

Abstract
The ability of immune cells to survey tissues and sense pathologic insults and deviations makes them a unique platform for interfacing with the body and disease. With the rapid advancement of synthetic biology, we can now engineer and equip immune cells with new sensors and controllable therapeutic response programs to sense and treat diseases that our natural immune system cannot normally handle. Here we review the current state of engineered immune cell therapeutics and their unique capabilities compared to small molecules and biologics. We then discuss how engineered immune cells are being designed to combat cancer, focusing on how new synthetic biology tools are providing potential ways to overcome the major roadblocks for treatment. Finally, we give a long-term vision for the use of synthetic biology to engineer immune cells as a general sensor-response platform to precisely detect disease, to remodel disease microenvironments, and to treat a potentially wide range of challenging diseases.
Keywords: synthetic biology, immunotherapy, engineered immune cells, T cells, synthetic Notch, chimeric antigen receptors
INTRODUCTION: IMMUNE CELLS ARE AN IDEAL PLATFORM FOR INTERFACING WITH DISEASE
Why Engineer Immune Cells?
Cells of the immune system play a unique role in the body, detecting and responding to pathologic insults and deviations (Figure 1a) (1). As such, these cells have exceptional properties. They are semiautonomous, often moving freely about the body to survey and infiltrate diverse tissues (2). Immune cells act as general sensor-response agents, detecting local problems and responding in diverse ways—sometimes executing powerful targeted actions like cell killing and phagocytosis, and at other times taking more subtle but broad actions, like secreting cytokines and chemokines that communicate with and mobilize other cells. Equally important, immune cells are relatively easy to remove, modify, and transfer back into a patient.
Figure 1.
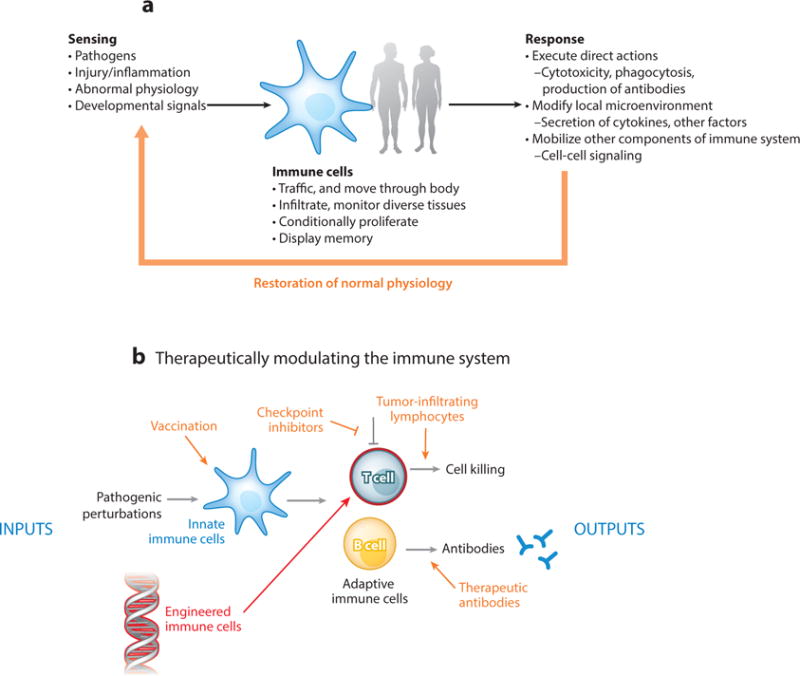
The immune system as a platform for interfacing with disease. Different modes of intervention to direct immune responses to disease are shown in orange. (a) Immune cells are an exquisite sensing and response system that monitors and responds to disease and loss of homeostasis. (b) Our endogenous immune system can be manipulated with biologics, but now immune cells can be directly, genetically reengineered to target and treat disease.
Given these unique properties, immune cells provide a remarkable platform for interfacing with and treating disease. There are many complex diseases, such as cancer and autoimmunity, that our natural immune systems either cannot handle or pathologically contribute to. Thus, there is a strong rationale to engineer new disease sensing and response behaviors in immune cells, especially given recent powerful advances in synthetic biology and genome editing, which give us unprecedented ability to modify and engineer cellular functions.
Synthetic immunology is an emerging strategy that applies the tools and approaches of systems and synthetic biology to reprogram and enhance the function of immune cells, thus also rewiring the overall capabilities of our immune system (3, 4).
The immune system comprises a diverse group of cells that survey the body for indications of disease. The major arms of the immune response can be categorized as innate and adaptive (Figure 1b). The innate response involves cells such as neutrophils, basophils, mast cells, natural killer (NK) cells, macrophages, and dendritic cells. As the early innate response ensues, a complex series of events leads to the activation of the adaptive immune response, which is mediated by T cells and B cells. Adaptive immunity is characterized by the exquisite specificity of T cells and B cells, which as a population theoretically have the ability to recognize any pathogen we encounter. This wide array of cell types offers diverse platforms and functional capabilities for therapeutic engineering.
Recent groundbreaking clinical studies using engineered T cells to treat blood cancers have definitively shown that immune cell function can be redirected and can be remarkably effective in treating disease. But our immune cells go beyond simply detecting cancer and other pathogenic insults and are involved in a variety of physiological processes. These include regulation of our microbiome (5); metabolism (6); and the homeostasis, development, and repair of various organs (1). Our immune system as a whole acts to coordinate both the sensing of significant changes in our bodies and the responses required to return tissues to homeostasis. Thus our immune cells have the potential to eventually serve as platforms for therapeutic engineering to address an even broader set of diseases beyond cancer, including autoimmunity, inflammation, and degeneration (7).
History of Therapeutically Harnessing the Immune System
Synthetic immunology follows in the footsteps of a long history of manipulating the immune system to stimulate and harness its capabilities (Figure 1b). Vaccination essentially involves convincing innate immune cells to initiate an immune response to a particular pathologic agent. Vaccination can involve a neutralized pathogen, or, in modern times, a complex nanoparticle that provides an optimized microenvironment for antigen presentation and immune system activation (8, 9). Therapeutic antibodies represent a way to directly provide the products of an adaptive B cell response. Expanding and adoptively transferring tumor-infiltrating lymphocytes (TILs) provides a way to enhance native antitumor responses, as does the deployment of checkpoint inhibitors [anti–programmed cell death protein 1 (PD-1), anti–PD ligand 1 (PD-L1), and anti–cytotoxic T lymphocyte–associated protein 4 (CTLA4)], which block immunosuppressive signals present in tumors. These approaches have generally involved providing new ways to influence immune cells to execute native responses (10, 11).
Direct Hacking of Immune Cells
The emerging field of synthetic immunology differs from these approaches in its direct and purposeful genetic hacking of immune cells to systematically generate new therapeutic capabilities (Figure 2a). The major goal of synthetic immunology is to manipulate immune cells such that they can be used as therapeutic agents with capabilities that extend significantly beyond those of the native immune system in order to generate a curative immune response to diseases that are difficult to treat, such as cancer and autoimmunity. This approach has emerged only recently with our increased ability and understanding of how to genetically manipulate cells so as to rationally change their functions.
Figure 2.
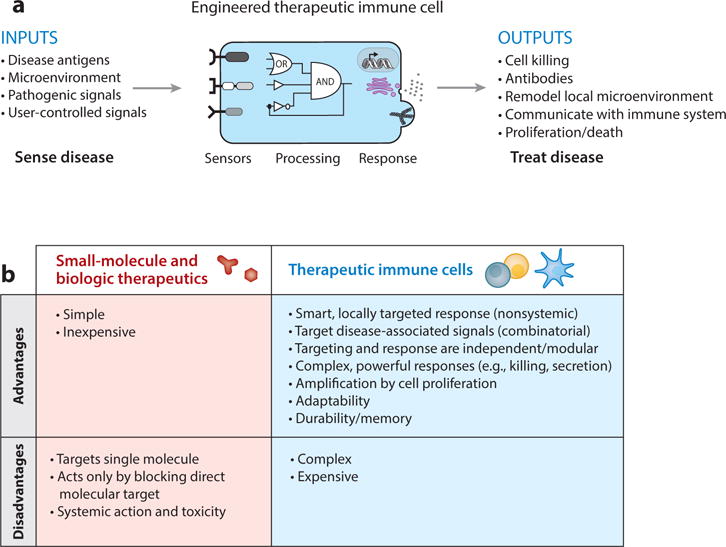
Therapeutic cells versus traditional therapeutic modalities. (a) Future therapeutic cells need multiple sensors, intrinsic information processing, decision-making logic, and controlled output programs for more effective and safe treatment of disease. (b) The pros and cons of cell therapies versus biologics and small-molecule therapeutics.
In this review, we describe the advantages of using living cells as a new therapeutic modality, with smart sensing-response capabilities that go beyond what individual therapeutic molecules can achieve. We describe the tool kit of genetically encoded components that can be used to engineer immune cells to respond to new disease-associated stimuli and how these cells can be used as control nodes to manipulate the broader immune system to address diverse diseases.
IMMUNE CELL THERAPEUTICS
Advantages of Cell Therapies Versus Small Molecules and Biologics
Engineered cells have the potential to operate as much smarter therapeutics compared to traditional small molecules and biologics. Small-molecule and macromolecular drugs are designed to disrupt the function of a specific target molecule. In contrast, living cells—particularly immune cells—can execute more comprehensive response programs. They are able to detect combinatorial environmental inputs and use this information to initiate equally complex, nuanced, and controlled therapeutic responses. In other words, a molecular drug is a tool that can do a single job, whereas a cell is a device that, like a computer, can be programmed to deploy the right set of tools, depending on the situation (Figure 2b).
Because of the distributed nature of the immune system, immune cells also have the potential to serve as connectors that communicate and influence the diverse cell types of the immune system. Engineered control of a particular immune cell type could serve as a way to influence broader aspects of the larger immune network through the production of cell-cell communication molecules, such as cytokines and chemokines.
Immune cells have several other unique properties that might make them particularly powerful therapeutics. First, immune cells can easily be harvested from patients, modified ex vivo, and transferred back into patients. Second, immune cells have the potential to survive in a persistent manner over the course of decades, thus potentially providing long-term memory for chronic diseases or diseases that have the potential for relapse. Third, many immune cells have the potential for adaptive function, changing as disease progresses. While we are far from directly harnessing and engineering this capability, it could be extremely powerful for complex diseases like cancer.
In the last few years, the explosion of this new field has led to many new examples of engineering new sensing and response capabilities into immune cells, with most of the work focused in the realm of cancer. In synthetic immunology, the goal is to endow therapeutic cells with new sensors (receptors), information-processing capabilities (cell signaling control), and therapeutic response programs that allow them to navigate, communicate, and effectively coordinate a safe and curative response to a wide variety of diseases (Figure 2a). Below we review what immune cells can be used in therapies, focusing particularly on how they are or could be used in treating cancer (Figure 3a). Here we primarily focus on cells with direct cytotoxic function, and not on antigen-presenting cells, such as dendritic cells engineered for cancer vaccination, which are the subject of other reviews (12).
Figure 3.

Engineering different immune cell types. (a) Immune cell types amenable to engineering for cell therapies. Here we focus on the use of T cells for targeting cancer (yellow box). (b) Engineered T cells, as well as naturally occurring, cancer-specific tumor-infiltrating lymphocytes, are of particular interest given the recent unprecedented clinical results.
T Cell Platforms
T cells have been a major focus in cell therapies as they are an immune cell type that exhibits targeted cytotoxicity and their killing specificity can be redirected to diseases such as cancer. They are also easily isolated from peripheral blood and can be modified and extensively expanded ex vivo, allowing for optimal dosing of cells during treatment. In addition, T cells are attractive because of their ability to survey the body and expand locally in response to disease-related antigens. The resultant population of responding T cells can in some cases drive clearance of tumors, and the generation of memory cells that are long-lived in patients (13–15). Several major classes of T cell therapies for cancer are being pursued (Figure 3b).
Tumor-infiltrating lymphocytes
Theoretically, our immune system has the intrinsic capability to respond to cancer. Cancer cells are by definition abnormal and acquire mutations as the disease progresses. Our bodies harbor T cells that can recognize mutant peptides that arise in cancers (16), but the immune response is often suppressed by the tumor. One approach to enhancing the T cell response is to excise parts of tumors and ex vivo expand the cancer-specific TILs. These T cells can then be infused back into the patient in greater numbers and in a more optimal cellular state that can effectively slow tumor progression (17–19). TILs can be found in melanoma, which is a relatively immunogenic cancer. But TILs have proven to be moderate or ineffective in cancer therapy, largely because of immunosuppression (20–23). It is possible that TIL therapy could greatly benefit from further engineering of the T cells during ex vivo processing as response rates could be improved through arming the cells to combat the often refractory tumor microenvironment. TILs are thus an important test bed for synthetic biology approaches discussed below.
Genetically modified T cell therapies: CARs and TCRs
The biggest advances in synthetic immunology have come from genetically modifying T cells from patients so that they directly recognize cancer cells. There are two major approaches for redirecting T cell recognition—chimeric antigen receptors (CARs) and engineered T cell receptors (TCRs). CARs are a type of synthetic T cell receptor that have at their core an artificial extracellular recognition domain—usually a single-chain antibody that recognizes a tumor-specific surface antigen—and the intracellular signaling domain of the T cell receptor (24–26). The most effective CARs also incorporate an intracellular costimulatory signaling domain, which helps to significantly drive the persistence and proliferation of the antigen-stimulated T cells (27–29). Several TCRs have been identified that recognize common tumor-specific antigens, and these can also be expressed in engineered T cells to attack cells that express this specific intracellular antigen (30–34).
Innate Immune Cell Platforms
NK cells are part of the innate immune system, but as of late they have shown several characteristics normally associated with adaptive immune cells, including capacity for memory responses (35). NK cells like cytotoxic T cells are able to recognize infected cells and directly kill target cells through release of cytolytic granules. Unlike T cells, NK cells do not have an antigen receptor that restricts them to a highly specific target. They use a series of receptors that have positive and negative effects on their activation status. Depending on the balance of engagement of the stimulatory and inhibitory receptors, the NK cell makes the decision whether to activate (36). Another differentiating factor of NK cells compared to T cells is their inability to activate and proliferate without cytokines derived from other immune cell types (37–39). Therefore, the major challenge with NK cell therapies may be keeping the cells alive long enough for effective treatment, owing to cell-extrinsic requirements for survival and activation. Cytokines like IL-12, IL-15, and IL-18 may be critical for maintaining the therapeutic NK cells over the course of therapy.
To date, most NK therapies are at the preclinical stage and have further to go than T cell therapies. However, there may be key control benefits like a short life span and the requirement for paracrine signals originating from other immune cell types to proliferate. These layers of added control when compared to T cells that produce their own stimulatory cytokine, IL-2, could make for more controlled cell therapies. At the same time, this control could come with a trade-off in cytotoxic efficacy.
In addition, NK cells have the signaling machinery to drive CAR signaling, and CAR NK cells are being studied as an alternative to CAR T cells. Currently, preclinical primary human CAR NK cell studies have been performed in various tumor models with CARs directed toward antigens such as Her-2 (40), CD244 (41), CD20 (42), and CD19 (43–45). NK cells are thus a unique platform for cell-based therapies and may have optimal characteristics for treatment of certain cancers.
SYNTHETIC BIOLOGY PRINCIPLES FOR ENGINEERING IMMUNE CELLS
Practical Constraints
Synthetic biology and evolution approach the task of controlling cell behavior in fundamentally different ways and with very different constraints. Evolution has the benefit of a long timescale to make large-scale changes to the genome and the overall architecture of signal transduction to derive subtle and sophisticated behaviors. In contrast, a major constraint on synthetic biology approaches is the size and number of changes one can incorporate into an immune cell chassis. Currently, the genetic payload that we can reliably deliver to primary T cells is on the order of 10 kb, or just a few genes. While some transposon systems have the potential for the delivery of larger genetic elements to cells (46), in reality there is precipitous decrease in transfection efficiency well before the theoretical payload limit (47). Thus, in practice, most examples of immune cell engineering at this point in time report the expression of one to five genes. What this means is that as synthetic biologists we must build compact multifunctional molecules that can reliably regulate cellular behavior, similar to how viruses maximize the functional capabilities of a small genome (48).
Gordian Knot Solutions: Simple Ways to Generate New Behavior
It is incumbent upon synthetic biologists to engineer molecular components to control cellular behavior that are robust and modular and have dominant effects when layered over the natural system. If these core considerations are ignored, the probability of tight and tunable control over cell behavior is low. Often this means creatively thinking about how to achieve a particular target immune cell function in a manner that is inspired by natural systems but that differs significantly in detailed mechanism. CARs are a beautiful example of this kind of a Gordian knot engineering solution—they are a simple, nonnatural molecule that provides a direct way to reliably target T cells to cancer cells in a manner that completely bypasses the complexities of antigen presentation and MHC recognition that are normally required by native T cells. The CAR is thus essentially a dominant bypass mutation (49). Another general strategy in synthetic biology is the use of orthogonal components—components that are not normally in the host immune cell. Synthetic Notch (synNotch) receptors, discussed below, are an example of orthogonal molecules that can allow more flexible rewiring of cellular behavior because they have fewer existing functional constraints (50).
Autonomous Versus User Controlled
A general goal of engineering immune cells is to generate new autonomous behaviors—behaviors that the cells will automatically execute when they are in the body, such as target cell killing by a CAR T cell. However, it is important to keep in mind that at this early stage of immune engineering, reprogramming efforts are imperfect. Thus, there is growing appreciation that in addition to autonomous behaviors, it is important to engineer certain functions that are user controlled. This is analogous to designing self-driving cars: Although the long-term goal may be completely autonomous function, we realize that a human at the steering wheel remains critical for many situations. As discussed below, immune cell functions that can be controlled by small-molecule inputs provide one mechanism of user control.
Autologous Versus Allogeneic Cells
Another important topic in immune cell engineering involves the source of the cells. Most current therapies involve autologous cells (i.e., modifying the patient’s cells), which avoids most issues of rejection. Nonetheless, it is well appreciated that allogeneic engineered cells would be a considerably more cost-effective and widespread therapy: Cells from a donor could be used for many patients in a relatively off-the-shelf manner. As such, how to develop allogeneic immune cells is a topic of very active research, including the use of gene-editing techniques to eliminate genes that are involved in either graft rejection or graft-versus-host disease (51). In this review, however, we do not focus on this issue of where the cells come from but rather focus on the process of engineering new therapeutic functionality into the cells. The advances that we discuss will determine the therapeutic capability of engineered cells and are essentially agnostic as to source (they would work equally well in autologous and allogeneic cells).
Engineering Challenges in Treating Cancer
Many of the advances we discuss below focus on efforts to engineer immune cells to treat cancer. Thus it is useful to consider the broad array of challenges to this goal.
Precision recognition. The immune cells must be able to precisely recognize the tumor cells and discriminate them from normal cells, particularly because many antigens are not absolutely cancer specific. We now know that mistargeting of T cells such that they lead to cross-reaction with critical normal tissues can lead to severe adverse effects, even death.
Trafficking. Immune cells need to be able to traffic to and infiltrate the tumor in order to exert their effects, including cytotoxicity.
Cell survival, expansion, and persistence. The cells need to survive, expand, and persist to kill the cancer cells.
Overcoming an immunosuppressive microenvironment. Because so many cancers, particularly solid cancers, involve a suppressive microenvironment, the cells must be able to overcome or disable this environment to both expand and launch a powerful cytotoxic response.
Safety controls. It is clear that many immunotherapies that harness the power of the immune system can lead to potent side effects, so mechanisms to control and regulate engineered immune cell function are critical for safety.
Many of the tools discussed below have been developed to address this set of challenges.
SYNTHETIC BIOLOGY TOOL KIT: GENETIC COMPONENTS FOR PROGRAMMING IMMUNE CELLS
Here we review the types of genetically encoded molecular building blocks that are becoming available for rationally modulating immune cell function.
Cellular Sensors
Essential to any engineered immune cell are novel sensor functions—the cell will need one or more altered receptors that allow it to detect a disease cell or a diseased tissue microenvironment and distinguish it from a healthy one.
CARs
CARs are the first truly successful synthetic immune receptor (13, 52–57). These synthetic receptors consist of (a) a single-chain variable fragment directed toward a cell surface tumor-specific or tumor-associated antigen and (b) the signaling domains from the TCR ζ chain (signal 1) and a costimulatory receptor (signal 2) (58). These receptors bind their target antigen and drive the necessary signaling for full T cell activation (Figure 4a). Depending on the costimulatory domain of the CAR, the T cells can show different phenotypes in regard to T cell effector functions such as cytokine secretion, cytotoxicity, and overall persistence in the patient (59). It is clear from certain clinical trials that that CAR T cell persistence can vary considerably in patients. CARs targeting the B cell antigen, CD19, containing either the costimulatory domain derived from CD28 or 4-1BB have shown different activity and persistence profiles in patients. In trials for chronic lymphocytic leukemia (CLL), α-CD19 CAR T cells with the CD28 costimulatory domain were short-lived, only lasting around a month in patients, whereas α-CD19 CAR T cells with the 4-1BB costimulatory domain have persisted up to four years (15, 56, 60). The signaling characteristics of the CAR thus can have profound effects on T cell biology and must be characterized and engineered to elicit the desired immune response to the particular cancer.
Figure 4.
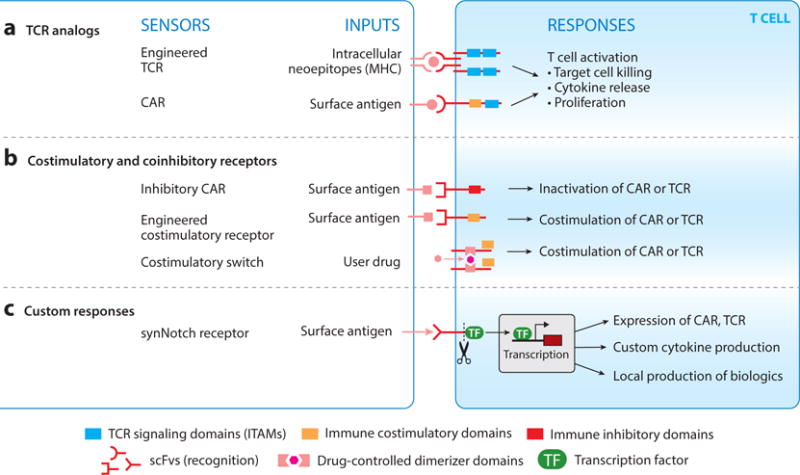
Basic T cell–sensing modules. (a) T cells can be engineered with T cell receptors (TCRs) and chimeric antigen receptors (CARs) to redirect them to cancer or other diseases. These receptors activate the entirety of the immune response. (b) T cells require multiple signals to activate. Synthetic costimulatory and coinhibitory receptors have been engineered that help to initiate or shut down an immune response. (c) synNotch receptors are a new class of receptors that detect environmental cues and directly regulate customized transcriptional circuits. Cells engineered with synNotch receptors can link environmental sensing to a variety of custom effector programs.
We are just now starting to understand the mechanisms underlying these differences and the set of parameters one should consider when engineering CAR T cells for applications beyond hematological malignancies. The specific T cell subset that is engineered, the signaling characteristics of the receptors, and the overall T cell effector functions that arise upon activation all need to be controlled to have optimal therapeutic effect. Often it is unclear how to engineer the T cells to robustly perform specific tasks. Therefore, it would be beneficial to develop more high-throughput approaches to test large libraries of CARs and identify the CARs that bias T cell behavior and effector functions toward ones that have potent effects on difficult to eradicate tumors.
Tumor-specific TCRs
Another major focus of T cell immunotherapy is the identification of TCRs that recognize tumor-specific, abnormally expressed antigens derived from fetal or developmental proteins, oncogenic viral components, tissue-specific differentiation proteins, or neoantigens that arise from mutations in tumors (Figure 4a). Cancers with higher mutational load often elicit a stronger immune response (61, 62); however, most tumors strongly suppress the immune cells once they have entered the tumor microenvironment. The major goal of this approach is to identify common cancer antigens that can be detected by T cells so that one can engineer a patient’s T cells with the appropriate tumor-specific TCR. These engineered T cells can then be infused in large numbers into patients in order to drive more potent tumor immunity. The underlying idea for these therapies is simple, but in reality it is difficult to find neoantigens that are shared by many cancer patients, and given the diversity of human leukocyte antigen (HLA) alleles, not all individuals are able to present the same neoantigens and stimulate a T cell response.
Several tumor antigen-specific T cell receptors have been identified and are being tested in clinical trials. These include TCRs recognizing melanocyte-derived antigens such as MART1 (30) and gp100 (31), carcinoembryonic antigen (CEA) (63), NY-ESO-1 (33), and MAGE-A3 (34). A major pitfall that has come to light is the catastrophic effects of off-target specificity of TCRs thought to be tumor specific. A recent example is the case of the MAGE-A3 TCR that binds an antigen that is normally relegated to the testis and gametogenesis but is abnormally expressed in a variety of cancers such as melanoma, non–small cell lung cancer, and certain hematological malignancies. The affinity-matured engineered TCR showed unanticipated cross-reactivity to a peptide derived from the muscle-specific protein titin, and patients that received the MAGE-A3 TCR T cells died of cardiac shock within a few days of treatment because of severe myocardial damage caused by massive T cell infiltration and activation (64). Adverse effects have also been observed in trials in patients treated with TCRs targeted with MART1, gp100, and CEA, due to on-target damage caused by antigen expression in normal tissue (65).
However, for antigens such as NY-ESO-1 that are not expressed in bystander tissue, there is little toxicity and the trials are showing promise. Patients with refractory synovial cell sarcoma or metastatic melanoma showed clinical responses (33, 66); this was also the first demonstration that a nonmelanoma tumor could be treated with T cells engineered with TCRs. Although these therapies have a long way to go, it is clear that they could be improved by enhanced control and boosted activity, given that the response rates are not optimal and few patients show long-term complete regression.
Engineered costimulatory and coinhibitory receptors
As described above, the current generation of CARs incorporate intracellular signaling elements from the TCR combined with elements from immune costimulatory receptors. Given the many coregulatory immune receptors—both costimulatory and coinhibitory—it is also possible to engineer chimeric coregulatory receptors that modulate key aspects of the T cell response (Figure 4b). Depending on the combination of coregulatory sequences utilized in the CAR, one can imagine controlling a spectrum of T cell activation parameters and effector functions, as the character and magnitude of signal transduction in T cells are known to have important implications for the immune response to disease (67).
Several efforts have been directed toward gaining control over costimulation. One example involves taking the costimulatory chains normally integrated into the CAR and expressing them as a separate membrane-tethered peptide. These stand-alone costimulatory chains are fused to small-molecule-regulated dimerization domains and can thus be clustered, driving dose-dependent activation of costimulatory signals (68). The overall goal of this approach is to have tighter control over the level of activity of the CAR T cells during treatment, potentially alleviating life-threatening toxicity.
As another approach to control toxicity, coinhibitory receptor signaling domains have been utilized in the context of CARs. For example, inhibitory CARs (iCARs) have been developed with extracellular antigen-recognition domains analogous to stimulatory CARs, but cytoplasmic signaling domains of these receptors are derived from coinhibitory receptors such as CTLA4 or PD-1 (the targets of checkpoint inhibitors) (69). iCARs allow recognition of specific antigens that then instruct the T cells to shut down their activity—a function that is useful for engineering more complex combinatorial antigen recognition (see below).
Nonnatural and synthetic cytokine and cytokine receptor platforms
A primary mode of communication between immune cells is the secretion of cytokines and chemokines that act in both an autocrine and a paracrine manner to orchestrate an immune response to disease. These secreted proteins are able to drive proliferation, differentiation, cell death, and recruitment of a highly specialized and coordinated army of immune cell types.
Synthetic controls over cytokine production or the development of completely orthogonal cytokines that provide privileged channels of communication between therapeutic cells are of much interest. Such engineered capabilities are yet another way to help therapeutic cells to elicit a curative response to disease. The underlying principles of cellular communication have been studied for decades, and we are beginning to understand how to build cellular communication systems that can regulate cellular behavior in defined ways (70, 71). Furthermore, we have sophisticated approaches for rational protein design and mutational screens of receptor:ligand pairs to build new communication channels that are orthogonal and/or have new and custom dynamic properties (72).
Garcia and colleagues have pioneered this approach to building new cytokines that will likely have impact both as biologics and in cell therapies. They identified a variant of IL-2 they call IL-2 superkine (Super-2) that bypasses the requirement for the high-affinity α chain of the IL-2 receptor, CD25 (73). This is important, as T cells do not express CD25 unless activated and suppressive regulatory T cells often have high levels of CD25, allowing them to titrate away IL-2 from activated T cells. This can be problematic in a tumor where immune suppression is strong and more often than not shuts down the T cell response. Thus, the administration of Super-2 or engineering cell therapies to produce Super-2 in tumors may allow for a potent response in solid tumors. Garcia and colleagues have also engineered variants of IL-2 that act as IL-2 receptor antagonist through mutagenesis and high-throughput screening (74). The use of Super-2 and other types of engineered IL-2 antagonist could be a powerful way to control the expansion and contraction of a synthetic T cell response, mimicking the way our natural immune system drives a highly controlled expansion and contraction phase, safely providing immunity to infection.
Several groups have also shown that synthetic cytokine receptors can be engineered by taking advantage of the fact that many cytokine receptors signal through heterodimerization of two chains that are brought together by cytokine association. For example, heterodimerization of the β chain and common γ chain of the IL-2 receptor can be mediated by other modalities if the ligand-binding domains are removed (75). Several groups have shown that the extracellular domains of the two chains can be replaced and alternative proteins can be used to drive heterodimerization and signaling through the receptor (76). While initial examples of synthetic cytokine receptors are clinically suboptimal, the general approach to engineering orthogonal cytokine receptors is effective and likely can be accomplished with clinically relevant dimerization modules. If this vision can be realized, clinicians could selectively titrate different aspects of T cell effector function such as proliferation and differentiation after the cells are infused into a patient. It would also be feasible to have therapeutic cells communicate with each other through such cytokine:receptor systems as long as the dimerizing agent could be produced and secreted similar to natural cytokines.
Chemokine receptors: enhancing therapeutic T cell migration
Migration of therapeutic T cells to solid tumors is often a limiting factor in treatment (77). Because of this, engineering T cells with chemokine receptors that allow them to better infiltrate tumors is an area of intensive research. Cancers can be tested for their chemokine signatures to determine which receptors would aid in T cell recruitment. There are many examples of enhancing therapeutic T cell activity by equipping the cells with chemokine receptors in a variety of tumor contexts. Di Stasi et al. (78) showed that anti-CD30 CAR T cells engineered with CCR4 were superior at clearing a Hodgkin lymphoma model that produces the chemokines CCL17 and CCL22. More recently, anti-GD2 (79) and anti-mesothelin (80) CAR T cells were engineered with the chemokine receptor CCR2b enhancing clearance of a xenograft neuroblastoma and mesothelioma model, respectively. Given these promising preclinical results, chemokine receptors may be an important addition to a variety of adoptive T cell therapies for cancer.
As an alternative approach to enhance migration of T cells to tumors, our group has engineered T cells capable of migrating toward bioinert small molecules that can be directly administered to tumors. This potentially relieves the requirement for knowledge of the chemokine signature of the tumor and may aid in scenarios where the tumor produces minimal chemoattractants. To accomplish this, Park et al. (81) utilized receptors activated solely by synthetic ligands (RASSLs), which are G protein–coupled receptors (GPCRs) that induce migration in response to orthogonal small molecules such as clozapine-N-oxide (CNO) (82). RASSL-engineered T cells showed enhanced migration and localization to tumors injected with CNO slow-release biodegradable microparticles. In the future it may be possible to administer nanoparticle drug delivery systems that can accumulate in tumors without the need for direct injection (83). This is an important proof-of-principle study showing how synthetic biology approaches can help to overcome the natural limits of our immune system.
synNotch receptors—transcriptional regulation coupled to environmental sensing
More recently, we have developed a new class of receptors called synNotch receptors that provide a more general platform for engineering novel sensor/response behaviors in immune cells (50, 84). synNotch receptors link recognition of a user-defined cell surface antigen to activation of transcription (Figure 4c). The native Notch receptor is made up of an extracellular, ligand-binding domain that binds the ligand, Delta, displayed in trans on an opposing cell surface. Upon ligand binding, two proteolytic cleavage events occur—one extracellular and one intramembrane— releasing the cytoplasmic domain transcriptional regulator (85–87). Unlike many other receptor classes, such as receptor tyrosine kinases (RTKs), the Notch receptor does not initiate a complex kinase signaling cascade (88). To build a fully customizable synNotch receptor platform, we first mapped a minimal region within the natural Notch receptor that controls the ligand-dependent cleavage of the receptor and release of the cytoplasmic tail (50, 89). The ligand-binding domain and the intracellular domain can then be replaced with different antigen-binding modalities, such as scFvs or nanobodies, and a transcriptional regulator of choice (e.g., Gal4-VP64 or tetR-VP64) can replace the natural cytoplasmic domain. Thus, one can build a receptor targeted to a cell surface ligand of interest, such as a disease-related or tissue-related antigen, and this environmental sensing event leads to the release of the transcriptional regulator and the initiation of a custom cellular response. synNotch receptors allow for unprecedented control over cellular sensing and response behaviors and can be used in a wide variety of cell types to help them sense their environment and locally modulate their own behavior or the surrounding microenvironment (50, 84, 90).
synNotch receptor circuits are a versatile and modular system to selectively regulate cellular responses and behavior in defined environmental contexts. These receptors are functional and control a variety of aspects of cellular function in fibroblasts, primary neurons, and T cells (50, 84). Since the transcriptional program controlled by synNotch receptors is user defined, the possibilities for the control of cells are vast, including the ability to drive cellular communication, differentiation, and direct killing of diseased cells, such as cancer cells. An important feature of the synNotch platform is the ability to equip cells to perform synthetic nonnatural behaviors. An example is the antigen-dependent production of genetically encodable therapeutic agents such as commercial antibodies. Thus, synNotch T cells can potentially be used to recognize and remodel a disease microenvironment. The ability to utilize synNotch receptor circuits to increase the landscape of antigen-dependent cellular response programs beyond the natural is an important, potentially transformative feature of this new class of synthetic receptors (90). Below we describe how synNotch, and other components, can be incorporated into more sophisticated therapeutic decision-making circuits.
Decision-Making Circuits: Increased Control and Discrimination
One of the major concerns with cell therapies is the lack of control over the cells once they have been administered to patients. Because of their powerful actions, T cells and other immune cells can rapidly cause severe damage to the body. Thus, it is important that the user (physician) be able to control cells after they have been infused into the body; basic cell therapies must provide improved control in the future. Below we discuss examples of using small-molecule drugs to regulate the ability of cells to persist and activate in patients.
Control over therapeutic cell death: kill switches
One way to make therapeutic use of T cells safer is to have the ability to eliminate them rapidly by engineering control over cell death pathways. There are a few ways that this problem has been approached. An early of example of this strategy was to modify T cells with the thymidine kinase gene from herpes simplex virus (HSV-TK) that sensitizes the cells to the antiviral medication ganciclovir (91, 92). This strategy has been tested in humans for both allogeneic transplants for the control of graft-versus-host disease (GVHD) and T cell therapies in patients with HIV (93). Although it is a promising strategy, it has several drawbacks: The viral protein is immunogenic, DNA synthesis must be active for the elimination to take effect, and mutations have been observed in the HSV-TK gene that render it resistant to ganciclovir (94). Because of this there has been considerable research to engineer alternatives.
There are two other prominent approaches to controlling the longevity of therapeutic immune cells. One is an engineered split caspase 9 (iCASP9) that is assembled in response to a heterodimerizing drug (Figure 5a). This system can rapidly drive therapeutic T cells into apoptosis upon addition of a drug with kinetics that may help to eliminate cell therapies that have become toxic or that are no longer needed after the patient is free of disease (95). A second approach to control the longevity of therapeutic cells is ectopic expression of a truncated epidermal growth factor receptor (tEGFR). The cells can be eliminated via antibody-dependent, cell-mediated cytotoxicity (ADCC) driven by treatment with cetuximab, an antibody specific to tEGFR (96).
Figure 5.
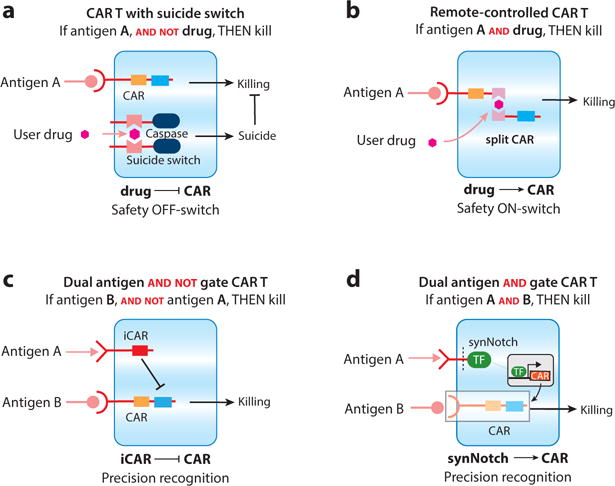
Synthetic combinatorial control therapeutic T cell circuits. (a) Split caspases have been engineered that allow for small-molecule control over therapeutic cell death. (b) ON-switch chimeric antigen receptors (CARs) have been engineered such that the ligand-binding domain and signaling chain are split. Receptor activity is regulated by antigen detection and a small-molecule heterodimerizer (AND logic), allowing for control over cellular activity after administration to the patient. (c) iCARs are inhibitory receptors that can overcome the positive activity of stimulatory receptors. These receptors can be utilized to sense bystander tissue and prevent the T cell from causing toxicity (AND-NOT logic). (d) synNotch receptors can be used to control the expression of CARs. synNotch → circuits allow for combinatorial antigen sensing and could help to confine CAR expression and T cell activation to the tumor (temporal AND logic).
All of these approaches to controlling cell death via drug or antibody delivery to engineered cells have their limitations. If any of the cells lose expression of the components, they are rendered insensitive to elimination. Given that T cells are capable of clonally expanding on rapid timescales, killing off 99% of the cells may not be enough to eliminate a cell therapy that has become dangerous to the patient. Finally, a kill switch leads to an all-or-none abortive outcome. Aborting this expensive treatment may not be ideal for patients who do not have a second chance for therapy. Thus, in some scenarios, the tunable regulation of cellular activity, as discussed below, may provide certain advantages over eliminating the cells altogether.
Remote control of therapeutic T cell activity with ON-switch cars
Another way to control cellular activity, less drastic than killing them, is to regulate the ability of the cell to transduce activation signals. We have developed a split CAR design that requires antigen recognition and a small-molecule heterodimerizer to drive signal transduction and activation of T cells (Figure 5b) (97). This is accomplished by separating the ligand-binding portion of the receptor from the cytoplasmic signaling chain, a common feature of many of the antigen receptors of the immune system. Where these synthetic split receptors differ from their natural counterparts is the addition of a chemically induced heterodimerization system. In the first iterations, the split ON-switch CAR was engineered where each half of the CAR contained either FK506 binding protein (FKBP) or rapamycin binding protein (Frb), proteins that inducibly heterodimerize in the presence of a small-molecule variant of rapamycin, AP21967 (rapalog) (98). When T cells are engineered with an ON-switch CAR, they act as a Boolean logic AND gate requiring the presence of the target antigen and drug to activate. Two other key features of ON-switch CAR T cells are (a) they can titrate activities such as killing, cytokine production, and proliferation by changing the concentration of drug and (b) the activity of the T cells can be rapidly halted or induced (on a scale of minutes), given that the drug works at the level of signal transduction. Thus, ON-switch CARs may allow for unprecedented control over therapeutic T cells, and versions of these receptors with clinically relevant dimerization systems are being actively pursued for rapid transition from proof-of-principle work in mice to clinical studies with cancer patients. Other small-molecule-regulated split CAR designs have recently been described with alternative approaches to dimerization, showing the high priority placed on safety and controllability of future CAR T cell therapies (99).
Sakemura et al. (100) recently developed an alternate drug-inducible CAR system based on the third generation tetracycline-inducible gene expression system (Tet-On-3G). Instead of controlling CAR signaling, as in the case of the ON-switch, addition of doxycycline simply drives transcription of the α-CD19 CAR. While this approach is simple and could prove useful, there are issues with leakage of CAR expression prior to drug treatment resulting in some activity of the engineered T cells when exposed to CD19+ cancer cells prior to drug treatment. Tet-On CAR T cells differ from ON-switch CAR T cells in several ways: (a) Tet-On CAR T cells do not necessarily require drug and antigen be present simultaneously to activate, (b) the ability to titrate cellular activity is less likely, owing to the inability to directly regulate signal transduction, and (c) the timescale for halting or inducing cellular activation is on the order of hours because of the inherent kinetics of the Tet-inducible promoter.
While regulated CARs are likely to become a major component of T cell therapies, all of these engineered ON-switch prototypes currently face similar optimization challenges—preventing basal leakiness, maintaining high levels of maximal ON state activity, tuning of dynamic range, and prevention of immunogenicity. Nonetheless, these controlled CAR T cells may go a long way to improve the safety profile of CAR T cells, allowing the treatment to become more widespread.
Boolean logic–gated therapeutic T cell activity: discrimination through multi-antigen recognition
The rarity of single antigens that distinguish a cancer cell from normal cells in our bodies is well established (101, 102). The single-antigen targeting of most current CAR T cell therapies is not viable long-term for multiple reasons, including off-target toxicity and loss of expression of targetable antigens that often allows tumors to escape recognition. Given this obvious challenge for cell therapy, one growing area of focus is developing T cells that are activated by a specific combination of antigen, a strategy that could significantly increase specificity of recognition. Below we discuss several of the current approaches to build multiple-antigen-triggered Boolean logic gate receptor systems in therapeutic cells.
Cooperative activity (and gate logic)
Many cells utilize cooperative receptor systems, where engagement of multiple receptors is necessary for full cellular activation. These sorts of systems force cells to recognize combinatorial signals, reducing the probability of spurious cellular responses, which can be damaging if deployed in the wrong context. T cells naturally require multiple inputs for activation. They need stimulation of the TCR (signal 1) and engagement of costimulatory receptors (signal 2) before they commit to full activation. In fact, TCR stimulation without the appropriate costimulatory signals can push T cells into a nonresponsive anergic state. NK cells also use activity of multiple receptors to activate, with different receptors positively stimulating activation and others inhibiting it (36). The relative level of signaling coming from these receptors determines whether the NK cell kills the target. As synthetic biologists, we have looked to these systems for inspiration in building new receptors with more layers of control over their activity for added safety.
Several groups have engineered cooperative synthetic receptor systems for improved cell autonomous control over CAR T cells with the goal of reducing off-target toxicity. These multiple CAR systems are akin to the natural T cell activation paradigm, where one CAR provides the primary TCR signal 1 and a second provides costimulation (signal 2). Because this is a two-receptor each receptor can be targeted to a different antigen, requiring the T cells to engage multiple antigens to activate (103–105). While these receptor systems can show AND gate behavior, careful engineering of each receptor is required to make a receptor system that shows binary activation— where T cell activity is minimal with either single-antigen input but maximal with dual-antigen inputs. Sadelain and colleagues were able to engineer T cells with a two-receptor cooperative system that recognize prostate stem cell antigen (PSCA) and prostate-specific membrane antigen (PSMA) (104). T cells engineered with their cooperative CAR system exhibited AND behavior, but considerable tuning of the strength of signaling through the receptors was required to achieve the AND gate logic. Thus, when trying to engineer cooperative receptor systems much attention must be paid to the contribution of each receptor to T cell activity, and it is exceedingly difficult to identify binary systems where neither receptor drives an appreciable T cell response (e.g., cytokine production or proliferation or cytotoxicity). In many cancer treatment scenarios, this may not be a prerequisite for safe treatment of disease, and something that approximates AND gate logic could reduce off-target toxicity to levels that are easily managed by conventional means.
Antagonistic receptor systems—AND/NOT gate logic
Sadelain and colleagues also developed negative regulatory receptors, called iCARs. Similar to the natural NK cell receptor system, iCARs act in a dominant fashion to shut down the activity of a coexpressed CAR that positively drives T cell activation (Figure 5c) (69). iCARs are engineered by replacing the stimulatory signaling domains on the cytoplasmic tail of the CAR with the inhibitory signaling chains derived from the coinhibitory receptors, PD-1 or CTLA4. The rationale for such a multireceptor system is that it could be used to selectively prevent killing of related bystander tissues (that share the CAR antigen), a major concern with many of the CARs tested in preclinical models and clinical trials. The preclinical results from the iCAR system show that T cells can distinguish target cells from bystander cells even when they are in a mixed population. As with other combinatorial antigen recognition approaches, the bottleneck for utilization of such an approach may be the determination of the negative antigens that the iCARs should target. This will require the further identification of antigens specific to potential off-target tissues.
synNotch gated CAR expression—temporal AND gate logic
An important demonstration of the utility of synNotch receptors in therapeutic T cells is to combine them with CARs. A simple but powerful way is to use synNotch receptors to induce the expression of a CAR, but where each receptor detects a distinct antigen (Figure 5d) (84). synNotch → CAR circuits are appealing in that they control when and where the T cell expresses the CAR, potentially relegating expression of toxic CARs to the tumor, away from bystander tissues. Under basal conditions the T cells are “unarmed”; i.e., they do not express the cytotoxic CAR. Since the two receptors can be targeted to different antigens, the T cells can be engineered to sense combinatorial antigen signatures. synNotch → CAR circuits can be engineered into primary human T cells, and these T cells are able to selectively express the CAR in the tumor microenvironment. In preclinical models, T cells with such a circuit can reliably target a tumor with a dual antigen signature, distinguishing them from bystander tumors with the single antigens. A critical and distinguishing feature of these combinatorially controlled synNotch → CAR T cells compared to the other Boolean logic–gated CAR systems discussed above is that the synNotch receptor is completely independent from and orthogonal to the T cell signaling machinery. Thus, no activation of the T cells is observed prior to CAR expression, making the therapeutic T cells completely inert (noncytotoxic) until the synNotch receptor is engaged by antigen present in the local tumor microenvironment. Such features may make this type of sequentially armed T cell a safer alternative for combinatorial antigen sensing.
Payloads and Output Activities
The natural progression of an immune response involves the local delivery of therapeutic payloads and coordinated cellular activities at the site of disease. A spectrum of payloads including cytokines, antibodies, and small molecules are made by the vasculature, tissue, and infiltrating immune cells to help eliminate disease and stimulate tissue repair. Future engineered cell therapies should have the ability to sense disease and initiate a response that includes the delivery of a custom set of therapeutic payloads and effector activities.
Beyond killing: enhancing T cell function to overcome an inhospitable tumor environment
In most cases, the goal of therapeutic T cell engineering is to retarget the key native outputs of T cells—cytotoxic responses and T cell proliferation. Nonetheless, it is becoming clear that nonnative enhancement of T cell function may be required to allow T cells to overcome multiple layers of immune suppression that exist in a variety of tumor types (106, 107).
Boosting therapeutic T cell activity by expressing inflammatory proteins
A general approach is to arm engineered T cells via constitutive expression of new functional proteins. For example, “armored” CAR T cells constitutively express the proinflammatory cytokine IL-12 and show enhanced ability to perform in a suppressive environment (108, 109). Alternatively, Morgan and colleagues engineered TILs to inducibly express IL-12 in response to TCR stimulation by putting the gene downstream of nuclear factor of activated T cells (NFAT) response elements (110, 111). These strategies represent a potential significant improvement over systemic infusion or production of cytokines like IL-12 or IL-2, which has been demonstrated to be highly toxic (112–115). Similarly, attaching nanoparticles infused with the proliferative and survival cytokine IL-2 to T cells can enhance their survival. In another example, expression of enzymes that remodel the extracellular matrix appears to increase the ability of T cells to infiltrate particular tumor models (116).
Local expression of new immune effectors via synNotch circuits
More recently, the synNotch receptor platform has been harnessed to induce the expression of these types of inflammatory modulators, but only in an antigen-triggered manner (90). This spatially targeted delivery of immune enhancer proteins has the potential to allow higher efficacy in response, without the cost of systemic side effects that could come with constitutive expression of these immune enhancers. synNotch receptors can be used to inducibly express a wide range of payloads, including stimulatory cytokines like IL-2 and IL-12 and chemokines. Moreover, synNotch systems can be used to deliver nonnative payloads such as antibodies (including checkpoint inhibitors), bispecific antibodies, and innate immune stimulatory molecules. Thus, a wide array of à la carte effector functions can in principle be engineered into therapeutic T cells, while maintaining the specificity of antigen-targeted activation (90). Such functions could be used to enhance CAR or TCR functions, or in independent cells that would act to recognize and remodel local microenvironments.
CONCLUSION: FUTURE OF THERAPEUTIC IMMUNE CELLS
Cell-Based Cancer Therapy
We are at a tremendously exciting point in the history of engineered immune cell cancer therapeutics. The emerging field of synthetic immunology is assembling a large arsenal of tools that can be used to enhance or reprogram T cell function in diverse ways and overcome the fundamental problems in treating cancer—how to recognize the tumor and discriminate it from normal cells and how to mount a potent cytotoxic response that overcomes local immunosuppression that is a feature of many solid tumors (Figure 6).
Figure 6.
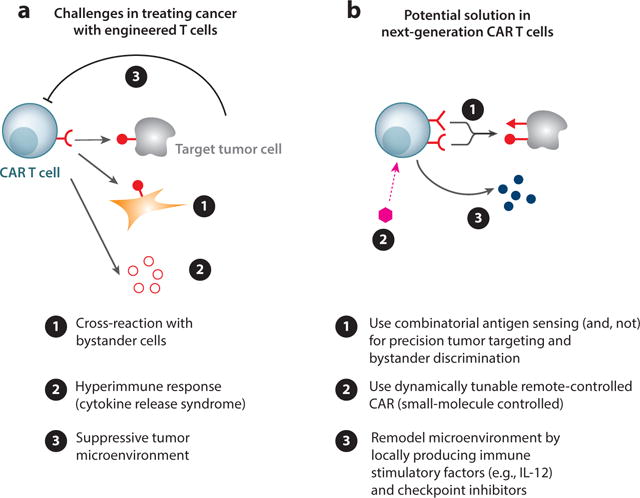
Overcoming the challenges of solid tumors. (a) T cell therapies can cause life-threatening damage due to ➊ cross-reaction with off-target tissue and ➋ excessive inflammation. On the other hand, tumor environments can be suppressive, ➌ preventing a curative immune response. (b) To overcome these pitfalls, the T cells can be engineered ➊ to sense combinatorial antigens, ➋ to be regulated with small molecules, and ➌ to produce therapeutic agents that prime the tumor environment for more effective treatment.
Abbreviation: CAR, chimeric antigen receptor.
As described here, precision recognition of tumors will perhaps be a routine option effected by deploying combinatorial antigen-sensing circuits. This includes cells that can use Boolean AND or NOT functionality both to more precisely recognize target tumor cells and to prevent cross-reaction with normal tissue with overlapping antigen profiles. The strategy of recognizing tumors via a set of antigen signatures is very promising, depending on whether clear signatures can be bioinformatically identified and robust recognition circuits developed.
The emerging tool kit of cell engineering also points to ways to overcome the immunosuppressive tumor microenvironment. We now have increased capacity to inducibly produce immune stimulatory factors, like IL-12, in the tumor, as well as to locally deliver checkpoint inhibitor antibodies. Thus, we may be able, in a tumor-specific manner, to induce the local expression of a cocktail of factors that robustly remodels the microenvironment to disable immunosuppression.
Although we can begin to more clearly imagine what ideal engineered immune cell cancer therapies would look like, there remain many challenges. Many of the components described here have only been prototyped in preclinical models, and how they function in a clinical setting (including issues such as immunogenicity) will present important hurdles. Moreover, as we consider building more complex circuits, the issues of genetic delivery payloads and costs loom larger. Issues like improved vectors and gene delivery, as well as allogeneic cells, will also be critical.
Beyond Cancer—Autoimmunity, Degeneration, and Inflammation
Although we have focused primarily on engineering T cell therapeutics for cancer, many of the tools and strategies outlined in this review have direct relevance to other diseases, such as autoimmunity and degeneration/regeneration (Figure 7). Recently, Payne and colleagues developed a modified CAR they call a chimeric autoantibody receptor that redirected T cells to recognize Pemphigus vulgaris–causing autoreactive B cells (117). This is a clever demonstration of how engineered cells can be used to eliminate pathogenic immune cells causing life-threatening autoimmunity. In the future, synNotch circuits could be utilized in T cells or other cell types to treat autoimmunity by eliciting production of immunosuppressive agents (anti-inflammatory cytokines or biologics) at sites of autoinflammation. Importantly, the treatment would be triggered locally by antigens unique to the disease environment, overcoming one of the major problems in treating inflammation and autoimmunity—the adverse effects of systemic immune suppression. Similarly, it is possible that antigens associated with degeneration or injury could be harnessed to trigger the precise delivery of anti-inflammatory and proregenerative factors.
Figure 7.
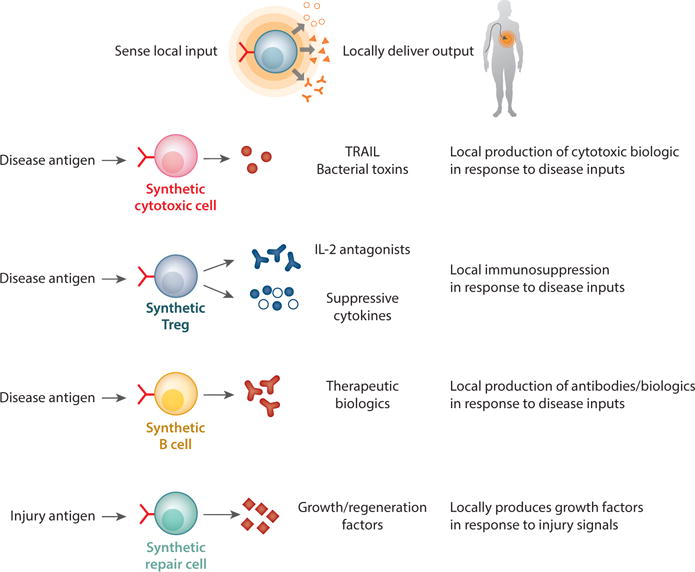
Engineering nonnatural functionalities of T cells. T cells can be engineered to sense disease microenvironments such as tumors and locally perform both natural and synthetic effector programs. T cells can produce cytotoxic agents, cytokines, commercial biologics such as antibodies, and factors involved in regeneration of tissues in a context-dependent manner.
It is also possible that engineered immune cells will emerge as a useful research tool. Like an antibody or drug, an immune cell that has the capability to recognize user-defined antigen signatures and remodel that microenvironment would provide a new and customizable mode of targeted perturbation. As we learn more about the underlying principles of tissues and systems comprising multiple cell types, a precision-engineered cell that can participate as an active but controllable node within complex, in vivo networks would be a powerful tool.
These strategies for therapy and research all hinge on harnessing the remarkable intrinsic ability of immune cells to survey and monitor the body for particular molecular signals. When combined with sophisticated genetic engineering, that capability would clearly enable many new therapeutic actions—especially for complex diseases where detection and remodeling of a microenvironment is essential. These are diseases in which cell therapeutics and their higher-order sense/response functions may particularly excel over drugs and biologics. Much remains to be seen about how the exciting emerging field of synthetic immunology will develop, and whether its remarkable promise can be fulfilled.
Footnotes
The Annual Review of Immunology is online at immunol.annualreviews.org
DISCLOSURE STATEMENT
W.A.L. is a founder of Cell Design Labs and a member of their advisory board. K.T.R. is a consultant for Cell Design Labs.
LITERATURE CITED
- 1.Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54(2):281–88. doi: 10.1016/j.molcel.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lämmermann T, Sixt M. The microanatomy of T-cell responses. Immunol Rev. 2008;221(1):26–43. doi: 10.1111/j.1600-065X.2008.00592.x. [DOI] [PubMed] [Google Scholar]
- 3.Spiegel DA. Grand challenge commentary: synthetic immunology to engineer human immunity. Nat Chem Biol. 2010;6(12):871–72. doi: 10.1038/nchembio.477. [DOI] [PubMed] [Google Scholar]
- 4.Geering B, Fussenegger M. Synthetic immunology: modulating the human immune system. Trends Biotechnol. 2015;33(2):65–79. doi: 10.1016/j.tibtech.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. 2016;535(7610):65–74. doi: 10.1038/nature18847. [DOI] [PubMed] [Google Scholar]
- 6.Brestoff JR, Artis D. Immune regulation of metabolic homeostasis in health and disease. Cell. 2015;161(1):146–60. doi: 10.1016/j.cell.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischbach MA, Bluestone JA, Lim WA. Cell-based therapeutics: the next pillar of medicine. Sci Transl Med. 2013;5(179):179ps7. doi: 10.1126/scitranslmed.3005568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12(11):978–90. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldberg MS. Immunoengineering: how nanotechnology can enhance cancer immunotherapy. Cell. 2015;161(2):201–4. doi: 10.1016/j.cell.2015.03.037. [DOI] [PubMed] [Google Scholar]
- 10.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol. 2016;34:539–73. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 11.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–14. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–77. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholler J, Brady TL, Binder-Scholl G, Hwang W-T, Plesa G, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4(132):132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter DL, Hwang W-T, Frey NV, Lacey SF, Shaw PA, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawakami Y, Robbins PF, Wang X, Tupesis JP, Parkhurst MR, et al. Identification of new melanoma epitopes on melanosomal proteins recognized by tumor infiltrating T lymphocytes restricted by HLA-A1, -A2, and -A3 alleles. J Immunol. 1998;161(12):6985–92. [PubMed] [Google Scholar]
- 17.Rosenberg SA, Yang JC, White DE, Steinberg SM. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: identification of the antigens mediating response. Ann Surg. 1998;228(3):307–19. doi: 10.1097/00000658-199809000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–39. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–57. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Investig. 2006;116(10):2777–90. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gros A, Turcotte S, Wunderlich JR, Ahmadzadeh M, Dudley ME, Rosenberg SA. Myeloid cells obtained from the blood but not from the tumor can suppress T-cell proliferation in patients with melanoma. Clin Cancer Res. 2012;18(19):5212–23. doi: 10.1158/1078-0432.CCR-12-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmadzadeh M, Felipe-Silva A, Heemskerk B, Powell DJ, Wunderlich JR, et al. FOXP3 expression accurately defines the population of intratumoral regulatory T cells that selectively accumulate in metastatic melanoma lesions. Blood Am Soc Hematol. 2008;112(13):4953–60. doi: 10.1182/blood-2008-06-163048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao X, Ahmadzadeh M, Lu Y-C, Liewehr DJ, Dudley ME, et al. Levels of peripheral CD4+FoxP3+ regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood. 2012;119(24):5688–96. doi: 10.1182/blood-2011-10-386482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. PNAS. 1989;86(24):10024–28. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64(5):891–901. doi: 10.1016/0092-8674(91)90314-o. [DOI] [PubMed] [Google Scholar]
- 26.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. PNAS. 1993;90(2):720–24. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161(6):2791–97. [PubMed] [Google Scholar]
- 28.Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat Biotechnol. 2002;20(1):70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 29.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–64. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–29. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan D-AN, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–26. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–51. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paust S, Andrian von UH. Natural killer cell memory. Nat Immunol. 2011;12(6):500–8. doi: 10.1038/ni.2032. [DOI] [PubMed] [Google Scholar]
- 36.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9(5):495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferlazzo G, Pack M, Thomas D, Paludan C, Schmid D, et al. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. PNAS. 2004;101(47):16606–11. doi: 10.1073/pnas.0407522101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koka R, Burkett P, Chien M, Chai S, Boone DL, Ma A. Cutting edge: Murine dendritic cells require IL-15R alpha to prime NK cells. J Immunol. 2004;173(6):3594–98. doi: 10.4049/jimmunol.173.6.3594. [DOI] [PubMed] [Google Scholar]
- 39.Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. 2007;26(4):503–17. doi: 10.1016/j.immuni.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, et al. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. PNAS. 2008;105(45):17481–86. doi: 10.1073/pnas.0804788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. 2009;15(15):4857–66. doi: 10.1158/1078-0432.CCR-08-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, et al. Targeting CD20+ aggressive B-cell non–Hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. 2015;3(4):333–44. doi: 10.1158/2326-6066.CIR-14-0114. [DOI] [PubMed] [Google Scholar]
- 43.Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83. doi: 10.1182/blood-2004-12-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Liu LN, Feller S, Allen C, Shivakumar R, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010;17(3):147–54. doi: 10.1038/cgt.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14(7):830–40. doi: 10.3109/14653249.2012.671519. [DOI] [PubMed] [Google Scholar]
- 46.Ivics Z, Izsvák Z. The expanding universe of transposon technologies for gene and cell engineering. Mob DNA. 2010;1(1):25. doi: 10.1186/1759-8753-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakazawa Y, Huye LE, Dotti G, Foster AE, Vera JF, et al. Optimization of the PiggyBac transposon system for the sustained genetic modification of human T lymphocytes. J Immunother. 2009;32(8):826–36. doi: 10.1097/CJI.0b013e3181ad762b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davey NE, Travé G, Gibson TJ. How viruses hijack cell regulation. Trends Biochem Sci. 2011;36(3):159–69. doi: 10.1016/j.tibs.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 49.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–47. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, et al. Engineering customized cell sensing and response behaviors using synthetic Notch receptors. Cell. 2016;164(4):780–91. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang Y, Jacoby E, Fry TJ. Challenges and opportunities of allogeneic donor-derived CAR T cells. Curr Opin Hematol. 2015;22(6):509–15. doi: 10.1097/MOH.0000000000000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RPT, Carpenter RO, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–49. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–90. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 60.Brentjens RJ, Davila ML, Rivière I, Park J, Wang X, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, et al. Cancer immunology: Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348(6230):124–28. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17(19):6287–97. doi: 10.1158/1078-0432.CCR-11-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–71. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kunert A, Straetemans T, Govers C, Lamers C, Mathijssen R, et al. TCR-engineered T cells meet new challenges to treat solid tumors: choice of antigen, T cell fitness, and sensitization of tumor milieu. Front Immunol. 2013;4:363. doi: 10.3389/fimmu.2013.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21(8):914–21. doi: 10.1038/nm.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–42. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Narayanan P, Lapteva N, Seethammagari M, Levitt JM, Slawin KM, Spencer DM. A composite MyD88/CD40 switch synergistically activates mouse and human dendritic cells for enhanced antitumor efficacy. J Clin Investig. 2011;121(4):1524–34. doi: 10.1172/JCI44327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hart Y, Alon U. The utility of paradoxical components in biological circuits. Mol Cell. 2013;49(2):213–21. doi: 10.1016/j.molcel.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 71.Hart Y, Reich-Zeliger S, Antebi YE, Zaretsky I, Mayo AE, et al. Paradoxical signaling by a secreted molecule leads to homeostasis of cell levels. Cell. 2014;158(5):1022–32. doi: 10.1016/j.cell.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 72.Spangler JB, Moraga I, Mendoza JL, Garcia KC. Insights into cytokine-receptor interactions from cytokine engineering. Annu Rev Immunol. 2015;33:139–67. doi: 10.1146/annurev-immunol-032713-120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484(7395):529–33. doi: 10.1038/nature10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, et al. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity. 2015;42(5):826–38. doi: 10.1016/j.immuni.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kawahara M, Ueda H, Nagamune T. Engineering cytokine receptors to control cellular functions. Biochem Eng J. 2010;48(3):283–94. [Google Scholar]
- 76.Sogo T, Kawahara M, Tsumoto K, Kumagai I, Ueda H, Nagamune T. Selective expansion of genetically modified T cells using an antibody/interleukin-2 receptor chimera. J Immunol Methods. 2008;337(1):16–23. doi: 10.1016/j.jim.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 77.Melero I, Rouzaut A, Motz GT, Coukos G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4(5):522–26. doi: 10.1158/2159-8290.CD-13-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33(8):780–88. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moon EK, Carpenito C, Sun J, Wang L-CS, Kapoor V, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17(14):4719–30. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park JS, Rhau B, Hermann A, McNally KA, Zhou C, et al. Synthetic control of mammalian-cell motility by engineering chemotaxis to an orthogonal bioinert chemical signal. PNAS. 2014;111(16):5896–901. doi: 10.1073/pnas.1402087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Conklin BR, Hsiao EC, Claeysen S, Dumuis A, Srinivasan S, et al. Engineering GPCR signaling pathways with RASSLs. Nat Methods. 2008;5(8):673–78. doi: 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sykes EA, Dai Q, Sarsons CD, Chen J, Rocheleau JV, et al. Tailoring nanoparticle designs to target cancer based on tumor pathophysiology. PNAS. 2016;113(9):E1142–51. doi: 10.1073/pnas.1521265113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164(4):770–79. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393(6683):382–86. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 86.Selkoe D, Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565–97. doi: 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- 87.Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, Blacklow SC. Structural basis for autoinhibition of Notch. Nat Struct Mol Biol. 2007;14(4):295–300. doi: 10.1038/nsmb1227. [DOI] [PubMed] [Google Scholar]
- 88.Bray SJ. Notch signalling: A simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7(9):678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 89.Gordon WR, Zimmerman B, He L, Miles LJ, Huang J, et al. Mechanical allostery: evidence for a force requirement in the proteolytic activation of Notch. Dev Cell. 2015;33(6):729–36. doi: 10.1016/j.devcel.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, et al. Engineering T cells with customized therapeutic response programs using synthetic Notch receptors. Cell. 2016;167(2):419–32.e16. doi: 10.1016/j.cell.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–24. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 92.Greco R, Oliveira G, Stanghellini MTL, Vago L, Bondanza A, et al. Improving the safety of cell therapy with the TK-suicide gene. Front Pharmacol. 2015;6:95. doi: 10.3389/fphar.2015.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, et al. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med. 1996;2(2):216–23. doi: 10.1038/nm0296-216. [DOI] [PubMed] [Google Scholar]
- 94.Frank O, Rudolph C, Heberlein C, von Neuhoff N, Schröck E, et al. Tumor cells escape suicide gene therapy by genetic and epigenetic instability. Blood. 2004;104(12):3543–49. doi: 10.1182/blood-2004-03-0852. [DOI] [PubMed] [Google Scholar]
- 95.Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang X, Chang W-C, Wong CW, Colcher D, Sherman M, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood Am Soc Hematol. 2011;118(5):1255–63. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu C-Y, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350(6258):aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bayle JH, Grimley JS, Stankunas K, Gestwicki JE, Wandless TJ, Crabtree GR. Rapamycin analogs with differential binding specificity permit orthogonal control of protein activity. Chem Biol. 2006;13(1):99–107. doi: 10.1016/j.chembiol.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 99.Juillerat A, Marechal A, Filhol J-M, Valton J, Duclert A, et al. Design of chimeric antigen receptors with integrated controllable transient functions. Sci Rep. 2016;6:18950. doi: 10.1038/srep18950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, et al. A Tet-On inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol Res. 2016;4(8):658–68. doi: 10.1158/2326-6066.CIR-16-0043. [DOI] [PubMed] [Google Scholar]
- 101.Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22(1):26–36. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gross G, Eshhar Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol. 2016;56:59–83. doi: 10.1146/annurev-pharmtox-010814-124844. [DOI] [PubMed] [Google Scholar]
- 103.Wilkie S, van Schalkwyk MCI, Hobbs S, Davies DM, van der Stegen SJC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32(5):1059–70. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 104.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1(1):43–53. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 108.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133–41. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70(17):6725–34. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 2011;19(4):751–59. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang L, Feldman SA, Zheng Z, Chinnasamy N, Xu H, et al. Evaluation of γ-retroviral vectors that mediate the inducible expression of IL-12 for clinical application. J Immunother. 2012;35(5):430–39. doi: 10.1097/CJI.0b013e31825898e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90(7):2541–48. [PubMed] [Google Scholar]
- 113.Margolin KA, Rayner AA, Hawkins MJ, Atkins MB, Dutcher JP, et al. Interleukin-2 and lymphokine-activated killer cell therapy of solid tumors: analysis of toxicity and management guidelines. J Clin Oncol. 1989;7(4):486–98. doi: 10.1200/JCO.1989.7.4.486. [DOI] [PubMed] [Google Scholar]
- 114.Ochoa JB, Curti B, Peitzman AB, Simmons RL, Billiar TR, et al. Increased circulating nitrogen oxides after human tumor immunotherapy: correlation with toxic hemodynamic changes. J Natl Cancer Inst. 1992;84(11):864–67. doi: 10.1093/jnci/84.11.864. [DOI] [PubMed] [Google Scholar]
- 115.Schwartz RN, Stover L, Dutcher J. Managing toxicities of high-dose interleukin-2. Oncology. 2002;16(Suppl. 13):11–20. [PubMed] [Google Scholar]
- 116.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–29. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353(6295):179–84. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]