residues of the Ccm components involved in these events. Future investigations will establish the occurrence of the reactions in vivo and further substantiate the validity of this model by isolation and characterization of appropriate intermediates, including CcmHCys-45–Cys-34apocyt c1Cys-37–CcmE, which plays a pivotal role during Ccm.
Experimental procedures
Bacterial strains, growth conditions, plasmid, and mutant constructions
E. coli and R. capsulatus strains used in this work are defined in Table 1. E. coli strains were grown aerobically at 37 °C, shaken at 200 rpm in Luria-Bertani (LB) broth medium, and supplemented with ampicillin (100 μg/ml) or chloramphenicol (50 μg/ml), as needed. Cultures were induced with 1 mm isopropyl β-d-1-thiogalactopyranoside as described elsewhere (29). R. capsulatus strains were grown chemoheterotrophically in the dark (i.e. by aerobic respiration) at 35 °C, shaking at 150 rpm on enriched medium (MPYE) supplemented with tetracycline (2.5 μg/ml), as appropriate.
R. capsulatus apocyt c1 mutants were produced using the QuikChange site-directed mutagenesis kit and the plasmid pMAM1 as a template. pMAM1 encodes a variant of apocyt c1 missing its last C-terminal 39 amino acids that constitute the TM helix and lacking the non-heme ligating Cys-144 and Cys-167 that form a structural disulfide bridge (Strep-apocyt c1WT) (30). The plasmids pMAM1C37S, pMAM1C34S, and pMAM1C34SC37S obtained by site-directed mutagenesis produced the single and double (indicated by *) Cys mutant derivatives Strep-apocyt c1Cys-34, Strep-apocyt c1Cys-37, and Strep-apocyt c1* of Strep-apocyt c1WT, respectively (Table 1).
A soluble variant of CcmH was constructed by digesting the plasmid pCS1604 with NdeI and BamHI, isolating the insert carrying a truncated CcmH and ligating it into the plasmid pFLAG-1 using the same sites to obtain the plasmid pAV10 (Table 1). This construct encodes an N-terminal FLAG-fused CcmH that lacks both its signal sequence and its 46 N-terminal amino acid residues acting as its membrane anchor (FLAG-CcmHWT). Plasmids pAV10C45S, pAV10C42S, and pAV10C42SC45S, producing the single FLAG-CcmHCys-42 and FLAG-CcmHCys-45 and the double FLAG-CcmH* Cys mutant derivatives of FLAG-CcmHWT, respectively, were obtained by site-directed mutagenesis as above, with plasmid pAV10 serving as a template (Table 1).
A soluble variant of CcmG was prepared by digesting with EcoRI and HindIII the plasmid pQE60-helX, isolating the DNA fragment corresponding to the C-terminal His6-tagged CcmG without its TM helix, and ligating it into pBSK using the same restriction sites to yield the plasmid pCS1555 producing His6-CcmGWT (Table 1). Plasmids pCS1553, pCS1552, and pCS1554 producing the single His6-CcmGCys-75 and His6-CcmGCys-78 and double His6-CcmG* Cys mutant derivatives of His6-CcmGWT, respectively, were obtained by site-directed mutagenesis as above, using plasmid pQE60-helX as a template. Accordingly, plasmids pCS1556, pCS1557, and pCS1558 were constructed like pCS1555 described above, by cloning into the EcoRI–HindIII sites of pBSK the His6-CcmG mutant derivatives carried by the plasmids pCS1552, pCS1553, and pCS1554, respectively (Table 1). All constructs were confirmed by DNA sequencing. The nomenclature used for the plasmids refers to the Cys residues that were mutated to Ser. Mutant proteins produced by these plasmids are referred to by the Cys residue that remained after mutagenesis (e.g. plasmid pAV10C45S specifies that Cys-45 of CcmH was mutated to Ser, thus it produces the FLAG-CcmHCys-42 mutant protein).
Protein purification and production of anti-CcmG polyclonal antibodies
E. coli cells overproducing each desired protein were resuspended in 25 mm Tris-HCl, pH 7.5, 150 mm NaCl buffer containing protease inhibitors mixture (Pierce), and disrupted using a French pressure cell. Crude cell extracts were centrifuged at 138,000 × g for 90 min at 4 °C. Purification of Strep-apocyt c1WT, Strep-apocyt c2WT, and their derivatives was performed using Strep-Tactin®–Sepharose® resin (IBA, Inc.) as done earlier (29). Purification of His6-CcmGWT and its derivatives was carried out using a nickel-Sepharose high performance column (GE Healthcare) equilibrated with 25 mm Tris-HCl, pH 7.5, 500 mm NaCl, 10 mm imidazole buffer. After extensive washing with the same buffer, elution was done at 100 mm imidazole. Purification of His10-CcmI used similar buffer conditions, except that the buffers contained 0.01% DDM, and elution was done at 500 mm imidazole, as described previously (29). Purification of FLAG-CcmHWT and its mutant derivatives used an anti-FLAG (DYKDDDDK) affinity gel (Biotool, Inc.) and 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.2 mm 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) buffer according to the manufacturer's instructions. Elution was carried out with 100 mm glycine, pH 2.75, 0.2 mm AEBSF buffer, and eluents were collected into tubes containing 1 m Tris-HCl, pH 8.0, buffer for immediate neutralization. All purified proteins were concentrated using Amicon-YM 3 (Millipore, Inc), desalted via a PD-10 column (GE Healthcare) equilibrated with 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA buffer, and kept at −20 °C until further use.
Detergent-dispersed membrane proteins of R. capsulatus strains MT-SRP1.r1 or MT-SRP1.r1/pNJ2 (Table 1) were prepared as described elsewhere (29). Solubilized membranes were loaded onto a Q-Sepharose ionic exchange resin (GE Healthcare) equilibrated with 25 mm Tris-HCl, pH 8.0, 50 mm NaCl, 0.1 mm aminocaproic acid, 1 mm EDTA, 1 mm PMSF, 0.02% DDM buffer. Elution was done with a NaCl gradient, and the CcmFGH proteins were co-eluted with 150 mm NaCl, as confirmed by immunoblots using specific anti-CcmF, anti-CcmH, and anti-CcmG polyclonal antibodies.
Purified R. capsulatus His6-CcmGWT (∼3 mg) was subjected to preparative SDS-PAGE, electro-eluted from the gel matrix, and used as an antigen for production of rabbit polyclonal antibodies, which was performed by Thermo Fisher Scientific.
Protein–protein interaction studies using co-purification assays
Protein–protein interactions between the double Cys mutants His6-CcmG*, FLAG-CcmH*, and Strep-apocyt c1* were determined in vitro using co-purification assays, as described below. Equimolar amounts (∼10 μm) of purified His6-CcmG* were mixed with FLAG-CcmH* or Strep-apocyt c1* in 50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10 mm imidazole buffer (final volume 60 μl) and incubated for 60 min at room temperature, with gentle shaking. Then, 10 μl of nickel-Sepharose HP resin previously equilibrated with the assay buffer was added, and incubation was continued for another 60 min with gentle shaking. At the end of incubation, the assay mixture was centrifuged at 8000 × g for 1 min, and the supernatant (i.e. flow-through (FT)) was removed. The resin was washed twice with 100 μl (10 resin volumes) of buffer supplemented with 80 mm imidazole, and the supernatants (i.e. washes (W)) were collected by centrifugation. His6-CcmG* and its partners were eluted with 50 μl of elution buffer containing 250 mm imidazole (i.e. elution (E)). FT and E fractions were concentrated and analyzed by SDS-PAGE. Protein–protein interaction assays using purified His6-CcmGWT, Strep-apocyt c2WT, and His10-CcmI were done, as described previously (29), using equimolar amounts of CcmG and apocyt c2 (1.5 μm) and substoichiometric amounts of CcmI (0.5 μm) in a final volume of 400 μl. Co-purification of Strep-apocyt c2WT with R. capsulatus-solubilized membranes was done as described previously (29). Co-purification of native CcmG from CcmFHG-enriched fractions (100 μg) was done using His10-CcmI (10 μg) and nickel-Sepharose resin as described elsewhere (29).
Protein–protein interactions determined by biolayer interferometry
Binding kinetics of CcmG and apocyt c1 were monitored quantitatively in real time by biolayer interferometry using an Octet RED96 instrument (ForteBio, Inc.) as described elsewhere (30). Briefly, streptavidin (SA)-coated biosensors were loaded with biotinylated Strep-apocyt c1WT (400 nm) in 50 mm Tris, pH 8.0, 100 mm NaCl, 0.05% DDM, 1% BSA buffer at 30 °C and at 1000 rpm. After washing, the SA sensors were incubated with increasing concentrations of His6-CcmGWT (from 1.28 to 20.4 μm) (association step). Subsequent washing of the biosensors with the assay buffer released CcmGWT from the immobilized apocyt c1WT (dissociation step). SA sensors without immobilized apocyt c1WT were incubated with CcmGWT as a control for unspecific binding of this protein to the sensors. In addition, an assay lacking CcmGWT was used as a negative control to confirm that the monitored shifts were due to the formation of CcmGWT–apocyt c1WT complexes. The assays were done in duplicate, and the kon and koff rates of binding measured and the KD values were determined by fitting the experimental data to a 1:1 homogeneous kinetic model describing bimolecular interactions, as done earlier (30).
Preparation of TNB adducts of purified proteins
Single Cys mutant derivatives CcmHCys-42, CcmHCys-45, apocyt c1Cys-34, and apocyt c1Cys-37 (50–100 μm) were reduced with large excess of DTT (25 mm) at room temperature for 60 min. Excess DTT was removed using a PD-10 desalting column and 100 mm potassium phosphate, pH 8.0 buffer. Reduced proteins were incubated with 15 mm DTNB in the dark for 120 min at room temperature, and excess of DTNB was also removed by desalting with a PD-10 column, using 50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA buffer. The yield of the protein–TNB adduct formed was determined by reducing a small aliquot with excess of DTT for a few minutes and measuring spectroscopically at 412 nm the amount of 2-nitro-5-thiobenzoic acid (TNB2−) ions released. The concentrations of the protein–TNB adducts were calculated using an extinction coefficient ϵ412 of 14,500 m−1 cm−1 for the TNB2− ions. For all protein–TNB adducts, the ratio of released TNB2− ion per protein was between 0.90 and 1.03, as expected for a single thiol group, confirming their full modification. Reduced forms of the CcmGCys-75, CcmGCys-78, CcmHCys-42, and CcmHCys-45 single Cys mutant derivatives were incubated immediately before use with an excess of DTT for 30 min at room temperature, and an excess of DTT was removed using a PD-10 column equilibrated with the assay buffer. In the case of the apocyt c1Cys-34 and apocyt c1Cys-37 derivatives, both reduction and DTNB conjugation were carried out in an anoxic chamber (COY Lab Products, Inc) to minimize their high tendency to form inter-molecular disulfide bonds in the presence of oxygen. Reduced samples of apocyt c1Cys-34 and apocyt c1Cys-37 were kept under anoxic conditions in sealed glass vials until use. An aliquot of each reduced sample was also incubated with 10 mm IOA for 30 min in the dark to control the extent of reduction and used as a negative control for the TNB2− release assays, to ensure that no reductant other than the thiol of the chosen protein was present during the assay. As needed, full reduction of the samples was confirmed by determining the amount of free thiol groups available just prior to the DTNB assays, using the Ellman's reagent protocol (47). All protein samples were concentrated using Amicon YM10 after desalting, and their final concentrations were determined using the BCA assay (Sigma).
Determination of bimolecular rate constants of thiol-disulfide exchange reactions between a TNB–protein adduct and another reduced protein with a single Cys residue
The bimolecular rate constant (k) of thiol-disulfide exchange reaction between a chosen pair of Cys residues from two proteins was determined using the DTNB assay, as described previously (12, 32, 33). Briefly, a given amount (1 μm) of TNB–protein (CcmHCys-42, CcmHCys-45, apocyt c1Cys-34, or apocyt c1Cys-37) adduct was added to a stirring cuvette containing variable concentrations (1–30 μm) of another fully reduced protein (CcmGCys-75, CcmGCys-78, CcmHCys-42, CcmHCys-45, apocyt c1Cys-34, and apocyt c1Cys-37) in 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA buffer. For each case, the time-dependent increase in A412 nm corresponding to release of the TNB2− ions was monitored. The reaction was carried out under pseudo-first order kinetics conditions, where the reduced protein was in excess relative to the TNB–protein adduct. All assays were performed at least in duplicate. For each Cys pair, the initial rate of release of TNB2− ions (product formation) was determined for a range of concentrations of reducing protein (e.g. CcmG78). These rates were converted to kobs values using the initial concentration of the protein–TNB adduct (1 μm CcmHCys-45-TNB in this case) and the absorption coefficient at 412 nm of TNB2−. Similar kobs values were also obtained by exponential fit to the saturation kinetics. The kobs values were then plotted against the concentrations of the reducing protein used (e.g. CcmGCys-78), where the slope of the curve is the bimolecular rate constant k (e.g. 23 × 102 m−1 s−1) of thiol-disulfide exchange reaction between the TNB–protein adduct (e.g. CcmHCys-45-TNB) and a fully reduced partner protein with a single Cys residue (e.g. CcmGCys-78).
Identification of mixed disulfides between CcmG and CcmH and nLC-MS/MS analysis
The stability and efficiency of mixed disulfide bond formation between each Cys pair of two proteins were tested by incubating 15 μm of a protein–TNB adduct with 2-fold (30 μm) excess of fully reduced single Cys mutant derivative of another protein in a final volume of 20 μl in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA buffer at room temperature for 2–16 h. Upon mixing of the two proteins, a slight yellow color was formed, indicating release of the TNB2− ions. At the end of incubation, the reaction was stopped by blocking the remaining free thiols with 5 mm IOA at room temperature in the dark for 15 min. SDS-PAGE loading buffer without any reducing agent was added to the samples, which were then submitted to 15% SDS-PAGE. SDS-polyacrylamide gel bands were excised and subjected to alkylation with iodoacetamide, followed by in-gel trypsin digestion (Promega, sequencing grade modified trypsin) overnight at 37 °C. Peptides eluted from the gel samples were dried and resuspended in 10 μl of 5% acetonitrile in 0.1% formic acid and analyzed with a nanospray LC-MS/MS Thermo LCQ Deca XP+ ion trap mass spectrometer coupled to a Thermo-Dionex LC Packings Ultimate Nano HPLC system controlled by Thermo Xcalibur version 2.0 software. A C18 nanocolumn (Thermo-Dionex, NAN-75-15-03-PM) was used to fractionate peptides, using a 60-min elution gradient (5–75% acetonitrile in 0.1% formic acid). MS/MS data were acquired in data-dependent analysis mode, where the top three precursor ions were trapped and fragmented using dynamic exclusion to maximize the detection of unique peptides. Collected spectra were searched against the R. capsulatus protein database, which included the mutated CcmH and CcmG sequences using Thermo Proteome Discoverer 1.4 software with standard settings.
Determination of the redox states of CcmG and CcmH in actively growing cells
To determine the in vivo redox state of CcmG and CcmH, we used a simple approach that consists of alkylating the free thiols of the Cys residues of the proteins of interest using AMS as described previously (48). Ten-ml cultures of R. capsulatus strains MD14 (lacking CcmH) and MD11 (lacking CcmG), complemented with plasmids pST6 (Strep-CcmHWT) and PCS1555 (His6-CcmGWT) (Table 1), respectively, were grown until reaching early exponential phase (A630 ∼0.3) at 35 °C, 150 rpm in enriched medium. Two 1.8-ml aliquots were collected (tubes A and B) and precipitated on ice with 10% ice-cold trichloroacetic acid (TCA) for 30 min. The remaining culture was reduced by addition of 10 mm DTT for 10 min at 35 °C and shaking at 150 rpm. Two additional aliquots of 1.8 ml were collected (tubes C and D) and precipitated with TCA, as before. After centrifugation for 5 min at 16,000 × g and 4 °C, the four pellets were washed twice with ice-cold acetone, and dried. Dry pellets of the tubes A and C were solubilized in 45 μl of 50 mm Tris-HCl, pH 7.5, 5 mm EDTA, 0.1% SDS, and 2 mm AEBSF buffer, and those of the tubes B and D were solubilized in the same buffer supplemented with 20 mm AMS, for 1 h at 37 °C, with shaking. SDS-PAGE loading buffer was added to the tubes and incubated at 95 °C for 15 min. Protein extracts were resolved in a 18% gel and analyzed by immunoblots using anti-CcmG- and anti-CcmH-specific polyclonal antibodies, as appropriate.
SDS-PAGE and immunoblot analyses
SDS-PAGE was performed using 15 or 18% gels according to Ref. 49. For immunodetection, gel-resolved proteins were electroblotted onto Immobilon-PVDF membranes (Millipore, Inc.) and probed with rabbit polyclonal antibodies specific for R. capsulatus CcmG, CcmF, and CcmH, except for CcmI for which rabbit polyclonal anti-FLAG antibody (Sigma) was used. In all cases, horseradish peroxidase-conjugated anti-rabbit IgG antibodies (GE Healthcare) were used as secondary antibodies, and detection was performed using the SuperSignal West Pico Chemiluminescent Substrate® from Thermo Scientific, Inc.
Author contributions
A. F. V. designed and conducted most of the experiments, analyzed the data, and wrote most of the manuscript. B. K. H. conducted experiments for the determination of the redox state of CcmH and CcmG and participated in data analysis. J. H. made and purified apocyt c1 Cys mutants. S. S. participated in data analysis and the preparation of Fig. 7. C. S. prepared the CcmG constructs and CcmG and CcmH complemented R. capsulatus strains. N. S. and C. K. participated in data analyses. F. D. managed the project, analyzed the data, and wrote the manuscript with A. F. V. All authors read, edited, and approved the final manuscript.
Supplementary Material
Acknowledgment
We thank the Core Facilities of Border Biomedical Research Center (BBRC) at the University of Texas at El Paso for use of equipment.
This work was supported primarily by the Division of Chemical Sciences, Geosciences and Biosciences, Office of Basic Energy Sciences of Department of Energy Grant DOE DE-FG02-91ER20052 (to F. D.) and in part by the National Institutes of Health Grants GM 38237 (to F. D.), which supported the production of mutant proteins, and Grant 5G12MD007592 (to C. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1 and Figs. S1 and S2.
- cyt
- cytochrome
- apocyt
- apocytochrome
- Ccm
- cyt c maturation
- HBS
- heme-binding site
- TM
- transmembrane
- IOA
- iodoacetamide
- DDM
- n-dodecyl β-d-maltoside
- DTNB
- 5,5′-dithiobis-(2-nitrobenzoic acid)
- TNB
- 2-nitro-5-thiobenzoic acid
- AEBSF
- 4-(2-aminoethyl) benzenesulfonyl fluoride
- SA
- streptavidin
- AMS
- 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- nLC-MS/MS
- nanoLC-tandem mass spectrometry
- FT
- flow-through
- W
- wash
- E
- elution.
References
- 1. Bertini I., Cavallaro G., and Rosato A. (2006) Cytochrome c: occurrence and functions. Chem. Rev. 106, 90–115 [DOI] [PubMed] [Google Scholar]
- 2. Verissimo A. F., and Daldal F. (2014) Cytochrome c biogenesis system I: an intricate process catalyzed by a maturase supercomplex? Biochim. Biophys. Acta 1837, 989–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kranz R. G., Richard-Fogal C., Taylor J. S., and Frawley E. R. (2009) Cytochrome c biogenesis: mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol. Mol. Biol. Rev. 73, 510–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mavridou D. A., Ferguson S. J., and Stevens J. M. (2013) Cytochrome c assembly. IUBMB Life 65, 209–216 [DOI] [PubMed] [Google Scholar]
- 5. Denks K., Vogt A., Sachelaru I., Petriman N.-A., Kudva R., and Koch H.-G. (2014) The Sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol. Membr. Biol. 31, 58–84 [DOI] [PubMed] [Google Scholar]
- 6. Shouldice S. R., Heras B., Walden P. M., Totsika M., Schembri M. A., and Martin J. L. (2011) Structure and function of DsbA, a key bacterial oxidative folding catalyst. Antioxid. Redox Signal. 14, 1729–1760 [DOI] [PubMed] [Google Scholar]
- 7. Inaba K., and Ito K. (2008) Structure and mechanisms of the DsbB–DsbA disulfide bond generation machine. Biochim. Biophys. Acta 1783, 520–529 [DOI] [PubMed] [Google Scholar]
- 8. Gao T., and O'Brian M. R. (2007) Control of DegP-dependent degradation of c-type cytochromes by heme and the cytochrome c maturation system in Escherichia coli. J. Bacteriol. 189, 6253–6259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Allen J. W., Barker P. D., and Ferguson S. J. (2003) A cytochrome b562 variant with a c-type cytochrome CXXCH heme-binding motif as a probe of the Escherichia coli cytochrome c maturation system. J. Biol. Chem. 278, 52075–52083 [DOI] [PubMed] [Google Scholar]
- 10. Ouyang N., Gao Y. G., Hu H. Y., and Xia Z. X. (2006) Crystal structures of E. coli CcmG and its mutants reveal key roles of the N-terminal β-sheet and the fingerprint region. Proteins 65, 1021–1031 [DOI] [PubMed] [Google Scholar]
- 11. Edeling M. A., Guddat L. W., Fabianek R. A., Thöny-Meyer L., and Martin J. L. (2002) Structure of CcmG/DsbE at 1.14 A resolution: high-fidelity reducing activity in an indiscriminately oxidizing environment. Structure 10, 973–979 [DOI] [PubMed] [Google Scholar]
- 12. Di Matteo A., Calosci N., Gianni S., Jemth P., Brunori M., and Travaglini-Allocatelli C. (2010) Structural and functional characterization of CcmG from Pseudomonas aeruginosa, a key component of the bacterial cytochrome c maturation apparatus. Proteins 78, 2213–2221 [DOI] [PubMed] [Google Scholar]
- 13. Monika E. M., Goldman B. S., Beckman D. L., and Kranz R. G. (1997) A thioreduction pathway tethered to the membrane for periplasmic cytochromes c biogenesis; in vitro and in vivo studies. J. Mol. Biol. 271, 679–692 [DOI] [PubMed] [Google Scholar]
- 14. Turkarslan S., Sanders C., Ekici S., and Daldal F. (2008) Compensatory thio-redox interactions between DsbA, CcdA and CcmG unveil the apocytochrome c holdase role of CcmG during cytochrome c maturation. Mol. Microbiol. 70, 652–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deshmukh M., Brasseur G., and Daldal F. (2000) Novel Rhodobacter capsulatus genes required for the biogenesis of various c-type cytochromes. Mol. Microbiol. 35, 123–138 [DOI] [PubMed] [Google Scholar]
- 16. Stirnimann C. U., Rozhkova A., Grauschopf U., Grütter M. G., Glockshuber R., and Capitani G. (2005) Structural basis and kinetics of DsbD-dependent cytochrome c maturation. Structure 13, 985–993 [DOI] [PubMed] [Google Scholar]
- 17. Katzen F., Deshmukh M., Daldal F., and Beckwith J. (2002) Evolutionary domain fusion expanded the substrate specificity of the transmembrane electron transporter DsbD. EMBO J. 21, 3960–3969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cho S. H., and Beckwith J. (2009) Two snapshots of electron transport across the membrane: insights into the structure and function of DsbD. J. Biol. Chem. 284, 11416–11424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di Matteo A., Gianni S., Schininà M. E., Giorgi A., Altieri F., Calosci N., Brunori M., and Travaglini-Allocatelli C. (2007) A strategic protein in cytochrome c maturation: three-dimensional structure of CcmH and binding to apocytochrome c. J. Biol. Chem. 282, 27012–27019 [DOI] [PubMed] [Google Scholar]
- 20. Ahuja U., Rozhkova A., Glockshuber R., Thöny-Meyer L., and Einsle O. (2008) Helix swapping leads to dimerization of the N-terminal domain of the c-type cytochrome maturation protein CcmH from Escherichia coli. FEBS Lett. 582, 2779–2786 [DOI] [PubMed] [Google Scholar]
- 21. Zheng X. M., Hong J., Li H. Y., Lin D. H., and Hu H. Y. (2012) Biochemical properties and catalytic domain structure of the CcmH protein from Escherichia coli. Biochim. Biophys. Acta 1824, 1394–1400 [DOI] [PubMed] [Google Scholar]
- 22. Deshmukh M., Turkarslan S., Astor D., Valkova-Valchanova M., and Daldal F. (2003) The dithiol:disulfide oxidoreductases DsbA and DsbB of Rhodobacter capsulatus are not directly involved in cytochrome c biogenesis, but their inactivation restores the cytochrome c biogenesis defect of CcdA-null mutants. J. Bacteriol. 185, 3361–3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sanders C., Turkarslan S., Lee D. W., Onder O., Kranz R. G., and Daldal F. (2008) The cytochrome c maturation components CcmF, CcmH, and CcmI form a membrane-integral multisubunit heme ligation complex. J. Biol. Chem. 283, 29715–29722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Setterdahl A. T., Goldman B. S., Hirasawa M., Jacquot P., Smith A. J., Kranz R. G., and Knaff D. B. (2000) Oxidation-reduction properties of disulfide-containing proteins of the Rhodobacter capsulatus cytochrome c biogenesis system. Biochemistry 39, 10172–10176 [DOI] [PubMed] [Google Scholar]
- 25. Reid E., Cole J., and Eaves D. J. (2001) The Escherichia coli CcmG protein fulfils a specific role in cytochrome c assembly. Biochem. J. 355, 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meyer E. H., Giege P., Gelhaye E., Rayapuram N., Ahuja U., Thony-Meyer L., Grienenberger J. M., and Bonnard G. (2005) AtCCMH, an essential component of the c-type cytochrome maturation pathway in Arabidopsis mitochondria, interacts with apocytochrome c. Proc. Natl. Acad. Sci. U.S.A. 102, 16113–16118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fabianek R. A., Hofer T., and Thöny-Meyer L. (1999) Characterization of the Escherichia coli CcmH protein reveals new insights into the redox pathway required for cytochrome c maturation. Arch. Microbiol. 171, 92–100 [DOI] [PubMed] [Google Scholar]
- 28. Netto L. E., de Oliveira M. A., Tairum C. A., and da Silva Neto J. F. (2016) Conferring specificity in redox pathways by enzymatic thiol/disulfide exchange reactions. Free Radic. Res. 50, 206–245 [DOI] [PubMed] [Google Scholar]
- 29. Verissimo A. F., Yang H., Wu X., Sanders C., and Daldal F. (2011) CcmI subunit of CcmFHI heme ligation complex functions as an apocytochrome c chaperone during c-type cytochrome maturation. J. Biol. Chem. 286, 40452–40463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Verissimo A. F., Shroff N. P., and Daldal F. (2015) During cytochrome c maturation CcmI chaperones the class I apocytochromes until the formation of their b-type cytochrome intermediates. J. Biol. Chem. 290, 16989–17003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Verissimo A. F., Mohtar M. A., and Daldal F. (2013) The heme chaperone ApoCcmE forms a ternary complex with CcmI and apocytochrome c. J. Biol. Chem. 288, 6272–6283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang P. F., Veine D. M., Ahn S. H., and Williams C. H. Jr. (1996) A stable mixed disulfide between thioredoxin reductase and its substrate, thioredoxin: preparation and characterization. Biochemistry 35, 4812–4819 [DOI] [PubMed] [Google Scholar]
- 33. Veine D. M., Mulrooney S. B., Wang P. F., and Williams C. H. Jr. (1998) Formation and properties of mixed disulfides between thioredoxin reductase from Escherichia coli and thioredoxin: evidence that cysteine-138 functions to initiate dithiol–disulfide interchange and to accept the reducing equivalent from reduced flavin. Protein Sci. 7, 1441–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ang S. K., Zhang M., Lodi T., and Lu H. (2014) Mitochondrial thiol oxidase Erv1: both shuttle cysteine residues are required for its function with distinct roles. Biochem. J. 460, 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Williamson J. A., Cho S. H., Ye J., Collet J. F., Beckwith J. R., and Chou J. J. (2015) Structure and multistate function of the transmembrane electron transporter CcdA. Nat. Struct. Mol. Biol. 22, 809–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schlecker T., Comini M. A., Melchers J., Ruppert T., and Krauth-Siegel R. L. (2007) Catalytic mechanism of the glutathione peroxidase-type tryparedoxin peroxidase of Trypanosoma brucei. Biochem. J. 405, 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vasas A., Dóka É., Fábián I., and Nagy P. (2015) Kinetic and thermodynamic studies on the disulfide-bond reducing potential of hydrogen sulfide. Nitric Oxide 46, 93–101 [DOI] [PubMed] [Google Scholar]
- 38. Di Silvio E., Di Matteo A., Malatesta F., and Travaglini-Allocatelli C. (2013) Recognition and binding of apocytochrome c to P. aeruginosa CcmI, a component of cytochrome c maturation machinery. Biochim. Biophys. Acta 1834, 1554–1561 [DOI] [PubMed] [Google Scholar]
- 39. San Francisco B., Sutherland M. C., and Kranz R. G. (2014) The CcmFH complex is the system I holocytochrome c synthetase: engineering cytochrome c maturation independent of CcmABCDE. Mol. Microbiol. 91, 996–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanders C., Deshmukh M., Astor D., Kranz R. G., and Daldal F. (2005) Overproduction of CcmG and CcmFH(Rc) fully suppresses the c-type cytochrome biogenesis defect of Rhodobacter capsulatus CcmI-null mutants. J. Bacteriol. 187, 4245–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rozhkova A., and Glockshuber R. (2007) Kinetics of the intramolecular disulfide exchange between the periplasmic domains of DsbD. J. Mol. Biol. 367, 1162–1170 [DOI] [PubMed] [Google Scholar]
- 42. Darby N. J., and Creighton T. E. (1995) Catalytic mechanism of DsbA and its comparison with that of protein disulfide isomerase. Biochemistry 34, 3576–3587 [DOI] [PubMed] [Google Scholar]
- 43. Moutevelis E., and Warwicker J. (2004) Prediction of pKa and redox properties in the thioredoxin superfamily. Protein Sci. 13, 2744–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng Z., Zhang J., Ballou D. P., and Williams C. H. Jr. (2011) Reactivity of thioredoxin as a protein thiol-disulfide oxidoreductase. Chem. Rev. 111, 5768–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sanders C., Turkarslan S., Lee D. W., and Daldal F. (2010) Cytochrome c biogenesis: the Ccm system. Trends Microbiol. 18, 266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mavridou D. A., Stevens J. M., Mönkemeyer L., Daltrop O., di Gleria K., Kessler B. M., Ferguson S. J., and Allen J. W. (2012) A pivotal heme-transfer reaction intermediate in cytochrome c biogenesis. J. Biol. Chem. 287, 2342–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aitken A., and Learmonth M. (2002) in The Protein Protocols Handbook (Walker J. M., ed.) 2nd Ed., pp. 595–596, Humana Press Inc., Totowa, NJ [Google Scholar]
- 48. Denoncin K., Nicolaes V., Cho S. H., Leverrier P., and Collet J. F. (2013) Protein disulfide bond formation in the periplasm: determination of the in vivo redox state of cysteine residues. Methods Mol. Biol. 966, 325–336 [DOI] [PubMed] [Google Scholar]
- 49. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 50. Deshmukh M., May M., Zhang Y., Gabbert K. K., Karberg K. A., Kranz R. G., and Daldal F. (2002) Overexpression of ccl1-2 can bypass the need for the putative apocytochrome chaperone CycH during the biogenesis of c-type cytochromes. Mol. Microbiol. 46, 1069–1080 [DOI] [PubMed] [Google Scholar]
- 51. Sanders C., Boulay C., and Daldal F. (2007) Membrane-spanning and periplasmic segments of CcmI have distinct functions during cytochrome c biogenesis in Rhodobacter capsulatus. J. Bacteriol. 189, 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sanders C., and Lill H. (2000) Expression of prokaryotic and eukaryotic cytochromes c in Escherichia coli. Biochim. Biophys. Acta 1459, 131–138 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.