

Abstract
The human gut microbiota utilizes complex carbohydrates as major nutrients. The requirement for efficient glycan degrading systems exerts a major selection pressure on this microbial community. Thus, we propose that this microbial ecosystem represents a substantial resource for discovering novel carbohydrate active enzymes. To test this hypothesis we screened the potential enzymatic functions of hypothetical proteins encoded by genes of Bacteroides thetaiotaomicron that were up-regulated by arabinogalactan proteins or AGPs. Although AGPs are ubiquitous in plants, there is a paucity of information on their detailed structure, the function of these glycans in planta, and the mechanisms by which they are depolymerized in microbial ecosystems. Here we have discovered a new polysaccharide lyase family that is specific for the l-rhamnose-α1,4-d-glucuronic acid linkage that caps the side chains of complex AGPs. The reaction product generated by the lyase, Δ4,5-unsaturated uronic acid, is removed from AGP by a glycoside hydrolase located in family GH105, producing the final product 4-deoxy-β-l-threo-hex-4-enepyranosyl-uronic acid. The crystal structure of a member of the novel lyase family revealed a catalytic domain that displays an (α/α)6 barrel-fold. In the center of the barrel is a deep pocket, which, based on mutagenesis data and amino acid conservation, comprises the active site of the lyase. A tyrosine is the proposed catalytic base in the β-elimination reaction. This study illustrates how highly complex glycans can be used as a scaffold to discover new enzyme families within microbial ecosystems where carbohydrate metabolism is a major evolutionary driver.
Keywords: carbohydrate processing, glycobiology, glycoside hydrolase, microbiome, X-ray crystallography
Introduction
The human gut microbiota (HGM)3 makes an important contribution to the health and physiology of its host (1). The major nutrients available to this microbial ecosystem are complex host and dietary glycans (2). To exploit these carbohydrate polymers as nutrients, members of the HGM, particularly bacteria from the Bacteroidetes phylum, have evolved extensive glycan degrading systems comprising enzymes, carbohydrate-binding proteins, transporters, and regulators (3–6). The genes encoding these proteins are physically linked within loci referred to as PULs (polysaccharide utilization loci) and transcriptionally up-regulated by specific components of the target glycan (2, 7, 8). Thus, the HGM represents a valuable microbial resource for discovering carbohydrate active enzymes (CAZymes), particularly glycoside hydrolases (GHs) and polysaccharide lyases (PLs), which cleave the glycosidic bonds that link the sugars in polysaccharides and oligosaccharides. CAZymes have been grouped into sequence-based families in the continuously updated CAZy database (9). The protein-fold, catalytic apparatus, and mechanism are generally conserved in each family (9), although substrate specificity can vary (10).
Arabinogalactan proteins (AGPs) are a structurally diverse but poorly characterized group of plant glycans. These molecules are highly glycosylated members of the hydroxyproline-rich glycoprotein superfamily of plant cell wall proteins, with 90% of their total mass being glycans. The core structure of the glycan component of AGPs comprises a backbone of β1,3-galactan that is substituted at O-6 with β1,6-galactooligosaccharides. These side chains can then be further decorated with arabinose, rhamnose, (methyl) d-glucuronic acid (GlcA), and less frequently, with fucose or xylose moieties (11–13). These additional decorations vary with respect to the plant species and cell type. Some AGPs, exemplified by the glycan from Acacia senegal (also known as gum arabic and defined henceforth as GA), are used extensively in the food industry as stabilizers and emulsifiers (e.g. in soft drink syrups, marshmallows, or gummy candies) (14). The common use of AGPs as food additives, and their widespread presence in plant species, explain why these glycans are common components of the human diet. Consistent with their exposure to AGPs, some members of the HGM utilize these complex carbohydrates as growth substrates. Indeed, Bacteroides thetaiotaomicron and Bacteroides ovatus, two prominent members of the HGM, were previously shown to grow on the simple AGP from larchwood, and the PULs activated by this glycan were identified (2). Dissecting the mechanisms by which AGPs are degraded will make an important contribution to our understanding of glycan utilization in the HGM. Such knowledge has the potential to underpin dietary probiotic- and prebiotic-based strategies to maximize the impact of this ecosystem on human health. Furthermore, enzymes that cleave specific linkages in AGPs will be invaluable in determining the structure of these complex but heterogeneous carbohydrates, which has the potential to deliver new products for the food industry.
α-l-Rhamnose (Rha) caps the side chains in several complex AGPs that are components of the human diet. There terminal Rha units create a significant enzymatic challenge for GHs (α-l-rhamnosidases) as unfavorable syn di-axial interactions, caused by the axial O-2, must be overcome during glycosidic bond cleavage (15, 16). As some AGPs, exemplified by GA, contain Rha units linked α1,4 to uronic acids (UAs) such as GlcA, it is possible that PLs also contribute to the removal of these 6-deoxy-sugars from the plant glycan. PLs perform glycosidic bond cleavage through a β-elimination reaction by acting on GlcA at the +1 subsite (the scissle glycosidic bond is between sugars bound at the −1 and +1 subsites of CAZymes (17)). In this instance PLs would cleave the Rha-α1,4-GlcA linkage without performing difficult rhamnosidase chemistry (see Ref. 18 for a review of the PL requirement for substrates containing UAs).
Here we have tested the hypothesis that the HGM provides a reservoir of new enzymes capable of cleaving α-l-rhamnosidic linkages. We show that PLs discovered in Bacteroides species within the HGM releases Rha from AGPs. We also demonstrate that the reaction product of the lyase, 4-deoxy-β-l-threo-hex-4-enepyranosyl-uronic acid (Δ4,5-GlcA)-linked β1,6 to d-galactose (Gal), is cleaved by a GH105-unsaturated glucuronidase. The crystal structures of these two enzymes provide insights into the catalytic apparatus and the structural basis for their substrate specificity.
Results
BT0263 releases l-rhamnose from AGP
B. thetaiotaomicron, a common member of the human gut microbiota, is capable of growing on the simple AGP from larchwood (2). The glycan up-regulates two PULs (BT0262-BT0290 and BT3674-BT3687) defined as AGP-PUL1 and AGP-PUL2, respectively. According to the prototypic PUL paradigm, this suggests that the proteins encoded by these loci contribute to the utilization of AGPs. As stated above the structure of AGPs are highly variable, which likely explains why these two PULs encode a large number of proteins. To explore the functional significance of AGP-PUL1 and AGP-PUL2, the proteins encoded by these loci were produced in recombinant form and their biochemical properties evaluated. Here we have investigated the role of BT0263 in the depolymerization of AGPs. The protein comprises 470 amino acids and has no sequence similarity with any known CAZyme or members of the CAZy database. The capacity of BT0263 to act on AGPs from larchwood, wheat, and GA was investigated. BT0263 only released Rha from GA indicating that the protein is an exo-acting enzyme (Fig. 1A, Table 1). BT0263 had a pH optimum of ∼5 (Fig. 1B) and experiments performed with BACCELL_00875 demonstrated the new family is likely not to be metal dependent (Table 2).
Figure 1.

Biophysical and biochemical properties of BT0263 and BT3687. A, HPLC chromatogram showing GA only control (blue) and GA with the addition of BT0263 (green). The experimental conditions were 20 mm sodium phosphate buffer, 150 mm NaCl, pH 7.0; concentration of GA was 10 mm (refers to available rhamnose units) and enzyme was at 1 μm. B, pH-profile of BT0263. The experimental conditions were the same as in A for the substrate and enzyme concentrations. The buffers used were 20 mm sodium acetate, pH 4.0–5.0, 20 mm MES, pH 5.5–6.5, 20 mm sodium phosphate, pH 6.5–7.5, and 20 mm CAPS buffer, pH 7.5–9.0. The assays were monitored at a wavelength of UV A235 nm. C and D are HPLC chromatograms of GA only control (blue); GA treated with the GH43 exo-β1,3-d-galactanase BT0265 (red); GA treated with BT0265 and then BT0263 (green); GA treated sequentially with BT0265, BT0263, and the GH105 enzyme BT3687 (black). C is measured by pulsed amperometric detection, whereas D is measured via UV A235 nm. X indicates specific products, in addition to Rha, generated by BT0263 that are susceptible to BT3687, which specifically targets Δ4,5-UA generated by polysaccharide lyases. The experimental conditions were as in A.
Table 1.
Kinetic parameters of BT0263, BACCELL_00875, BACFIN_07013, and BT3687
| Substratea | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | min−1 | min−1 m−1 | |
| BT0263 | |||
| GA | 450.3 ± 77.5 | 64.56 ± 4.94 | 143,400 ± 63,700 |
| Trisaccharide | 7,400 ± 200 | ||
| Tetrasaccharide | 5,400 ± 600 | ||
| Heptasaccharide | 24,100 ± 800 | ||
| BACCELL_00875 | |||
| GA | 31.29 ± 2.54 | 26.91 ± 3.2 | 860,019 ± 125,984 |
| Trisaccharide | 8,901 ± 450 | ||
| Tetrasaccharide | 7,888 ± 751 | ||
| Heptasaccharide | 26,390 ± 1,310 | ||
| BACFIN_07013 | |||
| GA | 26.87 ± 4.12 | 13.39 ± 0.61 | 498,235 ± 148,058 |
| Trisaccharide | 6,541 ± 421 | ||
| Tetrasaccharide | 7,989 ± 630 | ||
| Heptasaccharide | 25,213 ± 1,850 | ||
| BT3687 | |||
| Hexasaccharide | 71.87 ± 12.51 | 35.62 ± 3.40 | 49,5617 ± 271,782 |
| Δ4,5UA-GlcNAc | 947 ± 33 | ||
a The oligosaccharide substrates were derived from GA through cleavage of the galactan backbone by BT0265, a β1,3-galatanase. Details on how these oligosaccharides were generated, purifed, and their DP determined is provided under “Experimental procedures.”
Table 2.
Metal-dependent activity of BACCELL_00875 against GA
The experimental conditions were 20 mm sodium phosphate buffer, pH 7.0, Baccell_00875 = 0.5 μm, GA = 1 mm. The enzyme was assayed with no addition or after incubation with 50 mm EDTA, which was then removed by size exclusion chromatography. The lyase was then assayed in the absence (EDTA treated) or presence of the metals (1 mm) indicated.
| Metal | kcat/Km | Relative activity |
|---|---|---|
| min−1 m−1 | % | |
| No addition | 860019 ± 94602 | 100 |
| EDTA treated | 825618 ± 90818 | 96 |
| Mg2+ | 912018 ± 89872 | 95 |
| Ca2+ | 774017 ± 85142 | 90 |
| Mn2+ | 782617 ± 86087 | 91 |
| Zn2+ | 739616 ± 81358 | 86 |
BT0263 is a lyase that cleaves Rha-α1,4-GlcA linkages
Terminal Rha units in AGPs are reported to be linked α1,2, α1,4, or α1,6 to Gal and α1,4 to GlcA (12, 19). Recently BT3686 was shown to be an α-l-rhamnosidase specifically targeting Rha-α1,4-GlcA linkages in the side chains of GA (20). BT0263 was not active against GA treated with BT3686 indicating that both enzymes cleaved Rha-α1,4-GlcA linkages. To explore the origin of the Rha generated by BT0263, GA was treated with BT0265, which belongs to GH43 subfamily 24 that is populated exclusively with exo-β1,3-galactanases (21). The proposed exo-β1,3-galactanase activity of BT0265 is consistent with its capacity to generate oligosaccharide side chains from GA (Fig. 1C), which are attached to galactose residues released from the AGP β1,3-galactan backbone; a signature product profile for such enzymes (22, 23). When these oligosaccharide side chains were incubated with BT0263, only Rha was produced confirming its target substrates are the glycan decorations in AGPs and not Rha linked directly to the backbone (Fig. 1C). Significantly, the products generated by BT0263 from the BT0265-derived oligosaccharides showed absorbance in the near UV range at 235 nm, indicative of the formation of a carbon-carbon double bond conjugated to a carboxylate (Fig. 1D). Finally, mass spectrometry showed that the release of Rha from a GA-derived heptasaccharide, again generated through the action of BT0265, resulted in a decrease in mass of 164 consistent with the cleavage of the O1–C4 bond of the Rha-GlcA linkage and thus supporting the PL function of BT0263 (Fig. 2, A and B). The cleavage of glycosidic bonds by PLs is through a β-elimination mechanism. Briefly, this mechanism operates by a general base abstracting a proton from C5 of UA sugars, as this hydrogen is particularly acidic due to the presence of the carboxylic acid group also on C5. This results in an enolate transition state that collapses resulting in the formation of a conjugated double bond between the C4 and C5 and elimination of the glycosidic oxygen from C4, which is concomitantly protonated (24, 25). This results in the formation of unsaturated UA (Δ4,5-UA) products, which have the characteristic UV signal at A235 nm observed here (26).
Figure 2.
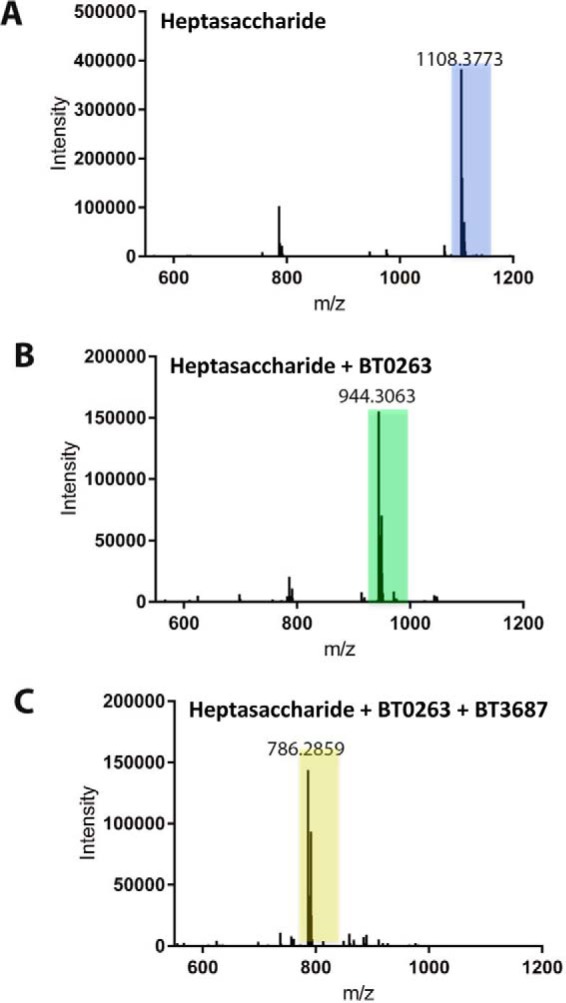
Mass spectrometry of the oligosacchacaride. A major GA-derived oligosaccharide generated by the exo-β1,3-galatanase BT0265 (Fig. 1C) was purified by size exclusion chromatography. The oligosaccharide were untreated (A), incubated with BT0263 (B), or treated with BT0263 and then BT3687 (C). The experimental conditions for enzyme treatment were 20 mm sodium phosphate buffer, pH 7.0. The concentrations of substrate and enzyme were 1 mm and 1 μm, respectively.
To further confirm the activity of BT0263 as a new PL, we recombinantly expressed BT3687, encoded by AGP-PUL2, as the enzyme is a member of family GH105 in the CAZy database (9). All members of GH105 characterized to date act exclusively on Δ4,5-UA-O-R (R is generally a sugar) generated by PLs that act on rhamnogalacturonan-I and ulvan. Briefly, this family operates through a mechanism where a general acid/base protonates the double bond, resulting in a hemiketal intermediate, which proceeds through either a glycosyl-enzyme, or an epoxide, intermediate causing glycosidic bond cleavage and loss of the C4–C5 double bond (27, 28). The activity of these enzymes can therefore be monitored by loss of the UV signal at 235 nm. When the products of oligosaccharides generated by BT0265 and then the lyase BT0263 were incubated with BT3687 the UV signal was lost (Fig. 1D). Furthermore, the reduction in mass of the BT0263-treated oligosaccharide after incubation with BT3687 was 158, which is entirely consistent with the loss of Δ4,5-UA with bond cleavage occurring between C1 and O4 of the scissile linkage (Fig. 2, B and C). These data confirm that BT0263 is a new PL cleaving the O4–C4 bond between Rha–α1,4-GlcA generating a Rha and Δ4,5-UA linked to an oligosaccharide. Thus, BT0263 is the founding member of a new PL family defined here as PL27.
Compared with the vast majority of PL27 members, BT0263 appears to be truncated lacking the N-terminal domain of ∼200 residues (see the phylogeny section). This may indicate that the 600-residue protein (termed full-length proteins) in this family display a different specificity or catalytic function to BT0263. To evaluate this possibility the biochemical properties of two PL27 full-length proteins, BACCELL_00875 and BACFIN_07013 derived from Bacteroides cellulosilyticus and Bacteroides finegoldii, respectively, were determined. Both proteins displayed the same activity as BT0263 (Table 1), showing that rhamno-glucurono lyase activity is a common feature of PL27. These data also indicate that the catalytic domain in PL27 enzymes comprises the 400-residue C-terminal domain. The potential role of the N-terminal domain is discussed below.
Crystal structure of members of PL27
To explore the structure-function relationships of enzymes in PL27, crystallization of BT0263 and the two homologs BACCELL_00875 and BACFIN_07013 was attempted. Only crystals of BACCELL_00875 could be generated. The structure of the enzyme was initially solved by single-wavelength anomalous dispersion using a selenomethionine (SeMet)-derivatized protein to a resolution of 2.2 Å. A higher resolution structure of the native enzyme was then solved to 1.7 Å by molecular replacement using the SeMet-BACCELL_00875 structure as the search model. The SeMet and native proteins were crystallized in space groups P41212 and P3221, respectively, with both having two molecules in the asymmetric unit. The structure of BACCELL_00875 comprises three domains, two β-sandwich domains and a C-terminal (α/α)6 barrel (Fig. 3A). The N-terminal domain, extending from Phe27 to His243, displays a twisted β-sandwich-fold. This domain contains 13 antiparallel strands arranged in two β-sheets. The order of the strands in β-sheets 1 and 2 is β1-β2-β7-β12-β11 and β3-β4-β5-β6-β13-β10-β9-β8, respectively. The second domain, extending from Ser257 to Ser336, is a classical β-sandwich domain in which the seven antiparallel strands are arranged in the order β1-β2-β5-β4 in β-sheet 1 and β7-β6-β3 in β-sheet 2. Some of the β-strands are interrupted and four small β-strands are not components of the two β-sandwich domains. A small α-helix stretching from Asn245 to Gly256 separates the β-sandwich domains and makes numerous apolar and polar contacts with β-sheet 2 of the N-terminal domain and a hydrophobic contact with Leu270 of the second domain. The loop connecting β8 and β9 of the N-terminal domain makes several apolar interactions with β7 of the second sandwich domain. The C-terminal domain comprises residues Asn337 to Leu694. This domain displays an (α/α)6 barrel-fold where a central barrel of 6 α-helices (helices 2, 4, 6, 8, 10, and 12) are spiraled by a further 6 α-helices (helices 1, 3, 5, 7, 9, and 11). The central barrel forms the hydrophobic core of the protein. This domain shows the highest levels of conservation within the PL27 family and is where the active site is housed, see below. Residues in the extended loop between helixes 9 and 10 (Pro561, Ser562, Tyr563, and Leu624) make apolar contacts with Tyr243, Val238, Val244, and Leu311 in the central domain and abuts on the top and base of helices 1 and 12, respectively. The (α/α)6 barrel also makes extensive aromatic-mediated hydrophobic interactions with the N-terminal β-sandwich domain. Specifically, Phe578, Val590, Ile592, Phe610, Phe581, and Tyr661, which are on elongated loops that connect helices 9, 10, 11, and 12, make apolar interactions with His164, His166, Trp168, Tyr175, Ile179, and Tyr200 in the N-terminal domain. A single salt bridge, between Arg215 and Asp608, also stabilizes the inter-domain association. The tight association between the catalytic domain and the two β-sandwich domains indicates that the N-terminal region of the protein contributes to stabilization of the C-terminal (α/α)6 barrel.
Figure 3.

The crystal structure of BACCELL_00875. A, schematic representation, ramped blue to red from the N to C terminus, of BACCELL_00875 showing the multidomain structure. B, active site of Baccell_00875 with residues implicated in substrate binding and catalysis shown in stick format. C, surface representation of the active site of Baccell_00875 in both conformations, loop closed (left) and loop open (right). In both cases, the residues implicated in stabilizing the conformation of this loop are shown as is the active site histidine whose position is dependent on the ionic lock. For reference the proposed catalytic tyrosine is also shown. D, surface representation of sequence conservation at the anterior and posterior sites of Baccell_00875. Residues were colored relative to their conservation within the PL27 family. Dark blue signifies amino acids that are invariant and cyan identifies residues that are 40–60% conserved. Red asterisk signifies the location of the active site.
Active site of BACCELL_00875
Based on sequence conservation, the active site is likely located in the (α/α)6-barrel domain (Fig. 3, B and C). This hypothesis is supported by the observation that although BT0263 lacks the N-terminal domains the enzyme had catalytic activity similar to BACCELL_00875 (Table 1). Furthermore, the (α/α)6-barrel-fold is often associated with CAZymes, both GHs and PLs (9, 29, 30). A multiple sequence alignment of the members of PL27 show a significant number of invariant and highly conserved residues(Fig. 3D and supplemental Fig. S1). When mapped onto the crystal structure of BACCELL_00875, many of these amino acids are localized at the center of (α/α)6-barrel, forming part of a deep pocket (Fig. 3C). It is interesting to note that the top of this deep pocket is formed by the closure of the loop between α-helices 7 and 8. The conformation of the loop is locked by a salt bridge between Arg447 and Glu537 and through aromatic amino acid interactions, with His536 sitting in a hydrophobic pocket formed by Phe489, Tyr490, and Pro534. This loop differs between the two molecules in the asymmetric unit of both the SeMet structure, and the higher resolution native structure, undergoing a ∼9 Å shift that results in His536 and Glu537 pointing into solution (Fig. 3C). These different conformations reflect the presence of MES and a single glycerol in the locked conformation versus no MES and two glycerol molecules in the open conformation (supplemental Fig. S2). Based on the sequence alignment and structural data, a targeted set of mutations was designed to elucidate residues that make a significant contribution to catalytic activity. These mutants were E537Q, R593A, D596A, D596N, W599A, H612A, and Y613F. The effect of all of these mutations was significant with E537Q, R593A, D596A, D596N, and H612A substitutions causing a ∼4,000- to ∼10,000-fold reduction in kcat/Km, whereas the loss of Trp613 at the top of the pocket resulted in a more dramatic 50,000-fold reduction (Table 3). The only mutation that completely inactivated the enzyme was Y613F, suggesting that this residue, sitting deep in the pocket (Fig. 3, B and C), is likely a significant component of the catalytic apparatus.
Table 3.
Activity of mutants of BACCELL_00875 and BT3687 against GA
| kcat/Km | Relative activity | |
|---|---|---|
| min−1 m−1 | % | |
| BACCELL_00875 | ||
| Wild type | 860019 ± 125984 | 100 |
| E537Q | 91.9 ± 1.1 | 0.01 |
| R593A | 199.1 ± 1.4 | 0.02 |
| D596A | 67.3 ± 0.94 | 0.008 |
| D596N | 204.1 ± 19.8 | 0.02 |
| W599A | 17.1 ± 5.5 | 0.002 |
| H612A | 83.9 ± 2.45 | 0.01 |
| Y613F | Inactive | 0 |
| R445A | 958.3 ± 103.1 | 0.11 |
| BT3687 | ||
| Wild type | 495617 ± 271782 | 100 |
| D160A | Inactive | 0 |
| D116A | Inactive | 0 |
A Dalilite search (31) was performed to find relatives to BACCELL_00875 that display a similar fold. This search returned a N-acyl-d-glucosamine 2-epimerase, 7% identity (PDB ID 1FP3), a cellobiose 2-epimerase, 10% identity (PDB ID 3WKF), and an α-1,6-d-mannanase, 10% identity, belonging to the GH76 family (PDB ID 4V1R). These homologs, however, had high root mean square deviations (r.m.s. deviations) >3.2 and Z-scores <26. The location of the active site in these enzymes, however, is similar to the position of the proposed catalytic center active of BACCELL_00875 but the catalytic residues do not overlay with Tyr613, and the ligands for these enzymes could not be satisfactorily accommodated in the deep pocket of the PL27 family. The closest characterized lyase homolog, a member of family PL15, displays 5% identity (PDB ID 3AFL (29)) and has r.m.s. deviations >4.0 and Z-score <11. Again, the location of the active site of the PL15 lyases and the deep pocket (the proposed catalytic center) of BACCELL_00875 are conserved. However, the catalytic residues of the PL15 enzyme, a histidine and tyrosine acting as the general base and acid, respectively, do not overlay with Tyr613 in BACCELL_00875. These results suggest that the structurally related proteins may not be functionally or evolutionarily related to BACCELL_00875.
Crystal structure of BT3687
The unsaturated glucuronidase BT3687 was also crystallized and its structure solved by molecular placement using PDB code 4CE7 as the search model (32). The enzyme also has an (α/α)6-barrel-fold, as has been shown previously for other members of the GH105 family (33). The active site location within GH105 enzymes is conserved and allows identification of the putative catalytic residues of BT3687 as Asp116 and Asp160 (Fig. 4A), which overlay well with catalytic apparatus of other structurally characterized members (YteR, YteR2, and Nu_GH105 (28, 32)) of family GH105 (Fig. 4B). Mutation of Asp116 and Asp160 to Ala in BT3687 completely ablated catalytic activity in the B. thetaiotaomicron enzyme (Table 3). The −1 active site pocket of BT3687 is lined with the hydrophobic amino acids Trp56, Trp158, Met164, Trp225, and Trp231, and appears to be a general feature of GH105 enzymes with YteR, YteR2, and Nu_GH105 all containing the equivalent residues (Fig. 4B). This may ensure protonation of the catalytic acid/base (Asp160) at physiological pH. Arg227, which coordinates with the carboxylate of the substrate is also conserved. This active site conservation also largely extends to GH88 enzymes, the only other family to perform catalysis by protonation of the double bond of PL products.
Figure 4.

The crystal structure of BT3687. A, schematic representation of BT3687 (beige) with the amino acids implicated in catalysis. The yellow amino acids, Asp116 and Asp160, are the proposed catalytic residues. The residues colored green and cyan are predicted to contribute to substrate binding. B, the conservation of residues in the −1 subsite (active site) between the GH105 enzymes BT3687 (green), NUGH105 (magenta), YTER (blue), YTER2 (beige), and the GH88 UGL (purple). The active site Trp56 of BT3687 (C) and the equivalent residue (Tyr41) in YTER (D) show how they are likely to interact with 4,5-UA-α1,2-l-Rha (left panel) and 4,5-UA-β-1,3-d-GalNAc (right panel), which were overlaid from structures with PDB codes 2GH4 and 2AHG, respectively.
Within the GH105 family, YteR and YteR2 cleave Δ4,5-UA-α1,2-Rha linkages, whereas the Bacillus subtilis enzymes UGL and NuGH105 hydrolyze Δ4,5-UA–β1,3GalNac and Δ4,5-UA–β1,4Rha3S glycosidic bonds, respectively. BT3687 targets Δ4,5UA–β1,6-Gal but was also shown to hydrolyze Δ4,5UAβ1,4–GlcNac glycosidic bonds (Table 1). When these enzymes are overlaid with their cognate ligands bound, where possible, the loops that form the +1 subsite (the scissile bond links the sugars bound in the −1 and +1 subsites (17)) appear highly variable and there seems to be no obvious rationale in driving specificity. It is interesting to note, however, that BT3687, UGL, and NuGH105 all have a conserved Trp at their −1 subsite (Trp56, Trp43, and Trp45, respectively) (Fig. 4B). This aromatic residue is <2.0 Å from an α-linked Rha causing steric hindrance, whereas a β-linked substrate lies parallel, pointing into solution (Fig. 4C). In YTER and YTER2, however, this residue is a Tyr, which forms a hydrogen to the O3 of Rha (Fig. 4D). This suggests that the nature of an aromatic residue equivalent to Trp56 in BT3687 may be the driver for β- versus α-linked substrates.
Phylogeny of the new PL family
The spread of the new PL family PL27 was investigated through iterative BLASTP and HMM searches. The limited sequence diversity, even between prokaryote and eukaryote sequences, and the absence of any distantly related family in the CAZy database, facilitated the delineation of this family and the construction of a phylogenetic tree (Fig. 5). Members are present in several bacterial phyla, particularly the Bacteroidetes (∼50 sequences) where ∼80% are in the Bacteroides genus. Interestingly, within a single species that contains the lyase, this family is not widely distributed in the different strains, a feature that is not shared with other CAZy families. Also noteworthy is the split of the homologs into two distinct proteins in B. thetaiotaomicron. In ∼50% of the strains in this species the lyase lacks the N-terminal 200 residues comprising the β-sandwich domains (these lyases are labeled as FUSED in Fig. 5). For example, in B. thetaiotaomicron strain VPI-5482 the lyase is BT0263 and the β-sandwich protein is BT0262. The biological rationale for the presence or absence of the N-terminal domain in the lyases is unclear although this region of the enzyme is not required for catalytic activity (Table 1), and likely contributes to the stabilization of the enzyme. Members of PL27 are also commonly found in Actinobacteria, where at least four different bacterial orders contain the lyase (∼10 sequences in Fig. 5) and in lower numbers in Firmicutes (mainly in the Lachnospiracea family of the Clostridia class, ∼10 sequences) and a single sequence from the Spirochaetes phylum. In the eukaryotes ∼60 fungal sequences have been identified, all belonging to the filamentous ascomycete fungus Pezizomycotina subphylum. The absence of any known distantly related families suggests that the new family may have evolved from a progenitor sequence that displayed no evolutionary link to current PL and GH families.
Figure 5.

Phylogenetic tree of the PL27 family. Four subgroups exhibit large evolutionary distances: Bacteroidetes (green subtree); Firmicutes and one Spirochaetes (orange subtree); Actinobacteria and a cohort pf Ascomycota (purple subtree); and Ascomycota-only (blue subtree). Labels on leaves indicate for each protein its species, its phylum, and the type of residue aligned to the catalytic Y. Split gene models that were manually fused for this analysis are indicated with the tag FUSED.
Of the 126 sequences that currently compose this new family the proposed catalytic Tyr is conserved in 117 sequences, with six having a His and one a Phe. His is a common base used by PLs (29, 34) and these variants could well be active, whereas the Phe variant probably represents a loss of function, exemplified by the BACELL_00875 mutant Y613F. It is interesting to note that in most species (∼120) a single copy of the PL gene is observed, whereas eight species have two copies. In these eight organisms there was either very little sequence divergence in the two copies of the two genes, suggesting the generation of these paralogs was a fairly recent event, In the fungi Clonostachys rosea and Colletotrichum fioriniae PJ7, however, high divergence of the paralog occurred, and in one version the catalytic Tyr was replaced with a His. Together, these observations support the hypothesis that only a single copy of the lyase has been necessary during the evolution of these organisms.
Discussion
This study provides the characterization of a new PL family that cleaves Rha-α1,4-GlcA glycosidic bonds. The family contains a limited number of sequences, is present in fungal and bacterial organisms, and is well conserved across taxa. This may suggest that the family has specifically evolved to target AGPs containing the Rha-α1,4-GlcA linkage and may be a relatively recent evolutionary event. Intriguingly, in a proportion of the Bacteroides species the N-terminal domain contains a stop codon at its C terminus resulting in two proteins in these organisms, a functional lyase and a protein containing two β-sandwich domains. This has the consequence, in these strains, of presumably localizing the lyases to the cytoplasm as they now lack a signal peptide, which is retained by the β-sandwich proteins. The loss of the N-terminal domain does not affect catalytic efficiency of the enzyme and this domain shows much more sequence diversity within the PL27 family than the C-terminal catalytic domain. The rationale for the loss of this domain in the lyase expressed by the B. thetaiotaomicron strains is unclear. Polysaccharide degradation occurs in extra-cytoplasmic locations with only monosaccharides being transported into the cytoplasm, so localization of BT0263 to the cytoplasm is counter-intuitive. It is formally possible, however, that BT0263 is secreted to the periplasm by an unidentified mechanism.
The catalytic mechanism of this new PL family likely proceeds through a classical lyase β-elimination mechanism, with the leaving group and the abstracted proton both being in the syn chemical space, and generating the signature Δ4,5-UA and Rha as its products (Fig. 6). Tyr613 was identified as being crucial to its function (the mutant Y613F is completely inactive, Table 3) and is highly conserved in the family. Tyr613 is therefore the candidate catalytic base abstracting the C5 proton, generating the enolate anion intermediate that leads to elimination of the glycosidic oxygen and the production of Δ4,5-UA. After cleavage the glycosidic oxygen, now being the O1 of Rha, requires protonation as the pKa values of secondary alcohols are >16. A catalytic acid is therefore required to protonate the glycosidic oxygen to facilitate leaving group departure. Apart from Tyr613, however, there are no candidate polar residues that could fulfill this function in the proximal region of the active site pocket. It would appear, therefore, that Tyr613 likely functions as the catalytic acid-base. Indeed, there are a number of PL families, such as PL1, PL8, and PL9, where a single catalytic residue has been proposed to function as the acid/base (25, 34–36). Usually, there is also an amino acid or metal ion that increases the negative charge of the carboxylic acid of the substrate. This helps to both further acidify the C5 proton, enhancing its ability to accept electrons from the base, and to stabilize the enolate anion at the transition state. Without a ligand complex it is hard to unambiguously assign a residue in BACCELL_00875 that interacts with the carboxylate of GlcA, however, Arg447 and Arg593 are potential candidates. These residues are invariant in the family and are ∼7 Å from the catalytic tyrosine, a distance consistent with this proposed function. An additional feature of the PL is the importance of the loop between helices, which close to form the active site pocket. The mutant E537Q shows a marked decrease in catalytic competence presumably due to the loss of the ionic lock that is needed to secure the loop in place and form the active site. The residual activity of the mutant without the “ionic lock” likely reflects the stochastic conformation adopted by the loop, which may infrequently allow productive binding of substrate or facilitate departure of the cleaved products.
Figure 6.

Proposed catalytic mechanism for PL27. The proposed catalytic mechanism, proceeding through an enolate transition state, with tyrosine acting as the catalytic acid/base and an arginine serving to stabilize the negative charge that is developed.
The structure of BT3687 further demonstrated that the −1 subsite of this class of enzyme contains a residue that is a tyrosine or tryptophan in α- and β-glycosidases, respectively. A tyrosine at this critical position creates the required space for α-configured sugars at the +1 subsite. The phenol also makes a hydrogen bond with the +1 sugar, which likely contributes to substrate binding. It is unusual for a GH family to contain enzymes that act on α- and β-linked substrates, although this is also observed in the GH4 family (37), which also departs significantly from classical acid-base catalysis.
Conclusions
This study describes the discovery of a new PL family that targets Rha-α1,4-GlcA and its GH105 enzyme partner, which cleaves the product of the lyase. The new PL family, of which BT0263, BACCELL_00875, and BACFIN_07013 are founding members, is highly conserved. The structure of BACCELL_00875 revealed that the closure of a dynamic loop forms the active site pocket and is secured in place by a salt bridge forming an ionic lock. The enzyme family displays a canonical β-elimination mechanism utilizing a single catalytic tyrosine. The active site of the GH105 enzyme, BT3687, displayed the expected characteristics of GH105 members, with a highly conserved −1 subsite and no obvious conservation of the +1 subsite architecture.
AGPs are important polysaccharides in plant biology and in the food industry. Their structures, however, are very diverse. The novel activities and structural details described here makes an important contribution to the toolbox of biocatalysts available to dissect the structure of these complex glycans and to generate bespoke industrially relevant AGP-derived oligosaccharides particularly for the food industry.
Experimental procedures
Materials
4-Nitrophenyl-α-l-rhamnopyranoside (4NP-Rha), 4-nitrophenyl-β-d-glucuronide (4NP-GlcA), GA, EDTA, l-rhamnose, and d-glucuronic acid were purchased from Sigma. Larch arabinogalactan was from Megazyme (Dublin, Ireland). All reagents were of analytic grade.
Cloning, expression, and purification
BT0263 and BT3687 were amplified from B. thetaiotaomicron genomic DNA and cloned into pET28a with an N-terminal His6 tag using NheI and XhoI restriction sites. To generate DNA encoding BACCELL_00875 and BACFIN_07013, the sequence of the protein was used as template for gene synthesis with codon optimization for Escherichia coli heterologous production (Biomatik, Cambridge, Canada) and was subsequently cloned into the pET28a vector. The genes were then expressed in E. coli BL21, or Tuner cells, transformed with the appropriate recombinant plasmids. The recombinant E. coli strains were cultured in Luria broth (LB) supplemented with 50 μg/ml of kanamycin. Cultured cells were grown at 37 °C to mid-log phase and induced with 1 mm isopropyl β-d-1-thiogalactopyranoside at 16 °C overnight. Cells were pelleted by centrifugation at 5,000 rpm for 10 min and resuspended in 20 mm Tris-HCl buffer, pH 8.0, containing 300 mm NaCl. For selenomethionine-derivatized protein the above procedure was used but adjusted as follows: E. coli B834 cells were transformed with the appropriate recombinant plasmid. Overnight 5-ml cultures, in LB, were then used to inoculate 100 ml of LB culture in a 250-ml flask, which was then grown to an O.D. of 0.4. A methionine-deficient media was prepared using the Molecular Dimensions SelenoMetTM Medium Base (MD12-501) and SelenoMetTM Nutrient mixtures (MD12-502) and was used to wash the cultured B834 cells. The cells were then inoculated into 1 liter of methionine-deficient media to which selenomethione was added to a final concentration of 5 mg/ml. Cells were collected and disrupted by sonication, and the cell-free extract was recovered by centrifugation at 15,000 rpm for 30 min. Recombinant proteins were purified from the cell-free extract using immobilized metal affinity chromatography using TalonTM, a cobalt-based matrix. Proteins were eluted from the column in Buffer A containing 100 mm imidazole. For crystallographic studies, BT0263 and BT3687 were further purified by size exclusion chromatography using a Superdex S200 16/600 column equilibrated with Buffer A on a fast protein liquid chromatography system (ÄKTA FPLC; GE Healthcare). All proteins were purified to electrophoretic homogeneity as judged by SDS-PAGE.
Mutagenesis
Site-directed mutagenesis was conducted using the PCR-based QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions, using the plasmid encoding BACCELL_00875 and BT3687 as the template and appropriate primer pairs.
Purification of oligosaccharides
GA (10 mg/ml) was treated with an exo-β1,3-d-galactanase from B. thetaiotaomicron (BT0265), which cleaves the β1,3-galactan backbone of the polysaccharide. The active site of the enzyme can bind galactose residues in the β1,3-galactan backbone that are decorated at O6 with oligosaccharide side chains. Thus, upon glycosidic bond cleavage of the backbone the galactose residues released carry their oligosaccharide side chains. The different oligosaccharide side chains generated by BT0265 were purified by size exclusion chromatography using a P-2 Gel filtration column (Bio-Rad). The column was equilibrated with 50 mm acetic acid and the same buffer was used in the chromatographic separation. To identify the targets, the samples were digested with BT0263 (1 μm, overnight at 37 °C) and the reactions were run on high-pressure anion exchange chromatography with pulsed amperometric and UV-visible detectors. Each sample was dried and stored at −20 °C until use.
Enzyme assays
All enzyme assays unless otherwise stated were carried out in 20 mm sodium phosphate buffer, pH 7.0, containing 150 mm NaCl and performed by triplicate. Assays were carried out with 1 μm BT0263, BACCELL_00875, BACFIN_07013, and BT3687 against 1–10 mg/ml of substrate at 37 °C. Aliquots were taken over a 16-h time course, and samples and products were assessed by TLC and high-pressure anion exchange chromatography with pulsed amperometric detection. Sugars were separated on a Carbopac PA1 guard and analytical column in an isocratic program of 100 mm sodium hydroxide and then with a 40% linear gradient of sodium acetate over 60 min. Sugars were detected using the carbohydrate standard quad waveform for electrochemical detection at a gold working electrode with an Ag/AgCl pH reference electrode. Kinetic parameters were determined using the l-rhamnose detection kit from Megazyme International, measuring the release of rhamnose by absorbance of 340 nm. To determine kinetic parameters, 1 μm of the appropriate enzyme was assayed against varying concentrations of polysaccharide or oligosaccharides between 0.1 and 3 mm. l-Rhamnose release was measured, and the values were plotted using linear regression giving kcat/Km as the slope of the line. Mutants were assessed for activity against GA at 1 mg/ml with varying enzyme concentrations between 1 and 10 μm with assays running for minutes (wild type) up to days (mutants displaying very little activity).
Mass spectrometry
BT0263 was incubated with the GA-derived side chain oligosaccharides overnight at 37 °C. To identify the size of each oligosaccharide the enzymatic reaction products and the original untreated glycans were per-O-methylated to improve the mass spectrometric response (38). Excess protein was removed by filtering the reaction mixture over a 0.5-ml Dowex anion-cation exchange resin mixture (Sigma). Permethylated glycans were redissolved in 10 μl of methanol. One microliter of the permethylated glycan solution was mixed with 1 μl of a solution of 10 mg/ml of 2,5-dihydroxybenzoic acid in acetonitrile; 0.7 ml of this mixture was spotted onto a stainless steel MALDI plate to air dry. Positive-ion MALDI mass spectra were obtained using an Ultraflex III mass spectrometer (Bruker) in reflectron mode, equipped with a Nd:yttrium/aluminum garnet SmartbeamTM laser. Mass spectra were acquired over the m/z range 250–3,000 with ion suppression below 700. The laser power setting varied around 50% of maximum with each spectrum acquired using between 1,500 and 4,000 laser shots in total, using Bruker flexControl software. Mass spectra were externally calibrated against an adjacent spot containing six peptides (des-Arg1-Bradykinin, 904.681; Angiotensin I, 1,296.685; Glu1-Fibri-nopeptide B, 1,750.677; adrenocorticotropic hormone fragment (ACTH) (1–17 clip), 2,093.086; ACTH (18–39 clip), 2,465.198; ACTH (7–38 clip), 3,657.929 (Sigma)). Bruker flexAna-lysis software (version 3.3) was used to perform the spectral processing.
Crystallization, data collection, structure solution, and refinement
For crystallization trials, immobilized metal affinity chromatography-purified protein was concentrated and further purified by gel filtration chromatography using a Superdex S200 16/600 column equilibrated in Buffer A. BT3687, at 15 mg/ml, was crystallized using the sitting drop vapor diffusion method with 200 mm sodium malonate and 20% polyethylene glycol (PEG) 3350. SeMet-BACCELL_00875 and native BACCELL_00875, at 10 mg/ml, were crystallized from 25% PEG1500, 0.1 m MMT buffer (dl-malic acid, MES, and TRIS (1:2:2 molar ratio)), pH 4.0. For data collection, the samples were transferred to cryo-protecting solution consisting of mother liquor supplemented with 15–20% (v/v) PEG 400. Alternatively Paratone-N oil was used to replace the mother liquor before cooling the sample in liquid nitrogen. Diffraction data were collected using synchrotron radiation at the Diamond Light Source on beamlines I02 and I04. The data were integrated using XDS (39) or iMosflm (40); scaled and merged with Aimless (41). The phase problem for BACCELL_00875 was solved by SeMet-single-wavelength anomalous dispersion using Hkl2map (42) and the Shelx suite (43). Native data of BACCELL_00875 was solved using Phaser (44) using the SeMet-solved structure as search model. BT3687 was solved by molecular replacement using MrBump (45). The best solution from MrBump was with search model 4CE7 prepared with Chainsaw (46) and solved with Phaser (44). Buccaneer (47) and/or Arp-warp (48) were used for automated model building where needed. Recursive cycles of manual model building in COOT (47) and automatic refinement in Refmac5 (49) were performed to produce the final model; 5% of the observations were randomly selected for the Rfree set. All models were validated using Molprobity (50). The data statistics and refinement details are reported in Table 4.
Table 4.
Crystallography statistics table
| Data collection | BACCELL_00875_SeMet | BACCELL_00875 | BT3687 |
|---|---|---|---|
| Date | 31/07/16 | 31/07/16 | 24/04/14 |
| Source | Diamond I02 | Diamond I02 | Diamond I04 |
| Wavelength (Å) | 0.98 | 0.98 | 0.98 |
| Space group | P41212 | P3221 | H32 |
| Cell dimensions | |||
| a, b, c (Å) | 116.7, 116.7, 228.5 | 117.6, 117.6, 202.0 | 88.4, 88.4, 215.8 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 120 | 90, 90, 120 |
| No. of measured reflections | 3,309,700 (174,829)a | 1,490,439 (177,287) | 771,167 (15,963) |
| No. of independent reflections | 76,640 (4,464) | 73,190 (8,697) | 87,238 (4,048) |
| Resolution (Å) | 47.48–2.24 (2.29–2.24) | 45.45–1.70 (1.73–1.70) | 21.13–1.26 (1.28–1.26) |
| CC1/2 | 0.997 (0.707) | 0.998 (0.496) | 0.998 (0.524) |
| Mean I/σI | 10.6 (1.7) | 10.8 (1.5) | 13.0 (2.4) |
| Completeness | 100 (100) | 100 (100) | 99.5 (94.3) |
| Redundancy | 43.2 (39.2) | 8.4 (8.4) | 8.8 (3.9) |
| Anomalous completeness | 100 (99.8) | ||
| Anomalous redundancy | 22.5 (19.8) | ||
| Refinement | |||
| Rwork/Rfree | 20.0/25.0 | 16.0/18.0 | 12.1/14.8 |
| No. atoms | |||
| Protein | 10,688 | 10,920 | 2,914 |
| Water | 244 | 1,019 | 299 |
| B-factors | |||
| Protein | 41.5 | 20.9 | 12.0 |
| Water | 36.8 | 32.0 | 23.5 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.0110 | 0.0107 | 0.0127 |
| Bond angles (°) | 1.48 | 1.45 | 1.52 |
| PDB code | 5NOK | 5NO8 | 5NOA |
a Values in parentheses represent outer shell.
Family delineation and phylogeny
BT0263 was used as an initial query against all available genomes in GenBankTM. Proteins with >60% identity were aligned to identify domain boundaries based on sequence conservation, and then used to build an HMM with HMMER (55). This model capturing the position-specific signal of conserved/unconstraint residues was used to detect remote family members and iteratively rebuilt until family convergence, which occurred immediately. The sequences of family members were then aligned with Multalin (51) and the resulting alignment was used to construct a neighbor joining tree using BLOSUM62 substitution parameters (52) and BIO-NJ (53) and the tree was visualized using Dendroscope (54).
Author contributions
J. L. M.-M. preformed biochemistry as described in the paper; A. C. preformed the crystal structure of Baccell_00875; A. B. preformed the crystal structure of BT3687; N. T. and B. H. performed the phylogeny and H. J. G. supervised the work of J. L. M.-M. and A. C. and contributed to writing the manuscript.
Supplementary Material
Acknowledgment
We thank Diamond Light Source for access to beamline I02 and I04 (mx9948 and mx13587) that contributed to the results presented here.
This work was supported by the European Research Council (ERC) Grant Agreement 322820. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1 and S2.
The atomic coordinates and structure factors (codes 5NOK, 5NO8, and 5NOA) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HGM
- human gut microbiota
- PUL
- polysaccharide utilization locus
- GH
- glycoside hydrolase
- PL
- polysaccharide lyase
- AGP
- arabinogalactan protein
- GA
- gum arabic GlcA, d-glucuronic acid
- Rha
- α-l-rhamnose
- UA
- uronic acid
- SeMet
- selenomethionine
- r.m.s.
- root mean square
- PDB
- Protein Data Bank
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid.
References
- 1. Lozupone C. A., Stombaugh J. I., Gordon J. I., Jansson J. K., and Knight R. (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martens E. C., Lowe E. C., Chiang H., Pudlo N. A., Wu M., McNulty N. P., Abbott D. W., Henrissat B., Gilbert H. J., Bolam D. N., and Gordon J. I. (2011) Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLos Biol. 9, e1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cuskin F., Lowe E. C., Temple M. J., Zhu Y., Cameron E. A., Pudlo N. A., Porter N. T., Urs K., Thompson A. J., Cartmell A., Rogowski A., Hamilton B. S., Chen R., Tolbert T. J., Piens K., et al. (2015) Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature 517, 165–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larsbrink J., Rogers T. E., Hemsworth G. R., McKee L. S., Tauzin A. S., Spadiut O., Klinter S., Pudlo N. A., Urs K., Koropatkin N. M., Creagh A. L., Haynes C. A., Kelly A. G., Cederholm S. N., Davies G. J., Martens E. C., and Brumer H. (2014) A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506, 498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ndeh D., Rogowski A., Cartmell A., Luis A. S., Baslé A., Gray J., Venditto I., Briggs J., Zhang X., Labourel A., Terrapon N., Buffetto F., Nepogodiev S., Xiao Y., Field R. A., et al. (2017) Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature 544, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rogowski A., Briggs J. A., Mortimer J. C., Tryfona T., Terrapon N., Lowe E. C., Baslé A., Morland C., Day A. M., Zheng H., Rogers T. E., Thompson P., Hawkins A. R., Yadav M. P., Henrissat B., et al. (2015) Glycan complexity dictates microbial resource allocation in the large intestine. Nat Commun 6, 7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koropatkin N. M., Cameron E. A., and Martens E. C. (2012) How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 10, 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martens E. C., Koropatkin N. M., Smith T. J., and Gordon J. I. (2009) Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J. Biol. Chem. 284, 24673–24677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aspeborg H., Coutinho P. M., Wang Y., Brumer H. 3rd, Henrissat B. (2012) Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol. Biol. 12, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaspar Y., Johnson K. L., McKenna J. A., Bacic A., and Schultz C. J. (2001) The complex structures of arabinogalactan-proteins and the journey towards understanding function. Plant Mol. Biol. 47, 161–176 [PubMed] [Google Scholar]
- 12. Nie S. P., Wang C., Cui S. W., Wang Q., Xie M. Y., and Phillips G. O. (2013) The core carbohydrate structure of Acacia seyal var. seyal (gum arabic). Food Hydrocolloids 32, 221–227 [Google Scholar]
- 13. Inaba M., Maruyama T., Yoshimi Y., Kotake T., Matsuoka K., Koyama T., Tryfona T., Dupree P., and Tsumuraya Y. (2015) l-Fucose-containing arabinogalactan-protein in radish leaves. Carbohydr. Res. 415, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verbeken D., Dierckx S., and Dewettinck K. (2003) Exudate gums: occurrence, production, and applications. Appl. Microbiol. Biotechnol. 63, 10–21 [DOI] [PubMed] [Google Scholar]
- 15. Speciale G., Thompson A. J., Davies G. J., and Williams S. J. (2014) Dissecting conformational contributions to glycosidase catalysis and inhibition. Curr. Opin. Struct. Biol. 28, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu Y., Suits M. D., Thompson A. J., Chavan S., Dinev Z., Dumon C., Smith N., Moremen K. W., Xiang Y., Siriwardena A., Williams S. J., Gilbert H. J., and Davies G. J. (2010) Mechanistic insights into a Ca2+-dependent family of α-mannosidases in a human gut symbiont. Nat. Chem. Biol. 6, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Davies G. J., Wilson K. S., and Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garron M. L., and Cygler M. (2014) Uronic polysaccharide degrading enzymes. Curr. Opin. Struct. Biol. 28, 87–95 [DOI] [PubMed] [Google Scholar]
- 19. Nie S. P., Wang C., Cui S. W., Wang Q., Xie M. Y., and Phillips G. O. (2013) A further amendment to the classical core structure of gum arabic (Acacia senegal). Food Hydrocolloids 31, 42–48 [Google Scholar]
- 20. Munoz-Munoz J., Cartmell A., Terrapon N., Henrissat B., and Gilbert H. J. (2017) Unusual active site location and catalytic apparatus in a glycoside hydrolase family. Proc. Natl. Acad. Sci. U.S.A. 114, 4936–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mewis K., Lenfant N., Lombard V., and Henrissat B. (2016) Dividing the large glycoside hydrolase family 43 into subfamilies: a motivation for detailed enzyme characterization. Appl. Environ. Microbiol. 82, 1686–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sakamoto T., Tanaka H., Nishimura Y., Ishimaru M., and Kasai N. (2011) Characterization of an exo-β-1,3-d-galactanase from Sphingomonas sp. 24T and its application to structural analysis of larch wood arabinogalactan. Appl. Microbiol. Biotechnol. 90, 1701–1710 [DOI] [PubMed] [Google Scholar]
- 23. Ichinose H., Kotake T., Tsumuraya Y., and Kaneko S. (2006) Characterization of an exo-β-1,3-d-galactanase from Streptomyces avermitilis NBRC14893 acting on arabinogalactan-proteins. Biosci. Biotechnol. Biochem. 70, 2745–2750 [DOI] [PubMed] [Google Scholar]
- 24. Charnock S. J., Brown I. E., Turkenburg J. P., Black G. W., and Davies G. J. (2002) Convergent evolution sheds light on the anti-β-elimination mechanism common to family 1 and 10 polysaccharide lyases. Proc. Natl. Acad. Sci. U.S.A. 99, 12067–12072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garron M. L., and Cygler M. (2010) Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 20, 1547–1573 [DOI] [PubMed] [Google Scholar]
- 26. Preiss J., and Ashwell G. (1962) Alginic acid metabolism in bacteria. I. Enzymatic formation of unsaturated oligosac-charides and 4-deoxy-l-erythro-5-hexoseulose uronic acid. J. Biol. Chem. 237, 309–316 [PubMed] [Google Scholar]
- 27. Jongkees S. A., Yoo H., and Withers S. G. (2014) Mechanistic investigations of unsaturated glucuronyl hydrolase from Clostridium perfringens. J. Biol. Chem. 289, 11385–11395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Itoh T., Hashimoto W., Mikami B., and Murata K. (2006) Crystal structure of unsaturated glucuronyl hydrolase complexed with substrate: molecular insights into its catalytic reaction mechanism. J. Biol. Chem. 281, 29807–29816 [DOI] [PubMed] [Google Scholar]
- 29. Ochiai A., Yamasaki M., Mikami B., Hashimoto W., and Murata K. (2010) Crystal structure of exotype alginate lyase Atu3025 from Agrobacterium tumefaciens. J. Biol. Chem. 285, 24519–24528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson A. J., Cuskin F., Spears R. J., Dabin J., Turkenburg J. P., Gilbert H. J., and Davies G. J. (2015) Structure of the GH76 α-mannanase homolog, BT2949, from the gut symbiont Bacteroides thetaiotaomicron. Acta Crystallogr. D Biol. Crystallogr. 71, 408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Collén P. N., Jeudy A., Sassi J. F., Groisillier A., Czjzek M., Coutinho P. M., and Helbert W. (2014) A novel unsaturated β-glucuronyl hydrolase involved in ulvan degradation unveils the versatility of stereochemistry requirements in family GH105. J. Biol. Chem. 289, 6199–6211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Itoh T., Ochiai A., Mikami B., Hashimoto W., and Murata K. (2006) A novel glycoside hydrolase family 105: the structure of family 105 unsaturated rhamnogalacturonyl hydrolase complexed with a disaccharide in comparison with family 88 enzyme complexed with the disaccharide. J. Mol. Biol. 360, 573–585 [DOI] [PubMed] [Google Scholar]
- 34. Huang W., Boju L., Tkalec L., Su H., Yang H. O., Gunay N. S., Linhardt R. J., Kim Y. S., Matte A., and Cygler M. (2001) Active site of chondroitin AC lyase revealed by the structure of enzyme-oligosaccharide complexes and mutagenesis. Biochemistry 40, 2359–2372 [DOI] [PubMed] [Google Scholar]
- 35. Seyedarabi A., To T. T., Ali S., Hussain S., Fries M., Madsen R., Clausen M. H., Teixteira S., Brocklehurst K., and Pickersgill R. W. (2010) Structural insights into substrate specificity and the anti-β-elimination mechanism of pectate lyase. Biochemistry 49, 539–546 [DOI] [PubMed] [Google Scholar]
- 36. Jenkins J., Shevchik V. E., Hugouvieux-Cotte-Pattat N., and Pickersgill R. W. (2004) The crystal structure of pectate lyase Pel9A from Erwinia chrysanthemi. J. Biol. Chem. 279, 9139–9145 [DOI] [PubMed] [Google Scholar]
- 37. Rye C. S., and Withers S. G. (2000) Glycosidase mechanisms. Curr. Opin. Chem. Biol. 4, 573–580 [DOI] [PubMed] [Google Scholar]
- 38. Ciucanu I., and Kerek F. (1984) A simple and rapid method for the permethylation of carbohydrates. Carbohydr. Res. 131, 209–217 [Google Scholar]
- 39. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pape T., and Schneider T. R. (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr. 37, 843–844 [Google Scholar]
- 43. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Keegan R. M., and Winn M. D. (2007) Automated search-model discovery and preparation for structure solution by molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 63, 447–457 [DOI] [PubMed] [Google Scholar]
- 46. Stein N. (2008) CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 [Google Scholar]
- 47. Cowtan K. (2008) Fitting molecular fragments into electron density. Acta Crystallogr. D Biol. Crystallogr. 64, 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., and Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 50. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Corpet F. (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Henikoff S., and Henikoff J. G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 89, 10915–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gascuel O. (1997) BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 [DOI] [PubMed] [Google Scholar]
- 54. Huson D. H., Richter D. C., Rausch C., Dezulian T., Franz M., and Rupp R. (2007) Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics 8, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Finn R. D., Clements J., and Eddy S. R. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.