

Abstract
Purpose
It has been suggested that patients with traumatic insults are resuscitated into a state of an early systemic inflammatory response. We aimed to evaluate the influence of hemorrhagic shock and resuscitation (HSR) upon the inflammatory response capacity assessed by overall TNF-α secretion capacity of the host compared to its release from circulating leukocytes in peripheral circulation.
Methods
Rats (8/group) subjected to HS (MAP of 30–35 mmHg for 90 min followed by resuscitation over 50 min) were challenged with Lipopolysaccharide (LPS), 1 μg/kg intravenously at the end of resuscitation (HSR-LPS group) or 24 h later (HSR-LPS24 group). Control animals were injected with LPS without bleeding (LPS group). Plasma TNF-α was measured at 90 min after the LPS challenge. In addition, whole blood (WB) was obtained either from healthy controls (CON) immediately after resuscitation (HSR), or at 24 h post-shock (HSR 24). WB was incubated with LPS (100 ng/mL) for 2 h at 37 °C. TNF-α concentration and LPS binding capacity (LBC) was determined.
Results
Compared to LPS group, HSR followed by LPS challenge resulted in suppression of plasma TNF-α in HSR-LPS and HSR-LPS24 groups (1835 ± 478, 273 ± 77, 498 ± 200 pg/mL, respectively). Compared to CON the LPS-induced TNF-α release capacity of circulating leukocytes ex vivo was strongly declined both at the end of resuscitation (HSR) and 24 h later (HSR24) (1012 ± 259, 313 ± 154, 177 ± 63 ng TNF/mL, respectively). The LBC in WB was similar between CON and HSR and only moderately enhanced in HSR24 (57 ± 6, 56 ± 6, 71 ± 5 %, respectively).
Conclusion
Our data suggest that the overall inflammatory response capacity is decreased immediately after HSR, persisting up to 24 h, and is independent of LBC.
Keywords: Injuries, Endotoxins shock, Immunosuppression
Introduction
Despite the advances made in critical care therapeutic protocols, severely injured patients that reach hospital after an immediate resuscitation frequently develop a life-threatening failure of one or more organs in the subsequent days of their ICU stay. The failure of essential organ systems has been alleged to result from an uncontrolled immune inflammatory response triggered by the injury/bleeding.1, 2 The current consensus has been that patients with traumatic insults are resuscitated into a state of an early systemic inflammatory response (SIRS). SIRS is initiated immediately after trauma by activation of polymorphonuclear leukocytes (PMNs), macrophages, lymphocytes, and the release of various inflammatory mediators including cytokines, responsible for orchestrating the normal physiologic responses to trauma and hemorrhage via complex cellular and molecular events.1, 3 While an immediate and excessive SIRS frequently leads to an acute organ failure and early death, an excessive counter-regulatory anti-inflammatory response (CARS) may induce a prolonged immunosuppressed state that can also be deleterious to the host at later stage.4 However, there is increasing evidence that significant overlap exists in the pro- and anti-inflammatory phase.5
Several authors have demonstrated various immunosuppressive effects of trauma experimentally and clinically. These include, among others, a significant correlation between severely traumatized patients with ISS of 16 or greater and elevated plasma levels of the anti-inflammatory cytokines and their correlation with mortality from trauma.6, 7 The down regulation of the immune response was also demonstrated by the work of Keel et al8 when compared to healthy volunteers, PMNs obtained in injured patients with an average ISS of 39 secreted markedly less pro-inflammatory cytokines following stimulation with LPS. Overall, these studies indicate that severe injury influences leukocyte function and cytokine formation producing both immune-activation and immune-suppression.
Circulating leukocytes represent a relatively narrow source of the overall inflammatory mediators released to the systemic circulation during the post-traumatic phase.9, 10, 11 Given that a post-traumatic inflammatory response maybe compartmentalized,10 the inflammatory capacity readout obtained from the circulating blood cells stimulated ex vivo may not necessarily reflect the global immune status of the host. We aimed to evaluate the influence of hemorrhagic shock upon a) the early secretion capacity of TNF-α by comparing its release from circulating leukocytes and in peripheral circulation, and b) the endotoxin-binding capacity (LBC).
Materials and methods
Animals
Male Sprague–Dawley rats (n = 24), weighing 440–460 g, were used. Animals were kept under controlled temperature (22 °C), humidity, and lighting (12-h light–dark cycles) with free access to standard laboratory chow and water. Prior to the experiment, animals were fasted overnight but allowed water ad libitum. All animal procedures were approved (MA58-955/94) by the Viennese (Austria) Legislative Committee and conducted according to National Institute of Health Guidelines.
Surgical procedure
Anesthesia was induced by ketamine/xylazine (112/15 mg/kg, intramuscularly). The animals were kept on a temperature controlled surgical board (38 °C ± 1 °C) and allowed to breathe spontaneously. The left femoral artery was cannulated under aseptic conditions and connected to a pressure transducer. A second catheter was inserted to the level of right atrium via the right jugular vein.
Hemorrhagic shock and resuscitation (HSR)
The rats were bled through the jugular vein catheter to mean arterial pressure (MAP) of 30–35 mmHg over 5 min and maintained at that level for 90 min (Fig. 1). The shed blood was then returned followed by the infusion of Ringer's solution, twice the shed blood volume, over 50 min. Next the catheter was removed, the femoral artery ligated, the groin incision closed, and the animals were returned to their cages for 24 h observation.
Fig. 1.

Experimental procedure: The rats were bled to mean arterial pressure of 30–35 mmHg over 5 min and maintained for 90 min. Shed blood was then infused followed by Ringer's solution, twice the shed blood volume, over 50 min. Blood samples for ex vivo experiments were obtained prior to the In vivo endotoxin (LPS: 1 μg/kg BW) challenge. LPS control animals received the same LPS dose but without bleeding.
In vivo endotoxin (lipopolysaccharide: LPS) challenge
Animals subjected to HSR received a bolus injection of E. coli 026:B6 LPS (Difco, Detroit, MI) at a dose of 1 μg/kg of body weight via penis vein at the end of reperfusion (140 min, HSR-LPS group, n = 8) or at 24 h post shock (HSR24-LPS group, n = 8). Control animals received the same LPS dose but without bleeding (LPS group, n = 8).
Ex vivo studies
Whole blood (WB) obtained from controls (CON), at the end of resuscitation (HSR = 140 min), and at 24 h post shock (HSR 24) prior to LPS challenge in vivo (Fig. 1) was incubated with LPS (100 ng/mL) for 2 h at 37 °C.
Samples
Blood samples of 1 mL volume, replaced by 2 mL saline, were obtained for ex vivo studies prior to shock, at 140 min, and 24 h post-shock (Fig. 1). Similarly, blood samples were obtained for TNF measurements at 230 min and 25.5 h following shock (90 min post-LPS challenge). All plasma samples were stored at −30 °C before analysis. Circulating cell counts were determined using a leukocyte counter (TOA Sysmex PL-110, Japan).
TNF analysis
A custom made enzyme-linked immunosorbent assay (ELISA) was performed as previously described.12, 13 Briefly, 96-well immunoplates were coated with guinea pig anti-mouse TNF monoclonal antibody TN3 (Celltech, Slough, Berkshire, United Kingdom) and blocked with albumin. After serum samples were incubated for 2 h at 37 °C the plates were washed, incubated with rabbit anti-mouse TNF immunoglobulin (Genzyme, Cambridge, Massachusetts) and peroxidase-conjugated goat anti-rabbit immunoglobulin G. After washing, o-phenylenediamine dihydrochloride substrate was added. The presence of TNF was detected at A492, using recombinant mouse TNF (Genzyme, Cambridge, Massachusetts) as the standard.
Endotoxin determination
The endotoxin content of plasma samples was assayed by the limulus amebocyte lysate test using a chromogenic substrate (Chrogenix, Molndal, Sweden) with a kinetic modification of the test kit procedure.13
Endotoxin-binding capacity
Whole blood spiked with 100 ng/mL LPS was incubated for 2 h at 37 °C and the LPS concentration in plasma determined. LPS-binding capacity (LBC %) = 100 – LPS-Recovery. LPS-Recovery = [(plasma LPS concentration/spiked LPS concentration) × 100].
Statistics
Statistical analysis was performed using GraphPad Prism 5.02 (GraphPad Software, California, USA). Data was tested for normal distribution, transformed when necessary and compared using analysis of variance (ANOVA) or a Kruskal–Wallis test as appropriate. The data are presented as means and deviations as standard error of the means. Significance level was set at p = 0.05.
Results
Body weight and blood cell counts did not differ between groups prior to the experiment (Table 1). Similarly, shed blood volume and mean arterial blood pressure were identical between the shock groups (Table 1). HS alone resulted in a moderate but significant increase in plasma TNF-α concentration at the end of resuscitation (41.0 pg/mL ± 10.3 pg/mL). At 24 h post shock, prior to LPS challenge, TNF-α concentration in the circulation declined (22.9 pg/mL ± 8.5 pg/mL). HS prior to the endotoxin challenge resulted in a significant reduction of plasma TNF-α in HSR-LPS and HSR-LPS24 (6 and 3-fold, respectively) rats as compared to the control LPS group, without HSR (Fig. 2).
Table 1.
Body weight, shed blood volume, blood cell counts, and mean arterial blood pressure (Data are presented as mean ± SEM).
| Group | Body weight (g) | Shed blood (mL) | White blood cells (K/μl) |
Platelets (K/μl) |
Mean arterial pressure (mmHg) |
||
|---|---|---|---|---|---|---|---|
| Pre – LPS | 0 min | 90 min | 140 min | ||||
| LPS | 455 ± 2 | 10.2 ± 0.7 | 548 ± 34 | ||||
| HSR-LPS | 448 ± 1 | 15.2 ± 0.2 | 8.9 ± 0.5 | 443 ± 50 | 110 ± 2 | 31 ± 1 | 106 ± 2 |
| HSR-LPS24h | 452 ± 3 | 15.6 ± 0.2 | 17.4 ± 1.2 | 381 ± 34 | 111 ± 2 | 31 ± 1 | 109 ± 2 |
Fig. 2.
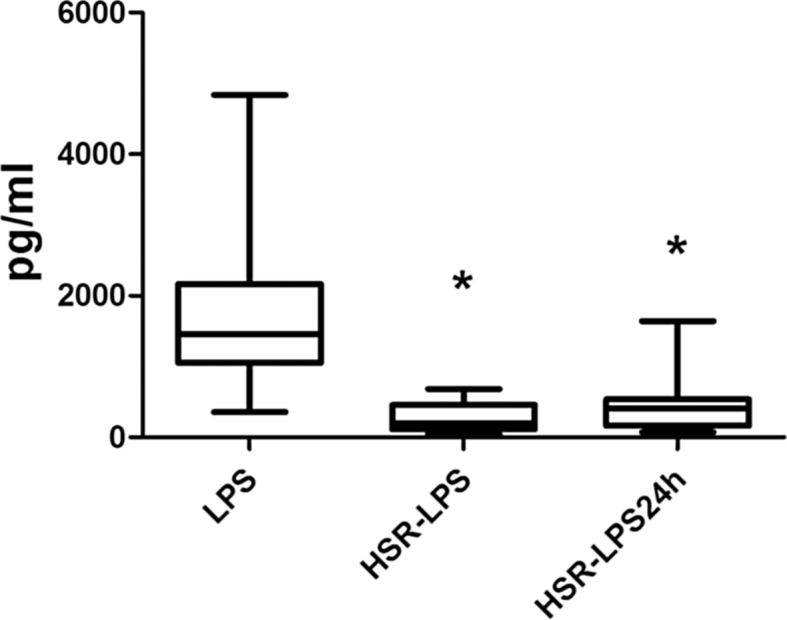
Plasma TNF-α concentrations 90 min after LPS challenge in vivo. LPS: Animals received 1.0 μg/kg LPS intravenously without hemorrhage; HSR-LPS: Animals received 1.0 μg/kg LPS intravenously immediately after hemorrhage and resuscitation; HSR-LPS24: Animals received 1.0 μg/kg LPS intravenously 24 h after hemorrhage and resuscitation; *p < 0.05 vs. LPS control group.
The ex vivo LPS-induced TNF release capacity of circulating leukocytes was very strongly suppressed by 3 and 5-fold in WB obtained at the end of resuscitation (HSR) and at 24 h (HSR24) compared to controls (Fig. 3). The LPS-binding capacity was not affected in WB obtained at the end of resuscitation if compared to the control values (57% ± 6%, 56% ± 6%), but it was slightly increased (to 71% ± 5 %) at 24 h after shock.
Fig. 3.
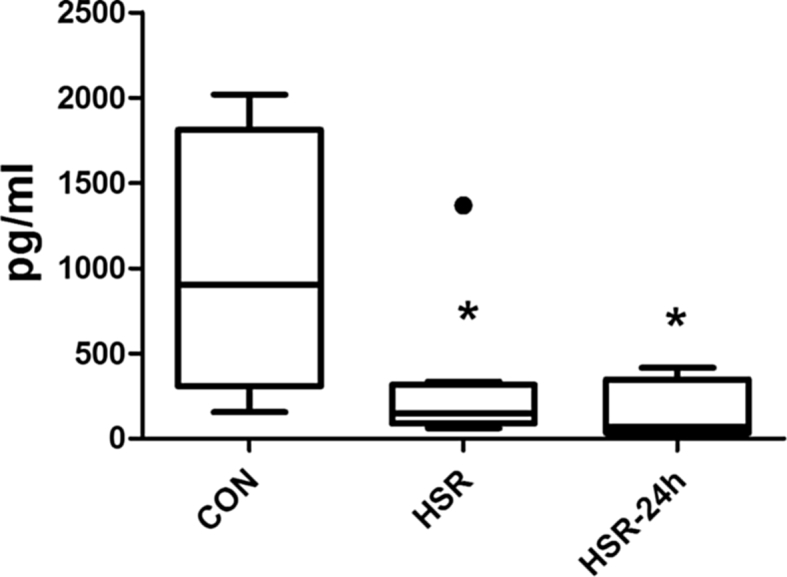
LPS-induced TNF-α formation ex vivo. The whole blood was obtained from controls immediately after hemorrhage and resuscitation (HSR = 140 min) or 24 h thereafter (HSR 24) prior to LPS challenge in vivo. The heparinized whole blood was incubated with LPS (100 ng/mL) for 2 h at 37 °C. *p < 0.05 vs. control.
Discussion
Traumatic insults are typically accompanied by a blood loss of different magnitude and in more severe cases, frequently followed by a hemorrhagic shock. The systemic response after trauma may be categorized into three phases14: immediate cardiovascular, delayed (hours to days) immunological, and late (days to weeks) metabolic. Although the inflammatory and/or immunological consequences of trauma/hemorrhage are usually detectable several hours or days after the initial insult, they may be triggered by the very early phase with consequences in the development of late multiple organ failure and death. The existing literature strongly suggest that the overwhelming release of inflammatory cytokines, such as TNF-α, IL-1, IL-6, and IL-10 is an initiating event in the posttraumatic acute phase response.1, 2, 15 Moreover, despite its transient appearance in circulation, TNF-α is considered the most important triggering mediator of the downstream cytokine cascade. Consequently, in the present study we selected TNF-α as surrogate marker of the early inflammatory response capacity elicited by HS.16, 17, 18
The main knowledge regarding the inflammatory response in trauma patients comes from the measurements of inflammatory mediators performed in systemic circulation.19 Considering that in critically ill patients virtually all body systems are activated, inflammatory markers detected in the blood have been likely synthesized in multiply organs and compartments. The current data are in line with our previous findings12, 13, 20 and those by others2, 21 collectively demonstrating that an early and transient increase in plasma TNF-α concentration occurring immediately after hemorrhage and resuscitation is irrespective of the cellular origin. In order to evaluate the global TNF-α response capacity of the host, we injected a low dose of endotoxin, to not exceed the maximum response capacity, into the rats either immediately after resuscitation or 24 h later. Our results clearly demonstrate that TNF-α formation capacity is suppressed in animals immediately after hemorrhage and resuscitation when compared to their naïve counterparts. Surprisingly, the magnitude of the early suppression remained unchanged within the initial 24 h of HS. This is an important observation, given that the early phase is typically referred to as the hyper-inflammatory phase due to the presence of circulating inflammatory mediators22, 23, 24 compartmentalized synthesis and release of inflammatory cytokines has been suggested being equally important in inflammation10, 25, 26 and that compartments other than circulating leukocytes may be responsible for morbidity and mortality in trauma.9, 26 Studies have also shown that differences exist in peripheral blood cells responses to hemorrhage that do not correlate with changes that occurs within organs.27, 28, 29, 30 It has been reported that an immediate hyper-activation of circulating monocytes is rapidly followed by a paralysis of cell function and that the capacity of de novo TNF-α synthesis in monocytes significantly correlated with the MODS score.31, 32 In the present study the TNF-α formation capacity of the circulating leukocytes obtained immediately after resuscitation and stimulated ex vivo was suppressed in a similar manner as the overall host response. The down regulated TNF-α release capacity did not recover within the first 24 h of shock.
One may argue that both HSR and HSR24 groups that showed an attenuated TNF-α release after LPS were infused with shed blood plus Ringer's solution in contrast to the LPS control group. Thus, the reduction of TNF-α observed in those two groups (vs. LPS control) may in part be attributed to the dilution effect (i.g. fewer number of circulating cells). In this respect, it is important to note that the cell counts (WBC, platelet counts prior to LPS challenge) are not significantly different between the LPS control group without HSR and HSR-LPS groups. Thus, the reduced TNF-α formation capacity by 3–5-fold following HSR in our experiments does not appear to be solely due to a dilution effect.
Given that the reduced TNF-α response could have been caused by an enhanced inactivation of LPS in the blood. Our results clearly show that the LPS binding capacity of blood was not significantly changed at the early phase after resuscitation - the time-point of the most pronounced TNF-α release down regulation. The LPS binding capacity was only moderately increased at 24 h post shock.
In general, endotoxemia in rodents is induced by a LPS dose in the range of 10 mg/kg BW. Our previous studies in rats have shown, however, that the TNF-α formation induced by 100 μg LPS/kg BW reaches a plateau similar to that induced by 10 mg LPS/kg BW.33 In order to avoid a maximal response and plateau level, which could have mask the inhibitory effect of hemorrhage and resuscitation on the inflammatory response capacity, we chose a small LPS dose of 1 μg/kg BW.
Shortcomings of our study include that pro-/anti-inflammatory cytokines other than TNF-α were not evaluated. However, considering the limited blood sample volume and TNF-α as an accepted surrogate marker of general inflammatory response, we decided to rely on it to examine the early inflammatory response to the HSR challenge. Moreover, our experiments focused on the first 24 h following HSR, which makes TNF-α an appropriate early phase marker. Further studies should examine for how long the response capacity remains down regulated after HSR.
In conclusion, our data suggest that the overall TNF-α release capacity decreases immediately after HSR and remains down regulated within the next 24 h. We additionally demonstrate that the LPS-inactivating capacity of the peripheral blood is enhanced 24 h after hemorrhagic shock.
Footnotes
Peer review under responsibility of Daping Hospital and the Research Institute of Surgery of the Third Military Medical University.
References
- 1.Jastrow K.M., 3rd, Gonzalez E.A., McGuire M.F. Early cytokine production risk stratifies trauma patients for multiple organ failure. J Am Coll Surg. 2009;209:320–331. doi: 10.1016/j.jamcollsurg.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Namas R., Ghuma A., Torres A. An adequately robust early TNF-alpha response is a hallmark of survival following trauma/hemorrhage. PLoS One. 2009;4:e8406. doi: 10.1371/journal.pone.0008406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao Y.M., Redl H., Bahrami S. The inflammatory basis of trauma/shock-associated multiple organ failure. Inflamm Res. 1998;47:201–210. doi: 10.1007/s000110050318. [DOI] [PubMed] [Google Scholar]
- 4.Moore F.A., Moore E.E. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin N Am. 1995;75:257–277. doi: 10.1016/s0039-6109(16)46587-4. [DOI] [PubMed] [Google Scholar]
- 5.Osuchowski M.F., Craciun F., Weixelbaumer K.M. Sepsis chronically in MARS: systemic cytokine responses are always mixed regardless of the outcome, magnitude, or phase of sepsis. J Immunol. 2012;189:4648–4656. doi: 10.4049/jimmunol.1201806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ertel W., Keel M., Bonaccio M. Release of anti-inflammatory mediators after mechanical trauma correlates with severity of injury and clinical outcome. J Trauma. 1995;39:879–885. doi: 10.1097/00005373-199511000-00011. [DOI] [PubMed] [Google Scholar]
- 7.Tan L.R., Waxman K., Scannell G. Trauma causes early release of soluble receptors for tumor necrosis factor. J Trauma. 1993;34:634–638. doi: 10.1097/00005373-199305000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Keel M., Ecknauer E., Stocker R. Different pattern of local and systemic release of proinflammatory and anti-inflammatory mediators in severely injured patients with chest trauma. J Trauma. 1996;40:907–912. doi: 10.1097/00005373-199606000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Lenz A., Franklin G.A., Cheadle W.G. Systemic inflammation after trauma. Injury. 2007;38:1336–1345. doi: 10.1016/j.injury.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Cavaillon J.M., Annane D. Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin Res. 2006;12:151–170. doi: 10.1179/096805106X102246. [DOI] [PubMed] [Google Scholar]
- 11.Corvol H., Fitting C., Chadelat K. Distinct cytokine production by lung and blood neutrophils from children with cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003;284:L997–L1003. doi: 10.1152/ajplung.00156.2002. [DOI] [PubMed] [Google Scholar]
- 12.Bahrami S., Yao Y.M., Leichtfried G. Significance of TNF in hemorrhage-related hemodynamic alterations, organ injury, and mortality in rats. Am J Physiol. 1997;272:H2219–H2226. doi: 10.1152/ajpheart.1997.272.5.H2219. [DOI] [PubMed] [Google Scholar]
- 13.Jiang J., Bahrami S., Leichtfried G. Kinetics of endotoxin and tumor necrosis factor appearance in portal and systemic circulation after hemorrhagic shock in rats. Ann Surg. 1995;221:100–106. doi: 10.1097/00000658-199501000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foex B.A. Systemic responses to trauma. Br Med Bull. 1999;55:726–743. doi: 10.1258/0007142991902745. [DOI] [PubMed] [Google Scholar]
- 15.Hegeman M.A., Hennus M.P., Heijnen C.J. Ventilator-induced endothelial activation and inflammation in the lung and distal organs. Crit Care. 2009;13:R182. doi: 10.1186/cc8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaudry I.H., Ayala A., Ertel W. Hemorrhage and resuscitation: immunological aspects. Am J Physiol. 1990;259:R663–R678. doi: 10.1152/ajpregu.1990.259.4.R663. [DOI] [PubMed] [Google Scholar]
- 17.Redl H., Schlag G., Bahrami S. Cytokine network in trauma and sepsis I: TNF and IL-8. In: Schlag G., Redl H., editors. vol. I. Springer Verlag; Germany: 1993. pp. 468–490. (Pathophysiology of Shock, Sepsis and Organ Failure). [Google Scholar]
- 18.Schlag G., Redl H., Bahrami S. Cytokine network in trauma. In: Schlag G., Redl, Siegel J.H., editors. Shock, Sepsis, and Organ Failure. Second Bernard Wiggers Conference. Springer Verlag; Germany: 1993. pp. 128–133. [Google Scholar]
- 19.DeLong W.G., Jr., Born C.T. Cytokines in patients with polytrauma. Clin Orthop Relat Res. 2004;422:57–65. doi: 10.1097/01.blo.0000130840.64528.1e. [DOI] [PubMed] [Google Scholar]
- 20.Bahrami S., Zimmermann K., Szelenyi Z. Small-volume fluid resuscitation with hypertonic saline prevents inflammation but not mortality in a rat model of hemorrhagic shock. Shock. 2006;25:283–289. doi: 10.1097/01.shk.0000208808.03148.ea. [DOI] [PubMed] [Google Scholar]
- 21.Relja B., Hohn C., Bormann F. Acute alcohol intoxication reduces mortality, inflammatory responses and hepatic injury after haemorrhage and resuscitation in vivo. Br J Pharmacol. 2012;165:1188–1199. doi: 10.1111/j.1476-5381.2011.01595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Botha A.J., Moore F.A., Moore E.E. Postinjury neutrophil priming and activation states: therapeutic challenges. Shock. 1995;3:157–166. doi: 10.1097/00024382-199503000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Flohe S.B., Flohe S., Schade F.U. Invited review: deterioration of the immune system after trauma: signals and cellular mechanisms. Innate Immun. 2008;14:333–344. doi: 10.1177/1753425908100016. [DOI] [PubMed] [Google Scholar]
- 24.Wutzler S., Lustenberger T., Relja B. Pathophysiology of multiple trauma: intensive care medicine and timing of treatment. Chirurg. 2013;84:753–758. doi: 10.1007/s00104-013-2477-0. [DOI] [PubMed] [Google Scholar]
- 25.Raeven P., Feichtinger G.A., Weixelbaumer K.M. Compartment-specific expression of plasminogen activator inhibitor-1 correlates with severity/outcome of murine polymicrobial sepsis. Thromb Res. 2012;129:e238–e245. doi: 10.1016/j.thromres.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Cavaillon J.M., Adib-Conquy M., Cloez-Tayarani I. Immunodepression in sepsis and SIRS assessed by ex vivo cytokine production is not a generalized phenomenon: a review. J Endotoxin Res. 2001;7:85–93. [PubMed] [Google Scholar]
- 27.Schneider C.P., Schwacha M.G., Chaudry I.H. Influence of gender and age on T-cell responses in a murine model of trauma-hemorrhage: differences between circulating and tissue-fixed cells. J Appl Physiol (1985) 2006;100:826–833. doi: 10.1152/japplphysiol.00898.2005. [DOI] [PubMed] [Google Scholar]
- 28.Lederer J.A., Brownstein B.H., Lopez M.C. Comparison of longitudinal leukocyte gene expression after burn injury or trauma-hemorrhage in mice. Physiol Genom. 2008;32:299–310. doi: 10.1152/physiolgenomics.00086.2007. [DOI] [PubMed] [Google Scholar]
- 29.Brownstein B.H., Logvinenko T., Lederer J.A. Commonality and differences in leukocyte gene expression patterns among three models of inflammation and injury. Physiol Genom. 2006;24:298–309. doi: 10.1152/physiolgenomics.00213.2005. [DOI] [PubMed] [Google Scholar]
- 30.Zhu X.L., Zellweger R., Zhu X.H. Cytokine gene expression in splenic macrophages and Kupffer cells following haemorrhage. Cytokine. 1995;7:8–14. doi: 10.1006/cyto.1995.1002. [DOI] [PubMed] [Google Scholar]
- 31.Kirchhoff C., Biberthaler P., Mutschler W.E. Early down-regulation of the pro-inflammatory potential of monocytes is correlated to organ dysfunction in patients after severe multiple injury: a cohort study. Crit Care. 2009;13:R88. doi: 10.1186/cc7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villarroel J.P., Guan Y., Werlin E. Hemorrhagic shock and resuscitation are associated with peripheral blood mononuclear cell mitochondrial dysfunction and immunosuppression. J Trauma. 2013;75:24–31. doi: 10.1097/TA.0b013e3182988b1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bahrami S., Redl H., Leichtfried G. Similar cytokine but different coagulation response to lipopolysaccharide injection in D-galactosamine-sensitized versus nonsensitized rats. Infect Immun. 1994;62:99–105. doi: 10.1128/iai.62.1.99-105.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]