

ABSTRACT
Enhanced L-type Ca2+ channel (LTCC) activity in arterial myocytes contributes to vascular dysfunction during diabetes. Modulation of LTCC activity under hyperglycemic conditions could result from membrane potential-dependent and independent mechanisms. We have demonstrated that elevations in extracellular glucose (HG), similar to hyperglycemic conditions during diabetes, stimulate LTCC activity through phosphorylation of CaV1.2 at serine 1928. Prior studies have also shown that HG can suppress the activity of K+ channels in arterial myocytes, which may contribute to vasoconstriction via membrane depolarization. Here, we used a mathematical model of membrane and Ca2+ dynamics in arterial myocytes to predict the relative roles of LTCC and K+ channel activity in modulating global Ca2+ in response to HG. Our data revealed that abolishing LTCC potentiation normalizes [Ca2+]i, despite the concomitant reduction in K+ currents in response to HG. These results suggest that LTCC stimulation may be the primary mechanism underlying vasoconstriction during hyperglycemia.
KEYWORDS: arterial myocytes, diabetes, ion channels, mathematical modeling, sensitivity analysis
Introduction
Diabetes is a devastating disease that affects millions worldwide, and is a major risk factor for stroke, hypertension, heart disease, and other pathological conditions.1,2 Hyperactivity of L-type Ca2+ channels (LTCCs) in arterial myocytes of small resistance vessels contributes to vascular complications during diabetes.3-7 This is in part attributed to the effects of hyperglycemia (elevated blood glucose). We recently gained novel mechanistic insights into this phenomenon and demonstrated that stimulation of vascular LTCCs in response to elevated glucose (HG) is dependent upon phosphorylation of the LTCC pore forming subunit CaV1.2 at serine 1928 (S1928) via anchoring of the cAMP-dependent protein kinase (PKA) by the scaffolding protein AKAP150.7 Glucose-mediated potentiation of LTCCs led to a significant increase in intracellular Ca2+ concentration ([Ca2+]i) and constriction in cells and arteries from wild-type (WT) mice. Conversely, in arterial myocytes and arteries from knock-in mice expressing a CaV1.2 with S1928 mutated to alanine (S1928A) to prevent phosphorylation at this site,8 glucose-mediated changes in LTCC activity, [Ca2+]i and excessive vasoconstriction were prevented.7
An increase in extracellular glucose also impacts the function of other ion channels in arterial myocytes. Indeed, acute and chronic exposure to HG has been shown to suppress the expression and/or activity of K+ channels in coronary,9 mesenteric,10 and cerebral arterial myocytes.11-13 K+ channels are key regulators of arterial myocyte excitability and vascular reactivity due to their influence on the membrane potential (Em).14 Inhibition of K+ channel function depolarizes arterial myocyte Em, which in turn increases LTCC activity and Ca2+ influx, leading to global raises in [Ca2+]i that induces arterial myocyte contraction and vasoconstriction. Thus, it is conceivable that reduced K+ channel activity contributes to enhanced myogenic tone in the presence of HG. However, observations of HG-induced K+ channel suppression contrast with our finding that HG failed to evoke changes in [Ca2+]i in arterial myocytes or vasoconstriction in arteries from S1928A mice.7 The lack of a glucose effect in myocytes and arteries from S1928A mice is unlikely due to differences in basal K+ channel density, as protein abundance for voltage-gated K+ (KV) KV2.1 subunit, and large-conductance Ca2+-activated BKCa α and β1 subunits, which regulate arterial myocyte excitability and are glucose-sensitive,12-15 was similar between WT and S1928A arteries.7 Changes in CaV1.2 expression are also unlikely, as the S1928A point mutation itself did not alter the abundance of CaV1.2.8 Consistent with this, pressure-induced constriction, which is highly dependent on LTCC and K+ channel function,14,16 resulted in a vascular tone of equal magnitude in WT and S1928A arteries under normal extracellular glucose concentrations (i.e. 10 mM D-glucose).7 Overall, our previous data suggest a relatively small influence of glucose-induced K+ channel inhibition and consequent Em depolarization to arterial myocyte hypercontractility in response to HG. These findings point toward a key role for CaV1.2 phosphorylation at S1928 in global Ca2+ increases and vasoconstriction in response to HG that is, at least partly, independent of changes in Em. Here, we used an established mathematical model of arterial myocyte electrophysiology and Ca2+ dynamics to quantitatively compare the relative contribution of K+ channels and LTCCs in modulating Em and [Ca2+]i changes in response to HG. Our analysis supports a predominant role of LTCC potentiation (vs. K+ channel inhibition) in HG-mediated increase in [Ca2+]i leading to vasoconstriction of cerebral arteries.
Results and discussion
We used the Kapela et al. model of an arterial myocyte 17 to assess changes in [Ca2+]i when simulating an acute elevation in glucose. Based on previous experimental data,3,7,9-13 HG targets both Ca2+ and K+ channels to enhance LTCC currents and suppress K+ currents. Under these conditions, our model predicted that HG would lead to a ∼2-fold increase in [Ca2+]i at steady-state in WT arterial myocytes (Fig. 1A and B). Indeed, this result is similar to our experimental data in isolated arterial myocytes from WT mice (Fig. 1C and D).7 When potentiation of LTCC activity in response to HG is ablated to simulate lack of S1928 phosphorylation, as in S1928A cells, the predicted increase in [Ca2+]i is limited to ∼20% (Fig. 1A and B), which again is in agreement with our experimental observations (Fig. 1C and D). These results suggest that changes in [Ca2+]i in arterial myocytes in response to HG are minimal in the absence of S1928 phosphorylation and LTCC potentiation, despite a reduction in K+ channel activity and subsequent Em depolarization, which is expected to enhance Ca2+ influx.
Figure 1.
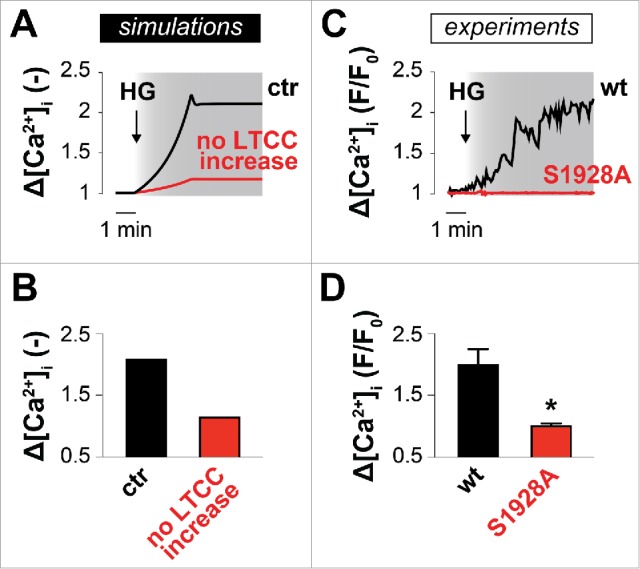
LTCCs play a key role in [Ca2+]i regulation during acute hyperglycemic conditions. Simulated (A, B) and experimental (C, D) HG-mediated effects on [Ca2+]i in WT and S1928A cells. To simulate gradual increase of glucose concentration over time, full modification of HG-dependent model parameters is reached after 180 s from the initial administration (indicated by the arrow). Fig. 1C and D are from our previously published study.7 © AAAS. Reproduced by permission of AAAS. Permission to reuse must be obtained from the rightsholder.
To dissect the relative contribution of HG-induced modifications of LTCCs and K+ channels to [Ca2+]i and Em, we compared the effects of HG on each target in isolation (Fig. 2). Under normal conditions (i.e., in the baseline model assuming 10 mM D-glucose), predicted [Ca2+]i and Em are 68 nM and −59.4 mV, respectively. When specific glucose-mediated alterations in K+ channels are simulated Em underwent a ∼5 mV depolarization (Fig. 2B), due to the reduction in outward currents, whereas the increase in [Ca2+]i was limited (∼10 nM, Fig. 2A). This limited change in Em is similar to that observed by Rainbow et al. in response to elevations in extracellular glucose.10 When only glucose-induced LTCC potentiation was simulated Em was unchanged (Fig. 2B), as expected.16 However, LTCC potentiation caused a pronounced increase in [Ca2+]i (∼30 nM, Fig. 2A). When both effects were included (Figs 1 and 2), HG-induced elevations in [Ca2+]i and Em were further enhanced (∼75 nM and ∼6 mV, Fig. 2A-B). Notably, the effect of HG on [Ca2+]i when alterations of all targets were simulated was larger than the sum of the effect at each individual target (Fig. 2B), suggesting potential synergy between LTCC activation and K+ channel inhibition. When simulations were repeated in voltage-clamped cells at different Em (e.g. −60 mV or −40 mV) to identify the effect of HG on [Ca2+]i independently of Em variations (Fig. 2C-D), K+ channel inhibition did not affect global Ca2+. However, holding Em at a more depolarized voltage augmented the HG- (and LTCC-) mediated increase in [Ca2+]i.
Figure 2.
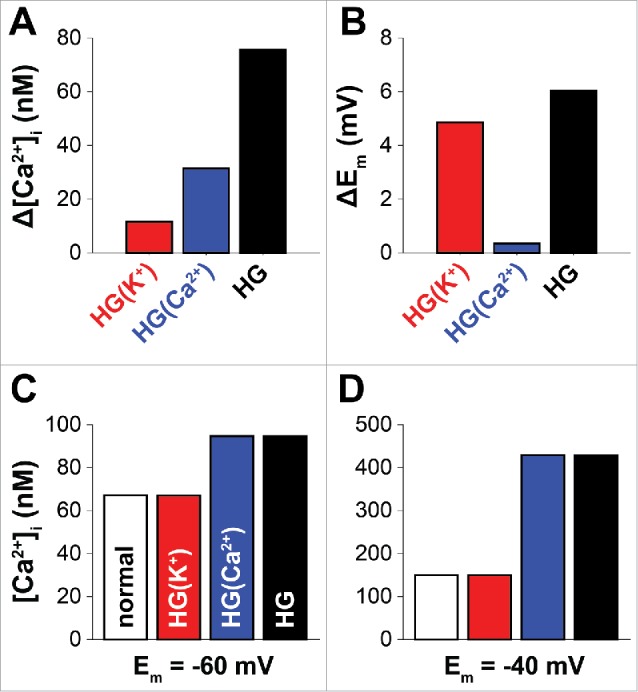
Effects of glucose-induced modifications on [Ca2+]i and Em. Predicted variations of [Ca2+]i (A) and Em (B) (compared with control) obtained at rest (i.e., absence of external stimuli) when glucose-mediated modifications are included for K+ channels (BKCa and Kv2.1) only (red), for LTCC only, or for both. When glucose concentration is normal resting [Ca2+]i and Em are 68 nM and −59.4 mV, respectively. (C, D) Steady-state [Ca2+]i values obtained by clamping the virtual cell at the indicated Em with normal glucose or when HG-dependent modifications are included as described in the text.
We also analyzed how modulating the activity of all ion currents and transport mechanisms affect [Ca2+]i and Em in response to HG, following the methodology described by Sobie.18 The results of this statistical process are summarized in the regression coefficients reported in Table 1, which can be used to estimate changes in [Ca2+]i and Em due to given variations in each model parameter. Importantly, our analysis on the HG targets (Fig. 3) showed that variations in [Ca2+]i are positively correlated with LTCC activity, while Em is insensitive to this model parameter. Inhibition of K+ channels, which is weakly correlated with an increase in [Ca2+]i, has a strong depolarizing effect. Interestingly, modulation of KV channel activity has a more prominent effect on [Ca2+]i and Em regulation compared with BKCa channels. We also found that [Ca2+]i is strongly affected by the activity of non-selective cation channels (NSCs), which also strongly regulate Em, causing depolarization,19 and plasma membrane Ca2+-ATPase (PMCA). Thus, this analysis revealed new mechanistic information that warrants further experimental studies to investigate the role of these fluxes in response to HG and their impact on arterial myocyte excitability and vascular reactivity. Overall, the sensitivity analysis confirmed our mechanistic interpretation of the predominant role of LTCC potentiation in HG-mediated increase in [Ca2+]i, and the secondary role of K+ channels in this outcome.
Table 1.
Regression coefficients. Sensitivity analysis was performed to assess the consequences of changes in all ion channels and transport mechanisms in the model: L-type Ca2+ channels (LTCC), large-conductance Ca2+-activated K+ channels (BKCa), voltage-gated K+ channels (KV2.1), unspecified K+ leak channels (Kleak), Ca2+-activated Cl− channels (ClCa), Na+-K+-Cl− cotransporter (NaKCl), Na+-K+ ATPase (NaK), Na+-Ca2+ exchanger (NCX), plasma membrane Ca2+-ATPase (PMCA), store-operated Ca2+-permeable non-selective cation channels (SOC), non-selective cation channels (NSC), IP3 receptor (IP3R), SR passive Ca2+ leak (SRleak), ryanodine receptor (RyR), SR Ca2+-ATPase pump (SERCA). A positive coefficient indicates positive correlation between changes in the observed outcome (Ca2+ or Em) and a given parameter, and vice versa. Given these coefficients, one can quantify how a change in a certain parameter affects a certain output as explained in the caption of Fig. 3.
| Output |
||
|---|---|---|
| Ion current or transport mechanism | [Ca2+]i | Em |
| LTCC | 0.220 | −0.021 |
| BKCa | −0.025 | 0.135 |
| KV2.1 | −0.054 | 0.298 |
| Kleak | −0.087 | 0.481 |
| ClCa | 9.00e-3 | −0.038 |
| NaKCl | 1.44e-3 | 0.012 |
| NaK | −0.190 | −0.097 |
| NCX | −0.048 | −0.044 |
| PMCA | −0.566 | 0.060 |
| SOC | 0.100e-3 | 0.51e-3 |
| NSC | 0.753 | −0.795 |
| IP3R | 2.00e-3 | 0.35e-3 |
| SRleak | 1.45e-3 | 2.96e-3 |
| RyR | −0.76e-3 | 1.65e-3 |
| SERCA | 1.31e-3 | 1.52e-3 |
Figure 3.

Effects of perturbations in HG-sensitive target parameters on [Ca2+]i and Em. Bar graphs show the regression coefficients indicating how perturbations in the conductance of LTCCs, BKCa and KV2.1 channels affect [Ca2+]i (A) and Em (B). (C) and (D) show the variations induced in these outputs when modulating (±30%) only one conductance at a time, as predicted by the regression model. Given the coefficient BP-O (correlating the parameter P) to the output O), one can quantify the variation of O (with respect to the baseline value Obaseline) induced by a change in P (with respect to the baseline value Pbaseline) using the following formulation: O/Obaseline = (P/Pbaseline) ˆ BP-O.
Our recent study demonstrated that acute elevations in extracellular glucose causes a significant increase in [Ca2+]i and constriction in cerebral arterial myocytes and arteries from WT mice.7 These changes were dependent on increased LTCC activity mediated by phosphorylation of CaV1.2 at S1928. The computational analysis performed here indicates that glucose-induced inhibition of K+ channels in the absence of LTCC hyperactivation (i.e., as it may occur in S1928A myocytes) does not cause appreciable changes in [Ca2+]i. Yet, LTCC potentiation in response to HG drives significant [Ca2+]i enhancement, as it seems to occur in WT myocytes. Note that during chronic elevations in extracellular glucose, as in diabetes, both Em- independent (e.g., PKA-mediated upregulation of LTCC activity7) as well as Em-dependent (e.g., downregulation of K+ channel activity leading to depolarization and further LTCC activation9,12,13) mechanisms synergize to contribute to modulate [Ca2+]i in arterial myocytes. Indeed, our simulation studies showed that the increase in [Ca2+]i seen with concomitant inhibition of K+ currents and LTCC potentiation is attenuated when Em depolarization is prevented. Our results also suggested a predominant role of KV2.1 (vs. BKCa) in the HG-induced Em depolarization. To summarize, our findings confirmed an essential role for CaV1.2 phosphorylation at S1928 in stimulating LTCC activity and vasoconstriction in response to HG. We also demonstrated the power of computational models and population-based statistical approaches in quantifying the relative roles of many elements that interact non-linearly to regulate such complex processes in arterial myocytes.
Methods
The Kapela et al. mathematical model of electrophysiology and Ca2+ dynamics in arterial myocytes17 served as the basis for our simulations. This model is a system of non-linear ordinary differential equations describing the main transmembrane fluxes of Ca2+, K+, Na+ and Cl− involved in Em regulation and the mechanisms controlling intracellular (cytosol and sarcoplasmic reticulum, SR) Ca2+ homeostasis. We modified the baseline model to include glucose-dependent effects on LTCCs, BKCa and KV2.1 conductances that recapitulate previous experimental observations.3,12,13 Specifically, elevated glucose caused a 2-fold increase in LTCC conductance, whereas BKCa and KV2.1 currents were reduced by 58 and 63%, respectively.
We performed sensitivity analysis to investigate the influence of the various currents and transporters in the regulation of global Ca2+ and Em using an established population-based approach.18 The baseline model (i.e., with normal glucose at 10 mM) was used to simulate a family of 1000 model variants, generated by randomly varying conductances or maximal transport rates of all ion currents and transport mechanisms (Table 1). Specifically, the baseline value of each parameter was independently varied with a log-normal distribution (standard deviation of 0.1), that approximately allows changes within the range −30% to +50%. We assessed the resulting values of Em and [Ca2+]i for each model variant, and performed multivariable regression (non-linear iterative partial least squares method) on log-transformed values to correlate the variation in each parameter to the consequent effect on these 2 outputs.18
All simulations were performed in MATLAB (The MathWorks, Natick, MA, USA) using the stiff ordinary differential equation solver ode15s. Our codes are freely available for download at:
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Institute of Health grants R01-HL131517 (to EG) and R01-HL098200 and R01-HL121059 (MFN), the American Heart Association grant 15SDG24910015 (EG), and the Heart Rhythm Society post-doctoral fellowship 16OA9HRS (SM).
References
- [1].National high blood pressure education program working group report on hypertension in diabetes. Hypertension 1994; 23:145-58; discussion 59-60; PMID:8307622; https://doi.org/ 10.1161/01.HYP.23.2.145 [DOI] [PubMed] [Google Scholar]
- [2].Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:837-53; PMID:9742976; https://doi.org/ 10.1016/S0140-6736(98)07019-6 [DOI] [PubMed] [Google Scholar]
- [3].Navedo MF, Takeda Y, Nieves-Cintron M, Molkentin JD, Santana LF. Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. Am J Physiol Cell Physiol 2010; 298:C211-20; PMID:19846755; https://doi.org/ 10.1152/ajpcell.00267.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Arun KH, Kaul CL, Ramarao P. AT1 receptors and L-type calcium channels: functional coupling in supersensitivity to angiotensin II in diabetic rats. Cardiovasc Res 2005; 65:374-86; PMID:15639476; https://doi.org/ 10.1016/j.cardiores.2004.10.010 [DOI] [PubMed] [Google Scholar]
- [5].Ungvari Z, Pacher P, Kecskemeti V, Papp G, Szollar L, Koller A. Increased myogenic tone in skeletal muscle arterioles of diabetic rats. Possible role of increased activity of smooth muscle Ca2+ channels and protein kinase C. Cardiovasc Res 1999; 43:1018-28; PMID:10615429; https://doi.org/ 10.1016/S0008-6363(99)00106-6 [DOI] [PubMed] [Google Scholar]
- [6].White RE, Carrier GO. Vascular contraction induced by activation of membrane calcium ion channels is enhanced in streptozotocin-diabetes. J Pharmacol Exp Ther 1990; 253:1057-62; PMID:1694242 [PubMed] [Google Scholar]
- [7].Nystoriak MA, Nieves-Cintron M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, Grandi E, Fernandes JD, Forbush K, Hofmann F, et al.. Ser1928 phosphorylation by PKA stimulates L-type Ca2+ channel Cav1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci Signal 2017; 10:eaaf9647; PMID:28119464; https://doi.org/ 10.1126/scisignal.aaf9647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, Lenhardt P, Hofmann F, Moosmang S. Unchanged beta-adrenergic stimulation of cardiac L-type calcium channels in Cav1.2 phosphorylation site S1928A mutant mice. J Biol Chem 2008; 283:34738-44; PMID:18829456; https://doi.org/ 10.1074/jbc.M804981200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu Y, Terata K, Rusch NJ, Gutterman DD. High glucose impairs voltage-gated K+ channel current in rat small coronary arteries. Circ Res 2001; 89:146-52; PMID:11463721; https://doi.org/ 10.1161/hh1401.093294 [DOI] [PubMed] [Google Scholar]
- [10].Rainbow RD, Hardy ME, Standen NB, Davies NW. Glucose reduces endothelin inhibition of voltage-gated potassium channels in rat arterial smooth muscle cells. J Physiol 2006; 575:833-44; PMID:16825302; https://doi.org/ 10.1113/jphysiol.2006.114009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Straub SV, Girouard H, Doetsch PE, Hannah RM, Wilkerson MK, Nelson MT. Regulation of intracerebral arteriolar tone by K(v) channels: effects of glucose and PKC. Am J Physiol Cell Physiol 2009; 297:C788-96; PMID:19605735; https://doi.org/ 10.1152/ajpcell.00148.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nieves-Cintron M, Nystoriak MA, Prada MP, Johnson K, Fayer W, Dell'Acqua ML, Scott JD, Navedo MF. Selective downregulation of Kv2.1 function contributes to enhanced arterial tone during diabetes. J Biol Chem 2015; 290:7918-29; PMID:25670860; https://doi.org/ 10.1074/jbc.M114.622811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nystoriak MA, Nieves-Cintron M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell'acqua M, et al.. AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res 2014; 114:607-15; PMID:24323672; https://doi.org/ 10.1161/CIRCRESAHA.114.302168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol 1995; 269:H348-55. PMID:7631867 [DOI] [PubMed] [Google Scholar]
- [15].Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 1992; 256:532-5; PMID:1373909; https://doi.org/ 10.1126/science.1373909 [DOI] [PubMed] [Google Scholar]
- [16].Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 1998; 508:199-209; PMID:9490839; https://doi.org/ 10.1111/j.1469-7793.1998.199br.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kapela A, Bezerianos A, Tsoukias NM. A mathematical model of Ca2+ dynamics in rat mesenteric smooth muscle cell: agonist and NO stimulation. J Theoretical Biol 2008; 253:238-60; PMID:18423672; https://doi.org/ 10.1016/j.jtbi.2008.03.004 [DOI] [PubMed] [Google Scholar]
- [18].Sobie EA. Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys J 2009; 96:1264-74; PMID:19217846; https://doi.org/ 10.1016/j.bpj.2008.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev 2015; 95:645-90; PMID:25834234; https://doi.org/ 10.1152/physrev.00026.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]