

ABSTRACT
Potassium channels play important roles in microglia functions and thus constitute potential targets for the treatment of neurodegenerative diseases like Alzheimer, Parkinson and stroke. However, uncertainty still prevails as to which potassium channels are expressed and at what levels in different species, how the expression pattern changes upon activation with M1 or M2 polarizing stimuli compared with more complex exposure paradigms, and - most importantly - how these findings relate to the in vivo situation. In this mini-review we discuss the functional potassium channel expression pattern in cultured neonatal mouse microglia in the light of data obtained previously from animal disease models and immunohistochemical studies and compare it with a recent study of adult human microglia isolated from epilepsy patients. Overall, microglial potassium channel expression is very plastic and possibly shows species differences and therefore should be studied carefully in each disease setting and respective animal models.
KEYWORDS: K+ channel, KCa3.1, Kir2.1, KV1.3, microglia
Introduction
Microglia are the resident immune cells of the central nervous system and have many properties in common with peripheral macrophages, including expression of the cytochemical marker Iba-1 (Fig. 1). In the unperturbed brain these cells exist in a “not activated” or “surveillant” state, in which they scout neurons and tissues for invading pathogens and damage via a ramified network of fine cellular processes equipped with receptors for chemical “danger signals,” such as Toll-like receptors for bacteria, purinergic receptors for ATP, and other phosphorylated nucleotides released from damaged or over-activated neurons (Fig. 2). Upon stimulation, microglia can attain various activated states characterized by both morphological and functional changes, such as rounding up and retraction of processes, directed migration along stimuli gradients, increased proliferation and phagocytosis, as well as polarization to phenotypes with distinct patterns of cytokine and chemokine production.1 A correctly orchestrated chain-of-events in a microglia response thus leads to identification and destruction of pathogens, removal of cellular and tissue debris, and eventually initiates healing processes involving other cells in the damaged area.
Figure 1.
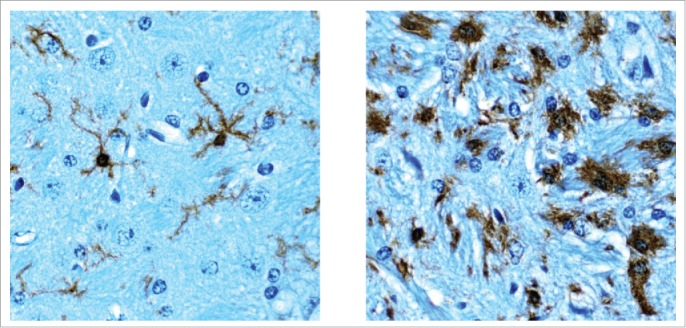
“Surveillant” microglia in normal adult mouse brain (left) and activated microglia in the border of an ischemic infarct (right) stained for the cytochemical marker Iba-1. The ischemic infarct was induced by middle cerebral artery occlusion as described.22
Figure 2.

Cartoon showing the regulation of Ca2+ signaling in microglia by a selected number of ion channels and receptors. (Abbreviations: KV, voltage-gated K+ channel; Kir, inward-rectifier K+ channel; KCa, calcium-activated K+ channel; IP3, ionositol-triphosphate; Orai1, calcium release-activated calcium channel protein 1; P2R, purinergic receptor; SERCA, sarco/endoplasmic reticulum Ca-ATPase; STIM, stromal interaction molecule; TRP, transient receptor potential channel). Activation of K+ channels hyperpolarizes the microglia membrane and thus facilitates Ca2+-influx through Ca2+ and cation channels.
The action of microglia can also in some instances become detrimental and promote neuronal death via excessive secretion of noxious substances, such as nitrous oxide (NO) and superoxide (O2−), or excessive synaptic pruning. For example, in Alzheimer disease (AD) activation of microglia by soluble amyloid-β oligomers is widely viewed as an early and central event in the pathogenesis.2-4 While microglial phagocytosis of amyloid-β aggregates is impaired in AD, oligomeric amyloid-β stimulates microglia to produce inflammatory cytokines, disrupt synaptic plasticity,5 and to kill neurons in organotypic hippocampal slices.6 In ischemic stroke and traumatic brain injury, inflammation induced in the wake of neuronal death causes secondary damage and significantly expands the primary insult,7,8 while in multiple sclerosis9,10 and some degenerative eye diseases, activated microglia contribute to the demyelination of axons or phagocytosis of photoreceptors.11
A popular concept claims that, similar to macrophages,12 microglia polarized to the “classical” pro-inflammatory M1-phenotype by stimulation with bacterial cell wall constituents, like lipopolysacccarides (LPS) and/or the cytokine interferon-γ (IFN-γ), are predominantly neurotoxic based on their production of IL-1β, TNF-α, IL-12, IL-6, and NO. In contrast, microglia stimulated to the M2 phenotype with IL-4 are thought to suppress inflammation and promote tissue repair by secreting anti-inflammatory mediators and neurotrophic factors (Fig. 2).13,14 It is increasingly becoming clear, however, that the presence of distinct M1/M2 subpopulations is an oversimplification,15 and it has been proposed to abandon this concept in favor of a stimulus based terminology (e.g. M(LPS) or M(IL-4)) and to assume no fixed, stable phenotype in vivo.16 Despite this consideration, the M1/M2 concept is helpful for hypothesis generation based on in vitro experiments, since it represents well-defined stimuli conditions, eliciting opposite extremes in a spectrum of activated microglia phenotypes, which can be compared with more complex stimulation paradigms or to the conditions prevailing in vivo. From a therapeutic perspective, the ultimate goal is of course to understand how to strengthen restorative properties and how to avoid microglia-mediated neurotoxicity in patients. Since microglial potassium channels have been suggested as relevant pharmaceutical targets, it is important to discuss their expression and function in relation to microglia polarization, and also to investigate possible species differences.
Potassium channels in microglia
Potassium (K+) channels are important in microglia since their activation can induce membrane hyperpolarizations, which are essential for driving Ca2+ influx trough inward rectifying Ca2+-Release-Activated-Ca2+-channels (CRAC),17,18 ATP-activated P2X receptors,19 and other Ca2+-permeable cation channels (Fig. 2).1 K+ channels facilitate refilling of intracellular Ca2+ stores following ATP or UTP activation of metabotropic P2Y-receptors,20 thus maintaining high Ca2+ and timing of intracellular signaling events important for microglia activation and proliferation. K+ channels can also participate in microglia volume regulation (working together with Cl− channels in setting up local osmotic gradients) and thereby in important cellular functions, such as shape changes, phagocytosis, and migration toward chemotaxic stimuli.1
Various specific inhibitors of KCa3.1 and KV1.3 channels have proven efficient in rodent models of neurodegenerative diseases, and conditions such as stroke,21,22 multiple sclerosis,23 retinal degeneration,24 and radiation induced brain damage.25 K+ channels are therefore considered a highly relevant target class for pharmacological intervention in microglia-derived pathologies. Based primarily on classical molecular techniques, it has been reported that isolated rodent microglia can express a plethora of voltage-gated (KV1.3, KV1.1, KV1.5, KV3.1), Ca2+ activated (KCa3.1, KCa2.3, KCa1.1), and inward rectifying K+ channels (Kir2.1).1 In addition, large gene array studies have identified KV4.1 and K2P13.1 as microglia signature genes in mice with experimental autoimmune encephalomyelitis and human microglia.26
Surprisingly, however, reports are quite inconsistent concerning which K+ channel types can be identified functionally by electrophysiological and pharmacological approaches. This may be hypothesized to relate to a combination of experimental variables, such as species differences, different isolation procedures and culturing methods, quantification methods, or age and sex of animals. However, systematic investigations of the relative importance of these factors for channel expression and function in rodent microglia are uncommon in the literature, and so is the translational aspect comparing data from neonatal and adult microglia with human data. This is obviously problematic for attempts to use isolated microglia for drug screening, and for predicting which specific K+ channel targets to pursue in drug discovery programs.
We have recently investigated some aspects of K+ channel expression in isolated microglia, to delineate this variability and better understand the expression of microglia K+ channels and their function under different conditions: 1) Nguyen et al.27 for the first time performed a systematic investigation of differential K+ channel expression and functional importance in neonatal cultured mouse and foetal human microglia activated under “classically” pro-inflammatory vs. “alternatively” activating conditions. 2) In parallel, Blomster et al.28 investigated the functional expression of K+ channels - with specific focus on quantifying the KCa3.1 component - in unstimulated vs. LPS and IL-4 polarized cultured human microglia from adult epilepsy patients, who received therapeutic surgery to control their seizures.
KV1.3, Kir2.1, and KCa3.1 are the main K+ channels expressed in mouse neonatal microglia
Unstimulated cultured mouse microglia express very low total K+ currents typically consisting of one to 3 distinct biophysical components: An inwardly rectifying current, a linear current, and finally a classical voltage-dependent current showing use-dependent inactivation upon repeated stimulation (Fig. 3). These biophysically defined components are also clearly pharmacologically distinguishable, since Ba2+ selectively abolishes the inward rectifier, and the KCa3.1 inhibitor TRAM-3429 and the KV1.3 inhibitors PAP-130 and ShK-18631 eliminate the linear and voltage-dependent components, respectively. In support of this biophysical and pharmacological evidence, KV1.3, Kir2.1, and KCa3.1 are also clearly identified by qPCR and immunohistochemistry, whereas other candidates (see above) are expressed at much lower levels or not detectable.
Figure 3.
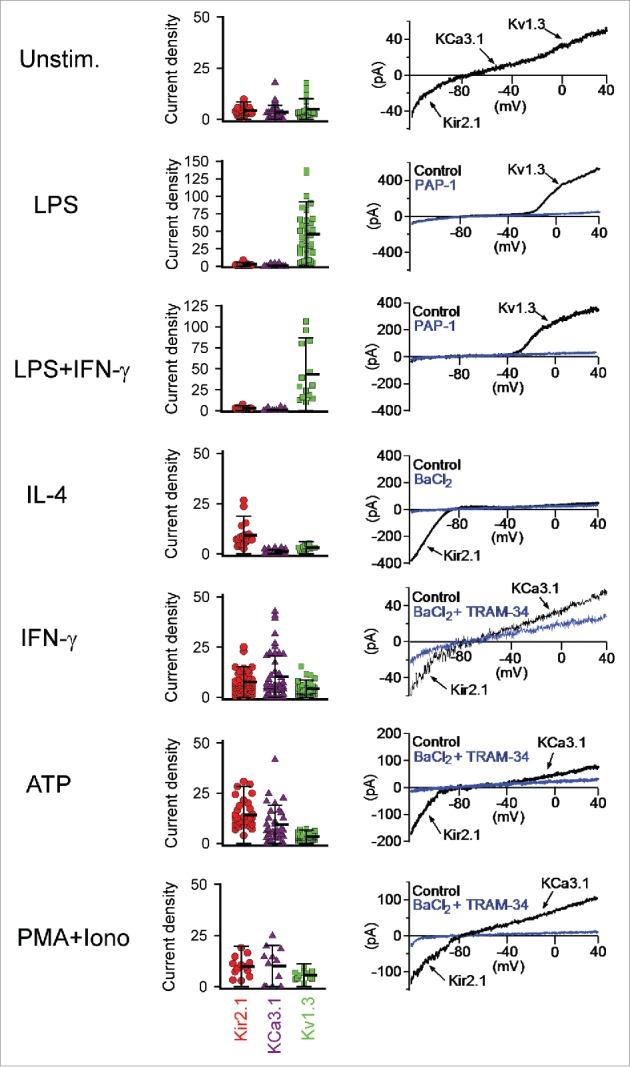
Scatterplots of the functional KV1.3, KCa3.1 and Kir2.1 expression levels measured by whole-cell patch-clamp in unstimulated neonatal mouse microglia and in microglia 40–48 h after stimulation with LPS, LPS plus IFN-γ, IL-4, IFN-γ, ATP or PMA plus ionomycin. Redrawn from Nguyen et al.27
Stimulation of neonatal mouse microglia with ATP or differentiation into M1/M2 phenotypes with LPS/IFN-γ or IL-4, respectively, differentially modulates expression of these K+ channels in a highly specific way (Fig. 3): LPS, the classical M1 “invading bacteria-stimuli,” selectively upregulates KV1.3, but causes no increase in either Kir2.1 or KCa3.1 currents. However, the exact opposite pattern is seen for stimulation with ATP, the pro-inflammatory cytokine IFN-γ, or the receptor-bypassing pro-inflammatory stimulus PMA+ionomycin, which all leave KV1.3 expression unaltered, but instead increase the expression of both Kir2.1 and KCa3.1. Noteworthily, combining IFN-γ with LPS completely abolishes the IFN-γ effect on Kir2.1 and KCa3.1, and exerts the same stimulating effect on KV1.3 as LPS alone. In contrast to the various pro-inflammatory stimuli, the M2 cytokine IL-4 yields yet another distinct profile with specific upregulation of Kir2.1, leaving KCa3.1 and KV1.3 at their pre-stimulation levels. These results provide an excellent reference for discussion of in vivo data as well as comparisons to other species, but first we will briefly address the functional importance of microglia K+ channels.
The importance of specific microglia K+ channels for expression, synthesis and secretion of mediators
One aspect of K+ channel subtypes in microglia is their expression as determined by electrophysiological experiments, another is the functional impact of specifically inhibiting these channels on pro- and anti-inflammatory responses from differentially activated microglia cells. To elucidate this question, we used the selective KCa3.1 inhibitor TRAM-3429 and the KV1.3 inhibitors PAP-130 and ShK-186,31 at concentrations previously determined sufficient to ascertain full inhibition of the target channels, while staying well below values causing non-selective actions on other channels (an aspect often neglected when using pharmacological tools) or having non-specific cytotoxic effects.29–31 Thus, the specific effect of Kir2.1 blockade could unfortunately not be determined due to the high cytotoxicity of the available pharmacological tools in our in vitro system.
LPS stimulation induces the standard M1 response pattern, characterized by very strong increases in the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α, as well as upregulation of iNOS and a corresponding increase in NO release.27 As expected from the strongly increased expression of KV1.3 by LPS, both the small molecule inhibitor PAP-1,30 and the peptide inhibitor ShK-18631 significantly reduce both expression and release of these mediators. More surprisingly, considering that LPS polarization did not significantly increase KCa3.1 expression above basal, undifferentiated levels, TRAM-3429 had similar or even stronger effects on IL-1β than the KV1.3 inhibitors, which may well reflect a critical role of the relatively few KCa3.1 channels present under these conditions. Interestingly, the effect sizes of both K+ channel inhibitors are at par with the standard microglia activation blocker minocycline (which acts by a very different mechanism). However, none of the inhibitors (including minocycline) causes a complete block of cytokine and NO production. Whether the blockers possibly might show additive or synergistic effects was not investigated in this study.
IL-4 stimulation selectively up-regulates the cancer promoting enzyme Arginase 1, the membrane saccharide receptors CD206 and YM1, as well as the insulin-like growth factor, IGF1. In contrast to the overall similar inhibiting effects of KCa3.1 and KV1.3 blockers on secretion of pro-inflammatory cytokines, the effect on these M2 markers were much more variable, with only TRAM-34 showing a significant inhibition of CD206 and YM1.27
Comparisons between K+ channel expression in cultured neonatal microglia and acutely isolated adult microglia from normal and inflamed mouse brains
It is of course often questioned, how findings made with cultured neonatal microglia translate to microglia in the adult brain. While in vitro studies typically show very clear distinctions between a LPS-induced M1-like state and an IL-4 induced M2-like state,32 studies in human inflammatory and neurodegenerative diseases often report heterogeneous microglia phenotypes co-expressing M1 and M2 markers.33,34 In a previous publication, we therefore acutely isolated CD11b+ microglia/macrophages from the brains of mice subjected to either ischemic stroke or intraventricular LPS injection using magnetic beads, and immediately afterwards studied their K+ channel expression by whole-cell patch clamp.22 Microglia acutely isolated from non-infarcted, normal brains exhibited very small K+ currents, which on average were even smaller than what was observed in unstimulated cultured neonatal microglia, but clearly consisted of combinations of KV1.3, KCa3.1 and Kir2.1. In contrast, microglia isolated from the infarcted brain area 8 d after an ischemic stroke showed greatly increased functional K+ channel expression. Interestingly, while some cells predominantly expressed Kir2.1 currents similar to IL-4 stimulated neonatal microglia, other cells exhibited large Kv1.3 currents similar to LPS stimulated neonatal microglia. However, many acutely isolated cells from the infarcted area had no “clean” phenotype, but were found to possess K+ currents consisting of various combinations of KV1.3, KCa3.1 and Kir2.1.22 In contrast to these findings from an ischemic stroke model, where microglia activation likely occurs in response to a combination of released ATP, neuronal debris, inflammatory cytokines and ischemia/reperfusion injury, microglia isolated from LPS injected mouse brains typically expressed either large KV1.3 currents or a combination of KV1.3 and KCa3.1, but virtually no Kir currents. The increase in KCa3.1 expression, which was not observed following LPS stimulation in vitro, in our opinion is probably a response to neuronal injury following the LPS injection.
Comparison to functional K+ channel expression in isolated prenatal and adult human microglia
Using commercially available human microglia derived from human fetuses, we demonstrated prominent expression of KV1.3 currents and a minor Kir component, irrespective of LPS or IL-4 stimulation (KCa3.1 was not quantified due to the need of using F− based pipette solutions for successful patch-clamping of these cells).27
In contrast, functional K+ channel expression in adult neocortical microglia from epilepsy patients varies significantly from the pattern observed with mouse microglia - both cultured neonatal cells and acutely isolated ones from adults - as well as the prenatally derived human microglia.28 Isolated adult human microglia have much larger cell capacitances (20–25 pF compared with 4–5 pF for both neonatal and adult mouse as well as prenatal human microglia), and much lower input resistances reflecting a 5–6-fold larger cell size, as well as a significantly higher K+ channel conductance at negative membrane potentials. At first glance, the summed K+ channel whole-cell ramp current mimics quite well the neonatal phenotype with distinguishable inward rectifying, linear, and voltage-dependent components. Quantitatively, however, the linear current typically dominates in the isolated adult microglia, often obscuring the other components. The linear conductance is activated by low concentrations of the potent KCa3.1/KCa2.x activator NS309 and fully inhibited by the KCa3.1 blocker NS6180,35 indicating a high functional expression of KCa3.1. KCa2 channels did not contribute significantly to the Ca2+ activated and voltage-insensitive current. Importantly, the high expression level of KCa3.1 also translates to a dominating effect on the membrane potential (Fig. 4), since activation/block of the channel immediately switched the membrane potential between −20 and −75 mV, thus predicting KCa3.1 channel expression as a powerful mechanism for amplification of conductive Ca2+-influxes via changes in membrane potential.
Figure 4.
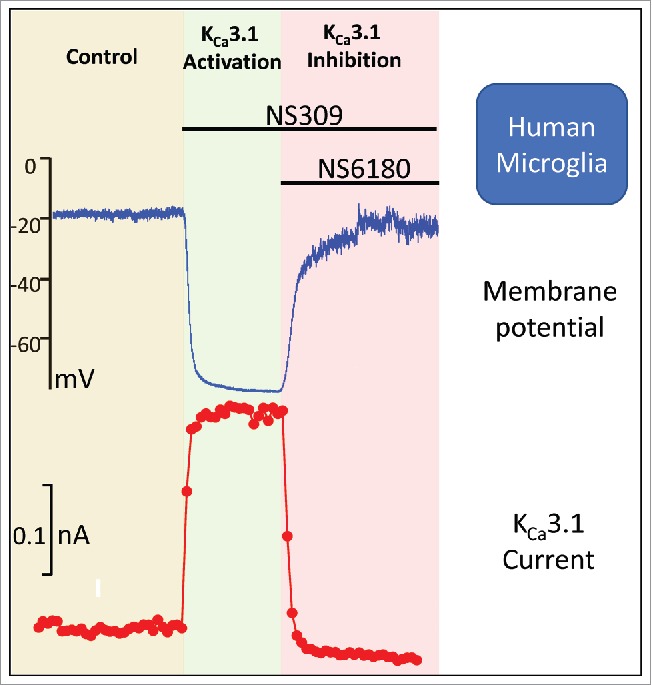
Effects of sequential activation and inhibition of KCa3.1 in a cultured human microglia cell isolated from an epilepsy patient. The upper panel shows the effect on membrane potential and the lower depicts the corresponding effect on the K+ currents. The activator is the positive modulator NS309 and the inhibitor the highly selective NS6180. Redrawn from Blomster et al.28
The inward rectifying K+ current (Kir) seems biophysically undistinguishable from its mouse counterpart, but formal identification as hKir2.1 has not yet been achieved. In contrast, the dominating voltage-dependent outward component activates at membrane potentials of 0 mV or higher, exhibits relatively large “gating noise” (indicative of a high single channel conductance), does not appreciably inactivate, and is modulated positively by NS1619,36 and negatively by the tremorgenic alkaloid paxilline,37 all features that identify the current as the big conductance Ca2+ and voltage-activated KCa1.1 channel. BK channels have previously been reported from human and bovine microglia in culture,38,39 as well as human macrophages,40 but are usually not found in rodent microglia under any stimulation condition (but can occasionally be seen as a small residual current in microglia from adult KV1.3 KO mice; Heike Wulff, unpublished). The highly surprising finding from this study was, however, that neither KV1.3 nor any other “classic” KV current, could be detected. Based on the mouse experiments it is no surprise, that M2 polarization with IL-4 did not upregulate KV1.3, but one might have expected an effect of M1 polarization with LPS (which significantly upregulated KV1.3 in the mouse microglia, see Fig. 3).
These results make us conclude that cultured human adult microglia from epilepsy patients predominantly exists in a state where KCa3.1 is very strongly expressed, and where Kir and KCa1.1 provide the main - if not the only - accompanying K+ conductances. It is worth noting that despite these cells being able to respond “normally” to LPS-stimulation in vitro through increased secretion of pro-inflammatory cytokines, neither LPS nor IL-4 caused a significant change in functional expression of any of the K+ channels probably reflecting that adult microglia cells are less plastic.
Future directions toward an integrated view of K+ channel expression and function in microglia
As seen from the preceding chapters, microglia are remarkably plastic in their expression of K+ channels as summarized for neonatal microglia in Fig. 5 (left): M1 polarizing stimuli (LPS, IFN-γ) selectively up-regulate KV1.3, M2 polarizing stimuli (IL-4) up-regulate Kir2.1, whereas IFN-γ and ATP induce a KCa3.1/Kir2.1 profile. Microglia in brains in animal models of stroke, Alzheimer or Parkinson disease cannot be expected to fall exactly into the well-defined in vitro phenotypes as demonstrated by other combinations of Kir2.1, KCa3.1, KV1.3 in microglia acutely isolated from adult stroke mice.22 This is easily rationalized from their individual, and probably complex, in vivo “stimulation history.” However, the K+ channel “players” are the same as in the in vitro experiments. The situation is somewhat different in human microglia from epilepsy patients (Fig. 5, right): The KCa3.1 current is by far the most prominent, no matter if cells are stimulated with LPS or IL-4 or not.28 Kir channels are also expressed, but the noteworthy finding is the lack of KV1.3 expression and presence of KCa1.1 instead. What is particularly surprising about the missing functional KV1.3 expression, is that several immunohistochemistry studies have previously shown strong KV1.3 specific staining on activated microglia in human ischemic infarcts,22 microglia surrounding amyloid-plaques in Alzheimer disease,41 as well as on cells with a glia morphology in active multiple sclerosis lesions.42 Combined with our finding that cultured prenatal human microglia clearly express KV1.3, and that microglia exhibit such a pronounced and stimuli-dependent expression plasticity, a reasonable assumption is that the “adult epilepsy microglia phenotype” carries a unique set of K+ channels dictated by their specific aging and/or epilepsy/medication history. High expression of KCa3.1 and Kir combined with low KV1.3 may, for example, be the result of a “stimulation milieu” dominated by an augmented purinergic drive, likely to be the case in epilepsy because of increased synaptic release of ATP/UTP, or secretion from astrocytes.43 In contrast, we have no idea what may drive KCa1.1 expression and suspect it could relate to a species difference.
Figure 5.
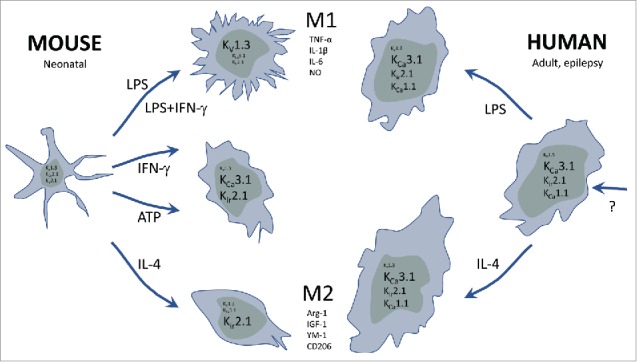
Cartoon summarizing the observed functional expression profiles of various K+ channels in cultured microglia isolated from prenatal mouse and adult human epilepsy patients, respectively. The cultured microglia were activated by classical M1(LPS, IFN-γ) and alternative M2(IL-4) stimulation paradigms, compared with stimulation with the damage or danger signal, ATP.
To settle the question of plasticity vs. species differences in the expression of microglia K+ channels in health and disease, we recommend the following approaches: 1) Differentiation of cultured microglia cells: There is a huge need for extending the in vitro polarization studies of cultured microglia using multiple well-defined stimuli and combinations thereof to other species. Even taxonomically closely related species or different strains of the same species are worth investigating in detail, as clearly demonstrated by papers describing M2(IL-4) mediated upregulation of KCa3.1 in cultured neonatal rat microglia44 and expression of KCa2.3;45 both findings that we have not been able to corroborate using mouse and human microglia. Additionally, microglia senescence during aging or in prolonged in vitro culture can alter their responsiveness to stimuli.46 Thus, particular attention should be paid to the age of the animals, as well as to the length of time, microglia are kept in culture for differentiation studies to avoid unintentional skewing of microglia activation and the resulting K+ channel expression. 2) Acutely isolated microglia cells and slices from animal models: Functional K+ channel expression in microglia should be studied more thoroughly in both mouse and rat models of diseases accompanied by neuroinflammation, such as ischemic stroke, Alzheimer disease, Parkinson disease, traumatic brain injury, and epilepsy. Compared to isolated cells acute slices obviously offer the advantage that microglia can be studied in their natural 3D-environment. So far, the only electrophysiological data available in literature is our characterization of KV, KCa and Kir channel expression in acutely isolated microglia from mice subjected to middle cerebral artery occlusion (MCAO),22 2 studies from the year 2000 reporting small Kir currents under normal conditions and increased KV channel expression following facial nerve axotomy or 48-hours after MCAO in acute rat brain slices,47,48 and a study showing increased KV1.3 expression in microglia in hippocampal slices prepared from mice 48 hours after kainate induced status epilepticus.49 Similarly, it would be very interesting to study K+ channel expression in acute microglia isolated from electrically kindled rats or from genetic epilepsy models such as Frings audiogenic seizure (AGS)-susceptible mice.50 3) Acute and cultured microglia from human diseased tissue: Despite the technical and practical difficulties involved, there is a need for confirming our current results from cultured cells from epilepsy patients28 by comparing to the functional K+ channel expression profile in acutely isolated microglia. Using one of the currently available electrophysiological HTS platforms for “instant parallel profiling” of microglia as fast as possible after surgery is a realistic way forward for such studies. Furthermore, the currently investigated microglia are all isolated from the supposedly “healthy” neocortical tissue, which unavoidably had to be removed as part of the amygdahippocampectomy (the position of the drug-resistant epileptic focal point). Therefore, it would also be highly interesting to compare the currently obtained data, with the K+ channel expression in microglia from the much more seriously affected limbic structures. We also need to investigate, if inhibition of specific K+ channels has the same impact on human microglia functions (secretion, migration etc.) as in mouse microglia. Finally, we believe that the recent success in obtaining microglia-like cells from human pluripotent stem cells51 should greatly aid this translational work, and even make it possible to investigate changes in channel expression in a broader disease specific context.
Taken together, such studies would help to inform decisions about whether to target microglial K+ channels to suppress detrimental microglia functions in neurologic diseases. So far there is only limited in vivo target validation available. Pharmacological KCa3.1 inhibition has been demonstrated to reduce infarct area and neurologic deficit in mouse and rat models of ischemic stroke,21,22 reverse tactile allodynia in rats with peripheral nerve injury,52 and reduce glioma associated microglia activation in a mouse model.53 Based on this data 2 groups have already proposed to repurpose the KCa3.1 blocker senicapoc for the treatment of Alzheimer disease,6,54 but so far, no data validating the therapeutic hypothesis has been published. KV1.3, which based on the in vitro data showing KV1.3 expression in “M1-like” microglia,27 might seem an even more attractive target but, as discussed above, many questions about the conditions of KV1.3 expression in human microglia remain, and only one in vivo study has shown that KV1.3 inhibition can reduce microglia mediated radiation induced brain damage.25
Abbreviations
- Iba-1
Ionized Ca2+ binding adaptor molecule
- NS309
6,7-dichloro-1H-indole-2,3-dione 3-oxime
- NS6180
4-[[3-(trifluoromethyl)phenyl]methyl]-2H-1,4-benzothiazin-3(4H)-one
- PAP-1
5-(4-phenoxybutoxy)psoralen
- PMA
phorbol-12-myristate-13-acetate
- ShK-186
Stichodactyla toxin-186
- TRAM-34
1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole
Disclosure of potential conflicts of interest
Linda Blomster and Palle Christophersen are full time employees of Saniona A/S.
Funding
This work was supported by the National Institute of Neurological Disease and Stroke under Award NS098328 and by the European Union's Seventh Framework Program (FP7/2007–2013) under Grant HEALTH-F2–2011–278850 (INMiND) from the Strategic Research Council (COGNITO).
References
- [1].Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011; 91:461-553; PMID:21527731; https://doi.org/ 10.1152/physrev.00011.2010 [DOI] [PubMed] [Google Scholar]
- [2].Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al.. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 1998; 95:6448-6453; PMID:9600986; https://doi.org/ 10.1073/pnas.95.11.6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tejera D, Heneka MT. Microglia in Alzheimer's disease: the good, the bad and the ugly. Curr Alzheimer Res 2016; 13:370-380; PMID:26567746; https://doi.org/ 10.2174/1567205013666151116125012 [DOI] [PubMed] [Google Scholar]
- [4].Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science 2016; 353:777-783; PMID:27540165; https://doi.org/ 10.1126/science.aag2590 [DOI] [PubMed] [Google Scholar]
- [5].Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al.. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008; 14:837-842; PMID:18568035; https://doi.org/ 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Maezawa I, Jenkins DP, Jin BE, Wulff H. Microglial KCa3.1 channels as a potential therapeutic target for Alzheimer's disease. Int J Alzheimers Dis 2012; 2012:868972; PMID:22675649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med 2011; 17:796-808; PMID:21738161; https://doi.org/ 10.1038/nm.2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Macrez R, Ali C, Toutirais O, Le Mauff B, Defer G, Dirnagl U, Vivien D. Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol 2011; 10:471-480; PMID:21511199; https://doi.org/ 10.1016/S1474-4422(11)70066-7 [DOI] [PubMed] [Google Scholar]
- [9].Mishra MK, Yong VW. Myeloid cells - targets of medication in multiple sclerosis. Nat Rev Neurol 2016; 12:539-551; PMID:27514287; https://doi.org/ 10.1038/nrneurol.2016.110 [DOI] [PubMed] [Google Scholar]
- [10].Ciccarelli O, Barkhof F, Bodini B, De Stefano N, Golay X, Nicolay K, Pelletier D, Pouwels PJ, Smith SA, Wheeler-Kingshott CA, et al.. Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. Lancet Neurol 2014; 13:807-822; PMID:25008549; https://doi.org/ 10.1016/S1474-4422(14)70101-2 [DOI] [PubMed] [Google Scholar]
- [11].Zhao L, Zabel MK, Wang X, Ma W, Shah P, Fariss RN, Qian H, Parkhurst CN, Gan WB, Wong WT. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med 2015; 7:1179-97; PMID:26139610; https://doi.org/ 10.15252/emmm.201505298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol 2005; 5:953-964; PMID:16322748; https://doi.org/ 10.1038/nri1733 [DOI] [PubMed] [Google Scholar]
- [13].Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, Bar-Or A, Antel JP. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012; 60:717-727; PMID:22290798; https://doi.org/ 10.1002/glia.22298 [DOI] [PubMed] [Google Scholar]
- [14].Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol 2010; 6:193-201; PMID:20234358; https://doi.org/ 10.1038/nrneurol.2010.17 [DOI] [PubMed] [Google Scholar]
- [15].Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al.. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41:14-20; PMID:25035950; https://doi.org/ 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 2015; 16:358-372; PMID:25991443; https://doi.org/ 10.1038/nrn3880 [DOI] [PubMed] [Google Scholar]
- [17].Kraft R. STIM and ORAI proteins in the nervous system. Channels (Austin) 2015; 9:245-52; PMID:26218135; https://doi.org/ 10.1080/19336950.2015.1071747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ohana L, Newell EW, Stanley EF, Schlichter LC. The Ca2+ release-activated Ca2+ current (I(CRAC)) mediates store-operated Ca2+ entry in rat microglia. Channels (Austin) 2009; 3:129-139; PMID:19411837; https://doi.org/ 10.4161/chan.3.2.8609 [DOI] [PubMed] [Google Scholar]
- [19].Burnstock G. Physiopathological roles of P2X receptors in the central nervous system. Curr Med Chem 2015; 22:819-844; PMID:25005189; https://doi.org/ 10.2174/0929867321666140706130415 [DOI] [PubMed] [Google Scholar]
- [20].Michaelis M, Nieswandt B, Stegner D, Eilers J, Kraft R. STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia 2015; 63:652-663; PMID:25471906; https://doi.org/ 10.1002/glia.22775 [DOI] [PubMed] [Google Scholar]
- [21].Chen YJ, Raman G, Bodendiek S, O'Donnell ME, Wulff H. The KCa3.1 blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J Cereb Blood Flow Metab 2011; 31:2363-2374; PMID:21750563; https://doi.org/ 10.1038/jcbfm.2011.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen YJ, Nguyen HM, Maezawa I, Grossinger EM, Garing AL, Kohler R, Jin LW, Wulff H. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J Cereb Blood Flow Metab 2016; 36:2146-2161; PMID:26661208; https://doi.org/ 10.1177/0271678X15611434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Reich EP, Cui L, Yang L, Pugliese-Sivo C, Golovko A, Petro M, Vassileva G, Chu I, Nomeir AA, Zhang LK, et al.. Blocking ion channel KCNN4 alleviates the symptoms of experimental autoimmune encephalomyelitis in mice. Eur J Immunol 2005; 35:1027-1036; PMID:15770697; https://doi.org/ 10.1002/eji.200425954 [DOI] [PubMed] [Google Scholar]
- [24].Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci 2007; 27:234-244; PMID:17202491; https://doi.org/ 10.1523/JNEUROSCI.3593-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Peng Y, Lu K, Li Z, Zhao Y, Wang Y, Hu B, Xu P, Shi X, Zhou B, Pennington M, et al.. Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro Oncol 2014; 16:528-539; PMID:24305723; https://doi.org/ 10.1093/neuonc/not221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al.. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 2014; 17:131-143; PMID:24316888; https://doi.org/ 10.1038/nn.3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nguyen HM, Grossinger EM, Horiuchi M, Davis KW, Jin LW, Maezawa I, Wulff H. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017; 65:106-121; PMID:27696527; https://doi.org/ 10.1002/glia.23078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Blomster LV, Strobaek D, Hougaard C, Klein J, Pinborg LH, Mikkelsen JD, Christophersen P. Quantification of the functional expression of the Ca2+ -activated K+ channel KCa3.1 on microglia from adult human neocortical tissue. Glia 2016; 64:2065-2078; PMID:27470924; https://doi.org/ 10.1002/glia.23040 [DOI] [PubMed] [Google Scholar]
- [29].Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci USA 2000; 97:8151-8156; PMID:10884437; https://doi.org/ 10.1073/pnas.97.14.8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schmitz A, Sankaranarayanan A, Azam P, Schmidt-Lassen K, Homerick D, Hansel W, Wulff H. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol Pharmacol 2005; 68:1254-1270; PMID:16099841; https://doi.org/ 10.1124/mol.105.015669 [DOI] [PubMed] [Google Scholar]
- [31].Chi V, Pennington MW, Norton RS, Tarcha EJ, Londono LM, Sims-Fahey B, Upadhyay SK, Lakey JT, Iadonato S, Wulff H, et al.. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 2012; 59:529-546; PMID:21867724; https://doi.org/ 10.1016/j.toxicon.2011.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Franco R, Fernandez-Suarez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol 2015; 131:65-86; PMID:26067058; https://doi.org/ 10.1016/j.pneurobio.2015.05.003 [DOI] [PubMed] [Google Scholar]
- [33].Vogel DY, Vereyken EJ, Glim JE, Heijnen PD, Moeton M, van der Valk P, Amor S, Teunissen CE, van Horssen J, Dijkstra CD. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation 2013; 10:35; PMID:23452918; https://doi.org/ 10.1186/1742-2094-10-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 2011; 14:1227-35; PMID:21952260; https://doi.org/ 10.1038/nn.2923 [DOI] [PubMed] [Google Scholar]
- [35].Strobaek D, Brown DT, Jenkins DP, Chen YJ, Coleman N, Ando Y, Chiu P, Jorgensen S, Demnitz J, Wulff H, et al.. NS6180, a new K(Ca) 3.1 channel inhibitor prevents T-cell activation and inflammation in a rat model of inflammatory bowel disease. Br J Pharmacol 2013; 168:432-444; PMID:22891655; https://doi.org/ 10.1111/j.1476-5381.2012.02143.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Olesen SP, Munch E, Moldt P, Drejer J. Selective activation of Ca2+-dependent K+ channels by novel benzimidazolone. Eur J Pharmacol 1994; 251:53-59; PMID:8137869; https://doi.org/ 10.1016/0014-2999(94)90442-1 [DOI] [PubMed] [Google Scholar]
- [37].Knaus HG, McManus OB, Lee SH, Schmalhofer WA, Garcia-Calvo M, Helms LM, Sanchez M, Giangiacomo K, Reuben JP, Smith AB 3rd, et al.. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry 1994; 33:5819-5828; PMID:7514038; https://doi.org/ 10.1021/bi00185a021 [DOI] [PubMed] [Google Scholar]
- [38].McLarnon JG, Sawyer D, Kim SU. Cation and anion unitary ion channel currents in cultured bovine microglia. Brain Res 1995; 693:8-20; PMID:8653424; https://doi.org/ 10.1016/0006-8993(95)00664-C [DOI] [PubMed] [Google Scholar]
- [39].McLarnon JG, Xu R, Lee YB, Kim SU. Ion channels of human microglia in culture. Neuroscience 1997; 78:1217-1228; PMID:9174088; https://doi.org/ 10.1016/S0306-4522(96)00680-X [DOI] [PubMed] [Google Scholar]
- [40].Blunck R, Scheel O, Muller M, Brandenburg K, Seitzer U, Seydel U. New insights into endotoxin-induced activation of macrophages: involvement of a K+ channel in transmembrane signaling. J Immunol 2001; 166:1009-1015; PMID:11145680; https://doi.org/ 10.4049/jimmunol.166.2.1009 [DOI] [PubMed] [Google Scholar]
- [41].Rangaraju S, Gearing M, Jin LW, Levey A. Potassium channel Kv1.3 is highly expressed by microglia in human Alzheimer's disease. J Alzheimers Dis 2015; 44:797-808; PMID:25362031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rus H, Pardo CA, Hu L, Darrah E, Cudrici C, Niculescu T, Niculescu F, Mullen KM, Allie R, Guo L, et al.. The voltage-gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc Natl Acad Sci USA 2005; 102:11094-11099; PMID:16043714; https://doi.org/ 10.1073/pnas.0501770102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rassendren F, Audinat E. Purinergic signaling in epilepsy. J Neurosci Res 2016; 94:781-793; PMID:27302739; https://doi.org/ 10.1002/jnr.23770 [DOI] [PubMed] [Google Scholar]
- [44].Ferreira R, Lively S, Schlichter LC. IL-4 type 1 receptor signaling up-regulates KCNN4 expression, and increases the KCa3.1 current and its contribution to migration of alternative-activated microglia. Front Cell Neurosci 2014; 8:183; PMID:25071444; https://doi.org/ 10.3389/fncel.2014.00183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schlichter LC, Kaushal V, Moxon-Emre I, Sivagnanam V, Vincent C. The Ca2+ activated SK3 channel is expressed in microglia in the rat striatum and contributes to microglia-mediated neurotoxicity in vitro. J Neuroinflammation 2010; 7:4; PMID:20074365; https://doi.org/ 10.1186/1742-2094-7-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Caldeira C, Oliveira AF, Cunha C, Vaz AR, Falcão AS, Fernandes A, Brites D. Microglia change from a reactive to an age-like phenotype with the time in culture. Front Cell Neurosci 2014; 8:152; PMID:24917789; https://doi.org/ 10.3389/fncel.2014.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lyons SA, Pastor A, Ohlemeyer C, Kann O, Wiegand F, Prass K, Knapp F, Kettenmann H, Dirnagl U. Distinct physiologic properties of microglia and blood-borne cells in rat brain slices after permanent middle cerebral artery occlusion. J Cereb Blood Flow Metab 2000; 20:1537-1549; PMID:11083228; https://doi.org/ 10.1097/00004647-200011000-00003 [DOI] [PubMed] [Google Scholar]
- [48].Boucsein C, Kettenmann H, Nolte C. Electrophysiological properties of microglial cells in normal and pathologic rat brain slices. Eur J Neurosci 2000; 12:2049-2058; PMID:10886344; https://doi.org/ 10.1046/j.1460-9568.2000.00100.x [DOI] [PubMed] [Google Scholar]
- [49].Menteyne A, Levavasseur F, Audinat E, Avignone E. Predominant functional expression of Kv1.3 by activated microglia of the hippocampus after Status epilepticus. PLoS One 2009; 4:e6770; PMID:19707551; https://doi.org/ 10.1371/journal.pone.0006770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Frings H, Frings M. Development of strains of albino mice with predictable susceptibilities to audiogenic seizures. Science 1953; 117:283-4; PMID:13048659; https://doi.org/ 10.1126/science.117.3037.283 [DOI] [PubMed] [Google Scholar]
- [51].Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai LH, Aubourg P, Ransohoff RM, et al.. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med 2016; 22:1358-1367; PMID:27668937; https://doi.org/ 10.1038/nm.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Staal RG, Khayrullina T, Zhang H, Davis S, Fallon SM, Cajina M, Nattini ME, Hu A, Zhou H, Poda SB, et al.. Inhibition of the potassium channel KCa3.1 by senicapoc reverses tactile allodynia in rats with peripheral nerve injury. Eur J Pharmacol 2016; 795:1-7; PMID:27876619; https://doi.org/ 10.1016/j.ejphar.2016.11.031 [DOI] [PubMed] [Google Scholar]
- [53].D'Alessandro G, Catalano M, Sciaccaluga M, Chece G, Cipriani R, Rosito M, Grimaldi A, Lauro C, Cantore G, Santoro A, et al.. KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis 2013; 4:e773; PMID:23949222; https://doi.org/ 10.1038/cddis.2013.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dale E, Staal RG, Eder C, Moller T. KCa 3.1-a microglial target ready for drug repurposing? Glia 2016; 64:1733-41; PMID:27121595; https://doi.org/ 10.1002/glia.22992 [DOI] [PubMed] [Google Scholar]