

Abstract
HIV-1 Env protein is essential for host cell entry, and targeting Env remains an important antiretroviral strategy. We previously found that a peptide triazole thiol KR13 and its gold nanoparticle conjugate AuNP-KR13 directly and irreversibly inactivate the virus by targeting the Env protein, leading to virus gp120 shedding, membrane disruption and p24 capsid protein release. Here, we examined the consequences of targeting cell-surface Env with the virus inactivators. We found that both agents led to formation of non-infectious virus from transiently transfected 293T cells. The budded non-infectious viruses lacked Env gp120 but contained gp41. Importantly, budded virions also retained the capsid protein p24, in stark contrast to p24 leakage from viruses directly treated by these agents and arguing that the agents led to deformed viruses by transforming the cells at a stage before virus budding. We found that the Env inactivators caused gp120 shedding from the transiently transfected 293T cells as well as non-producer CHO-K1-gp160 cells. Additionally, AuNP-KR13 was cytotoxic against the virus-producing 293T and CHO-K1-gp160 cells, but not untransfected 293T or unmodified CHO-K1 cells. The results obtained reinforce the argument that cell-surface HIV-1 Env is metastable, as on virus particles, and provides a conformationally vulnerable target for virus suppression and infectious cell inactivation.
Keywords: HIV-1, gold nanoparticles, infected cell inactivation, multivalency, peptide triazoles
While the receptor-induced conformational change program built into the HIV-1 Env spike complex is intrinsic to the protein on the virus surface and used for virus-cell infection, it also can function with Env on infected cell surfaces to arm newly forming infectious viruses and to promote cell-to-cell fusion (Herschhorn et al., 2011; Moore et al., 2006). Hence, one may predict that the mature gp120/gp41 complex formed on infected cells possesses similar metastability as on viruses and could be targeted by agents that can trigger inactivating rearrangements of Env protein.
We previously identified a peptide triazole (PT) class of entry inhibitors that binds to HIV-1 Env gp120 with nanomolar affinity, inhibits cell infection by a broad range of virus subtypes and inactivates virus before host cell encounter by triggering gp120 shedding (Bastian et al., 2013; Bastian et al., 2011; McFadden et al., 2012). The ability of PTs to inhibit both CD4 and co-receptor binding sites in gp120, and the virus-inactivation functions of this class of inhibitors, appear to derive from entrapment of Env gp120 in a conformationally-disrupted and functionally-inactivated state. Importantly, we recently found that peptide triazole thiols and a multivalent form of the PT thiol KR13 displayed on gold nanoparticles (AuNP-KR13) were able to disrupt the virus particles, causing leakage of the internal protein p24 (Bastian et al., 2013; Bastian et al., 2011). The Env-inactivating functions of KR13 and AuNP-KR13 rely on the ability to disrupt the intrinsically metastable Env gp120/gp41 complex on the virus surface (Bastian et al., 2013; Bastian et al., 2015).
In view of the ability to bind and inactivate Env gp120 on HIV-1 virions at high potency, we investigated here whether PTs could be used to inactivate cell-surface gp120.
Effect of KR13 and AuNP-KR13 on infectious HIV-1 production
For this study, peptide triazole thiol KR13 (PTT), with C-terminal Cys-SH, was prepared by solid phase synthesis (Bastian et al., 2013; Bastian et al., 2011; Bastian et al., 2015). Gold nanoparticles were synthesized using citric acid reduction (Frens, 1973) followed by AuNP-KR13 (AuNP-PT) conjugation (Bastian et al., 2013; Bastian et al., 2011; Bastian et al., 2015). We evaluated whether PT-based virus inactivators could suppress cellular production of infectious virus particles. KR13 and AuNP-KR13 treatments of virus-producing transfected 293T cells were conducted by transfecting Human Embryonic Kidney cells (HEK293T) with envelope (Env) and backbone viral DNA (Bastian et al., 2013; McFadden et al., 2012; Montefiori, 2005). Either wildtype (WT) HIV-1 BaL-01 Env DNA or DNA with S375W Env mutation to suppress peptide triazole binding (Biorn et al., 2004) was used. Envelope-deficient pNL4-3-Luc+Rev-Env−provirus was used for backbone DNA(Bastian et al., 2011; McFadden et al., 2012). Infected cells were re-seeded 24 hours after transfection and serial dilutions of KR13 or AuNP-KR13 were added to each well and incubated for 24 hours. Cell supernatants were collected at 24 hours after media change/treatment and gradient-purified (Bastian et al., 2013; Bastian et al., 2011). Remaining cells were measured for viability using WST-1 (Takara). The treated cells, once the supernatant was separated and collected, were washed 3 times with PBS to remove debris and other nonspecifically attached proteins, and then treated with 0.1% Triton X in order to release the gp120 from the cell surface without damaging the cells (Miranda et al., 2002). Collected fractions (virus-containing and non-virus containing) were validated for p24 and gp41 contents using capture ELISA, for gp120 content using western blot, and for virus infectivity using luciferase reporter (Bastian et al., 2013; Bastian et al., 2011; Bastian et al., 2015; McFadden et al., 2012).
Virions produced from cells treated with both KR13 and AuNP-KR13 showed dose-dependent loss of infectivity (Figure 1A) and virus gp120 (Figure 1B) but retention of gp41 (Figure 1C). Importantly, the virions produced from treated cells retained luminal p24 (Figure 1D), in stark contrast to the previously observed dose- and time-dependent loss of p24 from cell-free virus treated with these agents (Bastian et al., 2013; Bastian et al., 2011; Bastian et al., 2015). The p24 in the lumen of the virions treated with KR13 and AuNP-KR13 were in the mature form as judged by size in the western blot analysis (Supporting Figure S1A). We surmise that the reduction in total p24 content in the virion fraction obtained from cells in the presence of AuNP-KR13 is due to the reduction in total virus production from these treated cells. We speculate that the virion decrease in this case is caused by disruptive effects on the cell surface before virion budding, as reflected by the loss of cell viability (Figure 1F). Nonetheless, retention of substantial luminal p24 in the inactive viruses produced from infected cells in the presence of KR13 and AuNP-KR13 argues strongly that the agents remove gp120 from the cell surface before budding of p24-containing virions from treated cells. In turn, the consequent survival of p24-containing virions once formed in the presence of KR13 and AuNP-KR13 is due to the lack of gp120 on the “bald” budded virions from the treated cells. This fits with results in Figure 1F and previous reports that lysis by the PTs requires functionally active surface gp120 (Bastian et al., 2013; Bastian et al., 2011; Bastian et al., 2015). Additionally, in a separate control experiment, we found that isolated virions produced from cells in the presence of KR13 as expected became refractory to freshly added KR13, while virions produced in the absence of KR13 were not (Supporting Figure S1B). No impairment of produced virus infectivity (Supporting Figure S2A) or gp120 content (data not shown) was found from cells treated with unconjugated AuNP or AuNP conjugated to inactive sequence-scrambled KR13S peptide (RINNWXISEAMMβAQβAC-CONH2) (Bastian et al., 2015). The overall results argue that both KR13 and AuNP-KR13 cause formation of inactive virus particles upon interaction with cell-surface gp120.
Figure 1.
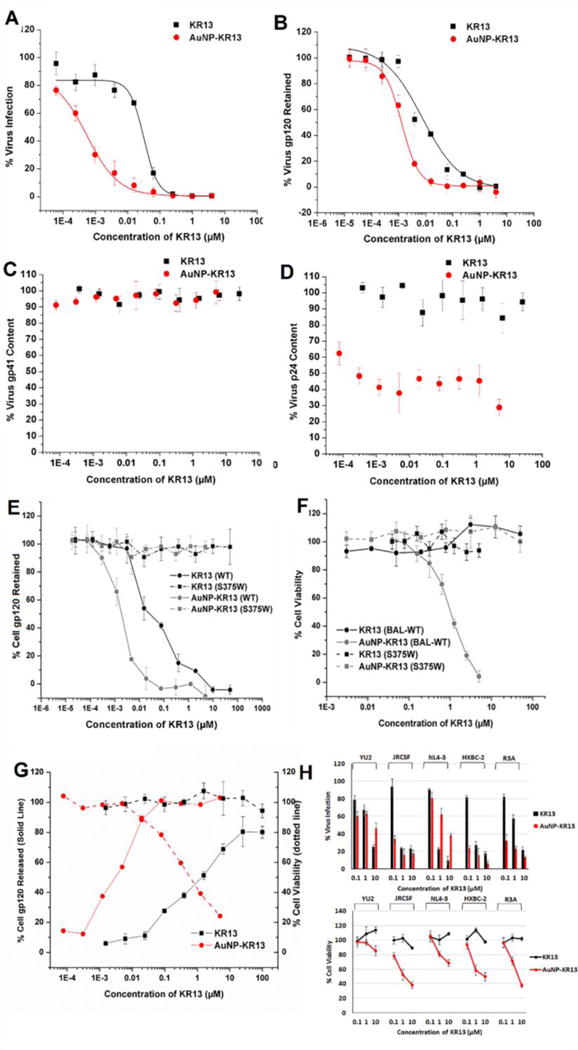
Properties of virions obtained from 293 T producer cells in the presence of KR13 or AuNP-KR13 treatments. (A) virus infectivity; (B) virus gp120 retention; (C) virus p24 content (D) virus gp41 content. Error bars represent standard deviation of the mean, n = 3. Effects of KR13 and AuNP-KR13 on cells expressing surface Env as either WT gp120 or the S375W peptide-resistant gp120 mutant: (E) Env gp120 retained on 293T cells 24 hours after beginning KR13 or AuNP-KR13 treatment; (F) Viability of transfected 293T cells 24 hours after beginning KR13 or AuNP-KR13 treatment. Positive and negative controls were cells treated with 0.1% triton X and cells treated with PBS respectively. Error bars represent standard deviation of the mean, n = 3. (G) Effects of KR13 and AuNP-KR13 on CHO-K1 cells stably expressing gp160 complex. (Left Y-Axis, Solid Line) Env gp120 released from the surface of CHO-K1-gp160 cells 24 hours after beginning KR13 or AuNP-KR13 treatment. (Right Y-Axis, Dotted Line) Cell viability of CHO-K1-gp160 cells 24 hours post KR13 or AuNP-KR13 treatment. Positive and negative controls were cells treated with 0.1% triton X and with PBS respectively. Error bars represent standard deviation of the mean, n = 3. (H) Breadth analysis of KR13 and AuNP-KR13 inhibition of infectious virus formation. Viruses tested included YU2 (clade B, tier 1, CCR5 tropic), JR-CSF (clade B, tier 2, CCR5 tropic), NL4-3 (clade B, tier 1, CXCR4 tropic), HXB2 (clade B, tier 1, CXCR4 tropic) and R3A (clade B, tier 2, dual tropic). (Top) Infectivity of the produced virions 24 hours after beginning KR13 or AuNP-KR13 treatment. (Bottom) Viability of transfected 293T cells 24 hours after beginning KR13 or AuNP-KR13 treatment. Positive and negative controls were cells treated with 0.1% triton X and with PBS respectively. Error bars represent standard deviation of the mean, n = 3.
Fate of HIV-1 virus-producing cells in the presence of KR13 and AuNP-KR13
Virus-producing 293T cells treated with KR13 or AuNP-KR13 were both found to shed gp120 (Figure 1E). In contrast, 293T cells transfected with the PT-resistant S375W mutant Env (Bastian et al., 2015) exhibited no gp120 shedding after either treatment, confirming that cell gp120 shedding requires specific gp120 binding. That gp120 shedding occurs upon PT binding has already been shown in a kinetic analysis of Env breakdown in HIV-1 pseudoviruses treated with KR13 (Bastian et al. 2013). Importantly, the viability of HIV-1 Env expressing cells treated with AuNP-KR13, but not with KR13, decreased precipitously (Figure 1F). The viability of S375W-Env expressing cells was unaffected by AuNP-KR13 (Figure 1F), verifying that AuNP-KR13 cytotoxicity was due to specific interaction with cell surface gp120. Also, no cytotoxicity was observed when cells not expressing Env were treated with AuNP-KR13 (Supporting Figure S2B). Cell viability was unaffected by either unconjugated AuNP or AuNP conjugated to the inactive variant KR13S (Supporting Figure S2C).
Effects on cells stably expressing gp160 in the absence of virion budding
CHO-K1 (engineered Chinese hamster ovary) cells stably transfected with gp160 (from Robert Blumenthal, NCI) were seeded at 100,000 cells/well onto a 24-well plate and treated for 24 hours with KR13 or AuNP-KR13 at the same concentrations used in the 293T cell assays. Cell supernatants were collected and spun at 1000 rpm for 5 min to pellet cell debris. Wash fractions were collected and quantified for the gp120 removed from the cell surfaces with Triton X, as described above for transfected 293T cell analyses. Treated cells were tested for cell viability using the WST-1 assay. As shown in Figure 1G, both KR13 and AuNP-KR13 caused gp120 shedding from CHO-K1-gp160 cells. That KR13 and AuNP-KR13 caused gp120 shedding from these cells reinforces the argument that Env spikes expressed on the cell surface are in a structurally vulnerable metastable state prior to virus-incorporation and hence can be exploited to inactivate Env spikes on the plasma membrane of infected cells before virion budding.
For AuNP-KR13, shedding showed a strong inverse correlation with viability of CHO-K1-gp160 cells (Figure 1G), further supporting that AuNP-KR13 interaction with gp120 is key to causing cytotoxicity. Prior observations argue that KR13 and AuNP-KR13 elicit viral lysis by different mechanisms, with KR13 prematurely triggering fusion-related membrane transformation (Bastian et al., 2013) versus simultaneous engagement of AuNP-KR13 with multiple Env spikes on the virus (Bastian et al., 2015).
Breadth of AuNP-KR13 and KR13 Functions in Cells Producing Replication-Competent Viruses
We evaluated the breadth of action of KR13 and AuNP-KR13 on cells producing replication-competent viruses. HEK293T cells were transfected with replication-competent virus DNA plasmids followed by re-seeding of infected cells and introduction of serial dilutions of KR13 and AuNP-KR13. Collected produced virus was quantified for p24 by capture ELISA as previously (Bastian et al., 2013; Bastian et al., 2011; Bastian et al., 2015). Infectivity was determined with a TZMB-1 assay (Montefiori, 2005) and compared to untreated, virus-only control. Treatment of infected cells with either KR13 or AuNP-KR13 led to production of noninfectious virions in all strains tested (Figure 1H). In addition, decreasing cell viability was seen across the range of Env subtypes treated with AuNP-KR13, with quantitative differences observed between strains. The overall finding of significant breadth in KR13 and AuNP-KR13 effects on cell-surface Env is consistent with PT breadth of action observed previously with virus Env, including with fully infectious virus (Bastian et al., 2013; McFadden et al., 2012).
Overall, the results reported here demonstrate that agents targeting the metastability of Env gp120/gp41 complex are capable of inactivating not only infectious virus but also infected cells producing the virus. In so doing, PT-based Env inactivators, can suppress virus proliferation from infected cells. This will open up exploring cellular effects of a recently-discovered class of metabolically-stable and high-potency macrocyclic PT Env inactivators (Chaiken and Rashad, 2015; Rashad et al., 2015).
Supplementary Material
Highlights for Paper Titled “Targeting cell surface HIV-1 Env protein to suppress infectious virus formation”.
Both peptide triazole thiols and their multivalent conjugates on AuNPs can inactivate HIV-1 Env.
HIV-1 Env protein inactivators can target Env gp120 proteins on virus-producing cells.
Targeting virus-producing cells suppresses new infectious virus formation.
Metastability of HIV-1 Env is a strategic vulnerability of both viruses and cells.
Env targeting by multivalent inactivators has potential for infected cell killing.
Acknowledgments
This work was funded by the National Institute of Health through the 1 R01 GM 111029-01. We thank Dr. Jeffrey Jacobson (Temple University) for his interest in our work and advice. We thank Drs. Julio Martin Garcia (Drexel), Chris Aiken (Vanderbilt University), Michele Kutzler (Drexel University) and Robert Blumenthal (National Cancer Institute) for materials used in this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bastian AR, Contarino M, Bailey LD, Aneja R, Moreira DR, Freedman K, McFadden K, Duffy C, Emileh A, Leslie G, Jacobson JM, Hoxie JA, Chaiken I. Interactions of peptide triazole thiols with Env gp120 induce irreversible breakdown and inactivation of HIV-1 virions. Retrovirology. 2013;10(1):153. doi: 10.1186/1742-4690-10-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian AR, Kantharaju, McFadden K, Duffy C, Rajagopal S, Contarino MR, Papazoglou E, Chaiken I. Cell-free HIV-1 virucidal action by modified peptide triazole inhibitors of Env gp120. ChemMedChem. 2011;6(8):1335–1339. 1318. doi: 10.1002/cmdc.201100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian AR, Nangarlia A, Bailey LD, Holmes A, Kalyana Sundaram RV, Ang C, Moreira DR, Freedman K, Duffy C, Contarino M, Abrams C, Root M, Chaiken I. Mechanism of multivalent nanoparticle encounter with HIV-1 for potency enhancement of peptide triazole virus inactivation. The Journal of Biological Chemistry. 2015;290(1):529–543. doi: 10.1074/jbc.M114.608315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biorn AC, Cocklin S, Madani N, Si Z, Ivanovic T, Samanen J, Van Ryk DI, Pantophlet R, Burton DR, Freire E, Sodroski J, Chaiken IM. Mode of action for linear peptide inhibitors of HIV-1 gp120 interactions. Biochemistry. 2004;43(7):1928–1938. doi: 10.1021/bi035088i. [DOI] [PubMed] [Google Scholar]
- Chaiken I, Rashad AA. Peptide triazole inactivators of HIV-1: how do they work and what is their potential? Future Med Chem. 2015;7(17):2305–2310. doi: 10.4155/fmc.15.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frens G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nature. 1973;241(105):20–22. [Google Scholar]
- Herschhorn A, Finzi A, Jones DM, Courter JR, Sugawara A, Smith AB, 3rd, Sodroski JG. An inducible cell-cell fusion system with integrated ability to measure the efficiency and specificity of HIV-1 entry inhibitors. PloS one. 2011;6(11):e26731. doi: 10.1371/journal.pone.0026731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden K, Fletcher P, Rossi F, Kantharaju, Umashankara M, Pirrone V, Rajagopal S, Gopi H, Krebs FC, Martin-Garcia J, Shattock RJ, Chaiken I. Antiviral breadth and combination potential of peptide triazole HIV-1 entry inhibitors. Antimicrobial Agents and Chemotherapy. 2012;56(2):1073–1080. doi: 10.1128/AAC.05555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda LR, Schaefer BC, Kupfer A, Hu Z, Franzusoff A. Cell surface expression of the HIV-1 envelope glycoproteins is directed from intracellular CTLA-4-containing regulated secretory granules. Proc Natl Acad Sci U S A. 2002;99(12):8031–8036. doi: 10.1073/pnas.122696599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefiori DC. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. In: Coligan John E, et al., editors. Current protocols in immunology. 2005. p. 11. Chapter 12, Unit 12. [DOI] [PubMed] [Google Scholar]
- Moore PL, Crooks ET, Porter L, Zhu P, Cayanan CS, Grise H, Corcoran P, Zwick MB, Franti M, Morris L, Roux KH, Burton DR, Binley JM. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. Journal of Virology. 2006;80(5):2515–2528. doi: 10.1128/JVI.80.5.2515-2528.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashad AA, Kalyana Sundaram RV, Aneja R, Duffy C, Chaiken I. Macrocyclic Envelope Glycoprotein Antagonists that Irreversibly Inactivate HIV-1 before Host Cell Encounter. Journal of medicinal chemistry. 2015;58(18):7603–7608. doi: 10.1021/acs.jmedchem.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.