

Abstract
Polycomb-group proteins control many fundamental biological processes, such as anatomical development in mammals and vernalization in plants. Polycomb repressive complex 2 (PRC2) is responsible for methylation of histone H3 lysine 27 (H3K27), and trimethylated H3K27 (H3K27me3) is implicated in epigenetic gene silencing. Recent genomic, biochemical, and structural data indicate that PRC2 is broadly conserved from yeast to human in many aspects. Here, we determined the crystal structure of an apo-PRC2 from the fungus Chaetomium thermophilum captured in a bona fide autoinhibited state, which represents a novel conformation of PRC2 associated with enzyme regulation in light of the basal and stimulated states that we reported previously. We found that binding by the cofactor S-adenosylmethionine mitigates this autoinhibited structural state. Using steady-state enzyme kinetics, we also demonstrated that disrupting the autoinhibition results in a vastly activated enzyme complex. Autoinhibition provides a novel structural platform that may enable control of PRC2 activity in response to diverse transcriptional states and chromatin contexts and set a ground state to allow PRC2 activation by other cellular mechanisms as well.
Keywords: crystal structure, enzyme, epigenetics, histone methylation, polycomb
Introduction
Polycomb-group proteins control many fundamental biological phenomena, such as formation of the body plan in mammals and vernalization in plants (1). Polycomb-group proteins form at least two types of distinct but interconnected histone-modifying enzyme complexes, polycomb repressive complexes 1 and 2 (PRC12 and PRC2), to influence the establishment, maintenance, and inheritance of the cell type-specific transcriptional program during development. PRC1 mediates monoubiquitination of histone H2AK119 and PRC2 catalyzes histone H3K27 methylation. PRC2 favors unmethylated H3K27 (H3K27me0) and, to a lesser extent, monomethylated H3K27 (H3K27me1) as the substrates and displays little activity on dimethylated H3K27 (H3K27me2) (2). Trimethylated H3K27 (H3K27me3) is a hallmark of chromatin that is transcriptionally silent. Dysregulation of the enzymatic activity of PRC2 is found in various cancers (3).
A canonical PRC2 contains Ezh2 (a SET domain methyltransferase), Suz12, Eed, and Rppb4. Ezh2 is inactive without Eed and Suz12 (4). In lower eukaryotes such as fungi, PRC2-like complexes have been found to mediate H3K27 methylation as well (5–8). Despite their limited sequence conservation, fungal and human PRC2 exhibit compositional and functional similarities, indicating that they may share common regulatory mechanisms.
The enzymatic activity of PRC2 is subjected to intricate cellular regulation. In particular, PRC2 catalysis is stimulated by H3K27me3, which is thought to account for spreading of the repressive H3K27me3 histone mark in formation of facultative heterochromatin (9, 10). Intriguingly, nascent RNAs bind to PRC2 and inhibit PRC2 catalysis, which may serve as a cellular mechanism to prevent gratuitous silencing of active genes by PRC2 (11–13). In addition, transcriptional states and histone modifications also influence PRC2 activity on chromatin. Transcriptional repression facilitates PRC2 recruitment to target loci (14–16). Active histone marks including H3K4me3 and H3K36me2/3 inhibit the enzymatic activity of PRC2 (15, 16).
We recently determined the crystal structures of an active PRC2 from Chaetomium thermophilum (referred to as “ct” hereinafter) in both the basal and H3K27me3-stimulated states, which provided the structural framework for understanding the mechanisms of PRC2 enzyme regulation (8). Human PRC2 bears a remarkable structural resemblance to ctPRC2 (17, 18). Unlike other families of lysine methyltransferases, Ezh2 contains an unusual split catalytic domain that consists of the SET activation loop (SAL) and the canonical SET domain from the N- and C-terminal portions of Ezh2, respectively (8, 17, 18). Additionally, in the H3K27me3-stimulated state, the flexible stimulation-responsive motif of Ezh2 binds to the H3K27me3 histone tail together with Eed and transmits the stimulating signal to the catalytic domain allosterically (8, 17).
Here, we report a novel stable conformational state of ctPRC2 that is distinct from the basal and H3K27me3-stimulated states and that exhibits an autoinhibited structure. The post-SET subdomain of the catalytic SET domain stably binds to the active site in the absence of the histone substrate and cofactor SAM, occupies the histone substrate-binding groove, blocks the SAM-binding pocket, and hence impedes catalysis. Additional structural evidence indicates that SAM binding partially alleviates the autoinhibition of ctPRC2. Disruption of the autoinhibited conformation results in overactivated ctPRC2 caused by increased histone substrate binding as shown by steady-state enzyme kinetics studies of the wild-type and a gain-of-function mutant ctPRC2. The crystal structure of the gain-of-function mutant reveals destabilization of the autoinhibited conformation of the post-SET and is in agreement with the increase in activity displayed by this mutant. The autoinhibition may not only set a threshold for the enzyme complex to sense the effective concentration of the histone substrate but may also serve as a structural foundation for enzyme activation by some cellular mechanisms.
Results
Apo-ctPRC2 adopts an autoinhibited conformation
The previously solved crystal structures of ctPRC2 provided a wealth of insight into the catalytic mechanism of this enzyme complex (8). The catalytic SET domain of Ezh2 is highly conserved in yeast species that encode PRC2-like complexes for H3K27 methylation, including pathogenic species in both plant (Fusarium graminearum) and human (Cryptococcus neoformans) (Fig. 1A). We determined the crystal structure of an apo-ctPRC2 at 2.2-Å resolution in which ctPRC2 adopted a stable autoinhibited conformation (supplemental Table S1 and Fig. 1, B and 1C). Compared with the two previously known states, the entire post-SET subdomain (residues 920–933 of ctEzh2) undergoes a dramatic conformational change in the current autoinhibited ctPRC2 (Fig. 1C). In the basal and stimulated states, the post-SET bends into a square shape to cage SAM and participate in histone substrate binding (Fig. 1C, green schematic, and detailed in supplemental Fig. S1) in a manner similar to that of other lysine methyltransferases. In stark contrast, the post-SET of the autoinhibited ctPRC2 is intimately associated with the substrate-binding groove, directly blocking histone substrate binding (Fig. 1C, gold schematic, and detailed in supplemental Fig. S1). The captured autoinhibited conformation is unlikely an artifact of crystal packing, which is located far away from the post-SET (supplemental Fig. S2).
Figure 1.

The autoinhibited conformation of ctPRC2. A, sequence alignment of Ezh2 from fungal species. nc, Neurospora crassa; cn, C. neoformans; fg, F. graminearum. The SET-I (cyan) and post-SET (gold) subdomains are indicated by dotted boxes. Selected residues are designated by symbols as follows: purple stars, aromatic residues from the lysine access channel; green upward triangles, gain-of-function mutations; red downward arrows, loss-of-function mutations. The secondary structures of the post-SET subdomain are also indicated by cylinders (α-helices) and lines (loops) above the sequence alignment. B, overall structure of the apo-ctPRC2 complex. Subunits and domains are color-coded, and zinc ions are shown as gray spheres. C, zoomed-in view of the SET domain of Ezh2 of apo-ctPRC2 aligned with the structure of ctPRC2 in the stimulated state (Protein Data Bank code 5KKL). The post-SET in apo-ctPRC2 (gold) clashes with the substrate peptide (magenta), whereas the same region of ctPRC2 (green) supports SAM and histone substrate binding in the stimulated state. Residue Phe-922 is shown as sticks and would clash with the substrate residue Lys-27 in apo-ctPRC2 but not in the stimulated-state structure. D, intramolecular interactions hold the post-SET and SET-I regions in an autoinhibited conformation. Critical residues of the post-SET (gold) and SET-I (cyan) are labeled. Water molecules are depicted as red spheres. Hydrogen bonds are shown as dashed black lines.
In the autoinhibited ctPRC2, the post-SET folds into an extended loop followed by a short α-helix, and an additional five residues on the C terminus are ordered compared with the basal and stimulated states (Fig. 1C, gold schematic, and detailed in supplemental Fig. S1). Residues 840–855 of SET-I are folded differently compared with those in the basal and stimulated states to accommodate the post-SET (supplemental Fig. S3). The autoinhibited conformation is largely maintained by extensive hydrophobic and hydrogen-bonding interactions between the post-SET and SET-I (Fig. 1D and zoomed-in view in supplemental Fig. S4). In particular, the benzene ring of residue Phe-922 from the loop region of the post-SET is positioned into the lysine access channel, clashes with the binding site of the substrate lysine H3K27 (Fig. 1C), and is surrounded by a number of aromatic residues from the active site (Fig. 1D). This aromatic cage is involved in orientating the substrate lysine for catalysis in the basal and stimulated states. Residue Lys-852 from SET-I mediates a series of direct and water-mediated hydrogen-bonding interactions with the main-chain atoms of residues Phe-922 and Pro-923 of the post-SET, contributing to the unique autoinhibited conformation as well (Fig. 1D). Intriguingly, residue Tyr-855 of SET-I packs with residue Phe-922, and the same residue also lies in parallel and engages in a hydrophobic interaction with the alkyl portion of the side chain of residue Lys-852 (Fig. 1D). Finally, the carboxylate side chain of residue Glu-840 of SET-I forms hydrogen bonds with the backbone amides of both Leu-925 and Thr-926 in the helix region of the post-SET (Fig. 1D). Glu-840 and Lys-852 are also coupled through water-mediated hydrogen bonds (Fig. 1D and zoomed-in view in supplemental Fig. S4). Thus, the entire post-SET appears to be locked into a stable autoinhibited conformation through multiple intramolecular interactions of Ezh2 that completely block the histone substrate-binding groove of the enzyme.
In the autoinhibited state, SAM binding is also largely hampered. The post-SET is inaccessible for SAM stabilization due to formation of the autoinhibited conformation, and the SAM-binding pocket observed in the basal and stimulated states becomes incomplete here (Fig. 1C). Furthermore, to accommodate the autoinhibited conformation of the post-SET, some residues within SET-I also change conformation, which disfavors SAM binding (supplemental Fig. S3). Notably, residue Tyr-855 of SET-I swings across the SAM-binding pocket from a SAM-interacting position in the basal and stimulated states to a Phe-922- and Lys-852-interacting position in the autoinhibited state, which would also clash with SAM physically (Fig. 1C). Taken together, a set of overlapping residues from the catalytic SET domain make the autoinhibited state of ctPRC2 refractory to both histone substrate and SAM binding, indicative of a coupled dual inhibition mechanism.
SAM binding partially alleviates the autoinhibition
To interact with the histone substrate, the autoinhibited ctPRC2 must undergo a dramatic conformational change to remove the post-SET from the substrate-binding groove. To understand whether SAM binding promotes histone substrate binding by ctPRC2, we determined the co-crystal structure of ctPRC2 bound to SAM at 2.6-Å resolution in which the electron density for SAM was clearly observed, which is suggestive of stable association (supplemental Table S1 and Fig. 2A). SAM retains the majority of its interactions with ctPRC2 residues displayed in the basal and stimulated states and adopts essentially the same conformation as in those two states (Fig. 2A and detailed view in supplemental Fig. S5). In particular, the side chain of residue Tyr-855 of SET-I that would clash with SAM in the autoinhibited state is repositioned such that it hydrogen bonds to the carboxylate of the methionine moiety of SAM (Fig. 2A). This is a critical interaction given that the Y855F single mutation resulted in a notable decrease of the enzymatic activity (Fig. 2B and supplemental Fig. S6A). Accompanying the movement of residue Tyr-855, the side chain of residue Lys-852 of SET-I, which contacts the former in the autoinhibited state, is rotated away from the post-SET by over 4 Å such that it no longer interacts with the backbones of residues Phe-922 and Pro-923 (Fig. 2A and detailed view in supplemental Fig. S5). Moreover, the main chain carbonyl oxygen of residue Lys-852 becomes coordinated with the hydroxyls of the sugar moiety of SAM through hydrogen bonding (Fig. 2A and detailed view in supplemental Fig. S5). Notably, in the SAM-bound structure, residues 922–927 of the post-SET stay in the same conformation as in the autoinhibited state and thus fail to line the SAM-binding pocket (Fig. 2A), indicating that these residues may not be essential for capturing SAM initially but may instead help trap SAM in a stable and favorable position for catalysis upon histone substrate binding.
Figure 2.

ctPRC2 in the SAM-bound state. A, SAM binding alters autoinhibition. Alignment of the autoinhibited (light gray) and SAM-bound (blue) ctPRC2 structures. Red dotted arrows depict the motion of key residues, Lys-852 and Tyr-855, upon conformational change from the apo to SAM-bound structures. Fo − Fc electron density, contoured at 3σ (green mesh), is shown for the bound SAM (orange sticks). B, methyltransferase activity of wild-type and Y855F mutant ctEzh2 as determined by incorporation of [3H]methyl group from 3H-labeled SAM into an H3K27me0 peptide substrate by a filter binding assay; dpm were measured by 3H scintillation counting. Error bars represent the standard deviation of three replicates. **, p value <0.01 as determined by an unpaired, two-tailed t test compared with wild type. C, detailed view of the SAM-bound structure in the SAL (green) and SET-I (light blue) regions of Ezh2 critical for interaction with SAM (orange). 2Fo − Fc electron density, contoured at 1σ, for SAM is represented by dark blue mesh. Black dashed lines indicate carbon-oxygen hydrogen bonds. Placement of the histone substrate-mimicking H3K27M peptide is based on structural alignment with the stimulated-state structure (Protein Data Bank code 5KKL). D, methyltransferase activity assay for the SAL and SET-I mutations conducted in the same manner as described in B. *, p value <0.05; **, p value <0.01 as determined by an unpaired, two-tailed t test compared with wild type.
The crystal structure of SAM-bound ctPRC2 also provides a glimpse into the catalytic mechanism for the enzyme complex and in particular that involving the essential SAL of Ezh2, a unique structural feature of both yeast and human PRC2. Similar to many other lysine methyltransferases, the partially positively charged methyl group of SAM resides in and is geometrically restricted by a partially negatively charged aperture at the active site of ctPRC2 (Fig. 2C). The substrate lysine is inserted from the opposite direction and resides directly above the donor methyl group (Fig. 2C). This partially negatively charged surface is believed to mediate critical carbon-oxygen (CH–O) hydrogen bonds with SAM (19), and in ctPRC2 it consists of the main chain carbonyl oxygens of residues Ser-854, Arg-877, and Ile-879 and the phenol oxygen of residue Tyr-918. In particular, the position of the main-chain carbonyl oxygen of residue Arg-877 is dictated by two pairs of hydrogen-binding interactions between residues Arg-877 and Tyr-878 of the SET and residues His-307 and Ala-308 of the SAL, suggesting that the latter two residues serve an indirect but vital structural role in stabilizing the partially negatively charged surface for SAM binding (Fig. 2C). In line with this structural observation, catalysis was hampered for ctPRC2 harboring a single H307A or R877A mutation that disrupts these important hydrogen bonds (Fig. 2D and supplemental Fig. S6B).
Previously, we proposed that the SAL of Ezh2 activates the SET domain in the context of PRC2 by mediating the rigid-body rotation of the SET-I subdomain to open up the self-blocked substrate-binding groove found in an isolated Ezh2 (8, 20, 21). Here, by analyzing the structure of SAM-bound ctPRC2, we showed that the SAL additionally buttresses the partially negatively charged SAM-binding surface. Residues from this surface then stabilize and orientate SAM in a favorable position to facilitate the methyl transfer process. Residue Ile-879 is a component of the partially negatively charged surface, and intriguingly a gain-of-function mutation at the equivalent position in human Ezh2, A687V, is frequently found in non-Hodgkin's lymphoma (22), highlighting the role of SAM binding in fine control of PRC2 activity.
Overall, SAM binding does not lead to a global structural alteration of the autoinhibited state of ctPRC2. However, it causes a dramatic local conformational change of SET-I in residues 852–855, disrupts some of the intramolecular interactions between the post-SET and SET-I, and thus alters the autoinhibited conformation.
Loss of the autoinhibition results in an overactivated ctPRC2
Our structural studies indicate that the post-SET and SET-I subdomains form extensive intramolecular interactions whereby the autoinhibited conformation of ctPRC2 is stably maintained (Fig. 1D). To investigate the functional importance of the autoinhibition in solution, we carried out a systematic structure-guided mutagenesis study to disrupt the intramolecular binding interface. Residues Glu-840 and Lys-852 from SET-I were found to play a critical role in the autoinhibition because single mutation of either of them resulted in at least a 2-fold activation of ctPRC2 (Fig. 3A and supplemental Fig. S6C). As controls, mutation of residue Thr-926 that is involved in the autoinhibition only via its main-chain atoms and residue Asn-921 that does not directly contact SET-I caused little change in the enzymatic activity (Fig. 3A and supplemental Fig. S6C and Thr-926 and Asn-921 highlighted in Fig. 1D and supplemental Fig. S4).
Figure 3.

Loss of the autoinhibition and gain-of-function mutations of ctPRC2. A, relief of autoinhibition results in increased enzyme activity. A methyltransferase assay for ctEzh2 variants with an H3K27me0 peptide substrate was performed. Error bars represent the standard deviation of three replicates in A, B, and C. **, p value <0.01; ***, p value <0.001; ****, p value < 0.0001 as determined by an unpaired, two-tailed t test compared with wild type. B, methyltransferase activity of ctEzh2 wild type and gain-of-function double mutant on H3K27me1 and H3K27me2 peptide substrates. C, histone methyltransferase assay of the wild-type and gain-of-function double mutant ctPRC2 holoenzyme on a recombinant nucleosomal substrate reconstituted with a 147-bp “601” DNA and Xenopus laevis core histones. D, enzyme kinetics analysis of ctEzh2 wild type and gain-of-function double mutant with a titration of H3K27me0 peptide substrate under 20 μm SAM. Error bars on the fitting curve represent the standard deviation of three replicates per titration point. Standard error of kinetic parameters is represented in the table. E, enzyme kinetics analysis of ctEzh2 wild type and gain-of-function double mutant with a titration of SAM under 50 μm H3K27me0 peptide substrate. Error bars on the fitting curve represent the standard deviation of three replicates per titration point. Standard error of kinetic parameters is represented in the table.
Notably, an E840A/K852D double mutant of ctPRC2 was about 4-fold more active than its wild-type counterpart (Fig. 3A and supplemental Fig. S6C). This gain-of-function mutant was also overactivated on the H3K27me1 and H3K27me2 peptide substrates, indicating that loss of the autoinhibition resulted in enhanced catalysis toward all the methylation states of H3K27 (Fig. 3B). Furthermore, the same double mutation in the context of a four-subunit ctPRC2 holoenzyme displayed a robust increase in enzymatic activity toward a nucleosomal substrate as well (Fig. 3C).
Residue Phe-922 from the post-SET is inserted into the lysine access channel of the active site (Fig. 1C) and may intuitively be important for the autoinhibition. However, mutation of residue Phe-922 led to a complete loss of activity (Fig. 3A and supplemental Fig. S6C), suggesting that this residue is important for binding of the H3K27 substrate when ctPRC2 is switched from the autoinhibited state to the basal and stimulated states for catalysis (8).
To obtain a more quantitative and mechanistic view, we next measured the steady-state enzyme kinetics of the wild-type and gain-of-function mutant ctPRC2 using a titration of the H3K27me0 substrate peptide and a saturating SAM concentration of 20 μm (Fig. 3D and supplemental Fig. S6D). An about 5-fold Km effect and an about 3-fold kcat effect were noted for the E840A/K852D double mutant, resulting in an about 15-fold increase in kcat/Km for the mutant compared with the wild-type complex (Fig. 3D). We also conducted a kinetic assay using a titration of SAM and a fixed concentration of 50 μm histone peptide substrate (Fig. 3E and supplemental Fig. S6D). In this case, the mutant displayed no Km effect but a 4-fold kcat effect, leading to an about 4-fold increase of the kcat/Km value for the mutant (Fig. 3E). Therefore, we conclude that enhanced histone substrate binding was primarily responsible for the observed overactivation of the mutant ctPRC2. This is also consistent with the structural observation that residues involved in histone substrate binding are sequestered in the autoinhibited state (Fig. 1, C and D).
To gain a structural insight into the consequence of disruption of autoinhibition, we solved the crystal structure of the ctPRC2 E840A/K852D double mutant at 2.1-Å resolution (supplemental Table S1). Although the post-SET of the wild-type ctPRC2 can be modeled up to residue Asn-933 (Fig. 4A), the E840A/K852D double mutation destabilized the post-SET and prohibited modeling beyond residue Lys-928 (Fig. 4B). Lack of electron density also prevented the K852D residue from being modeled and resulted in a loss of waters that bridge the interaction between SET-I and the post-SET in the wild-type ctPRC2 (Fig. 4, compare A and B). Overall, mutation of these two SET-I residues resulted in greater mobility of the post-SET as displayed by higher B-factors (supplemental Fig. S7). A more mobile post-SET is in accord with the loss of autoinhibition and subsequent tighter histone substrate binding as reflected by the enzymology results above (Fig. 3D).
Figure 4.
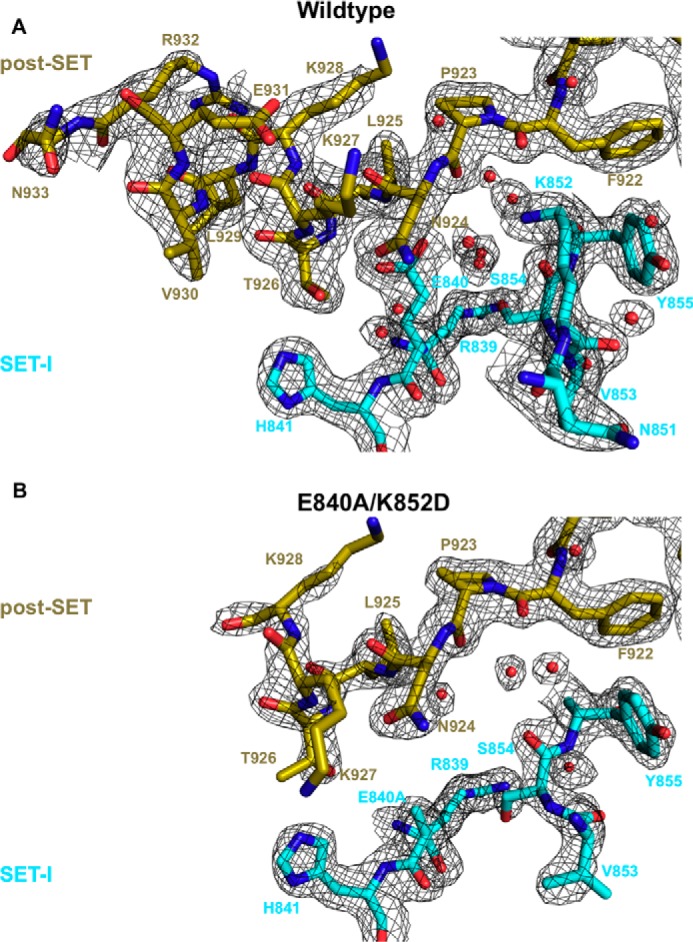
Structural comparison of apo wild-type (top) and gain-of-function double mutant (bottom) structures. Important residues of the post-SET (gold) and SET-I (cyan) subdomains are shown as sticks, and waters are shown as red spheres. 2Fo − Fc (black mesh) is contoured at 1σ.
Discussion
The autoinhibited (“A”) state described in this study represents a novel, stable conformational state of ctPRC2 that is likely present in PRC2 from other fungal species as well given the sequence conservation (Fig. 1A). SAM binding converts the A state to the “A′” state where the autoinhibited conformation is largely maintained. Together with the basal (“B”) and H3K27me3-stimulated (“S”) states revealed previously (8, 17, 18), these distinct stable conformational states provide interconnected structural platforms that may allow complex cellular regulation of the enzymatic activity of PRC2 in all stages of catalysis (Fig. 5).
Figure 5.

Schematic of the different stable conformational states of ctPRC2. Letter codes correspond to autoinhibited (A); SAM-bound, autoinhibited (A′), basal (B), and H3K27me3-stimulated (S) states based on the observed structural transitions of ctPRC2. A dotted black box indicates a destabilized conformation compared with that in a neighboring structural state. Note that although all the structural transitions are based on a minimally active PRC2 complex (i.e. Ezh2–Eed–Suz12(VEFS)), Suz12(VEFS) is omitted from all the schematics, and Eed is only shown in the S state for clarity. SRM, stimulation-responsive motif.
In the A state, PRC2 adopts a structural conformation that prevents both SAM and histone substrate binding and is therefore fully inhibited (Fig. 1D). The autoinhibition represents a widespread regulatory mechanism that prevents unwanted signaling pathways and is not unprecedented for lysine methyltransferases (23, 24). In the A′ state, SAM binding partially alleviates the autoinhibition, although ctPRC2 still remains largely autoinhibited when SAM is bound under the saturating SAM concentration (Fig. 2A). SAM is among the most regulated metabolites in different cell types, and the connection between metabolism and histone modification has recently started to emerge (25). Depending on the SAM concentration, the SAM-bound ctPRC2 may represent the major autoinhibited form. When restricted under extreme conditions, SAM availability may also regulate PRC2 activity due to the structurally observed partial relief of the autoinhibition.
Primary sequence analysis indicates that human Ezh2 is not conserved at residues Glu-840, Lys-852, and Phe-922 that are involved in autoinhibition in ctPRC2 (supplemental Fig. S8). Therefore, despite that human PRC2 displays a remarkable resemblance to ctPRC2 in the B and S states, it is unclear whether human PRC2 assumes a similar A state. Structural comparison suggests that when autoinhibited the SET domain of human Ezh2 shares some key features with its counterpart in the autoinhibited ctPRC2 (supplemental Fig. S9). SET-I and post-SET residues directly block histone substrate binding in the structure of an isolated SET domain of human Ezh2 and the structure of the human Ezh2 SET domain in an active drug-bound American chameleon/human PRC2 (18, 20, 21). In particular, in both cases, residue Phe-667 from SET-I directly clashes with the H3K27 substrate lysine residue in a manner similar to residue Phe-922 in ctPRC2 (supplemental Fig. S9). However, we have been unable to identify mutants of autoinhibitory residues of human Ezh2 that replicate the substantial increase in the enzymatic activity as observed for those in ctPRC2 (data not shown). Thus, it remains to be clarified whether autoinhibition in human PRC2 is as prominent of a feature as in ctPRC2.
The distribution of the repressive H3K27me3 histone mark on chromatin is determined by PRC2 recruitment and catalysis, which are profoundly impacted by transcriptional states and chromatin contexts in both human and fungi (7, 14–16, 26). The A state functions to set up a threshold to sense the effective concentration of the H3K27 substrate in different chromatin environments, which may be affected by active histone marks such as H3K4me3 and H3K36me2/me3 (15, 16). Furthermore, the enzymatic activity of human PRC2 is enhanced by Aebp2 and Jarid2 (4, 27). Similarly, fungal PRC2 may also be stimulated by other cellular factors. In this regard, the A state may serve to set a ground state for enzyme activation, and the stimulating cellular factors may act to release PRC2 from the A state. After the enzyme conformation is converted from the A state, H3K27 methylation takes place in the B state, and the resulting repressive H3K27me3 histone mark is propagated in the S state, which leads to formation of facultative heterochromatin and stable gene repression.
The gene repression function of PRC2 is directly correlated with its enzymatic activity. Cells have evolved to keep PRC2 activity in check. For example, RNAs and in particular nascent RNAs have been shown to inhibit PRC2 catalysis, which is thought to serve as a mechanism to prevent PRC2 from depositing the repressive H3K27me3 mark on active loci (11, 13). More generally, the A state increases the dynamic range of the enzymatic activity of PRC2 in cells, which may be best reflected in the long known biological complexity of gene expression during development.
Experimental procedures
Protein expression and purification
The PRC2 complex from C. thermophilum var. thermophilum DSM 1495 was expressed in Saccharomyces cerevisiae. A p416GAL1 vector (URA marker) contained a 2× Protein A-tagged fusion of Ezh2 (residues 191–950) and the Suz12 VEFS domain (residues 530–691). A tobacco etch virus protease cleavage sequence was contained between the Protein A tag and Ezh2. The Ezh2-Suz12(VEFS) fusion was co-transformed with a modified p416GAL1 vector (TRP marker) containing a 2× StrepII-tagged full-length Eed (residues 1–565) into S. cerevisiae CB010 strain (MATa pep4::HIS3 prb1::LEU2 prc::HISG can1 ade2 trp1ura3 his3 leu2–3,112), and transformants were selected on synthetic complete medium lacking uracil and tryptophan. Large-scale production was conducted in 30 liters of yeast peptone medium containing 2% galactose (YPG) in a fermentor at 30 °C overnight. Protein purification for ctPRC2 followed a similar procedure as detailed previously (8). Briefly, protein was purified by IgG-Sepharose followed by overnight cleavage with tobacco etch virus protease, purification over StrepTactin resin (IBA Life Sciences), and elution with d-desthiobiotin, concentration, and a final gel filtration step on Superdex 200 in 20 mm Tris-HCl, pH 8.0, 100 mm NaCl, and 2.5 mm DTT. Holo-ctPRC2 was expressed in S. cerevisiae and purified by an IgG affinity column as well.
Crystallization and data collection
The ctPRC2 complex at a concentration of 10 mg/ml was used for crystallization. Crystals of the apo, autoinhibited complex (wild type and E840A/K852D mutant) were produced by mixing an equal volume of protein solution with crystallization solution containing 100 mm sodium malonate, pH 7.0, and 16–20% PEG 3350 in hanging drop vapor diffusion trays. Diffraction quality crystals were grown by supplementing 43 mm 3-cyclohexyl-1-propylphosphocholine in drops. Crystals were cryoprotected in mother solution supplemented with 20% ethylene glycol and flash frozen in liquid nitrogen. To obtain the SAM-bound ctPRC2 structure, protein was co-crystallized with a 10-fold molar excess of SAM (New England Biolabs) by hanging drop vapor diffusion in the same solution as the apo crystals. Crystals were then cryoprotected in mother solution supplemented with 1 mm SAM and 20% ethylene glycol and flash cooled in liquid nitrogen.
Data collection
All data were collected using beamline 19ID at the Advanced Photon Source of Argonne National Laboratory and beamline 9-2 of the Stanford Synchrotron Radiation Lightsource. Data were collected on frozen crystals at a temperature of −180 °C using a wavelength of 0.979 Å on a Pilatus detector with an oscillation angle of 0.2–0.25°. Data were indexed, integrated, and scaled with the HKL3000 software package and then converted to an mtz file with the CCP4 program suite for use in structural determination (28, 29).
Structure determination and refinement
All structures were solved by molecular replacement with the program Phaser in the PHENIX software suite (30, 31). The structure of the apo-ctPRC2 complex was solved by using the structure of ctPRC2 in the stimulated state (Protein Data Bank code 5KJH) as a search model. The SAM-bound structure was solved by molecular replacement with the autoinhibited structure from this work. Manual building and adjustment were then performed with the program Coot. The structures were refined with Refmac5 and autoBUSTER followed by final refinement with PHENIX that utilized torsion, libation, screw (TLS) refinement (30, 32, 33).
Histone methyltransferase activity assays
For single-substrate concentration assays, 100 ng of ctPRC2 (29 nm) was mixed with 1 μm histone H3K27me0 peptide (residues 21–44; AnaSpec catalogue number AS-64641) and 1 μl of [3H]SAM (1 mCi stock; specific activity, 78–85 Ci/mmol; PerkinElmer Life Sciences) in a final volume of 20 μl (final concentration of about 0.33 μm [3H]SAM per reaction) in buffer containing 25 mm Tris, pH 8.0, 10 mm NaCl, 1 mm EDTA, 2.5 mm MgCl2, and 5 mm DTT. Assays conducted with mono- and dimethylated peptides (AnaSpec, catalogue numbers AS-64365 and AS-64366) used 233 ng of ctPRC2 (67 nm) and 1 μm peptide. Assays conducted with holoenzymes used 200 ng of enzyme and 1 μg of nucleosome per reaction. After incubation for 1 h at 30 °C, the reaction was stopped with 1.1 mm unlabeled SAM, and 10 μl was spotted onto phosphocellulose filters (Reaction Biology Corp.) and allowed to dry. Filters were washed five times with 50 ml of 50 mm NaCO3/NaHCO2, pH 9.0; once with 30 ml of 100% acetone; and dried. Filters were immersed in 4 ml of scintillation fluid and counted with a scintillation counter set to output readings as disintegrations per minute (dpm). Samples were background-corrected by subtracting with a reaction that contained protein but not peptide or nucleosome substrate. Individual experimental runs always contained a wild-type control, and data are represented compared with wild-type samples from the same experiment. Data are represented directly as scintillation output in dpm. All experiments were performed in triplicate. t test analysis was performed with GraphPad Prism 7 (GraphPad Software, La Jolla, CA).
Enzyme kinetics analysis
All enzyme kinetics analyses utilized purified proteins that were free of aggregation and nucleic acid contamination. Kinetics studies used 21.7 nm (75 ng) enzyme. Assays utilized either a titration of H3K27me0 peptide and a saturating 20 μm SAM concentration or a titration of SAM and a saturating 50 μm peptide concentration. Assays used various ratios of unlabeled:3H-labeled SAM, and activities were corrected based on these ratios. Experiments were performed in a manner analogous to those in the previous section, and each peptide concentration point was performed in triplicate. Disintegrations per minute were converted to curies (Ci) and then converted to mmol based on the specific activity of the batch of [3H]SAM (78–85 Ci/mmol), and enzyme activity data were then represented as velocity in nm/h. Data points were fit to enzyme saturation curves, and kinetics parameters and standard errors were calculated with the program GraphPad Prism 7 using Michaelis-Menten analysis.
Author contributions
X. L. conceived the study. M. B. and X. L. designed the experiments. M. B. performed all X-crystallography and all biochemical experiments. X. Y. prepared proteins for use in experiments. M. B. and X. L. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Lianying Jiao and Siming Chen from the Liu laboratory for helpful advice regarding expression and purification of PRC2 complexes. This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States DOE under Contract DE-AC02-05CH11231. Use of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, is supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including Grant P41GM103393).
This work was supported in part by Welch Foundation Research Grant I-1790, Cancer Prevention & Research Institute of Texas Research Grant R1119, a Rita Allen Foundation research grant, the UT Southwestern Medical Center Endowed Scholar fund, National Institutes of Health Grants GM114576 and GM121662 (to X. L.), and the Cecil H. and Ida Green Center Training Program in Reproductive Biology Sciences Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S9 and Table S1.
The atomic coordinates and structure factors (codes 5BJS, 5TQR, and 5VK3) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PRC
- polycomb repressive complex
- H3K27
- H3 lysine 27
- H3K27me3
- trimethylated H3K27
- H3K27me0
- unmethylated H3K27
- H3K27me1
- monomethylated H3K27
- H3K27me2
- dimethylated H3K27
- SET
- Su(var)3-9, Enhancer-of-zeste and Trithorax
- SAM
- S-adenosylmethionine
- ct
- C. thermophilum
- SAL
- SET activation loop
- A
- autoinhibited
- B
- basal
- S
- H3K27me3-stimulated
- VEFS
- VRN2, EMF2, FIS2, Su(z)12.
References
- 1. Whitcomb S. J., Basu A., Allis C. D., and Bernstein E. (2007) Polycomb group proteins: an evolutionary perspective. Trends Genet. 23, 494–502 [DOI] [PubMed] [Google Scholar]
- 2. McCabe M. T., Graves A. P., Ganji G., Diaz E., Halsey W. S., Jiang Y., Smitheman K. N., Ott H. M., Pappalardi M. B., Allen K. E., Chen S. B., Della Pietra A. 3rd, Dul E., Hughes A. M., Gilbert S. A., et al. (2012) Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. U.S.A. 109, 2989–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim K. H., and Roberts C. W. (2016) Targeting EZH2 in cancer. Nat. Med. 22, 128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cao R., and Zhang Y. (2004) SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 15, 57–67 [DOI] [PubMed] [Google Scholar]
- 5. Jamieson K., Rountree M. R., Lewis Z. A., Stajich J. E., and Selker E. U. (2013) Regional control of histone H3 lysine 27 methylation in Neurospora. Proc. Natl. Acad. Sci. U.S.A. 110, 6027–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Connolly L. R., Smith K. M., and Freitag M. (2013) The Fusarium graminearum histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genet. 9, e1003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dumesic P. A., Homer C. M., Moresco J. J., Pack L. R., Shanle E. K., Coyle S. M., Strahl B. D., Fujimori D. G., Yates J. R. 3rd, Madhani H. D. (2015) Product binding enforces the genomic specificity of a yeast polycomb repressive complex. Cell 160, 204–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jiao L., and Liu X. (2015) Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 350, aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Margueron R., Justin N., Ohno K., Sharpe M. L., Son J., Drury W. J. 3rd, Voigt P., Martin S. R., Taylor W. R., De Marco V., Pirrotta V., Reinberg D., and Gamblin S. J. (2009) Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hansen K. H., Bracken A. P., Pasini D., Dietrich N., Gehani S. S., Monrad A., Rappsilber J., Lerdrup M., and Helin K. (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 10, 1291–1300 [DOI] [PubMed] [Google Scholar]
- 11. Kaneko S., Son J., Bonasio R., Shen S. S., and Reinberg D. (2014) Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev. 28, 1983–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cifuentes-Rojas C., Hernandez A. J., Sarma K., and Lee J. T. (2014) Regulatory interactions between RNA and polycomb repressive complex 2. Mol. Cell 55, 171–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaneko S., Son J., Shen S. S., Reinberg D., and Bonasio R. (2013) PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat. Struct. Mol. Biol. 20, 1258–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Riising E. M., Comet I., Leblanc B., Wu X., Johansen J. V., and Helin K. (2014) Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell 55, 347–360 [DOI] [PubMed] [Google Scholar]
- 15. Schmitges F. W., Prusty A. B., Faty M., Stützer A., Lingaraju G. M., Aiwazian J., Sack R., Hess D., Li L., Zhou S., Bunker R. D., Wirth U., Bouwmeester T., Bauer A., Ly-Hartig N., et al. (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42, 330–341 [DOI] [PubMed] [Google Scholar]
- 16. Voigt P., LeRoy G., Drury W. J. 3rd, Zee B. M., Son J., Beck D. B., Young N. L., Garcia B. A., and Reinberg D. (2012) Asymmetrically modified nucleosomes. Cell 151, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Justin N., Zhang Y., Tarricone C., Martin S. R., Chen S., Underwood E., De Marco V., Haire L. F., Walker P. A., Reinberg D., Wilson J. R., and Gamblin S. J. (2016) Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 7, 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brooun A., Gajiwala K. S., Deng Y. L., Liu W., Bolaños B., Bingham P., He Y. A., Diehl W., Grable N., Kung P. P., Sutton S., Maegley K. A., Yu X., and Stewart A. E. (2016) Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat. Commun. 7, 11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horowitz S., Dirk L. M., Yesselman J. D., Nimtz J. S., Adhikari U., Mehl R. A., Scheiner S., Houtz R. L., Al-Hashimi H. M., and Trievel R. C. (2013) Conservation and functional importance of carbon-oxygen hydrogen bonding in AdoMet-dependent methyltransferases. J. Am. Chem. Soc. 135, 15536–15548 [DOI] [PubMed] [Google Scholar]
- 20. Wu H., Zeng H., Dong A., Li F., He H., Senisterra G., Seitova A., Duan S., Brown P. J., Vedadi M., Arrowsmith C. H., and Schapira M. (2013) Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS One 8, e83737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Antonysamy S., Condon B., Druzina Z., Bonanno J. B., Gheyi T., Zhang F., MacEwan I., Zhang A., Ashok S., Rodgers L., Russell M., and Gately Luz J. (2013) Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PLoS One 8, e84147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Majer C. R., Jin L., Scott M. P., Knutson S. K., Kuntz K. W., Keilhack H., Smith J. J., Moyer M. P., Richon V. M., Copeland R. A., and Wigle T. J. (2012) A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 586, 3448–3451 [DOI] [PubMed] [Google Scholar]
- 23. Pufall M. A., and Graves B. J. (2002) Autoinhibitory domains: modular effectors of cellular regulation. Annu. Rev. Cell Dev. Biol. 18, 421–462 [DOI] [PubMed] [Google Scholar]
- 24. Boriack-Sjodin P. A., and Swinger K. K. (2016) Protein methyltransferases: a distinct, diverse, and dynamic family of enzymes. Biochemistry 55, 1557–1569 [DOI] [PubMed] [Google Scholar]
- 25. Janke R., Dodson A. E., and Rine J. (2015) Metabolism and epigenetics. Annu. Rev. Cell Dev. Biol. 31, 473–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jamieson K., Wiles E. T., McNaught K. J., Sidoli S., Leggett N., Shao Y., Garcia B. A., and Selker E. U. (2016) Loss of HP1 causes depletion of H3K27me3 from facultative heterochromatin and gain of H3K27me2 at constitutive heterochromatin. Genome Res. 26, 97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li G., Margueron R., Ku M., Chambon P., Bernstein B. E., and Reinberg D. (2010) Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 24, 368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 29. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 33. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., and Womack T. O. (2016) BUSTER, version 2.10.2, Global Phasing Ltd., Cambridge, UK [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.