

Abstract
Lecithin:retinol acyltransferase (LRAT) catalyzes the acyl transfer from the sn-1 position of phosphatidylcholine (PC) to all-trans-retinol, creating fatty acid retinyl esters (palmitoyl, stearoyl, and some unsaturated derivatives). In the eye, these retinyl esters are substrates for the 65 kDa retinoid isomerase (RPE65). LRAT is well characterized biochemically, and recent structural data from closely related family members of the NlpC/P60 superfamily and a chimeric protein have established its catalytic mechanism. Mutations in the LRAT gene are responsible for approximately 1% of reported cases of Leber congenital amaurosis (LCA). Lack of functional LRAT, expressed in the retinal pigmented epithelium (RPE), results in loss of the visual chromophore and photoreceptor degeneration. LCA is a rare hereditary retinal dystrophy with an early onset associated with mutations in one of 21 known genes. Protocols have been devised to identify therapeutics that compensate for mutations in RPE65, also associated with LCA. The same protocols can be adapted to combat dystrophies associated with LRAT. Improvement in the visual function of clinical recipients of therapy with recombinant adeno-associated virus (rAAV) vectors incorporating the RPE65 gene provides a proof of concept for LRAT, which functions in the same cell type and metabolic pathway as RPE65. In parallel, a clinical trial that employs oral 9-cis-retinyl acetate to replace the missing chromophore in RPE65 and LRAT causative disease has proven to be effective and free of adverse effects. This article summarizes the biochemistry of LRAT and examines chromophore replacement as a treatment for LCA caused by LRAT mutations.
Graphical abstract
1. INTRODUCTION
Leber congenital amaurosis (LCA) is a rare hereditary retinal dystrophy characterized by an early onset in the first decade of life.1–3 Originally described as absent vision with amaurotic pupils and wandering nystagmus accompanied by a pigmented retinopathy,4 LCA is a general definition of a hereditary childhood blindness that encompasses a heterogeneous group of gene defects. Analyses of gene candidates and linkages have identified at least 21 associated genes (Figure 1 and Table 1) [OMIM (http://www.omim.org) and Retinal Information Network (http://retnet.org)]. LRAT dysfunction (LCA14) is a form of LCA affecting <1% of all individuals with LCA.3 Although disorders such as LCA are rare, analysis of individual mutations and their respective phenotypes has helped elucidate the role that each gene product plays in the mechanistic basis of vision. Analysis of these molecular events in turn will provide opportunities for novel therapeutic interventions against LCA and other blinding disorders. Genes associated with LCA14 and LCA2 (encoding RPE65 or retinoid isomerase) are of special interest because their expression is restricted in the eye to the retinal pigmented epithelium (RPE), and both genes encode enzymes critical to the recycling of 11-cis-retinal,5,6 the ligand for the G protein-coupled receptor rhodopsin in the retina.7 Mutations of these genes also can be associated with retinitis pigmentosa (RP), challenging the idea that a monogenetic defect always leads to a distinctive retinal phenotype.
Figure 1.
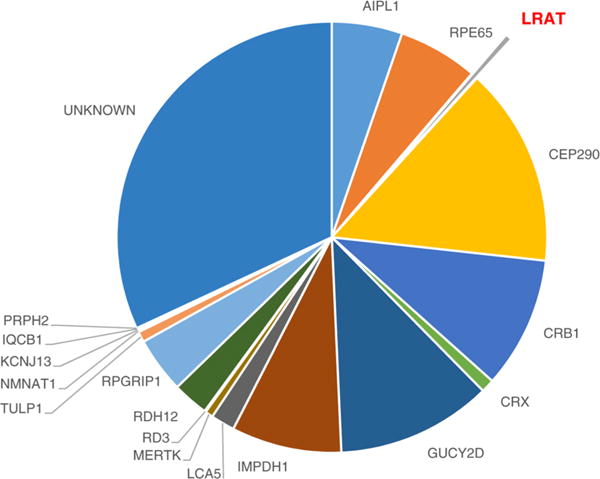
Estimated LCA prevalence associated with currently identified genes. LRAT is responsible for <1% of known LCA cases, most of which lack a causative gene. CEP290 and GUCY2D are the most frequently mutated genes in humans with LCA.3
Table 1.
List of LCA Causative Genes and Their Respective Functionsa
| gene | function |
|---|---|
| AIPL1 (LCA4), GUCY2D (LCA1), KCNJ13 (LCA16) | visual transduction |
| CEP290 (LCA10), IFT20, LCA5 (LCA5), RPGRIP1 (LCA6), TULP1 (LCA15), SPATA7 (LCA3) | ciliary transport |
| ALMS1, CRB1 (LCA8), CRX (LCA7), GDF6 (LCA17), PRPH2 (LCA18) | photoreceptor morphogenesis |
| IMPDH1 (LCA11), RD3 (LCA12) | guanine biosynthesis |
| MERTK | outer segment phagocytosis |
| RPE65 (LCA2), RDH12 (LCA13), LRAT (LCA14) | retinoid cycle enzymes |
| NMNAT1 (LCA9) | nucleotide metabolism |
The following genes encode proteins: AIPL, actin-interacting-like protein 1; GUCY2D, retinal guanylyl cyclase 1; KCNJ13, inward rectifier potassium channel 13; CEP290, centrosomal protein of 290 kDa; IFT20, intraflagellar transport protein 20; LCA5, lebercilin; RPGRIP1, RPGR-interacting protein-like 1; SPATA7, spermato-genesis-associated protein 7; TULP1, tubby-like protein 1; ALMS1, centrosome- and basal body-associated protein; CRB1, crumbs 1; CRX, cone-rod homeobox; GDF6, growth differentiation factor 6; PRPH2, peripherin; IMPDH1, IMP dehydrogenase; RD3, retinal degeneration 3; MERTK, receptor tyrosine kinase; RPE65, retinal pigmented epithelium isomerase 65 kDa; RDH12, retinol dehydrogen-ase 12; LRAT, lecithin:retinol acyltransferase; NMNAT1, nicotina-mide nucleotide adenylyltransferase 1.
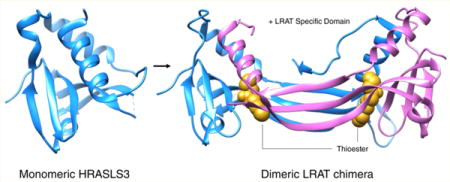
Recent crystallographic data describing a chimeric protein of LRAT and a homologous family member have provided a novel catalytic mechanism involving dimerization and domain swapping. Such structural studies can help determine how different mutations in the same protein cause heterogeneous phenotypes. The purpose of this review is to summarize the molecular mechanism by which LRAT acylates retinol to support the retinoid cycle and vision and discuss whether known human mutations of LRAT validate the putative molecular mechanism of acyl transfer revealed by X-ray diffraction. It also summarizes our current knowledge of LRAT-induced visual loss and retinal degeneration in animal models and human patients.
2. CHARACTERIZATION OF LRAT
2.1. Retinoid Cycle
The retina is the light-sensing neurovascular tissue in humans, located in the posterior portion of the eye adjacent to the apical processes of the RPE. Rod and cone photoreceptor cells of the retina are required to initiate vision under low illumination (rods) and perceive color under higher illumination (cones). Located within rod and cone cell outer segment membranes are the visual pigments, consisting of a protein moiety (opsin) and the vitamin A-derived chromophore, 11-cis-retinal. The two are covalently bound by a Lys side chain amino group via a protonated Schiff base.7 Photoisomerization of 11-cis-retinyldene to an all-trans conformation is the event that initiates the biochemical cascade constituting phototransduction. After conformational changes produce an active form of opsin, signaling is terminated by phosphorylation and the binding of arrestin.8 Importantly, the resulting pigment can no longer be activated by light. Work by Kühne and Wald9,10 laid the foundation for our understanding of visual photochemistry, which takes place within two integrated cellular systems, retinal photoreceptor cells and the RPE. The pathway required for isomerization of all-trans-retinal to 11-cis-retinal is termed the retinoid cycle, which recycles the chromophore needed to maintain vision (Figure 2A).5,11 The process initially requires dissociation of all-trans-retinal from opsin, accomplished by hydrolysis of the Schiff base linkage. All-trans-retinal is subsequently reduced in photoreceptor cells by NADPH-dependent retinol dehydrogenases (RDHs) to produce all-trans-retinol, also known as vitamin A. All-trans-retinol is then shuttled to the RPE by interphotoreceptor retinoid-binding protein (IRBP), where it is esterified by LRAT to form fatty acid retinyl esters.12 These fatty acid esters are substrates for isomerization by the RPE-specific 65 kDa retinoid isomerase (RPE65) that produces 11-cis-retinol, the conformation needed to bind opsin. Prior to transport from the RPE to photoreceptors, 11-cis-retinol is oxidized to 11-cis-retinal by 11-cis-retinol dehydrogenase (11-cis-RDH). Thus, the retinoid cycle represents a multienzyme pathway for the continuous replenishment of the visual chromophore.11 Mutations in nearly all genes contributing to the retinoid cycle can cause congenital and progressive defects in vision.13
Figure 2.
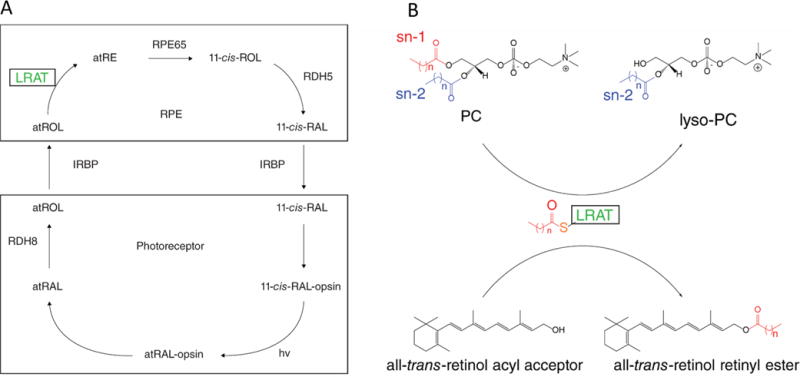
Schematics of the retinoid cycle and LRAT enzymatic reaction. (A) Retinoid cycle displaying the enzymatic conversion of all-trans-retinol (atROL) to 11-cis-retinal (11-cis-RAL). Absorption of a photon of light (hv) by the visual pigment (11-cis-RAL-opsin) causes isomerization of the active state of rhodopsin (atRAL-opsin). Hydrolysis of the Schiff base linkage then releases free all-trans-retinal (atRAL), which is subsequently reduced to atROL and transported across the interphotoreceptor matrix where it is esterified by LRAT in the RPE. All-trans-retinol can also be absorbed from the bloodstream via STRA6, providing a substrate for LRAT. (B) LRAT functions as an acyltransferase with sn-1 specificity, transferring an acyl moiety from phosphatidylcholine (PC) to the all-trans-retinol substrate. For more chemical and biochemical details, see refs 5 and 11.
2.2. LRAT Enzymology
LRAT was initially described as a retinyl ester synthase,14,15 an enzyme that can generate fatty acid retinyl esters from vitamin A in a cellular or solubilized free state.16,17 LRAT was subsequently characterized at the molecular level as a 25.3 kDa enzyme responsible for retinyl ester formation, needed for both vitamin A storage and a critical reaction in the retinoid cycle.18 LRAT is strongly expressed in the liver and eye, specifically in the RPE as the major storage site in the eye for vitamin A (or all-trans-retinol) fatty acid esters. The esters are stored in RPE subcellular lipid droplets, also called retinosomes.19–22 Vitamin A is delivered to the eye by plasma retinoid-binding protein (RBP4) via the RBP4 receptor, stimulated by retinoic acid 6 (STRA6), and expressed on the basolateral surface of the RPE.23,24 The clinical importance of LRAT was initially recognized for its role in the intestine and liver,14,15 with only minor appreciation for its function in the retinoid cycle until mutations in the gene were associated with early onset retinal dystrophy.25 The gene was later discovered to have three exons mapped to chromosome 4q31.2 (Figure 3A) (EC 2.3.1.135, OMIM 604863).26 The catalytic mechanism of LRAT was recently elucidated through kinetic and crystallization studies of the LRAT-like protein family.27,28
Figure 3.
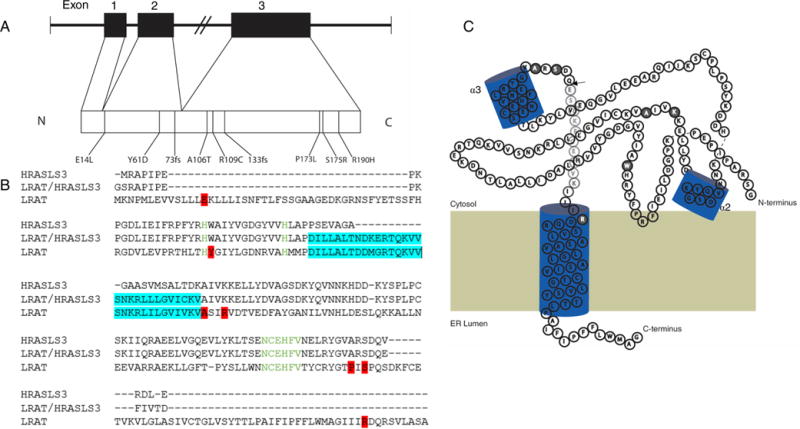
LRAT exons overlaid with known mutations. (A) LRAT exons mapped to chromosome 4, with known mutations associated with LCA. (B) Sequences of human HRASLS3, HRASLS3/LRAT, and LRAT chimera were aligned with T-Coffee multiple-sequence alignment available at the EMBL-EBI server. Both the conserved His residues and the six-amino acid stretch containing the catalytic Cys residue are colored green. The LRAT-specific domain is highlighted in teal. Point mutations associated with LCA are highlighted in red. (C) Two-dimensional model of an HRASLS3/LRAT protomer showing the sites of naturally occurring mutations associated with LCA. Residues in HRASLS3/LRAT occurring downstream of the arrowhead (→) are hypothetical due to truncation of the C-terminal α helix to promote solubility. Point mutations associated with LCA are highlighted in gray. The model is based on the crystal structure of the chimeric protein.28
LRAT-like proteins comprise a vertebrate subfamily of thiol proteases that function as phospholipid-metabolizing enzymes.29 The family consists of LRAT and HRAS-like tumor suppressors 1−5 (HRASLS 1−5, respectively). The subfamily shares a common structural fold and a Cys-His-His catalytic triad responsible for transferring an acyl group from the sn-1 or sn-2 position of phosphatidylcholine (PC) to an acyl acceptor (Figure 2B). LRAT catalyzes the acyl transfer only from the sn-1 position of PC, donating a fatty acid group from this position to a retinoid acceptor. Also unique to LRAT with respect to other acyl transferases is the fact that it does not require activation of a fatty acyl group attached to coenzyme A via a thioester bond. High-resolution data from HRASLS2 and HRASLS3 led to the mechanistic characterization of both enzymes as well as LRAT and other LRAT-like family members27,30 and confirmed the double-displacement reaction mechanism established by Shi et al.,17,31 in which the LRAT catalytic Cys residue is acylated and subsequently transfers the acyl intermediate to retinol to create retinyl esters.
2.3. High-Resolution Structures of LRAT Chimera and LRAT-like Proteins Confirm Membrane Binding and Dimerization
High-resolution crystal structures of HRASLS2 and HRASLS3 provided insight into the common catalytic triad as well as the two conformational states of the catalytic Cys-113 within the active site of the LRAT-like protein family. To facilitate crystallization, the C-terminal α helix was removed. This helix anchors the protein onto the ER membrane, ensuring the proximity of the active site to this cellular structure.
Homology modeling identified a stretch of primary sequence unique to LRAT hypothesized to be a crucial structural motif needed for the acyl-receiving specificity of retinol (Figure 3B). The X-ray crystal structure confirmed the hypothesis, as the unique primary sequence folds into a structural motif responsible for domain swapping, allowing LRAT to retain its sn-1 regiospecificity for the PC acyl donor that produces esters with retinol.28 Domain swapping is the process by which two identical proteins exchange parts of their structure to form a highly ordered oligomer.32 Removal of this primary sequence prevented LRAT from catalyzing the fatty acid esterification of retinol. To further confirm the importance of this motif, insertion of the sequence into HRASLS3 allowed that enzyme to adopt LRAT specificity, capable of sn-1-specifc transfer of acyl moieties from PC to retinol. The structure confirmed homodimerization as well, displaying two globular catalytic domains separated by a pair of three-stranded antiparallel β strands. The LRAT-specific domain exists within these antiparallel β strands as a β hairpin motif, allowing them to interact with each other. This result provides firm evidence that the specificity of LRAT is determined by this LRAT-specific domain. The finding also supports the concept that LRAT functions as a homodimer,33 as the HRASLS3-LRAT fusion protein shares active sites between its two individual subunits (Figure 4). Comparison with the monomeric HRASLS3 structure confirms that α helix 3 translocates to complete the α/β fold of the second protomer. Catalytic Cys-113 is embedded in an α helix; thus, each active site in both protomers is formed by residues from the partner polypeptide. The chimeric protein catalytic sites are embedded in a hydrophobic pocket containing Leu-45, -62, and -120, Val-65, Tyr-22, and Pro-39, whereas the active site of monomeric HRASLS3 is exposed to the aqueous environment of the cytoplasm. This exposure of the active site explains why HRASLS3 can transfer acyl acceptors to water, creating a fatty acid. Thus, analysis of the structure proves that domain swapping between the two monomers provides access to the substrate directly from the lipid interface, expelling water and allowing an acyl group to be transferred to retinol.
Figure 4.
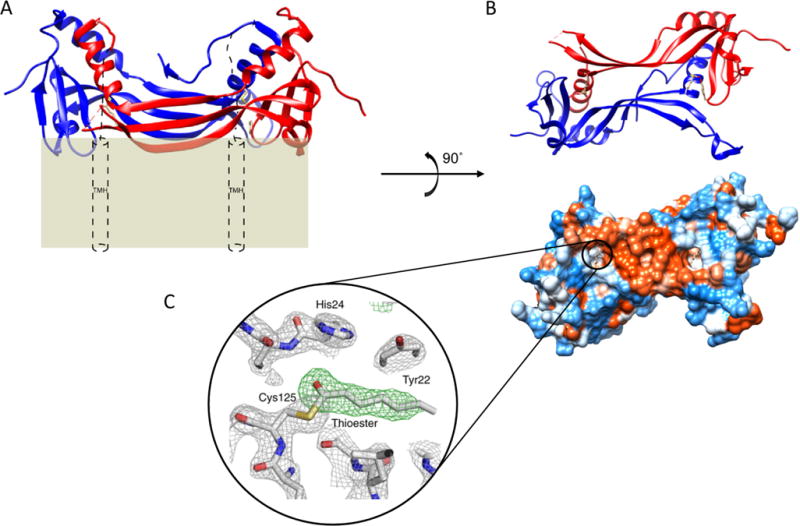
Membrane topology of a HRASLS3/LRAT chimera with hydrophobic exposed active sites. (A) Ribbon representation of a HRASLS3/LRAT chimera viewed in parallel with a plasma membrane. Red and blue ribbons represent single protomers. Theoretical placement of the C-terminal transmembrane α helices (TMH) spanning the lipid membrane is represented by cylinders. (B) Two perpendicular views from underneath dimeric HRASLS3/LRAT. The bottom view presents a hydrophobic surface map with the proposed membrane interaction surface colored red. Surfaces of the molecules are colored according to their hydrophobicity. Red corresponds to hydrophobic residues that largely comprise the LRAT-specific domain and membrane-interacting surface. (C) Electron density of the acylated form of HRASLS3/LRAT embedded within the hydrophobic active site. The acyl moiety is heptanoic acid bound to Cys-125. Gray mesh represents a 2.2 Å resolution σA-weighted 2Fo − Fc electron density map contoured at 1.6σ. The green mesh represents the unbiased σA-weighted Fo − Fc electron density map contoured at 3.5σ. The figures are adapted from the crystal structure of the chimeric protein.28
The known mutations associated with LCA14 are distributed throughout the primary sequence (Figure 3B), which may explain the wide range of phenotypes observed in this disease (Table 2). For example, frameshift mutations tend to have a more obvious impact on protein dysfunction. Alternatively, visualizing the two-dimensional structure of a single gene transcript allows one to understand how specific mutations might impact the function of LRAT by correlating the location of the defect(s) to the active site (Figure 3C). There are two point mutations close to the catalytic Cys-113, which might disrupt the catalytic triad, preventing proper nucleophilic activation and a relative increase in the pKa of the residue. In addition, domain swapping between two molecules of LRAT can provide clues about why mutations outside the active site severely impact the activity of LRAT. Further investigation of these molecular mechanisms will require combined muta-genesis and functional studies as well as the corresponding crystal structures of the mutants.
Table 2.
Known Mutations in LRAT That Cause LCA
| nucleotide change | AA mutation | no. of exons | phenotype | source |
|---|---|---|---|---|
| 40_41delGAinsTT | E14L | 2 | 34 | |
| Met73fs | 3 | |||
| A181T | Y61D | 2 | 34 | |
| 217_218delAT | 73 fs | 2 | LCA type 2 (RPE65) | 36 |
| A316G | A106T | 2 | 34 | |
| A342G | E144E | 3 | LCA type 14 (LRAT) | 36 |
| C371T | R109C | 3 | 67 | |
| 396delAA | 133 fs | 3 | 25 | |
| 427_428delCG | R143VfsX3 | 3 | 68 | |
| C518T | P173L | 3 | 67 | |
| 519delG | I173SfsX12 | 3 | ||
| A525T | S175R | 3 | retinal degeneration | 25 |
| 605G→A | R190H | 3 | 67 |
3. HUMAN RETINOPATHIES CAUSED BY LRAT MUTATIONS
3.1. Clinical Features of LRAT-LCA
Retinal dystrophies make up a chronic and progressive group of visual disorders often categorized as either isolated or syndromic diseases. LCA caused by mutations in the LRAT gene (LCA14; OMIM 613341) is often termed an early onset retinal dystrophy leading to debilitating blindness with an autosomal recessive inheritance.34–36 Congenital or early infantile blindness, the key characteristic of LCA, is defined by early vision loss, nystagmus, amaurotic pupils, and a greatly diminished or absent electro-retinogram (ERG). The phenotypic heterogeneity associated with LCA14 is striking (Table 2). Typically, severe forms of the disease become evident in the first year of life,37 whereas less severe forms can be categorized as juvenile RP, with patients displaying initial visual acuity of 20/70 with progressive degeneration.35,38
LRAT mutations structurally affect the human retina. Retinas often reveal a normal optic disc, narrowing of the vasculature, and “salt and pepper” RPE changes.25 However, much like other clinical forms of LCA, patients with LRAT mutations may have retinas that initially appear normal, but a wide variety of abnormalities have been observed. These include macular coloboma due to chorioretinal degeneration, foveal atrophy, “bone spicule” retinal pigmentation, subretinal flecks that appear like retinitis punctate albescens, a “marbled” fundus, pigmented nummular patches combined with pigmented epithelial atrophy, and several optic disc abnormalities, including drusen, papilledema, vessel attenuation, and neo-vascularization.3,35,37
ERG responses of patients with LCA14 also vary depending on the severity and length of existing disease. Thompson et al.25 described three patients with retinal dystrophy, one of whom (396delAA) was diagnosed at the age of 3 and had no ERG responses by the age of 15. Patients with early and severe forms of LCA14 typically have delayed ERG threshold responses and reduced amplitudes.
3.2. Diagnosis
Congenital or early infantile blindness due to LCA is a clinical diagnosis with the following features: marked visual impairment presenting in infancy, a severely reduced photopic ERG, a positive oculo-digital sign, and a family history consistent with autosomal recessive inheritance.37 Although these findings are not diagnostic, individuals with LCA also may exhibit sluggish pupillary reactions, a pendulous nystagmus, more than five diopters of hyperopia, photophobia, and keratoconus, a non-inflammatory-mediated ectasia of the central cornea.
Genomic testing and clinical manifestations have led to flowchart proposals using the presence or absence of photophobia, night blindness, hyperopia, retinal abnormalities, and visual acuity that aid in the diagnosis of LCA.39 However, there is no relationship between disease severity and genotype. Reproducible and quantifiable measures of disease severity in patients, combined with a predicted structural basis of disease, could help establish a predictable clinical course. For example, visual acuity in patients with LRAT mutations covers a wide range from minimal light perception to 20/50 vision. Over the past 10 years, molecular genetic techniques have improved as high-throughput technologies have become more precise, promoting further characterization and easier diagnosis of genetic diseases. Multigene testing panels can now reveal known mutations in genes associated with LCA, but unfortunately, a large portion of patients lack a known specific mutation (Figure 1). Arrayed-primer extension (APEX) analysis has been used to identify new causative loci in LRAT,34 indicating that even more unknown mutations remain to be discovered.40 APEX exemplifies an approach that can help reconcile the heterogeneity in LCA gene candidates with the wide range of phenotypes.
4. RETINAL DEGENERATION CAUSED BY LRAT DEFICIENCY
4.1. Murine Models
Similar to the Rpe65−/− mouse, the Lrat−/− mouse does not generate 11-cis-retinal (Figure 5A), and cis-retinal administration can restore both rod responses and cone loss.41–45 LRAT gene expression was initially disrupted by targeted recombination, generating a homozygous knockout mouse.46 Despite these Lrat−/− mice possessing only trace amounts of fatty acid retinyl esters in liver, lung, and kidney, they displayed increased concentrations of these esters in adipose tissue as compared with wild-type mice, due to an acyl-CoA-dependent mechanism.47,48
Figure 5.
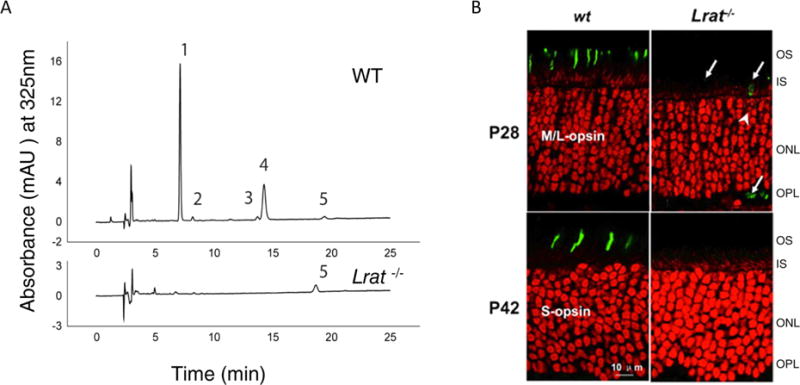
LRAT knockout murine models lack functional retinoids and display photoreceptor degradation. (A) Retinoids were extracted from whole mouse eye and separated by normal phase high-performance liquid chromatography. The peaks shown correspond to the following retinoids: (1) 11-cis- and 13-cis-retinyl esters, (2) all-trans-retinyl esters, (3) 4′-syn- and anti-all-trans-retinal oximes, (4) 3′-syn- and anti-11-cis-retinal oximes, and (5) all-trans-retinol. The chromatogram was generated as described by Batten et al.46 with permission from the American Society for Biochemistry and Molecular Biology. Mice were dark adapted for 48 h prior to ocular extraction. (B) P28 and P42 WT and Lrat−/− mutant mouse retinas were stained for cone pigments: middle- and long-wavelength sensitive (M/L) opsin and short-wavelength (S) opsin are located only within cone cells.69 A perinuclear ring (white arrows) indicates the endoplasmic reticulum where M/L opsin is synthesized. The absence of these opsins in Lrat−/− models indicates degeneration of functional cone outer segments. Abbreviations: OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer. This figure is adapted from ref 49 with approval from The Association for Research Vision and Ophthalmology.
Lrat−/− knockout mice develop retinal degeneration, measured by the lengths of rod outer segments that were ∼35% shorter than those of wild-type mice by 8 weeks of age. These mice also had fast-progressing degeneration49 (Figure 5B). Such mice exhibited severely diminished levels of fatty acid retinyl esters in the eye (in contrast to overaccumulation of these esters in Rpe65−/− mice) and blood, whereas the level of the substrate for LRAT, all-trans-retinol, was reduced only slightly.48 Selective ablation of the LRAT gene in the RPE of mice also resulted in a significant reduction in retinyl ester production.50 Lrat−/− mice were studied by electrophysiological methods under multiple conditions, and the data demonstrated that ERG amplitudes had elevated thresholds and smaller amplitudes, suggesting severely attenuated rod and cone function probably due to a lack of visual chromophore and loss of photoreceptor cells.
A different Lrat−/− mouse model, generated in cases in which the neo/loxP cassette was removed to analyze the susceptibility to vitamin A deficiency, featured a 10-fold decrease in serum retinol levels as well as undetectable retinol levels in most tissues.51 Retinyl ester levels in the livers and lungs of these Lrat−/− mice were <0.2% of those measured in wild-type mice.51 Both mouse models provide evidence that retinol metabolism depends on LRAT function, and loss of LRAT activity in the RPE results in retinal degeneration from a lack of functional chromophore production.
Interestingly, recent work on short-wavelength opsin (S-opsin) in the Lrat−/− mouse revealed a possible mechanism underlying photoreceptor degeneration. Fu et al. demonstrated that cone photoreceptor degeneration in the model is dependent not only upon the presence of LRAT and a functioning retinoid cycle but also upon the presence of S-cone opsin.52,53 Ablation of S-opsin actually slowed cone degeneration.54
5. CURRENT THERAPIES AND DIAGNOSTICS
5.1. Oral Pharmacologic Intervention
The current goal for pharmacological treatment of LRAT-associated LCA involves the replacement of the missing chromophore, 11-cis-retinal. Murine and canine models of LCA treated with 9-cis-retinyl acetate display partial preservation of photoreceptor architecture.42,45,55,56 The 9-cis-retinyl acetate compound is preferred over 11-cis-retinal, because the 11-cis isomer is an unstable aldehyde making laboratory manipulation difficult. 9-cis-Retinyl acetate is converted in the body to 9-cis-retinal, which eventually combines with rhodopsin and can photo-isomerize to initiate the phototransduction cascade.57 Phase 1b clinical trial results were published in 2014 wherein replacement of the chromophore was tested with 9-cis-retinyl acetate administered orally for 7 days at either 10 or 40 mg/m2 each day for 1 week. Of the 14 patients treated, 10 (71%) exhibited improvement in their Goldmann visual fields, 6 (43%) displayed improved visual acuity, and self or parent reports revealed an improvement in daily living.58 Two years after 9-cis-retinoid administration had been discontinued, 11 subjects (79%) had returned to their baseline Goldmann visual field test results and 10 (71%) to their former baseline visual acuity. The results of this study are promising because 9-cis-retinyl acetate appeared to be effective, well-tolerated, and safe, as evidenced by the improved visual function and absence of adverse side effects.
5.2. Gene Therapy
Although nonrandomized gene replacement to treat LCA-related RPE65 deficiency resulted in improved visual function in multiple clinical trials, retinal degeneration persisted. Initial optimism was due to successful short-term effects resulting from recombinant adeno-associated viral vector (rAAV-hRPE65)-mediated gene transfer in canines affected with RPE65 deficiency.59 Initiated in 2007, clinical trials assessed the safety of using rAAV-hRPE65 to replace a dysfunctional RPE65 gene. Besides monitoring toxicity and side effects, multiple parallel experiments at several institutions measured the effects of RPE65 gene transfer on visual acuity, visual field, nystagmus, pupillary light reflexes, dark adaptation, microperimetry, and full field sensitivity testing.60–62
Clinical trials with a follow-up of at least 4 years in adults demonstrated modest efficacy with high safety. Both rod and cone photoreceptor-based vision improved in the treated areas. However, such visual improvement proved to be short-term as two independent studies revealed that retinal degeneration still progressed with optical coherence tomography (OCT), demonstrating thinning of the photoreceptor nuclear layer,63,64 and within 6 years of treatment, areas of reported improvement had diminished in all three patients first treated. The authors determined that degeneration continued at the same rate as in untreated areas, and that the improvement seen soon after treatment was transient.
rAAV gene therapy also was investigated in the same line of Lrat−/− mice that helped establish oral pharmacologic treatment with 9-cis-retinyl acetate.65 Through intraocular injection of rAAV carrying the LRAT gene, the authors successfully restored ERG responses in all treated mice to ∼50% of wild-type levels, comparable to their results with oral 9-cis-retinyl acetate treatment. Unfortunately, there currently is no human gene therapy for LRAT causative LCA. Before human gene therapy is begun in patients with LCA14, a thorough investigation of the natural history of LCA14 is required. Analysis of gene replacement therapy should be initiated in a large animal model akin to those employed for RPE65 replacement therapy in the Swedish Briard/Briard-beagle model (see above); however, no equivalent Lrat−/− model exists.66 Perhaps via replacement of the dysfunctional enzyme prior to the onset of retinal degeneration, photoreceptor integrity and cell function could be preserved.
For gene therapy to be considered effective, it must stop photoreceptor degeneration to preserve remaining vision. Early therapies conducted in children are likely to be more successful. We speculate that recent progress in gene therapy for RPE65-related disease can serve as a proof of concept for LRAT-LCA. Considering the key roles that LRAT and RPE65 play in catalyzing subsequent retinoid transformations in the retinoid cycle, using shared techniques to treat either condition seems logical.
6. DISCUSSION
LRAT was first characterized as a retinyl ester synthase, catalyzing the formation of fatty acid retinyl esters from retinol and PC. The enzyme was initially discovered in the intestine and liver, and its clinical importance was attributed only to its role in the storage of dietary vitamin A. Soon thereafter, it was characterized at the molecular level as catalyzing a crucial step in the retinoid cycle. The protein is highly expressed in the liver and eye, where it is critical for dietary vitamin A storage and regeneration of the visual chromophore in the retinoid cycle. In the eye, it is specifically expressed in the RPE, and thus, lack of functional LRAT resulted in diminished visual chromophore and eventual retinal degeneration. Although LRAT is not expressed to the same extent as RPE65 or cellular retinaldehyde-binding protein (CRALBP) in both rod-dominated (rat) or cone-dominated species (ground squirrel) (Table 3), all three proteins are essential components of a functional retinoid cycle (Figure 2A).
Table 3.
Expression of Key Retinoid Metabolic Genesa in Different Mammals, Displayed in Fragments per Kilobase of Gene Product per One Million Reads (FPKM)
| rat | Nile rat | ground squirrel | macaque | human | |
|---|---|---|---|---|---|
| CRALBP | 144.93 | 431.91 | 83.38 | 667.92 | 323.31 |
| LRAT | 17.68 | 10.28 | 20.65 | 30.00 | 0.50 |
| RPE65 | 58.97 | 238.18 | 58.05 | 285.76 | 6.25 |
Abbreviations: CRALBP, cellular retinaldehyde-binding protein; LRAT, lecithin:retinol acyltransferase RPE65, 65 kDa retinoid isomerase.
LCA is a rare hereditary dystrophy, and LRAT (LCA14) is just one of 21 known LCA-causing genes. To truly understand how point mutations in the LRAT gene could cause such a debilitating blindness, the enzymology and substrate specificity of LRAT required greater elucidation. Crystal structures of members of the LRAT-like protein family and a chimera between HRASLS3 and LRAT identified a domain that allows LRAT to adopt its specificity for retinol as its acyl acceptor. Removal of this domain from LRAT prohibited retinol from accepting the acyl moiety, but addition of the domain to HRASLS3 allowed the heterologous enzyme to adopt specificity for retinol as an acyl acceptor.
Multiple point mutations that provide clues about how the enzyme might not function properly exist within the gene. Unfortunately, such mutations in the LRAT gene produce a wide range of disease phenotypes observed in patients with LCA, and it is not yet clear how these mutations are linked to a specific phenotype. Further insight into the structural features of LRAT may provide clues about how to compensate for these mutations with small molecular agents for more individualized medical care. At this point, however, gene therapy remains the greatest hope for preventing blindness from LRAT-associated LCA.
Acknowledgments
We thank Dr. Leslie T. Webster, Jr., for helpful comments regarding the manuscript.
Funding
This work was supported by funding from the National Institutes of Health (NIH) (EY021126). A.E.S. was supported in part by NIH training grants awarded to the Case Western Reserve University Medical Scientist Training Program (2T32GM007250) and the Visual Sciences Training Program (T32EY007157). K.P. is the John H. Hord Professor of Pharmacology.
ABBREVIATIONS
- CRALBP
cellular retinaldehyde-binding protein
- cGMP
cyclic GMP
- ERG
electroretinogram
- HRASLS2
HRAS-like tumor suppressor 2
- HRASLS3
HRAS-like tumor suppressor 3
- IRBP
interphotoreceptor retinoid-binding protein
- LCA
Leber congenital amaurosis
- LRAT, lecithin
retinol acyltransferase
- fRMI
functional magnetic resonance imaging
- OCT
optical coherence tomography
- PC
phosphatidylcholine
- PDE6b
phosphodiesterase 6b
- rAAV
recombinant adeno-associated virus
- RBP4
retinol-binding protein 4
- RPE
retinal-pigmented epithelium
- RPE65
65 kDa retinoid isomerase
- RP
retinitis pigmentosa
- RDH
retinol dehydrogenase
- STRA6
stimulated by retinoic acid 6
Footnotes
Notes
The authors declare the following competing financial interest(s): Treatment with 9-cis-retinoids was developed in K.P.’s laboratory at the University of Washington. The university licenced it to QLT. K.P. has no relationship with QLT.
References
- 1.Stone EM. Leber congenital amaurosis – a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 2007;144:791–811. doi: 10.1016/j.ajo.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 2.Schroeder R, Mets MB, Maumenee IH. Leber’s congenital amaurosis. Retrospective review of 43 cases and a new fundus finding in two cases. Arch Ophthalmol. 1987;105:356–359. doi: 10.1001/archopht.1987.01060030076030. [DOI] [PubMed] [Google Scholar]
- 3.den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retinal Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Leber T. Uber retinitis pigmentosa und angeborene amaurose. Graefes Archiv für Ophthalmologie. 1869;15:1–25. [Google Scholar]
- 5.Kiser PD, Golczak M, Palczewski K. Chemistry of the retinoid (visual) cycle. Chem Rev. 2014;114:194–232. doi: 10.1021/cr400107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cideciyan AV. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retinal Eye Res. 2010;29:398–427. doi: 10.1016/j.preteyeres.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polans A, Baehr W, Palczewski K. Turned on by Ca2+! The physiology and pathology of Ca(2+)-binding proteins in the retina. Trends Neurosci. 1996;19:547–554. doi: 10.1016/s0166-2236(96)10059-x. [DOI] [PubMed] [Google Scholar]
- 9.Wald G. Molecular basis of visual excitation. Science. 1968;162:230–239. doi: 10.1126/science.162.3850.230. [DOI] [PubMed] [Google Scholar]
- 10.Kuhne W. Chemical processes in the retina. Vision Res. 1977;17:1269–1316. doi: 10.1016/0042-6989(77)90114-6. [DOI] [PubMed] [Google Scholar]
- 11.Kiser PD, Golczak M, Maeda A, Palczewski K. Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim Biophys Acta, Mol Cell Biol Lipids. 2012;1821:137–151. doi: 10.1016/j.bbalip.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pepperberg DR, Okajima TL, Wiggert B, Ripps H, Crouch RK, Chader GJ. Interphotoreceptor retinoid-binding protein (IRBP). Molecular biology and physiological role in the visual cycle of rhodopsin. Mol Neurobiol. 1993;7:61–85. doi: 10.1007/BF02780609. [DOI] [PubMed] [Google Scholar]
- 13.Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacDonald PN, Ong DE. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. J Biol Chem. 1988;263:12478–12482. [PubMed] [Google Scholar]
- 15.MacDonald PN, Ong DE. A lecithin:retinol acyltransferase activity in human and rat liver. Biochem Biophys Res Commun. 1988;156:157–163. doi: 10.1016/s0006-291x(88)80818-0. [DOI] [PubMed] [Google Scholar]
- 16.Barry RJ, Canada FJ, Rando RR. Solubilization and partial purification of retinyl ester synthetase and retinoid isomerase from bovine ocular pigment epithelium. J Biol Chem. 1989;264:9231–9238. [PubMed] [Google Scholar]
- 17.Shi YQ, Hubacek I, Rando RR. Kinetic mechanism of lecithin retinol acyl transferase. Biochemistry. 1993;32:1257–1263. doi: 10.1021/bi00056a009. [DOI] [PubMed] [Google Scholar]
- 18.Ruiz A, Winston A, Lim YH, Gilbert BA, Rando RR, Bok D. Molecular and biochemical characterization of lecithin retinol acyltransferase. J Biol Chem. 1999;274:3834–3841. doi: 10.1074/jbc.274.6.3834. [DOI] [PubMed] [Google Scholar]
- 19.Imanishi Y, Gerke V, Palczewski K. Retinosomes: new insights into intracellular managing of hydrophobic substances in lipid bodies. J Cell Biol. 2004;166:447–453. doi: 10.1083/jcb.200405110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imanishi Y, Batten ML, Piston DW, Baehr W, Palczewski K. Noninvasive two-photon imaging reveals retinyl ester storage structures in the eye. J Cell Biol. 2004;164:373–383. doi: 10.1083/jcb.200311079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palczewska G, Dong Z, Golczak M, Hunter JJ, Williams DR, Alexander NS, Palczewski K. Noninvasive two-photon microscopy imaging of mouse retina and retinal pigment epithelium through the pupil of the eye. Nat Med. 2014;20:785–789. doi: 10.1038/nm.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palczewska G, Maeda T, Imanishi Y, Sun W, Chen Y, Williams DR, Piston DW, Maeda A, Palczewski K. Noninvasive multiphoton fluorescence microscopy resolves retinol and retinal condensation products in mouse eyes. Nat Med. 2010;16:1444–1449. doi: 10.1038/nm.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, Wiita P, Bok D, Sun H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315:820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 24.Amengual J, Golczak M, Palczewski K, von Lintig J. Lecithin:retinol acyltransferase is critical for cellular uptake of vitamin A from serum retinol-binding protein. J Biol Chem. 2012;287:24216–24227. doi: 10.1074/jbc.M112.353979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, Apfelstedt-Sylla E, Gal A. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet. 2001;28:123–124. doi: 10.1038/88828. [DOI] [PubMed] [Google Scholar]
- 26.Ruiz A, Kuehn MH, Andorf JL, Stone E, Hageman GS, Bok D. Genomic organization and mutation analysis of the gene encoding lecithin retinol acyltransferase in human retinal pigment epithelium. Invest Ophthalmol Visual Sci. 2001;42:31–37. [PubMed] [Google Scholar]
- 27.Golczak M, Kiser PD, Sears AE, Lodowski DT, Blaner WS, Palczewski K. Structural basis for the acyltransferase activity of lecithin:retinol acyltransferase-like proteins. J Biol Chem. 2012;287:23790–23807. doi: 10.1074/jbc.M112.361550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golczak M, Sears AE, Kiser PD, Palczewski K. LRAT-specific domain facilitates vitamin A metabolism by domain swapping in HRASLS3. Nat Chem Biol. 2014;11:26–32. doi: 10.1038/nchembio.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anantharaman V, Aravind L. Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol. 2003;4:R11. doi: 10.1186/gb-2003-4-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pang XY, Cao J, Addington L, Lovell S, Battaile KP, Zhang N, Rao JL, Dennis EA, Moise AR. Structure/ function relationships of adipose phospholipase A2 containing a cys-his-his catalytic triad. J Biol Chem. 2012;287:35260–35274. doi: 10.1074/jbc.M112.398859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi YQ, Furuyoshi S, Hubacek I, Rando RR. Affinity labeling of lecithin retinol acyltransferase. Biochemistry. 1993;32:3077–3080. doi: 10.1021/bi00063a019. [DOI] [PubMed] [Google Scholar]
- 32.Rousseau F, Schymkowitz J, Itzhaki LS. Implications of 3D domain swapping for protein folding, misfolding and function. Adv Exp Med Biol. 2012;747:137–152. doi: 10.1007/978-1-4614-3229-6_9. [DOI] [PubMed] [Google Scholar]
- 33.Jahng WJ, Cheung E, Rando RR. Lecithin retinol acyltransferase forms functional homodimers. Biochemistry. 2002;41:6311–6319. doi: 10.1021/bi015710b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dev Borman A, Ocaka LA, Mackay DS, Ripamonti C, Henderson RH, Moradi P, Hall G, Black GC, Robson AG, Holder GE, Webster AR, Fitzke F, Stockman A, Moore AT. Early onset retinal dystrophy due to mutations in LRAT: molecular analysis and detailed phenotypic study. Invest Ophthalmol Visual Sci. 2012;53:3927–3938. doi: 10.1167/iovs.12-9548. [DOI] [PubMed] [Google Scholar]
- 35.den Hollander AI, Lopez I, Yzer S, Zonneveld MN, Janssen IM, Strom TM, Hehir-Kwa JY, Veltman JA, Arends ML, Meitinger T, Musarella MA, van den Born LI, Fishman GA, Maumenee IH, Rohrschneider K, Cremers FP, Koenekoop RK. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Visual Sci. 2007;48:5690–5698. doi: 10.1167/iovs.07-0610. [DOI] [PubMed] [Google Scholar]
- 36.Senechal A, Humbert G, Surget MO, Bazalgette C, Bazalgette C, Arnaud B, Arndt C, Laurent E, Brabet P, Hamel CP. Screening genes of the retinoid metabolism: novel LRAT mutation in leber congenital amaurosis. Am J Ophthalmol. 2006;142:702–704. doi: 10.1016/j.ajo.2006.04.057. [DOI] [PubMed] [Google Scholar]
- 37.Weleber RG, Francis PJ, Trzupek KM. Leber Congenital Amaurosis. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews. University of Washington; Seattle: 1993. [Google Scholar]
- 38.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17:194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 39.Hanein S, Perrault I, Gerber S, Tanguy G, Barbet F, Ducroq D, Calvas P, Dollfus H, Hamel C, Lopponen T, Munier F, Santos L, Shalev S, Zafeiriou D, Dufier JL, Munnich A, Rozet JM, Kaplan J. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23:306–317. doi: 10.1002/humu.20010. [DOI] [PubMed] [Google Scholar]
- 40.Sweeney MO, McGee TL, Berson EL, Dryja TP. Low prevalence of lecithin retinol acyltransferase mutations in patients with Leber congenital amaurosis and autosomal recessive retinitis pigmentosa. Mol Vision. 2007;13:588–593. [PMC free article] [PubMed] [Google Scholar]
- 41.Maeda T, Dong Z, Jin H, Sawada O, Gao S, Utkhede D, Monk W, Palczewska G, Palczewski K. QLT091001, a 9-cis-retinal analog, is well-tolerated by retinas of mice with impaired visual cycles. Invest Ophthalmol Visual Sci. 2013;54:455–466. doi: 10.1167/iovs.12-11152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maeda A, Maeda T, Palczewski K. Improvement in rod and cone function in mouse model of Fundus albipunctatus after pharmacologic treatment with 9-cis-retinal. Invest Ophthalmol Visual Sci. 2006;47:4540–4546. doi: 10.1167/iovs.06-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maeda T, Cideciyan AV, Maeda A, Golczak M, Aleman TS, Jacobson SG, Palczewski K. Loss of cone photoreceptors caused by chromophore depletion is partially prevented by the artificial chromophore pro-drug, 9-cis-retinyl acetate. Hum Mol Genet. 2009;18:2277–2287. doi: 10.1093/hmg/ddp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan J, Rohrer B, Frederick JM, Baehr W, Crouch RK. Rpe65−/− and Lrat−/− mice: comparable models of leber congenital amaurosis. Invest Ophthalmol Visual Sci. 2008;49:2384–2389. doi: 10.1167/iovs.08-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Hooser JP, Aleman TS, He YG, Cideciyan AV, Kuksa V, Pittler SJ, Stone EM, Jacobson SG, Palczewski K. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci U S A. 2000;97:8623–8628. doi: 10.1073/pnas.150236297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, Possin D, Van Gelder RN, Baehr W, Palczewski K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J Biol Chem. 2004;279:10422–10432. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wongsiriroj N, Piantedosi R, Palczewski K, Goldberg IJ, Johnston TP, Li E, Blaner WS. The molecular basis of retinoid absorption: a genetic dissection. J Biol Chem. 2008;283:13510–13519. doi: 10.1074/jbc.M800777200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, Palczewski K, Blaner WS. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT) J Biol Chem. 2005;280:35647–35657. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan J, Rohrer B, Frederick JM, Baehr W, Crouch RK. Rpe65−/− and Lrat−/− mice: comparable models of leber congenital amaurosis. Invest Ophthalmol Visual Sci. 2008;49:2384–2389. doi: 10.1167/iovs.08-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruiz A, Ghyselinck NB, Mata N, Nusinowitz S, Lloyd M, Dennefeld C, Chambon P, Bok D. Somatic ablation of the Lrat gene in the mouse retinal pigment epithelium drastically reduces its retinoid storage. Invest Ophthalmol Visual Sci. 2007;48:5377–5387. doi: 10.1167/iovs.07-0673. [DOI] [PubMed] [Google Scholar]
- 51.Liu L, Gudas LJ. Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J Biol Chem. 2005;280:40226–40234. doi: 10.1074/jbc.M509643200. [DOI] [PubMed] [Google Scholar]
- 52.Zhang T, Zhang N, Baehr W, Fu Y. Cone opsin determines the time course of cone photoreceptor degeneration in Leber congenital amaurosis. Proc Natl Acad Sci U S A. 2011;108:8879–8884. doi: 10.1073/pnas.1017127108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang T, Fu Y. A Phe-rich region in short-wavelength sensitive opsins is responsible for their aggregation in the absence of 11-cis-retinal. FEBS Lett. 2013;587:2430–2434. doi: 10.1016/j.febslet.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang T, Enemchukwu NO, Jones A, Wang S, Dennis E, Watt CB, Pugh EN, Jr, Fu Y. Genetic deletion of S-opsin prevents rapid cone degeneration in a mouse model of Leber congenital amaurosis. Hum Mol Genet. 2015;24:1755–1763. doi: 10.1093/hmg/ddu588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maeda T, Maeda A, Casadesus G, Palczewski K, Margaron P. Evaluation of 9-cis-retinyl acetate therapy in Rpe65−/− mice. Invest Ophthalmol Visual Sci. 2009;50:4368–4378. doi: 10.1167/iovs.09-3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maeda T, Maeda A, Leahy P, Saperstein DA, Palczewski K. Effects of long-term administration of 9-cis-retinyl acetate on visual function in mice. Invest Ophthalmol Visual Sci. 2009;50:322–333. doi: 10.1167/iovs.08-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palczewski K. Retinoids for treatment of retinal diseases. Trends Pharmacol Sci. 2010;31:284–295. doi: 10.1016/j.tips.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koenekoop RK, Sui R, Sallum J, van den Born LI, Ajlan R, Khan A, den Hollander AI, Cremers FP, Mendola JD, Bittner AK, Dagnelie G, Schuchard RA, Saperstein DA. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase 1b trial. Lancet. 2014;384:1513–1520. doi: 10.1016/S0140-6736(14)60153-7. [DOI] [PubMed] [Google Scholar]
- 59.Acland GM, Aguirre GD, Bennett J, Aleman TS, Cideciyan AV, Bennicelli J, Dejneka NS, Pearce-Kelling SE, Maguire AM, Palczewski K, Hauswirth WW, Jacobson SG. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005;12:1072–1082. doi: 10.1016/j.ymthe.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 61.Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, Pang JJ, Sumaroka A, Windsor EA, Wilson JM, Flotte TR, Fishman GA, Heon E, Stone EM, Byrne BJ, Jacobson SG, Hauswirth WW. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bern C, Verastegui M, Gilman RH, Lafuente C, Galdos-Cardenas G, Calderon M, Pacori J, Del Carmen Abastoflor M, Aparicio H, Brady MF, Ferrufino L, Angulo N, Marcus S, Sterling C, Maguire JH. Congenital Trypanosoma cruzi transmission in Santa Cruz, Bolivia. Clin Infect Dis. 2009;49:1667–1674. doi: 10.1086/648070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Testa F, Maguire AM, Rossi S, Pierce EA, Melillo P, Marshall K, Banfi S, Surace EM, Sun J, Acerra C, Wright JF, Wellman J, High KA, Auricchio A, Bennett J, Simonelli F. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology. 2013;120:1283–1291. doi: 10.1016/j.ophtha.2012.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cideciyan AV, Jacobson SG, Beltran WA, Sumaroka A, Swider M, Iwabe S, Roman AJ, Olivares MB, Schwartz SB, Komaromy AM, Hauswirth WW, Aguirre GD. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci U S A. 2013;110:E517–525. doi: 10.1073/pnas.1218933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Batten ML, Imanishi Y, Tu DC, Doan T, Zhu L, Pang J, Glushakova L, Moise AR, Baehr W, Van Gelder RN, Hauswirth WW, Rieke F, Palczewski K. Pharmacological and rAAV gene therapy rescue of visual functions in a blind mouse model of Leber congenital amaurosis. PLoS Med. 2005;2:e333. doi: 10.1371/journal.pmed.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Veske A, Nilsson SE, Narfstrom K, Gal A. Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics. 1999;57:57–61. doi: 10.1006/geno.1999.5754. [DOI] [PubMed] [Google Scholar]
- 67.Preising MN, Paunescu K, Friedburg C, Lorenz B. Genetic and clinical heterogeneity in LCA patients. The end of uniformity. Ophthalmologe. 2007;104:490–498. doi: 10.1007/s00347-007-1533-x. [DOI] [PubMed] [Google Scholar]
- 68.Scholl HP, Moore AT, Koenekoop RK, Wen Y, Fishman GA, van den Born LI, Bittner A, Bowles K, Fletcher EC, Collison FT, Dagnelie G, Degli Eposti S, Michaelides M, Saperstein DA, Schuchard RA, Barnes C, Zein W, Zobor D, Birch DG, Mendola JD, Zrenner E. Safety and Proof-of-Concept Study of Oral QLT091001 in Retinitis Pigmentosa Due to Inherited Deficiencies of Retinal Pigment Epithelial 65 Protein (RPE65) or Lecithin:Retinol Acyltransferase (LRAT) PLoS One. 2015;10:e0143846. doi: 10.1371/journal.pone.0143846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mustafi D, Engel AH, Palczewski K. Structure of cone photoreceptors. Prog Retinal Eye Res. 2009;28:289–302. doi: 10.1016/j.preteyeres.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]