

Abstract
Recently, a single study revealed a new complex composed of Toll-like receptor 4 (TLR4), TLR6, and CD36 induced by fibrillary Aβ peptides, the hallmark of Alzheimer's disease. Unlike TLRs located on the plasma membrane that dimerize on the membrane after ligand binding to their extracellular domain, the TLR4-TLR6–CD36 complex assembly has been suggested to be induced by intracellular signals from CD36, similar to integrin inside-out signaling. However, the assembly site of TLR4-TLR6–CD36 and the domains participating in Aβ-induced signaling is still unknown. By interfering with TLR4-TLR6 dimerization using a TLR4-derived peptide, we show that receptor assembly is abrogated within the plasma membrane. Furthermore, we reveal that the transmembrane domains of TLR4 and TLR6 have an essential role in receptor dimerization and activation. Inhibition of TLR4-TLR6 assembly was associated with reduced secretion of proinflammatory mediators from microglia cells, ultimately rescuing neurons from death. Our findings support TLR4-TLR6 dimerization induced by Aβ. Moreover, we shed new light on TLR4-TLR6 assembly and localization and show the potential of inhibiting TLR4-TLR6 dimerization as a treatment of Alzheimer's disease.
Keywords: Alzheimer disease, inflammation, microglia, neurodegenerative disease, neuroinflammation, CD36, TLR4, TLR6, amyloid-β, toll-like receptor
Introduction
Toll-like receptors (TLRs)2 are type l transmembrane proteins that function as pattern recognition receptors that recognize pathogen-associated molecular patterns and danger-associated molecular patterns (1, 2). The activation of the TLRs requires a dimerization state that is specific to a particular ligand, e.g. lipopolysaccharide (LPS) from Gram-negative bacteria for TLR4 homodimerization and lipoteichoic acid (LTA) from Gram-positive bacteria for TLR2/6 heterodimerization. This is coordinated through ligand binding to the extracellular domain of the receptor leading to conformational changes throughout the protein (3). The conformational changes result in the induction of several intracellular signaling cascades leading to a secretion of a range of inflammatory cytokines and chemokines (4, 5). Recent studies proposed that the transmembrane domains (TMDs) of TLR2, TLR6, and TLR1 are involved in the regulation of the receptor's activity (6). Exogenously added peptides that correspond to the TMDs of either TLR2 or TLR6 were able to modulate the activity of these receptors in a mouse sepsis and acute colitis models (7, 8).
TLRs are highly associated with Alzheimer's disease (AD), especially TLR2 and TLR4, which have a main role in neuroinflammation after binding to fibrils of Aβ peptides (9–12). This is believed to be a major driving force that initiates the local inflammation response in AD (13–15). Extensive extracellular deposits of fibrillary Aβ peptides condense to form plaques in the brain, which leads to neurodegeneration. These structures are accompanied by activated microglia, the tissue resident macrophages of the central nervous system (CNS). The activation of the microglia is thought to be driven by a direct interaction of TLRs with Aβ (9–12, 16, 17), leading to the excessive secretion of proinflammatory mediators and neurotoxic factors, including reactive oxygen species, nitrogen oxide (NO), proteolytic enzymes, glutamate, inflammatory cytokines, chemokines, and complement factors (18). These proinflammatory factors cause progressive synaptic and neuritic injuries that eventually lead to dementia (19–22). On the other hand, studies showed that these cells could actually inhibit the progression of AD pathogenesis through phagocytic clearance of Aβ from the brain. However, a proinflammatory environment showed reduction in the phagocytic capacity of microglia cells (23, 24) and prolonged damage from microglia-mediated inflammatory response (18, 25), likely exacerbating disease pathogenesis. These findings indicate that activation of microglia through TLRs increases the levels of proinflammatory mediators but down-regulates their Aβ-clearance capacity in AD.
Recently, a new complex composed of the scavenger B receptor CD36, TLR4, and TLR6 was reported (26). TLR4 and TLR6 were shown to precipitate in the presence of CD36 in THP-1 monocyte and HEK397 cells induced by oxidized LDL or Aβ. As opposed to common TLR dimerization that occurs after binding of the ligand to the extracellular domain of TLRs (3), the assembly of this complex was suggested to be regulated by intracellular signals from CD36 (26), similar to integrin “inside-out” signaling (27). This raises the question of whether parts other than the intracellular domains, such as the TMDs, have a role in the receptor dimerization as well. We focused our investigation on microglia cells that are the major contributors of neuroinflammation during AD. To address the mode behind TLR4-TLR6 dimerization in these cells, we used a peptide interference approach by using peptides derived from the TMDs of TLR4 and TLR6 as inhibitors of receptor dimerization. We show that the TMDs of TLR4 and TLR6 form a heterodimer in the membrane and that peptides derived from TLR4 and TLR6 TMDs have the ability to interact with their corresponding receptor TMD. The C terminus of TLR4 displayed the strongest inhibition of microglia cells, suggesting that this region has an essential role in receptor dimerization and activation. Furthermore, we show that this inhibition results in abrogation of the inflammatory response and neuron rescue.
Besides shedding light on the mechanistic aspects of TLR4-TLR6 dimerization and their contribution to neurodegeneration, the peptides act as direct inhibitors of TLR4-TLR6 assembly and, therefore, may serve as therapeutic agents for treatment of AD or related TLR-mediated inflammatory diseases.
Results
TLR4 and TLR6 TMD form a heterodimer within membranes that can be inhibited by TMD-derived peptides
To test the capability of the TLR4 and TLR6 TMD to interact, we used the GALLEX system that enables measurement of hetero- and homodimerization within the Escherichia coli inner membrane (28). The TMD of TLR4 and TLR6 were divided into different segments (Table 1) and coupled to two LexA DNA-binding domains at their N terminus with different DNA sequence specificity, whereas the operator sequence contained one specific binding site for each. Each TMD was coupled to a maltose-binding protein (MBP) domain from E. coli at its C terminus. The hydrophobicity of the TMD functions as a membrane insertion signal, placing the LexA domain in the cytoplasm and the MBP domain in the periplasm of E. coli. If the transmembrane domains interact, the LexA cytoplasmic domains are in close proximity and can bind to the operator, repressing the expression of the β-galactosidase (β-gal; see the scheme in Fig. 1A). β-Gal activity was measured after the expression of the chimera protein, whereas the TMDs are those of TLR4 and TLR6 (Fig. 1B). As a positive control, we expressed the glycophorin A (GPA) TMD that is well characterized for its ability to homodimerize in the membrane (29) and as a negative control homodimerization-deficient mutant sequence of GPA, G83I. The data revealed that the TMDs of TLR4 and TLR6 have a high tendency to form heterodimers within the membrane, preferably from corresponding regions, e.g. TLR4 C terminus with TLR6 C terminus and TLR4 N terminus with TLR6 N terminus. The strongest assembly observed was between the TLR4C-2 and TLR6C-2 segments. The expression levels of the MBP protein were measured by Western blotting in order to normalize the β-gal expression level to those of the chimera protein. Additionally, a malE complementation assay was used for testing LexA-TMD-MBP orientation. All constructs exhibited the MBP in the periplasm, making them suitable for the GALLEX system (supplemental Fig. S1). Recently, we revealed that the TLR2 TMD has a key role in receptor activation and that peptides derived from this region may serve as regulators of TLR2 activation (7, 8). In light of these findings, we assessed whether TLR4 and TLR6 TMD-derived peptides could regulate their corresponding receptors. To this aim, we synthesized peptides with the same sequence as used in the GALLEX system (Table 1) and tested their ability to regulate activation of BV2 microglia cells that are activated through the CD36–TLR4-TLR6 receptor by the ligand Aβ (Fig. 1C). To increase peptide solubility, we added two lysine residues to either the C or the N terminus of each peptide in such a way that the Lys residues will be outside the membrane, except for TLR4C-2 peptide that already contains two Lys in its sequence (Table 1). Microglia cells pretreated with TLR4- or TLR6-derived peptides showed reduced levels in the secretion of IL-1β after stimulation with Aβ (Fig. 1C), whereas the most active peptide is TLR4C-2. The control peptide showed no inhibitory activity as expected. Overall, these results show that TLR4 and TLR6 form a heterodimer that can be regulated by their TMDs.
Table 1.
Peptides used in this study
Lysine residues were added at N and C termini for TLR6/4N and TLR4/6C peptides, respectively. Toxicity is represented by LD50 values for each peptide. Lysine residues (lowercase k) were added to the peptides to increase solubility.
| Designation | Sequence | Toxicity |
|---|---|---|
| μm | ||
| TLR-derived peptides | ||
| TLR4N1 | kkMYKTIISVSVVSVIVVS | >80 |
| TLR4N2 | kkVIVVSTVAFLIYHFYFH | >80 |
| TLR4C1 | AFLIYHFYFHLILIAGkk | >80 |
| TLR4C2 | HFYFHLILIAGCKKYS | >80 |
| TLR6N1 | kkSPLSCDTVLLTVTIGAT | >80 |
| TLR6N2 | kkTVLLTVTIGATMLVLAV | >80 |
| TLR6C1 | TIGATMLVLAVTGAFLCkk | >80 |
| TLR6C2 | GAFLCLYFDLPWYVRMLkk | >80 |
| Control peptides | ||
| scrTLR4C | SLICYKIYHAFKLHGF | >80 |
| scrTLR6C | MWLYKCPRLGFLKAYDVFL | >80 |
| scrTLR2C | kkGFGLHIRVWLHLLTLCkk | >80 |
Figure 1.

TLR4 and TLR6 TMDs form a heterodimer within the membrane that can be inhibited by TMD-derived peptides. A and B, TLR4 and TLR6 TMDs heterodimerize within the E. coli membrane. A, schematic illustration of the GALLEX heterodimerization system. The TMD anchors the chimera in the cytoplasmic membrane of E. coli with the MBP domain located in the periplasm and the LexA DNA-binding domain in the cytoplasm. Interaction of the TMDs leads to the formation of LexA heterodimers, which can bind to the operator region. The binding of the LexA dimer results in repression of the reporter gene (lacZ) and to reduced expression levels of β-gal. B, β-gal activity was measured after expression of the chimera proteins with either the N or C termini of TLR4 or TLR6 TMD. As a positive control we expressed the GPA TMD, which is well characterized for its ability to homodimerize in the membrane and as a negative control the homodimerization-deficient mutant sequence of GPA, G83I. β-Gal activity was normalized to the expression levels of the chimera proteins. Results are the mean ± S.E. of three independent experiments (6–8 repeats for each experiment). C, TLR-derived peptides inhibit the secretion of the proinflammatory cytokine, IL-1β, in BV2 microglia cells. Cells were incubated for 2 h with 20 μm concentrations of the indicated peptides, then washed and stimulated with 10 μm Aβ for 24 h. Secreted levels of IL-1β in the supernatant were assessed by ELISA. The results are normalized to Aβ only (absolute number: 106 ± 14pg/ml). Results are the mean ± S.E. of two independent experiments (*, p < 0.05; **, p < 0.01, 3 repeats for each experiment).
A peptide derived from the TLR4 TMD inhibits heterodimerization by interacting with its target receptor within the membrane
Based on the results obtained from the GALLEX assay and the inhibitory experiment, the most active peptide, TLR4C-2 (termed TLR4C), was further tested for its mode of action as an inhibitor of TLR4-TLR6 signaling. To evaluate whether the interference with the TLR4-TLR6 heterodimer activation is due to peptide-protein interaction within the membrane, we used a fluorescence resonance energy transfer (FRET) experiment (Fig. 2, A and B). TLR4C peptide was labeled with rhodamine (acceptor), and its counterpart TLR6C-2 peptide was labeled with NBD (donor). The interaction of the peptides was tested in phosphate-buffered saline (PBS) with 100 μm large unilamellar vesicles (LUVs) phosphatidylcholine and cholesterol (PC:Chol; 9:1) used to mimic the outer leaflet of the mammalian plasma membrane. Interaction between a pair of peptides is evidenced by a decrease in the emission of NBD-labeled peptide after the addition of successive amounts of rhodamine-labeled peptides. A strong decrease in NBD emission was observed when TLR4C peptide was added to TLR6C-2 (Fig. 2A) as opposed to the control peptide that showed no significant decrease in the fluorescence signal when paired with TLR4C peptide (Fig. 3B). To ensure that the interaction we observed occurs within the membrane, we performed a lipid-binding assay of TLR6C-2 (Fig. 2C). NBD fluorescence increased upon its insertion into a hydrophobic environment, thus enabling us to determine the affinity of the peptide toward the PC:Chol liposomes. TLR6C-2 showed strong affinity toward liposomes with Ka values of 5.4 × 105 m−1 (Fig. 2D). We further evaluated the specific interaction of TLR4C peptide with its reciprocal receptor, TLR6, in BV2 microglia cells by co-immunoprecipitation assay. Rhodamine-labeled TLR4C peptide showed strong interaction with TLR6, as opposed to the control peptide, that showed low interaction (Fig. 2E). This interaction is specific, as observed by low interaction with TLR2, which does not bind to TLR4, similar to the levels obtained with the control peptide. These results establish the specific interaction of TLR4C peptide with its corresponding receptor, TLR6, and provide mechanistic insights into the inhibitory activity of the TLR4C peptide.
Figure 2.

A peptide derived from the TLR4 TMD inhibited heterodimerization by interacting with its target receptor within the membrane. A and B, the TLR4 and TLR6 TMDs form a heterodimer within a membrane mimicking environment. 0.2 μm TLR6C2 peptide (A) scrTLR4C peptide (B) were labeled with NBD (donor) and added to 100 μm PC:Chol LUVs in PBS buffer. Changes in the intensity of the emission signal were monitored between 500 and 600 nm upon the addition of successive amounts of rhodamine labeled TLR4C peptide (acceptor). Acceptor peptide was added at ratios of 1:32 to 1:4. For each experiment the spectra was normalized to the value of the donor alone. Curves were calculated based on the average of three independent experiments. C and D, the TLR6C2 peptide interacts with the LUVs. C, 0.2 μm TLR6C2 peptide was labeled with NBD, and PC:cholesterol LUVs were added at different peptide lipid ratios: 1:2000, 1:1500, 1:1000, 1;750, 1:500, 1;250, 1:100. Changes in the intensity of the emission signal were monitored between 500 and 600 nm. D, the Kd of the TLR6C2 peptide was calculated. Curves were calculated based on the average of three independent experiments. E, the TLR4C peptide physically interacted with its corresponding receptor, TLR6. BV2 microglia cells were incubated with 20 μm concentrations of rhodamine-labeled peptide for 2 h at 37 °C. Then the cells were lysed using radio RIPA, and the soluble fraction was used for immunoprecipitation with antibodies against TLR6 or TLR2. As a negative control we used the scrTLR2C peptide. Protein samples were run on SDS-PAGE, and the presence of the peptide was detected with a fluorescence scanner (excitation at 532 nm and emission at 585 nm). Nonspecific binding of the peptides to G protein beads was subtracted. Subsequently, the gel was transferred to a membrane and subjected to Western blotting (W.B.) for TLR6 and TLR2 in the appropriate samples. Equal loading was measured by detecting of anti-tubulin in the cell lysate. Results are presented as three independent experiments. A.U., absorbance units.
Figure 3.
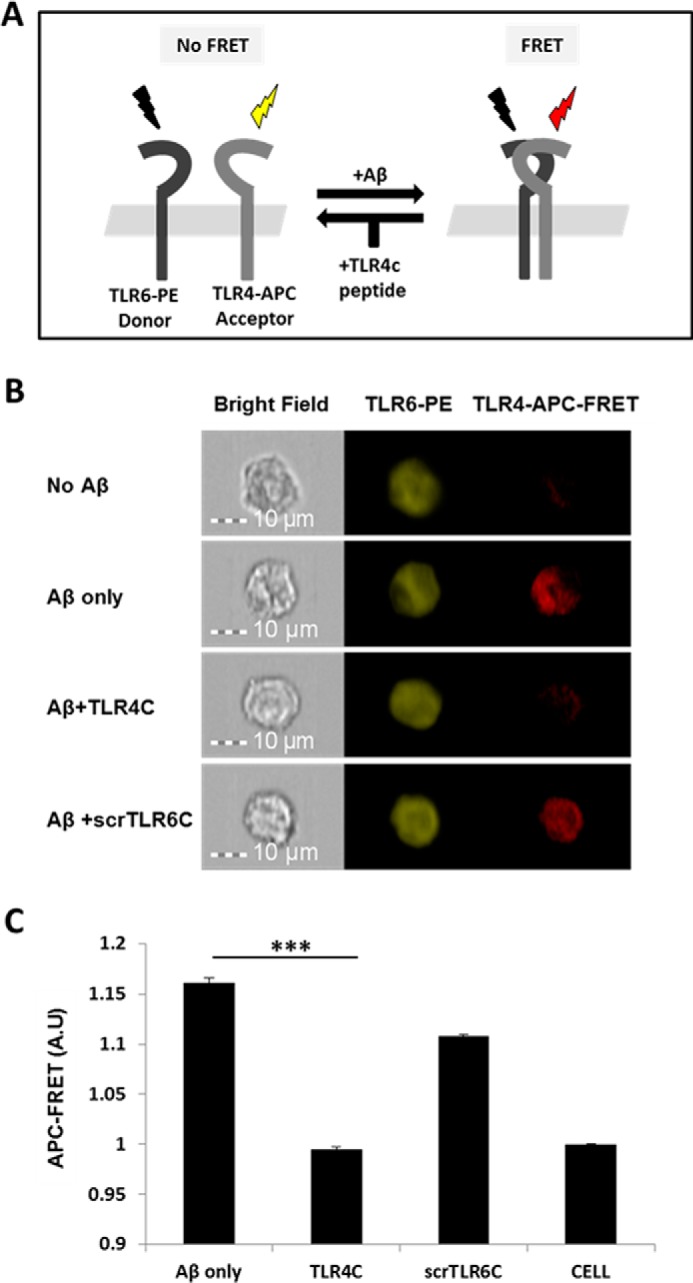
TLR4C peptide specifically blocked the TLR4-TLR6 heterodimer in BV2 microglia cells after Aβ activation. A, A scheme showing the FRET reaction. B, representative images of cellular interaction between TLR4 and TLR6 in BV2 microglia cells with the indicated treatments observed by FRET using ImageStreamX. Scale bars, 10 μm. Cells were incubated with 20 μm concentrations of the TLR4C or scrTLR6C peptide for 0.5 h and then washed and incubated with 10 μm Aβ for another 0.5 h at 37 °C. Cells were probed with an anti-TLR6-PE-conjugated antibody (donor) and an anti-TLR4 antibody followed by staining with an APC-labeled secondary antibody (acceptor). PE intensity (middle panel) and FRET intensity (right panel) were measured. C, a graphic summary of the FRET percentage normalized to cells only. Results are the mean ± S.E. of two independent experiments (***, p < 0.005, n ≥ 17,000 for each experiment). A.U., absorbance units.
TLR4C specifically attenuates TLR4-TLR6 heterodimer downstream signaling of BV2 microglia cells
After the previous results, we evaluated TLR4C's ability to influence downstream signaling. To begin, we assessed the heterodimerization of the full TLR4 and TLR6 proteins via a FRET experiment using the ImageStream imaging flow cytometer (Fig. 3). BV2 microglia cells were treated with anti-TLR6 antibody conjugated to PE, used as a donor (yellow, middle column), and antibody against TLR4 followed by staining with a secondary APC-labeled antibody, used as an acceptor (see scheme in Fig. 3A). The addition of Aβ to cells induced the APC-FRET signal (red, right column), indicating a TLR4-TLR6 heterodimer. Cells preincubated for 30 min with TLR4C peptide before Aβ induction showed reduced FRET levels as opposed to the control peptide that showed the same levels as Aβ-induced cells without peptide treatment (Fig. 3, B and C). We next tested the inhibitory activity of TLR4C peptide on the TLR downstream signaling cascade (Fig. 4). The transcription factor NF-κB is a critical regulator of inflammation induced by Aβ (30). We, therefore, tested the ability of the TLR4C peptide to regulate NF-κB translocation to the nucleus. BV2 cells pretreated with the peptide for 30 min after induction with Aβ showed reduction in NF-κB translocation to the nucleus (Fig. 4, A and B). The same cells, when treated with the control peptide after induction with Aβ, showed high levels of NF-κB in the nucleus, similar to the levels obtained in Aβ-induced cells. Treatment of the cells with TLR4C peptide reduced Aβ-induced ERK phosphorylation as well. However, the control peptide showed no inhibitory activity (Fig. 4C). We further examined the inhibitory activity of TLR4C peptide on the secretion of a variety of proinflammatory mediators (see Figs. 6 and 7). Cells activated by Aβ and pretreated with TLR4C peptide showed reduced levels in the secretion of the proinflammatory cytokines IL-6 (Figs. 5 and 6A) and TNF-α (Figs. 5 and 6B), the chemokine MCP-1 (Figs. 5 and 6C), and nitric oxide (Fig. 5D), whereas cells treated with the control peptide showed no reduction. To assess specificity of the peptides to TLR4-TLR6, cells were pretreated with TLR4C peptide after stimulation with either LTA (TLR2-TLR6 ligand), PAM3CSK (TLR2-TLR1 ligand), or LPS (TLR4-TLR4 ligand). The TLR4C peptide-treated cells showed no differences in their secreted TNF-α amounts (Fig. 5E), indicating the peptide's specificity toward the TLR4-TLR6 dimer. The same secretion levels were also shown after stimulation with half the maximal concentration of the ligands. To exclude the possibility that the effect we obtained is due to peptide toxicity, we performed a 2,3-bis-2H-tetrazolium-5-carboxanilide inner salt assay for cell viability. The peptide was shown to be not toxic to the BV2 microglia cells in a concentration 4-fold higher than used in the experiments (Table 1). Over all, TLR4C peptide inhibits downstream signaling of the TLR4-TLR6 heterodimer.
Figure 4.

The TLR4C peptide attenuated TLR4-TLR6 heterodimer downstream signaling of BV2 microglia cells. A and B, the TLR4C peptide inhibits NF-κB translocation to the nucleus upon activation with Aβ. A, representative images of cellular NF-κB staining after 20min in BV2 microglia cells. B, a graphic summary of cellular NF-κB translocation to the nucleus at different time points in BV2 microglia cells with the indicated treatments as observed by fluorescence resonance energy transfer using ImageStreamX. Cells were treatment with 10 μm Aβ (●) for the indicated times after incubation with 20 μm concentrations of either the TLR4C (□) or the scrTLR6C (♢) peptide for 1 h. For labeling of NF-κB, rabbit anti-NF-κB was added (1:50) overnight at 4 °C (Santa Cruz Biotechnology) followed by staining with a secondary anti-rabbit-APC (1:100) for 2 h at room temperature (Biolegend). For nuclear staining, Hoechst was added to the cells (1:1000) for 5 min. Results are the mean ± S.E. of six independent experiments (***, p < 0.005, n ≥ 1300 cells for each experiment). C, the TLR4C peptide inhibits ERK1/2 phosphorylation after activation with Aβ. Shown is a representative image from two independent experiments of ERK1/2 phosphorylation levels in BV2 microglia cells after Aβ exposure. Cells were pretreated for 0.5 h with 20 μm concentrations of the TLR4C peptide, scrTLR6C peptide, or untreated and then washed and incubated with 10 μm Aβ for the indicated times. ERK1/2 phosphorylation levels and total ERK1/2 levels were detected by Western blotting. Equal loading was detected by measuring tubulin.
Figure 6.

TLR4C inhibited the secretion of IL-6, TNF-α and MCP-1 in primary murine microglia cells. Cells were incubated for 2 h with 20 μm concentrations of the indicated peptides, then washed and stimulated with 10 μm Aβ for 24 h. Secreted levels of IL-6 (A), TNF-α (B), and MCP-1 (C) in the supernatant were assessed by ELISA. The results are the mean ± S.E. of 3 independent experiments and are normalized to Aβ for each cytokine as follows: IL-6, 304.98 ± 13.42 pg/ml; TNF-α, 732.137 ± 250pg/ml; MCP-1, 620.49 ± 20.57 pg/ml. ***, p < 0.005, 3 repeats for each experiment.
Figure 7.

The TLR4C peptide rescued neurons from death mediated by microglia-induced inflammation. A, graphic summary of neurons dead/live ratio caused by microglia-induced inflammation. CAD neuronal cells were differentiated for 4 days by serum withdrawal. Then microglia cells pretreated with 20 μm concentrations of either TLR4C or scrTLR6C for 2 h were seeded into a transwell insert on top of the neuronal cells. After 72 h of stimulation with 10 μm Aβ, neurons were collected and labeled with propidium iodide for detection of dead cells. Live and dead neurons were analyzed by FACS. The results are the ratio between the dead and the live neurons and are the mean ± S.E. of four independent experiments (***, p < 0.005). B, representative images from two independent experiments of neuron survival were observed using confocal microscopy. For labeling of neurons, rabbit anti-tubulin antibody was added (1:50) overnight at 4 °C followed by incubation with a secondary anti-rabbit-APC (1:100) for 2 h at room temperature (Biolegend). The nucleus was stained by Hoechst. Scale bar, 20 μm.
Figure 5.

TLR4C specifically inhibited the secretion of proinflammatory mediators in a BV2 microglia cell line. Cells were incubated for 2 h with 20 μm concentrations of the indicated peptides, then washed and stimulated with 10 μm Aβ for 24h. Secreted levels of IL-6 (A), TNF-α (B), MCP-1 (C), and NO− (D) in the supernatant were assessed by ELISA and Greiss kit. The results are the means ± S.E. of 2–4 independent experiments and are normalized to Aβ only for each cytokine as follows: IL-6, 559.47 ± 35 pg/ml; TNF-α, 671.12 ± 70 pg/ml; MCP-1, 4707.762 ± 802 pg/ml; NO, 95.6 ± 6.62 μm (***, p < 0.005, 3 repeats for each experiment). E, The specificity of the TLR4C peptide toward the TLR4-TLR6 heterodimer was evaluated by testing its inhibitory activity (white bars, μg/ml); 0.5 μg/ml and 0.25 μg/ml LTA for TLR2-TLR6 heterodimer, PAM3CSK for TLR2-TLR1 heterodimer, and LPS for TLR4-TLR4 homodimer (black bars). Results are the mean ± S.E. of two independent experiments (three repeats for each experiment). ns, not significant.
TLR4C peptide rescues neurons from death mediated by microglia-induced inflammation
Recognition of Aβ by CD36 triggers the assembly of a TLR4-TLR6 dimer that regulates the expression of neurotoxic mediators in microglia, resulting in the development of neurodegenerative diseases such as AD (31). Therefore, blocking TLR4-TLR6 activation may ameliorate neurodegeneration by rescuing neurons from death. We tested TLR4C's ability to rescue neurons after the onset of inflammation (Fig. 7). CAD cells were cultured and induced for differentiation for 4 days. Then, microglia cells pretreated/untreated with the peptide were seeded on a transwell (Corning) of 5 μm polycarbonate membrane. This allows the growth of two adherent cell types in the same well, so that they share the same growing media without coming in contact with one another. This allowed us to assess the effect of the secreted molecules on the neurons.
Then microglia cells were stimulated with Aβ for 72 h for induction of inflammation. Neuron death was evaluated by fluorescence-activated cell sorting (FACS) using propidium iodide for staining of the dead cells (Fig. 7A and supplemental Fig. S2, A–D) and by using confocal microscopy (Fig. 7B). The data revealed that cells treated with TLR4C peptide show a similar amount of dead cells relative to non-stimulated cells, suggesting that neurons were rescued from cells death (Fig. 7, A and B). To ensure that Aβ induces neuron death through stimulation of microglia cells and not due to direct interaction with neurons, we performed a similar experiment in the absence of microglia cells. As expected, no dead neurons were detected when directly stimulated with Aβ (supplemental Fig. S2E) (26). Altogether, these results reveal the mechanism behind TLR4C peptide inhibition of TLR4-TLR6 dimerization and activation and the peptides' potential to reduce inflammation mediated by microglia cells, consequently rescuing neurons from death.
Discussion
Recently, a new complex between the scavenger receptor CD36, TLR6, and TLR4 was reported in response to oxidized LDL and Aβ. The assembly of this complex was suggested to be regulated by intracellular signals of CD36 that require endocytosis (26). Aβ peptides condense to form plaques in the brain that initiate microglia-mediated inflammation, leading to neurodegeneration (18, 25). Here, we investigated the role of the TLR4-TLR6 heterodimer in microglia after Aβ activation and the mechanism behind their heterodimerization. We show that the TMDs of TLR4 and TLR6 play a pivotal role in receptor dimerization. Moreover, we show that by specifically interfering with TLR4-TLR6 TMD dimerization in microglia, neurodegeneration can be attenuated. This study establishes the critical role of the TLR4 and TLR6 TMD in receptor activation as well as the key role of the TLR4-TLR6 dimer in the generation of inflammation-mediated neuron death.
As opposed to TLR4-TLR6 heterodimerization, TLRs expressed on the plasma membrane dimerize after ligand binding to the extracellular domain of one receptor, leading to their lateral movement toward the partner molecule, resulting in conformational changes throughout the proteins (3). This leads to intracellular interactions and to the induction of a range of inflammatory mediators such as IL-6, TNF-α, and IL-1β (4). This process was shown to be regulated by the TMD of TLRs (6–8). Specifically, it was shown that exogenously added peptides derived from the TLR2 TMD can inhibit the assembly and activation of the receptor by binding to the corresponding TLR6 TMD and by that maintain its inactive monomeric form (7, 8). Here, the role of the TLR4 and TLR6 TMDs in receptor dimerization was investigated by using a peptide-interference approach. Specifically, we synthesized consecutive segments of the TLR4 and TLR6 TMDs and subjected them to activity assays. We found that a peptide derived from the C terminus of TLR4 (termed TLR4C) displayed strong binding to its corresponding TLR6 TMD and the strongest inhibition of BV2 microglia cells, suggesting that the TMD C terminus is important for TLR4-TLR6 dimerization. Moreover, these findings propose the TMD as a dimerization site for activated TLR4 and TLR6 proteins.
GALLEX results suggest that TLR4 interacts with TLR6 within the membrane. This was corroborated by FRET, which showed assembly of TLR4 and TLR6 TMDs in LUVs and co-immunoprecipitation revealing the interaction of the TLR4C peptide with TLR6 in microglia cells. These results suggest that the TMDs of TLR4 and TLR6 regulate receptor dimerization initiated by Aβ. We further tested the ability of the TLR4C peptide to inhibit dimerization of TLR4-TLR6 in microglia cells. TLR4C peptide inhibited the heterodimerization of TLR4-TLR6 after Aβ activation in microglia cells, suggesting that the external addition of a TMD-derived peptide can attenuate Aβ activation. These results reveal the importance of the TMD at the level of the full protein. To further test the extent of the inhibition, we investigated the effect of the TLR4C peptide on proinflammatory mediators secreted by microglia initiated by Aβ that ultimately lead to neuron death (16, 26). Aβ activation of microglia is characterized by activation of NFκB and ERK1/2. Therefore, we assessed their activation and found that treatment with the TLR4C peptide reduced NFκB translocation to the nucleus and mitigated ERK1/2 phosphorylation. This effect was also apparent in the reduction of cytokines, chemokines, and nitric oxide secretion after the admission of the TLR4C peptide. Aβ has also been shown to bind and activate TLR2 and its co-receptor CD14 (32). One might assume that because the TLR4 TMD interacts with TLR6, it will interfere with other TLR6-based dimers such as the TLR2-TLR6 couple. This promiscuity might be further extended to other TLR pairs such as TLR2-TLR1, which might also be effected, as the TLR6 TMD shows similarity to TLR1 (7). Therefore, we tested whether the TLR4C peptide can inhibit TLR2 activation. The peptide showed no inhibitory activity on microglia cells stimulated with TLR2 ligands, e.g. LTA for TLR2-TLR6 heterodimer and PAM3CSK for TLR2-TLR1 heterodimer, indicating that TLR4C peptide specifically inhibits TLR4-TLR6 signaling. These results highlight the potential of TMD regions to express specificity despite apparent similarity. Importantly, it might be possible that each region of the TLR's TMD might exhibit specificity to a different TLR, thus governing TLR pairing. However, future studies are needed to address this as a possible mode governing TLR dimerization.
Under normal non-pathogenic conditions, microglia cells exhibit a “resting” morphology. In this state they carry out tissue maintenance and immune surveillance (33). However, during AD, microglia surrounded by Aβ deposits shift their activation state to “active” mode where they act as immune effectors cells. This leads to chronic neuroinflammation, degradation of neurons, and functional decline. Therefore, by preventing TLR4-TLR6 heterodimerization, neuron death should be mitigated. To this aim we tested the extent of neuron death after Aβ stimulation of microglia coupled with treatment by the TLR4C peptide. As hypothesized, neurons were rescued by TLR4C peptide treatment, further demonstrating the therapeutic capability of TLR4C and highlighting the essential role of the TMD in TLR4-TLR6 dimerization leading to neurodegenerative disorders. Although not specifically addressed in this study, approaches that specifically interfere with microglia activation and transition into effector cells will, therefore, benefit from re-establishment of resting microglia and thus will preserve the protective activity of these cells. Inhibiting the activation of TLRs has been proposed to have a therapeutic potential in numerous CNS disorders (32). A proinflammatory environment shows a reduction in the phagocytic capacity of microglia cells, further contributing to the progression of the disease (23). Interfering with TLR signaling will be beneficial due to reduced inflammation as well as an increase in Aβ phagocytosis. Our results emphasize the potential of such an approach by showing that specific interference with TLR4-TLR6 signaling inhibits microglia cell activation and rescues neurons from death.
Taken together, our study identifies an essential role for the TLR4 TMD in receptor activation and highlights the therapeutic potential of the TLR4C peptide to treat AD. Furthermore, this study may have broader implications in other pathologies mediated by TLR4-TLR6 dimerization such as atherosclerosis (26).
Experimental procedures
Peptide synthesis, purification, and fluorescent labeling
Peptides were synthesized by a 9-fluorenylmethoxylcarbonyl (Fmoc) solid-phase method on Rink amide MBHA resin (Calbiochem-Novabiochem) by using an ABI 433A automatic peptide synthesizer (Applied Biosystems, Foster City, CA) as described previously (34). Then peptides were cleaved from the resin by incubation for 2 h with 95% trifluoroacetic acid, 2.5% H2O, and 2.5% triethylsilane. Purification of the crude peptides was performed by reverse phase HPLC (>98%) on a Vydac C4 column (Grace Discovery Sciences, Deerfield, Il) using a linear gradient of 20–90% acetonitrile in 0.1% trifluoroacetic acid for 40 min. The cleaning of the peptide was then identified by electrospray mass spectroscopy. Two lysine residues were added to the C termini or to the N termini of the peptides to confer water solubility to the hydrophobic TM domains. It was previously shown that hydrophobic peptides conjugated to lysine tags were correctly oligomerized and inserted into the membrane (35, 36). For assays with fluorescent peptides, the addition of rhodamine 5(6)-carboxytetramethylrhodamine N-succinimidyl ester (TAMRA, BioChemika) or NBD-F (2-fold excess) fluorescent probe to the N terminus of the peptides was performed by standard Fmoc chemistry (37).
Construction of the GALLEX chimera
The hetero-interactions of the TMDs in a natural membrane were studied using the GALLEX assay (28, 38). The expression vectors and E. coli strains required for the assay were kindly provided by Dr. Dirk Schneider. Briefly, GALLEX employs a chimeric protein in which the α-helical TM domain of interest is inserted between the gene for maltose-binding protein (MalE) and the N-terminal domain (residues 1–87) of the LexA protein from E. coli. Two plasmids are used in the assay containing either the wild-type LexA sequence (pBLM) or a mutated form of LexA (pALM). The desired TM sequences are inserted between these two protein sequences using standard cloning techniques. The resulting plasmids were both transformed into an SU202 E. coli strain, which then expressed the LexA-TM-MalE fusion proteins (after induction with 1 mm IPTG). Hetero-association of TM domains leads to the formation of LexAWT/LexAmut heterodimers that bind to a hybrid LexA promoter/operator in the reporter strain SU202 (which recognizes the mutant LexA domain) and repress the expression of β-gal. Furthermore, this hybrid operator does not recognize wild-type LexA homodimers; therefore, TM homo-oligomer formation does not interfere with the measurement of hetero-association. Sequences of the TLR TMDs GPA and G83I used are shown in Fig. 1 GPA was used as positive control, and its G83I mutant deficient in its ability to form dimer was used as a negative control. All the constructs were confirmed by DNA sequencing.
Measurements of dimerization (β-galactosidase assay)
The dimerization propensity of two constructs was measured by transforming an E. coli SU202 indicator strain with the relevant pALM and pBLM plasmids (each harboring a different TM sequence) simultaneously (39). The bacteria were grown overnight in the presence of 0.1 mm IPTG in LB medium with ampicillin and tetracycline in a 24-well plate. Then 4 samples of 10 μl were taken from each well to which as 90 μl of Z-buffer (60 mm Na2HPO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgSO4, pH 7.0, 10% chloroform, 1% β-mercaptoethanol). Then absorbance was measured at A590. The bacteria were added to 50 μl of 2% SDS in Z-buffer and given 5 min to disrupt. Then o-nitrophenyl-galactopyranose was added, and a kinetic reading of absorbance at A405 was taken at 20 min to measure the activity of β-gal. Vmax was calculated and divided by the absorbance at A590 to correct for bacterial growth. The stronger the dimerization, the stronger the inhibition of β-gal expression and less color. For competition assay with exogenous added peptides we used 20 μm peptides dissolved in 100% dimethyl sulfoxide (DMSO); the final concentration of DMSO was 2%. The peptides were added to the bacteria for overnight incubation in 24-well plates.
GALLEX-protein expression levels
We performed Western blotting analysis to determine whether the different TMDs affected the expression level of the chimera protein. We normalized the dimerization levels to the proteins levels. Aliquots of 10 μl of SU202 cells, each with a different plasmid, were mixed with a sample buffer, frozen for 1 h at −80 °C, boiled for 5 min, subjected to 12% SDS-PAGE, and then transferred to nitrocellulose. The primary antibody used was anti-maltose-binding protein (New England bioLabs, Ipswich, MA). The detection was done with Phototope-HRP Western blot Detection System from Cell Signaling Technology.
An assay for insertion and orientation (maltose complementation assay)
Membrane insertion and correct orientation were examined as previously described (28). Briefly, PD28 cells transformed with the different plasmids were cultured overnight. Only if the MBP, MalE, portion of the chimeric protein was present in the periplasm of the cells could we use maltose as the carbohydrate source, because this strain is deficient in endogenous maltose-binding protein. The cells were washed twice with PBS and were plated on an M9 minimal medium agar plates including 0.4% maltose, 1 mm IPTG, and the appropriate antibiotic to maintain the plasmid. The cell's growth is visually observed after overnight incubation.
FRET for NBD and rhodamine-labeled peptides
LUVs composed of PC and Chol (9:1, w/w) were prepared using the extrusion method as described previously (40), NBD-labeled peptides were added from a stock solution in DMSO to a solution containing 100 μm LUVs in PBS buffer. This was followed by the successive addition of rhodamine-labeled peptides. Fluorescence spectra were obtained before and after the addition of the rhodamine-labeled peptides and corrected by subtracting the changes in emission obtained by adding PBS-only and by reduction of the fluorescence of rhodamine alone.
Membrane-binding assays
PC:cholesterol LUVs were added to a 0.1 or 0.2 μm NBD-labeled peptide dissolved in 100% DMSO. The changes in NBD emission (530 nm; ΔF) were monitored as a function of the lipid/peptide molar ratio with excitation set at 465 nm (10-nm slit) until the system reached equilibrium (41). To account for the background, the emission of LUVs alone at the same wavelength was subtracted. The changes in the probe emission represent the amount of peptide bound to the hydrophobic interface because NBD is known to change its emission wavelength in a hydrophobic environment (41), thereby enabling us to evaluate the peptide binding to LUVs. The affinity constants determine by nonlinear least-squares analysis. The nonlinear least-squares analysis fitting was done using the GraphPad prism5 program.
Preparation of fibrillar Aβ
Synthetic lyophilized Aβ1–42 was purchased from Peptide 2.0 Inc. (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA). The peptide was dissolved in DMSO to a concentration of 2000 μm and sonicated for 20 s to avoid pre-aggregation. Aβ solutions were prepared by immediate dilution with 10 mm PBS (100 mm NaCl, 0.5 mm EDTA, pH 7.4) to a final concentration of 100 μm. The samples then incubated at 37 °C for at 6 days. The fibrils were viewed by transmission electron microscopy.
Transmission electron microscopy
15-μl samples from fibrillar Aβ assays were placed on 400-mesh copper grids covered by carbon-stabilized Formvar film (SPI Supplies, West Chester, PA). After 1 min excess fluid was removed, and the grids were negatively stained with 10 μl of 2% uranyl acetate solution for 40 s. Finally, excess fluid was removed, and the samples were examined by using a JEOL 100B electron microscope operating at 80 kV.
Cell culture
In vitro assays were performed on BV-2 murine microglia (kindly provide by Prof. Zvi Vogel laboratory from the Weizmann Institute of Science, Israel) and on CAD neuronal cells (42). Cells were grown in DMEM supplemented with 10% FBS, l-glutamine, sodium pyruvate, nonessential amino acids, and antibiotics (Biological Industries, Beit Haemek, Israel). The incubator was set at 37 °C with a humidified atmosphere containing 5% CO2.
Primary murine microglia
Cells were purchased from ScienCell Research Laboratories. Cells were grown in microglia medium supplemented with 5% FBS, 1% microglia growth supplement, and 1% of penicillin/streptomycin solution. The incubator was set at 37 °C with a humidified atmosphere containing 5% CO2.
2,3-Bis-2H-tetrazolium-5-carboxanilide inner salt (XTT) cytotoxicity assays
The cells were grown under the same conditions as in the cytokines secretion assays with peptide concentrations ranging from 5 μm to 80 μm. To each well was added 50 μl of XTT reaction solution (Biological Industries). Viability was determined after 2 h as described previously (43, 44). The LC50 was obtained from the dose-dependent cell viability curves.
Cytokines, chemokine, and NO secretion
100,000 BV-2 cells or 25,000 primary murine microglia cells were cultured overnight in a 96-well plate or 24-well plate, respectively. The following day the peptides were added to the cells at a final concentration of 20 μm (in 1% DMSO) and incubated for 2 h, then washed and incubated with fresh medium containing Aβ at a final concentration of 10 μm. The cells were incubated for 24 h at 37 °C. For control assay, the cells were stimulated with either LTA (TLR2-TLR6 ligand), PAM3CSK (TLR2-TLR1 ligand), or LPS (TLR4-TLR4 ligand) for 5 h (for TNF-α). Supernatants were collected and kept in −20 °C until assessment. Quantitative evaluations of IL-6, IL-1β, MCP-1, and TNF-α levels in supernatants were done using a DuoSet ELISA kit (R&D Systems). Nitric oxide was measured by a Griess reaction.
Immunoprecipitation of fluorescently labeled peptides with TLR
BV2 cells (2 × 106) were incubated with 1 μm rhodamine-labeled peptide for 1 h at 37 °C. Then the cells were washed and lysed with ice-cold radio immunoprecipitation (RIPA) assay buffer (50 mm Tris HCl pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS). After 15 min of centrifugation at 14,000 rpm at 4 °C, the supernatants were incubated with TLR antibodies including anti-TLR6 (Santa Cruz Biotechnology, catalogue no. sc-5662, lot A0906)) and anti-TLR2 (Santa Cruz Biotechnology, catalogue no. sc-10739, lot E0913) overnight at 4 °C. The supernatants were then added to protein G beads (Santa Cruz Biotechnology) for a second overnight incubation at 4 °C. The beads were then washed with cold PBS, diluted 1:2 with SDS Tricine buffer and boiled for 10 min at 95 °C. Proteins supernatants were subjected to 12% SDS-PAGE. and the co-immunoprecipitated peptide was detected with a Typhoon 9400 variable mode imager. Excitation was set at 532 nm, and emission at 585 nm. Western blotting analysis was performed with a secondary antibody conjugated to HRP. To verify equal loading amount, tubulin was blotted using an anti-tubulin antibody (Santa Cruz Biotechnology, catalogue no. sc-73242).
FRET between TLR4 and TLR6
BV2 microglia cells (4 × 106) were stimulated with 20 μm concentrations of TLR4C peptide or control peptide for 0.5 h at 37 °C, washed 3 times with PBS, and stimulated with 10 μm Aβ for another 0.5 h at 37 °C. Then cells were fixed with 3.7% paraformaldehyde for 20 min at room temperature and washed with PBS. The cells were then permeabilized and blocked for unspecific binding with 5% donkey serum, 1 mg/ml BSA, and 0.1% Triton in PBS at room temperature for 15 min. For labeling of TLR6, rabbit anti-TLR6-PE-conjugated polyclonal antibody was added (1:50) overnight at 4 °C (Bioss, inc. catalogue no. bs-2716R-PE, lot 9D17M81). For labeling of TLR4, mouse anti-TLR4 was added (1:25) overnight at 4 °C (Biolegend, catalogue no. 14502, lot B172069) followed by staining with a secondary anti-mouse-APC (1:100) for 2 h at room temperature (Biolegend, catalogue no. 405308, lot B175712). Cells were imaged using multispectral imaging flow cytometry (ImageStreamX Mark II, Amnis Corp.), objective 60×, NA:0.9 at room temperature. Cells in PBS were collected from each sample, and data were analyzed using image analysis software (dedicated software; Amnis). Images were compensated for fluorescent dye overlap by using single-stain controls. Cells were gated for single cells using the area and aspect ratio features and for focused cells using the Gradient RMS feature, as previously described (George et al. (45)). The Gradient RMS feature measures the sharpness quality of an image by detecting large changes of pixel values in the image and is useful for the selection of focused objects. The Gradient RMS feature is computed using the average gradient of a pixel normalized for variations in intensity levels. The instrument is composed of two cameras that allows separation of the signals; camera 1 for lasers 488 nm and 561 nm (channels 1–6) and camera 2 for the 405-nm and 642-nm lasers (channels 7–12). For FRET measurements, the samples were illuminated with the 488-nm laser. The PE staining was measured on channel 3 (560–595 nm), and the FRET-resulting fluorescence was collected on channel 5 (640–745 nm). As a control for staining intensity, the APC staining was also read using the 642-nm laser on channel 11 (640–745). The FRET was calculated for each cell by the FRET intensity. Results are normalized to untreated cells). Cells were imaged using multispectral imaging flow cytometry (ImageStreamX Mark II, Amnis Corp), objective 60×, NA 0.9 at room temperature.
NF-κB translocation to the nucleus
BV2 microglia cells (2 × 106) were treated with the peptides for 1 h (20 μm), washed with PBS, and stimulated with 10 μm Aβ at 37 °C for the indicated times. Cells were then fixed with 3.7% paraformaldehyde for 20 min at room temperature and washed with PBS. The cells were then permeabilized and blocked for unspecific binding with 5% donkey serum, 1 mg/ml BSA, and 0.1% Triton in PBS at room temperature for 15 min. For labeling of NF-κB, rabbit anti-NF-κB was added (1:50) overnight at 4 °C (Santa Cruz Biotechnology, catalogue no. cs-109, lot J2215) followed by staining with a secondary anti-rabbit-APC (1:100) for 2 h at room temperature (Biolegend). For nuclear staining, Hoechst was added to the cells (1:1000) for 5 min. Cells were imaged using multispectral imaging flow cytometry (ImageStreamX Mark II, Amnis), objective 60×, NA 0.9 at room temperature. Cells in PBS were collected from each sample, and data were analyzed using image analysis software (dedicated software; Amnis). Images were compensated for fluorescent dye overlap by using single-stain controls. Cells were gated for single cells using the area and aspect ratio features and for focused cells using the Gradient RMS feature as previously described (45). The nuclear localization of NF-κB was quantified using the similarity feature (the log-transformed Pearson's correlation coefficient in the two input images) between the NF-κB and Hoechst staining.
Phospho-ERK detection assay
BV2 microglia cells (1 × 106) were pretreated with growing medium containing 0.1% serum overnight. Then cells were treated with the peptides for 1 h (20 μm), washed with PBS, and stimulated with 10 μm Aβ at 37 °C for the indicated times. Then cell lysates were prepared in RIPA buffer containing 50 mm NaF, 2 mm Na3VO4, protease and phosphatase inhibitors (Sigma). Protein concentration was measured using the BCA protein assay kit (Pierce), and 50 μg of protein was loaded and separated on 12% SDS-PAGE. Afterword, the samples were transferred onto PVDF membrane. The immunoblot was incubated overnight with blocking solution (2% BSA in Tris-buffered saline and Tween 20 (TBST)) at 4 °C and then incubated with primary antibodies, phospho-ERK1/2, total-ERK1/2, or tubulin kindly provided by the laboratories of Prof. Rony Seger for 1 h at room temperature (Sigma). After washing with PBST, HRP-conjugated secondary antibody was applied for 2 h at room temperature. The blots were developed on an enhanced chemiluminescence (ECL) detection system.
Neurotoxicity assay
For assessment of microglia-induced neurotoxicity, 5 × 105 CAD mouse neuronal cells were seeded on poly-l-lysin-coated glass coverslips in F-12/DMEM medium. Neuronal differentiation was induced by differentiation medium containing F12/DMEM, 20 μg/ml transferin (Sigma), and 50 μg/ml sodium selenite (Sigma) for 4 days. 1 × 105 microglia cells with and without treatment of the peptide were seeded into Transwell inserts (5 μm polycarbonate membrane, Corning) on top of the neuronal cells. A pore size of 5 μm allowed medium transfer without the crossing of cells through it.
72 h after stimulation with 10 μm Aβ, neurons were fixed and stained with rabbit anti-neuronal class III β tubulin antibody (Jackson ImmunoResearch Laboratories Inc., catalogue no. 711-545-152) followed by anti-rabbit IgG Alexa Fluor (Covance Research Products, catalogue no. MRB-435P-100) and DAPI nuclear stain. Neuronal cells were imaged by confocal microscopy. For cell viability analysis, CAD cells were stained with propidium iodide, and the number of live/dead cells was analyzed by FACS.
Statistical analysis
Data were analyzed by analysis of variance test followed by Tukey's post hoc, two-tailed t test using GraphPad Prism 4 (San Diego, CA). Data presented as the mean ± S.E. values of p < 0.05 were considered statistically significant. *, p < 0.05; **, p < 0.01, ***, p < 0.005.
Author contributions
L. S.-G. conceived the study, designed and performed the experiments, and analyzed the data. M. C. and L. S.-G. performed the GALLEX experiments. B. Z. and L. S.-G. synthesized the peptides. Z. P. and L. S.-G. directed and analyzed the FRET and NF-κB translocation to the nucleus experiments. Y. S. directed the project. L. S.-G., Y. K., and Y. S. wrote the article.
Supplementary Material
Acknowledgments
We thank Vladimir Kiss for technical assistance with the confocal imaging, Dr. Rotem Ben Tov Perry and Dr. Ida Rishal for assistance with neurotoxicity experiments and the laboratory of Prof. Ehud Gazit for assistance with the preparation of Aβ fibrils. We also thank Dr. Ron Saar Dover, Dr. Eliran M. Reuven, and Liav Segev Zarko for insightful discussions throughout the study.
This work was supported by the Benoziyo Center for Neurological Diseases at the Weizmann Institute of science and partially by the Israel Science Foundation (Grant 1409/12). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1 and S2.
- TLR
- Toll-like receptor
- LTA
- lipoteichoic acid
- TMD
- transmembrane domain
- AD
- Alzheimer's disease
- Aβ
- amyloid β
- MBP
- maltose-binding protein
- GPA
- glycophorin A
- NBD
- 12-(N-methyl-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl))
- PC
- phosphatidylcholine
- Chol
- cholesterol
- LUV
- large unilamellar vesicle
- Fmoc
- 9-fluorenylmethoxylcarbonyl
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- XTT
- 2,3-bis-2H-tetrazolium-5-carboxanilide inner salt
- RIPA
- radio immunoprecipitation
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine (systematic)
- PE
- phosphatidylethanolamine.
References
- 1. Li J., Wang X., Zhang F., and Yin H. (2013) Toll-like receptors as therapeutic targets for autoimmune connective tissue diseases. Pharmacol. Ther. 138, 441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vadillo E., and Pelayo R. (2012) Toll-like receptors in development and function of the hematopoietic system. Rev. Invest. Clin. 64, 461–476 [PubMed] [Google Scholar]
- 3. Akira S., and Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 [DOI] [PubMed] [Google Scholar]
- 4. Kaisho T., and Akira S. (2006) Toll-like receptor function and signaling. J. Allergy Clin. Immunol. 117, 979–987 [DOI] [PubMed] [Google Scholar]
- 5. Fukata M., and Arditi M. (2013) The role of pattern recognition receptors in intestinal inflammation. Mucosal. Immunol. 6, 451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reuven E. M., Fink A., and Shai Y. (2014) Regulation of innate immune responses by transmembrane interactions: lessons from the TLR family. Biochim. Biophys. Acta 1838, 1586–1593 [DOI] [PubMed] [Google Scholar]
- 7. Fink A., Reuven E. M., Arnusch C. J., Shmuel-Galia L., Antonovsky N., and Shai Y. (2013) Assembly of the TLR2/6 transmembrane domains is essential for activation and is a target for prevention of sepsis. J. Immunol. 190, 6410–6422 [DOI] [PubMed] [Google Scholar]
- 8. Shmuel-Galia L., Aychek T., Fink A., Porat Z., Zarmi B., Bernshtein B., Brenner O., Jung S., and Shai Y. (2016) Neutralization of proinflammatory monocytes by targeting TLR2 dimerization ameliorates colitis. EMBO J. 35, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jin J. J., Kim H. D., Maxwell J. A., Li L., and Fukuchi K. (2008) Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J. Neuroinflammation 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Udan M. L., Ajit D., Crouse N. R., and Nichols M. R. (2008) Toll-like receptors 2 and 4 mediate Aβ(1–42) activation of the innate immune response in a human monocytic cell line. J. Neurochem. 104, 524–533 [DOI] [PubMed] [Google Scholar]
- 11. Walter S., Letiembre M., Liu Y., Heine H., Penke B., Hao W., Bode B., Manietta N., Walter J., Schulz-Schuffer W., and Fassbender K. (2007) Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol. Biochem. 20, 947–956 [DOI] [PubMed] [Google Scholar]
- 12. Landreth G. E., and Reed-Geaghan E. G. (2009) Toll-like receptors in Alzheimer's disease. Curr. Top. Microbiol. Immunol. 336, 137–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barger S. W., and Harmon A. D. (1997) Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388, 878–881 [DOI] [PubMed] [Google Scholar]
- 14. Heneka M. T., Golenbock D. T., and Latz E. (2015) Innate immunity in Alzheimer's disease. Nat. Immunol. 16, 229–236 [DOI] [PubMed] [Google Scholar]
- 15. Gold M., and El Khoury J. (2015) β-Amyloid, microglia, and the inflammasome in Alzheimer's disease. Semin Immunopathol. 37, 607–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arroyo D. S., Soria J. A., Gaviglio E. A., Rodriguez-Galan M. C., and Iribarren P. (2011) Toll-like receptors are key players in neurodegeneration. Int. Immunopharmacol. 11, 1415–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Okun E., Griffioen K. J., Lathia J. D., Tang S. C., Mattson M. P., and Arumugam T. V. (2009) Toll-like receptors in neurodegeneration. Brain Res. Rev. 59, 278–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cameron B., and Landreth G. E. (2010) Inflammation, microglia, and Alzheimer's disease. Neurobiol. Dis. 37, 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hardy J., and Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 20. Heneka M. T., and O'Banion M. K. (2007) Inflammatory processes in Alzheimer's disease. J. Neuroimmunol. 184, 69–91 [DOI] [PubMed] [Google Scholar]
- 21. Spangenberg E. E., and Green K. N. (2017) Inflammation in Alzheimer's disease: lessons learned from microglia-depletion models. Brain Behav. Immun. 61, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frank-Cannon T. C., Alto L. T., McAlpine F. E., and Tansey M. G. (2009) Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 4, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koenigsknecht-Talboo J., and Landreth G. E. (2005) Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 25, 8240–8249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zelcer N., Khanlou N., Clare R., Jiang Q., Reed-Geaghan E. G., Landreth G. E., Vinters H. V., and Tontonoz P. (2007) Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 10601–10606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Combs C. K. (2009) Inflammation and microglia actions in Alzheimer's disease. J. Neuroimmune Pharmacol. 4, 380–388 [DOI] [PubMed] [Google Scholar]
- 26. Stewart C. R., Stuart L. M., Wilkinson K., van Gils J. M., Deng J., Halle A., Rayner K. J., Boyer L., Zhong R., Frazier W. A., Lacy-Hulbert A., El Khoury J., Golenbock D. T., and Moore K. J. (2010) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kinashi T. (2005) Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 5, 546–559 [DOI] [PubMed] [Google Scholar]
- 28. Schneider D., and Engelman D. M. (2003) GALLEX, a measurement of heterologous association of transmembrane helices in a biological membrane. J. Biol. Chem. 278, 3105–3111 [DOI] [PubMed] [Google Scholar]
- 29. Gottschalk K. E., Adams P. D., Brunger A. T., and Kessler H. (2002) Transmembrane signal transduction of the αIIbβ3 integrin. Protein Sci. 11, 1800–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tan L., Schedl P., Song H. J., Garza D., and Konsolaki M. (2008) The Toll→NFκB signaling pathway mediates the neuropathological effects of the human Alzheimer's Aβ42 polypeptide in Drosophila. PLoS ONE 3, e3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alam Q., Alam M. Z., Mushtaq G., Damanhouri G. A., Rasool M., Kamal M. A., and Haque A. (2016) Inflammatory process in Alzheimer's and Parkinson's diseases: central role of cytokines. Curr. Pharm. Des. 22, 541–548 [DOI] [PubMed] [Google Scholar]
- 32. Gambuzza M. E., Sofo V., Salmeri F. M., Soraci L., Marino S., and Bramanti P. (2014) Toll-like receptors in Alzheimer's disease: a therapeutic perspective. CNS Neurol. Disord. Drug Targets 13, 1542–1558 [DOI] [PubMed] [Google Scholar]
- 33. Nimmerjahn A., Kirchhoff F., and Helmchen F. (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 [DOI] [PubMed] [Google Scholar]
- 34. Merrifield R. B., Vizioli L. D., and Boman H. G. (1982) Synthesis of the antibacterial peptide cecropin A (1–33). Biochemistry 21, 5020–5031 [DOI] [PubMed] [Google Scholar]
- 35. Rapaport D., Peled R., Nir S., and Shai Y. (1996) Reversible surface aggregation in pore formation by pardaxin. Biophys. J. 70, 2502–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu F., Lewis R. N., Hodges R. S., and McElhaney R. N. (2004) Effect of variations in the structure of a polyleucine-based α-helical transmembrane peptide on its interaction with phosphatidylglycerol bilayers. Biochemistry 43, 3679–3687 [DOI] [PubMed] [Google Scholar]
- 37. Rotem E., Reuven E. M., Klug Y. A., and Shai Y. (2016) The Transmembrane Domain of HIV-1 gp41 inhibits T-cell activation by targeting multiple T-cell receptor complex components through its GXXXG motif. Biochemistry 55, 1049–1057 [DOI] [PubMed] [Google Scholar]
- 38. King G., and Dixon A. M. (2010) Evidence for role of transmembrane helix-helix interactions in the assembly of the Class II major histocompatibility complex. Mol. Biosyst. 6, 1650–1661 [DOI] [PubMed] [Google Scholar]
- 39. Dmitrova M., Younès-Cauet G., Oertel-Buchheit P., Porte D., Schnarr M., and Granger-Schnarr M. (1998) A new LexA-based genetic system for monitoring and analyzing protein heterodimerization in Escherichia coli. Mol. Gen. Genet. 257, 205–212 [DOI] [PubMed] [Google Scholar]
- 40. He M. M., Sun J., and Kaback H. R. (1996) Cysteine-scanning mutagenesis of transmembrane domain XII and the flanking periplasmic loop in the lactose permease of Escherichia coli. Biochemistry 35, 12909–12914 [DOI] [PubMed] [Google Scholar]
- 41. Rapaport D., and Shai Y. (1991) Interaction of fluorescently labeled pardaxin and its analogues with lipid bilayers. J. Biol. Chem. 266, 23769–23775 [PubMed] [Google Scholar]
- 42. Qi Y., Wang J. K., McMillian M., and Chikaraishi D. M. (1997) Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci. 17, 1217–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Papo N., Braunstein A., Eshhar Z., and Shai Y. (2004) Suppression of human prostate tumor growth in mice by a cytolytic d-, l-amino acid peptide: membrane lysis, increased necrosis, and inhibition of prostate-specific antigen secretion. Cancer Res. 64, 5779–5786 [DOI] [PubMed] [Google Scholar]
- 44. Papo N., Seger D., Makovitzki A., Kalchenko V., Eshhar Z., Degani H., and Shai Y. (2006) Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res. 66, 5371–5378 [DOI] [PubMed] [Google Scholar]
- 45. George T. C., Fanning S. L., Fitzgerald-Bocarsly P., Fitzgeral-Bocarsly P., Medeiros R. B., Highfill S., Shimizu Y., Hall B. E., Frost K., Basiji D., Ortyn W. E., Morrissey P. J., and Lynch D. H. (2006) Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J. Immunol. Methods 311, 117–129 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.