

Abstract
Atypical protein kinase C ζ (PKCζ) forms an apico-basal polarity complex with Partitioning Defective (Pard) 3 and Pard6 to regulate normal epithelial cell apico-basolateral polarization. The dissociation of the PKCζ/Pard3/Pard6 complex is essential for the disassembly of the tight/adherens junction and epithelial-mesenchymal transition (EMT) that is critical for tumor spreading. Loss of cell polarity and epithelial organization is strongly correlated with malignancy and tumor progression in some other cancer types. However, it is unclear whether the PKCζ/Pard3/Pard6 complex plays a role in the progression of non-small-cell lung cancer (NSCLC). We found that hypoxia downregulated the PKCζ/Pard3/Pard6 complex, correlating with induction of lung cancer cell migration and invasion. Silencing of the PKCζ/Pard3/Pard6 polarity complex components induced lung cancer cell EMT, invasion, and colonization in vivo. Suppression of Pard3 was associated with altered expression of genes regulating wound healing, cell apoptosis/death and cell motility, and particularly upregulation of MAP3K1 and fibronectin which are known to contribute to lung cancer progression. Human lung adenocarcinoma tissues expressed less Pard6b and PKCζ than the adjacent normal tissues and in experimental mouse lung adenocarcinoma, the levels of Pard3 and PKCζ were also decreased. In addition, we showed that a methylation locus in the gene body of Pard3 is positively associated with the expression of Pard3 and that methylation of the Pard3 gene increased cellular sensitivity to carboplatin, a common chemotherapy drug. Suppression of Pard3 increased chemoresistance in lung cancer cells. Together, these results suggest that reduced expression of PKCζ/Pard3/Pard6 contributes to NSCLC EMT, invasion, and chemoresistance.
Keywords: epithelial-mesenchymal transition, hypoxia, invasion, lung cancer, PKCζ /Pard3/ Pard6 polarity complex
1. Introduction
Lung cancer is the most common cause of cancer-related death in the United States and worldwide [1]. Despite decades of efforts on research and smoking cessation, the 5-year survival rate of lung cancer patients remains poor at 15% [1, 2], and the number of adenocarcinoma cases in nonsmokers is rising [3, 4]. The main cause of death in patients with lung cancer is local invasion followed by metastasis [1, 2]. Effective therapeutic options for advanced lung cancers are limited and the response rates to second- and third-generations of chemotherapy regimens are poor (30% to 40%), plateauing at a median survival of 8 to 9 months [5]. Thus, there is an urgent need for novel therapeutic targets for advanced lung cancers.
During lung cancer growth, cancer cells obtain increased invasiveness and metastatic potential, which is the main cause of death[1, 2]. The first step of tumor progression is the detaching of tumor cells from the environment and the acquisition of motility and invasiveness, which correspond to the characteristics of epithelial-mesenchymal transition (EMT) [6-8]. During EMT, epithelial cells lose cell-cell connections and apico-basal polarity and acquire mesenchymal and migratory properties. EMT features changes on cell morphology and genetic markers including the disappearance of epithelial markers such as E-cadherin and acquisition of mesenchymal markers such as α smooth muscle actin (α-SMA) and vimentin [9-11]. Recently, researchers observed the loss of E-cadherin paired with the gain of α-SMA, vimentin, etc. in the invasive front in a few tumor models, pointing to a possible contribution of EMT in tumor progression [12, 13]. In non-small-cell lung cancer (NSCLC), EMT has been observed in surgically resected specimens [14, 15]. Clinical cases of NSCLC and NSCLC cell lines exhibiting EMT characteristics are more invasive and insensitive to chemotherapy [15-17], resulting in a lower survival rate for patients with EMT-positive tumors compared to those with EMT-negative tumors [15].
Recent evidence suggests that dissociation of the PKCζ/Pard3/Pard6 polarity complex is essential for disassembly of tight/adherens junction and EMT [18]. Normal epithelial cells display an asymmetric distribution of proteins along the internal apical-basal axis, which is known as apico-basal polarity [19, 20]. The apico-basolateral polarization is mediated by a protein complex named polarity complex, consisting of atypical protein kinase C ζ (PKCζ), Pard (partitioning defective) 3, and Pard6 [18, 21]. In mammalian cells, Pard3 contains multiple postsynaptic density, disc large and ZO-1 domains and is localized to tight junctions at the apical/lateral boundary [18, 21]. It binds to junction adhesion molecule and nectin-1/3 to promote junction assembly through a cofilin-mediated actin dynamics [22]. Thus, Pard3 provides an anchorage for the other components of the polarity complex (e.g. Pard6 and Pard6-associated proteins) at the apical-lateral border [22]. In addition to bind to Pard6 and PKCζ, it may also inhibits atypical PKC activity to regulate the location of Pard3 between apical domains and adherens junctions [23]. Pard6 is a scaffold protein that interacts with Pard3 and recruits PKCζ to the tight junctions [24]. Regulation of PKCζ activity has been the main focus of studies to understand mechanisms controlling intact cell polarity [19, 20]. PKCζ is a member of the atypical PKC subfamily and does not respond to second messenger diacylglycerol or phorbol-diesters. The kinase domain of PKCζ contains important threonine residues (Thr-410 and Thr-560) that are phosphorylated upon activation, which results in its ubiquitination and degradation via proteasome [25]. Transforming growth factor-β1 (TGF-β1), a prototypical cytokine for induction of EMT [9], enables the dissociation of the PKCζ/Pard3/Pard6 polarity complex, thus leading to disassembly of tight/adherens junction and EMT [18].
Recent evidence also suggests that cell polarity plays a role in tumor progression [18-20] and that loss of epithelial organization is strongly correlated with malignancy [22, 26]. PKCζ is downregulated or mutated in lung cancers [27-30], whereas PKCζ knockout enhances Ras-induced lung cancers [31], pointing out that PKCζ is a tumor suppressor [32, 33]. Indeed, majority of PKCζ mutations in cancer is loss-of-function [33]. However, it is worth to point out that another member of atypical PKC, PKCι is essentially an oncogene, opposing PKCζ's effects [32, 33]. Loss or inactivation of Pard3 correlates with invasion in prostate cancer and lung squamous cell carcinoma, likely via impaired STAT3 signaling [34, 35]. Furthermore, high levels of Pard6 are associated with a good prognosis of lung cancer [36]. Our previous reports have also demonstrated that hypoxia downregulates PKCζ and induces EMT in lung cancer cells [30, 37, 38]. This suggests that the PKCζ/Pard3/Pard6 polarity complex is clinically relevant in lung cancers and may serve as a tumor suppressor.
However, it remains unknown whether the PKCζ/Pard3/Pard6 polarity complex plays a role in lung adenocarcinoma invasion. We aim to test the hypothesis that decreased levels or loss of the PKCζ/Pard3/Pard6 polarity complex promotes EMT and invasion. Our results suggest that both human and mouse lung adenocarcinoma tissues express less PKCζ/Pard3/Pard6 proteins than the adjacent normal tissues and that loss of PKCζ/Pard3/Pard6 polarity complex results in EMT and increased invasion in vitro, colonization in vivo, and chemoresistance.
2. Materials and methods
2.1 Materials
Cisplatin and carboplatin were purchased from Sigma-Aldrich (St. Louis, MO) and dissolved in water. Bisindolylmaleimide I (Bis) was obtained from Cayman Chimicals and dissolved in DMSO.
2.2 Cell culture
Human lung adenocarcinoma cells (A549) were obtained from the American Type Tissue Collection and grown in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell cultures were routinely passaged when 85–90% confluent. Hypoxic conditions (1.5% O2) were achieved in a humidified workstation (Invivo2; Ruskinn Technologies, Leeds, UK). The hypoxia workstation contains an oxygen sensor and the workstation's oxygen tension was continuously monitored.
2.3 Western blot analysis
Western blot analysis was performed as previously described [39] using the following primary antibodies: PKCζ (C-20, SC-216, Santa Cruz Biotechnology, Santa Cruz, CA), pPKCζ (T410), E-cadherin (Santa Cruz Biotechnology, Santa Cruz, CA), Pard3 (Millipore, Temecula, CA), Pard6a, Par6b (Santa Cruz Biotechnology, Santa Cruz, CA), MAP3K1, CEACAM1, CEACAM6, FGFR2 (Sigma-Aldrich, St. Louis, MO), PKCiota (ab5282, Abcam, Cambridge, MA), and fibronectin (Millipore, Temecula, CA).
2.4 In vitro scratch assay
Cells were plated in 35-mm cell culture dishes to reach confluence. We scratched the cell monolayer with a 250-μl tip vertically in the center of the plate to create a wound and washed away the floating cells. We took images of the wound under microscope with Zeiss AxioCam LCC1 and measured the beginning width of each wound with AxioVision LE software. After experimental procedures, the image of each wound was taken and the final width of the wound was measured. We calculated the migration distance by subtracting the final width from the beginning width.
2.5 In vitro invasion analysis
2.5×104 cells were seeded on Matrigel-coated inserts (BD Biosciences, Franklin Lakes, NJ) and incubated for 24 or 48 hours. For the experiment with Bisindolylmaleimide I (Bis), we treated cells for one hour then exposed cells to normoxia or hypoxia for 48 hours. The cells that migrated to the other side of the Matrigel were fixed and stained. The total number of invaded cells was counted under the microscope. Five random microscopic fields at 200× magnification were counted in each filter using a calibrated ocular grid.
2.6 Small interfering RNA (siRNA) suppression of selected genes
A549 cells were plated in 60-mm dishes at ∼70-80% confluence and transfected with siRNAs (Santa Cruz Biotechnology, Santa Cruz, CA) using Lipofectamine 2000 (Invitrogen, Grand Island, NY) as described [40]. The siRNAs are pools of at least three different siRNA oligoes that are specific to the target. Negative control siRNA (siNeg, Santa Cruz Biotechnology) was used as a negative control. Two days after transfection, cells were lysed for Western blot analysis.
2.7 Establishment of stable cell lines with suppression of PKCζ, Pard3, and Pard6
As we described previously [38, 41], these stable cell lines were established utilizing lentiviral vectors encoding shRNA against PKCζ, Pard3, or Pard6 (Santa Cruz Biotechnology).
2.8 In vivo colonization assay
We injected cancer cells intravenously into the athymic nude mouse tail vein at the density of 2×106 cells in 0.1 ml of PBS. Five weeks after injection, mice were sacrificed and left lungs were stained with Bouin's solution for quantification of lung cancer nodules. Right lungs were formalin-fixed, paraffin-embedded for hematoxylin and eosin staining [42, 43].
2.9 Quantitative real-time RT-PCR and microarray analysis
We extracted total RNAs with the miRNeasy mini kit (Qiagen, Valencia, CA) and measured RNA concentrations with Nanodrop 2000 Spectrophotometer (Thermo Scientific, Waltham, MA). Complementary DNA (cDNA) was synthesized with the ABI High Capacity cDNA Reverse Transcription Kit (Applied Biosystems Inc., Foster City, CA). Quantitative real-time RT-PCR (qRT-PCR) was performed on the ABI StepOnePlus real-time PCR system with the ABI SYBR Green PCR master mix (Applied Biosystems Inc.). The amplification of the genes was normalized to the amplification of the mitochondrial ribosomal protein L19 (RPL19). The sequences of the primers used in the real time qRT-PCR are shown in Supplemental Table ST2. For microarray analysis, we extracted the total RNA from A549-sh-Neg and A549-sh-Pard3 and performed microarray analysis with the OneArray system (PhalanxBio Inc., Belmont, CA). Quality control of RNA samples was performed with Agilent BioAnalyzer (Agilent Technologies, Santa Clara, CA) and NanoDrop (Thermo Scientific, Waltham, MA). Raw data were quantile-normalized and summarized using the Robust Multi-array Average (RMA) [44]. We removed 12,887 genes not found in both A549-sh-Neg and A549-sh-Pard3, leaving us 16,300 genes for the final analysis set. ANOVA test was used to calculate significance of the differential expression between A549-sh-Neg and A549-sh-Pard3 cells. False discovery rate was controlled at 10%, i.e., nominal p < 6e-6, using the Bonferroni correction. Fold-change cutoff was set at 1.4. These criteria gave us 419 differential genes. Among these dysregulated genes, we evaluated the enriched canonical pathways based on KEGG (Kyoto Encyclopedia of Genes and Genomes) [45] and Gene Ontology[46] biological processes at the Benjamini-Hochberg [47] adjusted p < 0.25.
2.10 KRasG12D lung cancer model
KRasG12D mouse carries a point mutation (G12D) in the KRas gene with loxP-flanked stop codon[48]. Intranasal infection with an adenovirus encoding Cre (Ad-Cre) can excise the stop codon and permit the expression of KRasG12D, resulting in a very high frequency of lung tumors [48]. All animals were handled according to National Institutes of Health guidelines and the Institutional Animal Care and Use Committee-approved experimental protocols.
2.11 Human lung cancer samples, Tissue array, and Immunohistochemical staining
Human lung tissue samples were obtained from patients with suspected or proven NSCLC undergoing pulmonary resection. Informed consent was obtained from all subjects. The acquirement and handling of these samples were approved by the Scientific Review Committee and the Institutional Review Board of Northwestern University and the University of Illinois at Chicago, and complied with all relevant federal guidelines. Human Lung Adenocarcinoma Tissue microarray was purchased from tissue array network (Tissue-Array.net). PKCζ, Pard3, and Pard6 immunohistochemical staining was performed as we described previously with multiple antibody dilutions [49].
2.12 Exploring cytosine modification, gene expression and cytotoxicity in human lymphoblastoid cell lines
We obtained baseline cytosine modification data using the Illumina 450K array [50] (GSE39672) and baseline gene expression data using the Affymetrix Human exon array [51] (GSE7851) on the European (CEU: Caucasian residents from Utah, USA) and African (YRI: Yoruba people from Ibadan, Nigeria) HapMap lymphoblastoid cell lines (LCLs). Details of sample preparation and profiling assays were described in our previous publications [50, 51]. We obtained cytotoxicity data (IC50) generated for carboplatin on the same HapMap LCLs from Huang et al. [52]. Linear regression models were used to link gene expression and modification levels of local CpG sites (within 100 kb) as well as between cytosine modifications and cytotoxicity data, controlled for population identity and gender as described in our previous publications [50, 53-56].
2.13 Cell viability assay
Cells (1 × 104 cells/well) were plated in 96-well flat bottom plates and incubated overnight prior to the treatment of cisplatin or carboplatin at various doses for two days either in normoxic or hypoxic conditions. Cell viability was determined using the Cell Titer 96® AQueous One Solution Cell Proliferation Assay (Promega Corporation, Madison, WI), which measures the amount of modified tetrazolium salt (MTS) reduced to a colored formazan product in metabolically active cells [49]. The optical absorbance was read on a GloMax®-96 Microplate luminometer/plate reader (Promega, Madison, WI) at a wavelength of 450 nm.
2.14 Statistical analysis
Statistical analysis was performed with GraphPad Prism 4 (GraphPad Software, La Jolla, CA) and Microsoft Excel program when applicable. Data were expressed as mean ± SEM. p < 0.05 and 0.01 were selected as significance levels.
3. Results
3.1 Hypoxia downregulates PKCζ/Pard3/Pard6b in non-small-cell lung carcinoma cells
In solid tumors including lung cancer, tumor cells proliferate at a rate that exceeds the oxygen supply, resulting in regions of low oxygen tension (hypoxia) [57, 58]. Tumor cells adapt to hypoxia by inducing genes involved in angiogenesis or glucose metabolism [57-59]. Adaptation to hypoxia increases tumor cell invasiveness and metastatic potential, contributing to their resistance to radiation-induced cell death. Previously, we have shown that hypoxia promotes PKCζ degradation via the proteasome in normal and cancer cells [30]. More importantly, we have shown that hypoxia induces EMT in lung cancer cells [37, 38]. In epithelial cells, PKCζ forms a polarity complex with partitioning defective 3 (Pard3) and Pard6 to regulate EMT and maintain epithelial barrier integrity [18, 20, 21]. Since the abundance of these proteins is critical to maintain the polarity complex, we sought to investigate whether hypoxia induces EMT in lung cancer cells by altering the expression levels of PKCζ, Pard3, or the two common isoforms of Pard6 (Pard6a and Pard6b). We incubated A549 cells in normoxia (21% O2) or hypoxia (1.5% O2) for two days and then measured the expression levels of these proteins. We found that hypoxia decreased protein levels of PKCζ, Pard3, and Pard6b, but not Pard6a (Fig 1A-D). PKCζ shares high homology with PKCι and PKCζ antibodies cross-react with PKCι to some extent, thus we measured the expression levels of PKCι after hypoxic exposure. We found that that hypoxia did not alter protein levels of PKCι (Fig 1E). These results suggest the hypoxia-mediated downregulation of PKCζ/Pard3/Pard6b is specific.
Fig 1. Hypoxia-mediated downregulaion of PKCζ/Pard3/Pard6 correlates with the induction of migration and invasion in non-small-cell lung carcinoma cells.
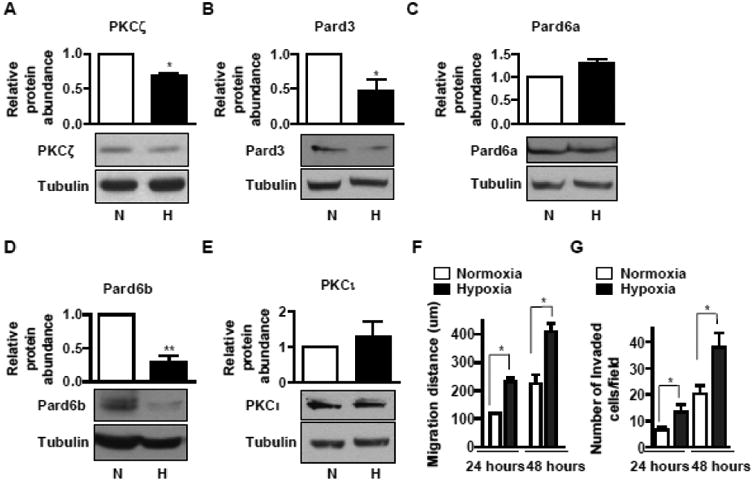
A-D) A549 cells were exposed to normoxia (N) or hypoxia (H, 1.5% O2) for two days. The whole cell lysates were prepared and aliquots with the same amount of protein were subjected to SDS-PAGE followed by Western blot analysis for PKCζ (A), Pard3 (B), Pard6a (C), Pard6b (D), and PKCι (E). The amount of tubulin was used as control for equal loading. Data were expressed as mean ± SEM. n = 4. *, p < 0.05; **, p < 0.01. F) A549 cells were cultured on 35-mm dishes overnight to reach confluence. Wounds were created by scratching a straight line with a 250-μl tip. After 24 hours or 48 hours of incubation in normoxia or hypoxia (1.5% O2), the widths of the wounds were measured and the difference between the width before and after migration was presented as the migration distance. Data were expressed as mean ± SEM. n = 5. *, p < 0.05. G) A549 cells were cultured on BD Matrigel invasion chambers for 24 or 48 hours in normoxia or hypoxia. Invaded cells were stained and the number of invaded cells in each field was counted under microscopic fields at 200x magnification. Experiments were carried out in triplicates and repeated three times. The results were compared to that of A549 cells and data are expressed as mean ± SEM. *, p < 0.05.
3.2 Silencing of the PKCζ/Pard3/Pard6 polarity complex increases lung cancer cell EMT and invasion
EMT is associated with increased migration and invasion [37, 38, 43]; therefore, we measured these parameters in A549 cells exposed to nomoxia or hypoxia. We found that hypoxia induced A549 cell migration and invasion in a time-dependent manner (Fig 1F-G). To examine whether hypoxia-mediated downregulation of the PKCζ/Pard3/Pard6b complex causes the increased migration and invasion during hypoxia, we sought to address whether loss of the polarity complex components is sufficient to induce EMT, migration, and invasion in normal conditions. As shown in Fig 2A-D, we knocked down the expression of PKCζ, Pard3, and Pard6 using siRNAs in A549 cells and found that suppression of PKCζ, Pard3, and Pard6 individually resulted in decreased expression of E-cadherin as well as other components of the polarity complex, suggesting that suppression of the PKCζ/Pard3/Pard6 complex indeed leads to EMT. Furthermore, suppression of PKCζ and Pard3 but not Pard6 increased invasion of A549 cells (Fig 2E). Previously, we have shown that in rat primary type II alveolar epithelial cells exposed to transient hypoxia PKCζ is phosphorylated and activated [60], which leads to downregulation of PKCζ in chronic hypoxia via enhanced ubiquitin-mediated PKCζ degradation [25, 30]. Thus, we speculate that inhibition of PKCζ may preserve PKCζ levels and therefore restore its function. To address that, we first validated the activation of PKCζ in A549 cells exposed to transient hypoxia. We exposed A549 cells to hypoxia for a short time period and measured the phosphorylation of PKCζ at Thr-410. As shown in Fig SF1A, hypoxia increased phosphorylation of PKCζ at Thr-410. We also measured the membrane fraction of PKCζ (active form), and as shown in Fig SF1B, short term exposure to hypoxia increased PKCζ in the membrane fraction. These results suggest that hypoxia indeed activates PKCζ. Next, we treated A549 cells with PKC inhibitor bisindolylmaleimide I (Bis) at two doses (1 μM, which inhibits classical PKC such as PKCα and β; 10 μM, which inhibits atypical PKC such as PKCζ) and exposed them to normoxia and hypoxia, followed with the measurement of cell invasion. We found that inhibition of PKC had no effects on cell invasion in normal conditions (Fig SF1C). In hypoxic condition, high dose, but not low dose, of Bis prevented hypoxia-induced invasion (Fig SF1C), suggesting that activation of PKCζ is required for enhanced cell invasion during hypoxia. These results suggest that the PKCζ/Pard3/Pard6 complex, particularly PKCζ and Pard3, are coordinately regulated and that the loss of the complex, leads to EMT.
Fig 2. Silencing of the PKCζ/Pard3/Pard6 polarity complex increases lung cancer cell EMT and invasion.
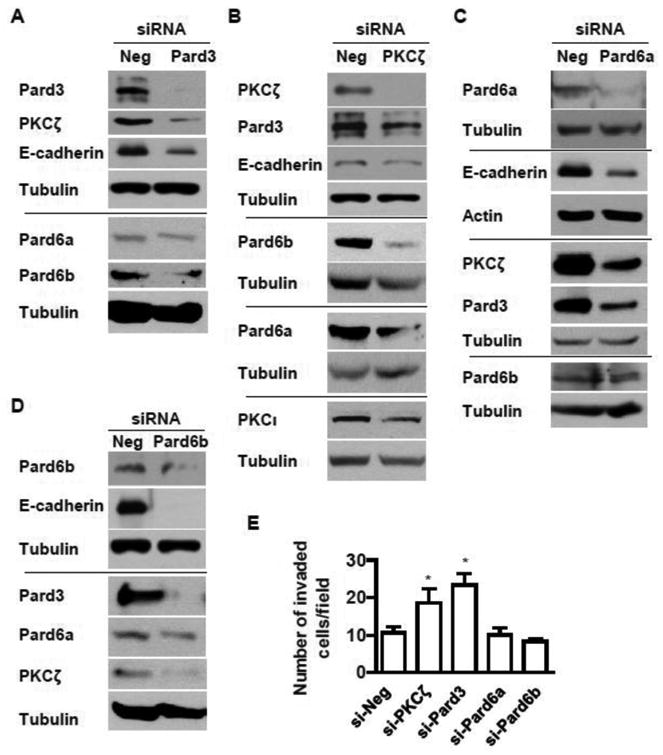
A-D) A549 were transfected with siRNAs for human Pard3 (A), PKCζ (B), Pard6a (C), and Pard6b (D). The expression of PKCζ, Pard3, Pard6a, Pard6b, and E-cadherin were measured by Western blot analysis with tubulin as control for equal loading. E) A549 cells were transfected with siRNAs to suppress the expression of PKCζ, Pard3, Pard6a, and Pard6b followed by the measurement of cell invasion by the Matrigel-coated invasion chambers as described above. The data are expressed as mean ± SEM. *, p < 0.05.
3.3 Silencing of PKCζ and Pard3 increases lung cancer cell colonization to lung in vivo
To address whether loss of PKCζ and Pard3 leads to enhanced cancer spreading in vivo, we generated stable cell lines with PKCζ- and Pard3-knockdown A549 using lentiviral particles encoding shRNA for PKCζ and Pard3(A549-sh-PKCζ and A549-sh-Pard3). A control cell line was established with empty lentiviral particles (A549-sh-Neg). We confirmed the suppression of these proteins and once again found that suppression of one component of this complex decreased the amount of other component concurrently (Fig 3A-B). We found that, in vitro, suppression of PKCζ but not Pard3 decreased cell proliferation rate (Fig 3C). In a tail-vein injection model of cancer spreading in athymic nude mice, we found that the numbers of lung tumor nodules were significantly increased in mice with injection of A549-sh-Pard3 but not A549-sh-PKCζ cells (Fig 3D-E), suggesting that suppression of the polarity complex, particularly Pard3, increases colonization of lung cancer cells in lungs in vivo.
Fig 3. Silencing of the PKCζ/Pard3/Pard6 polarity complex increases lung cancer cell colonization in lungs in vivo.
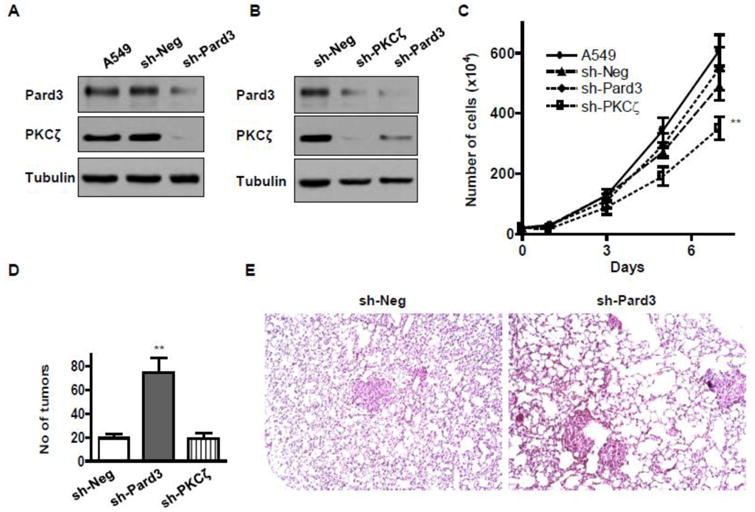
A, B) A549 cells were infected with control lentivirus (A549-sh-Neg) or a lentivirus encoding small hairpin RNA against Pard3 (A549-sh-Pard3) or PKCζ (A549-sh-PKCζ). Stable colonies were selected by incubation with media containing puromycin. C) 0.2 million of these cells were plated, and the number of cells were counted at day 1, 3, 5, and 7 of culture. D) 2 million of A549-sh-Neg, A549-sh-Pard3, or A549-sh-PKCζ cells were injected into the lateral tail vein of athymic nude mice, and after 5 weeks, the metastatic cancer colonies were counted in the left lungs. n > 6; *, p < 0.05; **, p < 0.01. The representative images of right lung sections of mice injected with A549-sh-Neg and A549-sh-Pard3 cells are shown in (E).
3.4 Suppression of Pard3 alters MAP3K1 and fibronectin signaling in lung cancer cells
To study how loss of the polarity complex alters cellular behavior, particularly cell proliferation and migration/invasion, we performed a microarray analysis using A549-sh-Neg and A549-sh-Pard3 cells. ANOVA test was used to calculate significance of the differential expression between A549-sh-Neg and A549-sh-Pard3 cells and we performed Bonferroni post-test. Through Gene Ontology (GO) analysis, we found that the suppression of Pard3 altered expression of genes that participate in cell proliferation, apoptosis, and motility, which is consistent with our results in Fig 1-3 (Supplemental Table ST1). Since many genes are shared by multiple GO Terms, we selected genes that are in GO:0009611∼response to wounding (35 genes) and GO:0010941∼regulation of cell death (32 genes), which contain the most genes, for validation by qRT-PCR. Our GO analysis also revealed altered expression of CEACAM1, which is involved in cell motility and localization (Supplemental Table ST1). Since two similar genes, CEACAM6 and CEACAM7, have been implicated in lung cancer [61, 62], we also examined the levels of these genes. As shown in Fig 4A, suppression of Pard3a significantly decreased the mRNA levels of APOE, CCL2, IL12A, SFRP5, SOD2, and VCAM1 while inducing mRNA levels of CEACAM1, CEACAM6, FGFR2, FN1, IGFBP1, MAP3K1, MMP7, NOX1, PAX7, and VCAN. Next, we examined protein levels of MAP3K1, CEACAM1, CEACAM6, FGFR2, and IGFBP1, as they are known to play roles in lung cancer [61-65]. We found that suppression of Pard3 induced MAP3K1 and fibronectin (FN1) protein levels, but had little effect on protein levels of IGFBP1 and CEACAM6 (Fig 4B). In both cell lines, we did not find detectable levels of CEACAM1 and FGFR2 proteins (Fig 4B). These results suggest that Pard3 may regulate lung cancer cell proliferation and mobility via MAPK3K1 and FN1.
Fig 4. Silencing of Pard3 leads to upregulation of MAP3K1 and fibronectin signaling in lung cancer cells.
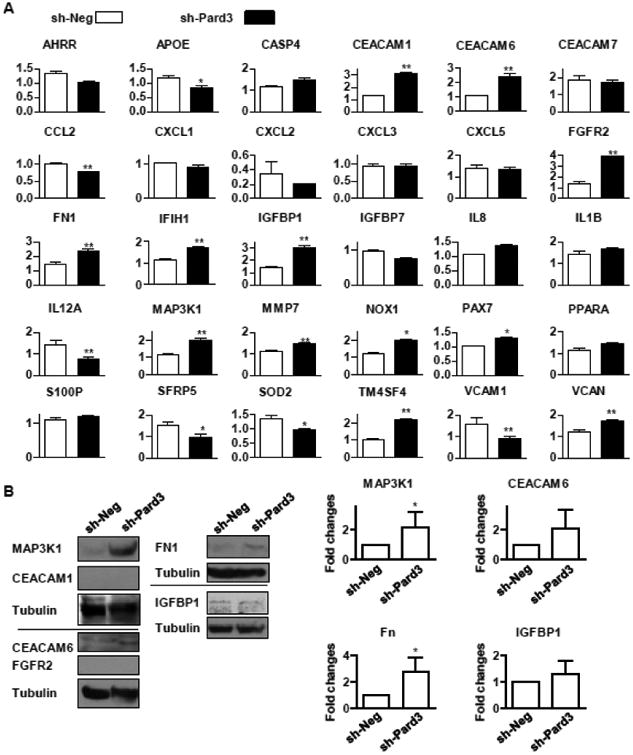
A) A549-sh-Neg and A549-sh-Pard3 were used for real time qRT-PCR for the expression levels of described genes. RPL19 gene was used as the internal control for qRT-PCR. n ≥ 5. B) A549-sh-Neg and A549-sh-Pard3 cells were lysed for the determination of proteins levels by Western blot analysis. n = 5. Quantification is shown in the right panel. *, p < 0.05; **, p < 0.01.
3.5 Downregulation of Pard3 and PKCζ in human lung adenocarcinoma samples and animal models of lung cancer
Next, we examined the clinical relevance of PKCζ/Pard3/Pard6 in lung cancer. A few laboratories have published studies in which they compared the gene expression profiles between lung adenocarcinoma and normal tissues [66-73]. We used Oncomine™ (Compendia Bioscience, Ann Arbor, MI) to access these datasets and analyzed the expression levels of PKCζ, Pard3, and Pard6 in lung adenocarcinoma and normal tissues. Seven out of 8 studies suggest that the expression of PKCζ and Pard3a is significantly reduced in lung adenocaricinomas, while Pard6 levels remained unchanged [66-72]. For example, in the study by Bhattacharjee et al., PKCζ and Pard3 are 3.65- or 2.67-fold lower in lung adenocarcinoma than in normal lung, respectively (Supplemental Fig SF2). Since we previously reported that in lung cancer, PKCζ is downregulated [30], we examined whether the protein expression levels of Pard3 and Pard6 are altered in lung adenocarcinoma. We obtained human lung adenocarcinoma samples and their self-matched normal lung tissues. We homogenized these samples and measured the levels of these proteins. We found that although there was no difference in the levels of Pard3 and Pard6a between normal and cancerous tissues, the levels of Pard6b was significantly lower in lung cancer than normal tissues (Fig 5A). In another approach, we obtained a human lung adenocarcinoma tissue array from Tissue Array Network (Tissue Array Network, Rockville, MD), which contains 48 cases of human lung adenocarcinoma tissue samples, 48 cases of self-matched adjacent normal tissues, and 4 cases of unmatched normal tissues (Fig 5B) for immunohistochemistry staining of these proteins. There is no difference of these protein expression between unmatched normal tissues and normal tissues adjacent to tumors. We did not observe any differences in PKCζ, Pard3, and Pard6a between normal tissues adjacent to tumors and cancer tissues; however, we found that lung adenocarcinoma cells expressed reduced staining of Par6b protein (Fig 5C-D). In a mouse lung cancer model, we instilled KRasG12D mice with Ad-Cre (108 pfu/mouse) to induce lung cancer, and these mice were maintained for up to 24 weeks. We compared the expression levels of PKCζ, Pard3, and E-cadherin in these mouse lungs by Western blot analysis, and we found that the expression levels of PKCζ, Pard3, and E-cadherin decreased gradually when the cancer develops, suggesting that expression levels of PKCζ, Pard3, and E-cadherin may be inversely correlated with lung cancer development (Fig 5E).
Fig 5. Lung adenocarcinoma cells express reduced levels of PKCζ/Pard3/Pard6b.
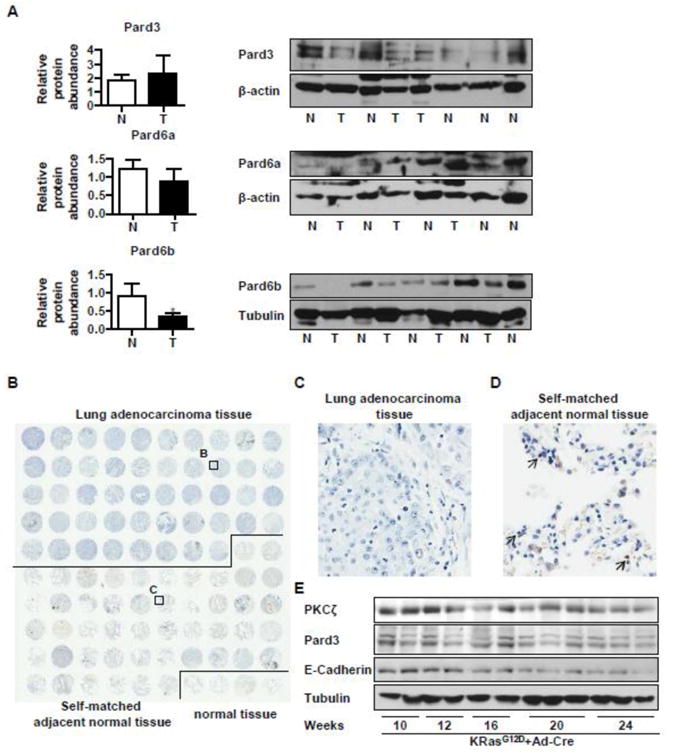
A) Human lung cancer samples (T) and their normal tissues (N) were homogenized for Western blot analysis for protein levels of Pard3, Pard6a, and Pard6b. The relative amount of these proteins was normalized to those of β-actin or Tubulin. The value of one normal tissue sample was set as 1 and others were normalized to that sample. *, p < 0.05; B, C, D) A tumor tissue array (48 cases of human lung adenocarcinoma with self-matched adjacent normal tissue and 4 cases of unmatched normal tissues) was used for immunohistochemistry staining of Pard6b. Brown color represents the staining of Pard6b. The image was obtained with Aperio ImageScope (B). A representative image of lung adenocarcinoma (C) and that of the self-matched adjacent normal tissue are shown in D (200× magnification). Arrows indicate representative cells with positive staining of Pard6b. E) KRasG12D mice was instilled with Ad-Cre (108 pfu/mouse) to induce lung cancer. These mice were maintained for up to 24 weeks, and the mouse lungs were harvested and the aliquots of tissue homogenates with equal amount of proteins were used to detect expression levels of PKCζ, Pard3, and E-cadherin by Western blot analysis, with tubulin or β-actin as loading control.
3.6 Suppression of Pard3 increases NSCLC resistance to cisplatin but not carboplatin
Other than environment factors, epigenetic factors can also regulate the gene expression in cancers. 5-methylcytosine occurs in CpG dinucleotides throughout the human genome and is known to regulate gene expression [74]. More importantly, alteration in gene expression affects resistance to chemotherapy. Thus, we want to address whether there is a correlation between epigenetic regulation of PKCζ/Pard3/Pard6 and resistance to chemotherapy. Since there are no such datasets available in lung adenocarcinoma, we analyzed 5-methylcytosine modifications and gene expression in lymphoblastoid cell lines (LCLs), including 73 unrelated African (YRI - Yoruba people from Ibadan, Nigeria) and 60 with European ancestry (CEU - Caucasians from Utah, US) (the International HapMap Project [75, 76]) and their correlation with the sensitivity to cisplatin and carboplatin, two common chemotherapy drugs. We found that there is a methylation locus in the gene body of Pard3 and methylation of Pard3 correlated with cellular sensitivity to carboplatin (Fig 6A) but not cisplatin (data not shown). Methylation of Pard3 also positively associated with the expression of Pard3 (Fig 6B). As it is not known whether Pard3 is downregulated via hypomethylation in NSCLC, we set out to investigate whether reduced expression of Pard3 contributes to selective resistance to cisplatin or carboplatin. We found that suppression of Pard3 had little effect on sensitivity to carboplatin in the normal condition but increased cell resistance in the hypoxic condition (Fig 6C and 6E). In response to cisplatin, suppression of Pard3 induced resistance in both normal and hypoxic conditions (Fig 6D and 6F).
Fig 6. Suppression of Pard3 increases NSCLC chemoresistance.
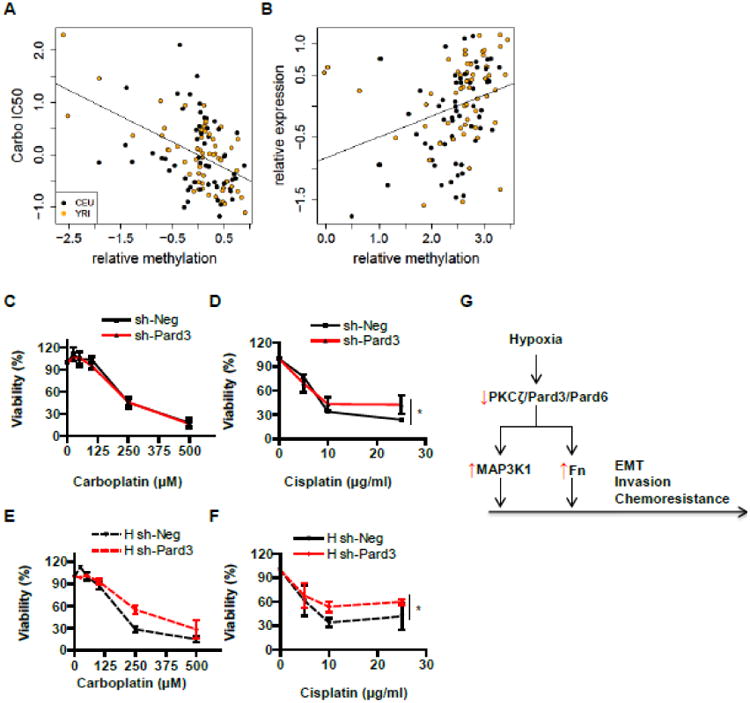
A-B) We analyzed 5-methylcytosine modifications and gene expression in HapMap LCLs, derived from 73 unrelated African (YRI - Yoruba people from Ibadan, Nigeria) and 60 European individuals (CEU-Caucasian residents from Utah, US). Relationships between cytosine modification and cytotoxicity to carboplatin (carbo IC50) (A) as well as between cytosine modification and gene expression (B) are shown. C-F) We cultured A549-sh-Neg and A549-sh-Pard3 in the presence of carboplatin (C, E) or cisplatin (D, F) in normal and hypoxic conditions and measured the viability of these cells. Data were expressed as mean ± SEM. n = 3; *, p < 0.05; **, p < 0.01. G) A schematic diagram on the role of PKCζ/Pard3/Pard6 on lung cancer cell EMT, invasion, and chemoresistance.
4. Discussion
Pard3 forms an apico-basal polarity complex with PKCζ and Pard6 to regulate normal epithelial cell apico-basolateral polarization. The dissociation of the Pard3/PKCζ/Pard6 complex is essential for the disassembly of the tight/adherens junction and epithelial-mesanchymal transition (EMT) that is critical for tumor spreading[18]. Loss of cell polarity and epithelial organization is strongly correlated with malignancy and tumor progression in several cancer types[18-20, 22, 26], whereas the role of Pard3/PKCζ/Pard6 in non-small-cell lung cancer (NSCLC) is less known. Hypoxia is also known to induce EMT and lung cancer progression; however, whether there is a link between hypoxia, polarity, and EMT has not been known in lung cancer. In the current study, we have shown that hypoxia degrades PKCζ to induce EMT and invasion by lung cancer cells. Suppression of the PKCζ/Pard3/Pard6 polarity complex causes EMT and cancer cell invasion and lung colonization. A recent study also suggests a loss-of-function mutation of Pard6G, another isoform of Pard6, in multiple epithelial cancer cells [77]. Thus, hypoxia may disrupt the polarity complex to induce EMT and lung cancer progression.
Previously, we have shown that hypoxia induces EMT in lung cancer cells via ROS/HIF/TGF-β1 pathway and activation of PKA [37, 38]. Interestingly, either HIF or PKA activation alone is not sufficient to induce EMT [37, 38], suggesting that there are other mechanisms involved in hypoxia-mediated EMT. In this study, we have shown that hypoxia downregulates PKCζ/Pard3/Pard6 and that reduction of PKCζ/Pard3/Pard6 leads to EMT, implicating PKCζ as well as its binding partners Pard3 and Pard6 as other key players in the hypoxia-mediated EMT.
Our results suggest that hypoxia reduces levels of PKCζ/Pard3/Pard6b, which is consistent with our previous study in which we report that, during hypoxia, PKCζ is ubiquitinated and degraded via the ubiquitin ligase HOIL-1L, a component of the linear ubiquitin chain assembly complex (LUBAC) [30]. HOIL-1L ubiquitinates PKCζ at Lys-48 for subsequent proteasomal degradation. Whether hypoxia-mediated downregulation of Pard3/Pard6b is LUBAC-dependent warrants further investigation. We also show that HOIL-1L is controlled by HIF [30]. Thus, the HIF transcription factor may be a key coordinator to simultaneously upregulate PKA [37] and downregulate PKCζ/Pard3/Pard6 (Fig 1) [30] for the induction of EMT. Given the evidence that activation of PKCζ results in its degradation, we speculate that inhibition of PKCζ may preserve PKCζ levels and therefore restore its function, as evidenced that treatment of bisindolylmaleimide I (Bis) at a higher dose normalized invasion in hypoxia (Fig SF1C). However, we have not measured the levels of total and activated PKCζ levels in these conditions and further investigations are warranted.
We show that the expression levels of the three polarity complex components are interrelated. Suppression of each component causes the reduction of the other two components (Fig 2-3). This suggests that an intact polarity complex is likely required for epithelial integrity. Intriguingly, the effect of the loss of each component on lung cancer cell proliferation and motility varies significantly (Fig 2-3). This could be due to the compensation of other polarity complex proteins such as PKCι, which can form a complex with Pard3 and three isoforms of Pard6 in place of PKCζ. Although both PKCι and PKCζ can contribute to the polarity complex, the role of PKCι and PKCζ appears to be opposite as PKCι drives a NOTCH-3-mediated stemness in lung adenocarcinoma cells [78]. Thus, loss-of-function of PKCζ may in part represent a gain-of-function of PKCι to drive lung cancer. Accordingly, PKCι is known to activate MEK/ERK signaling to promote lung cancer cell growth [79], and we observed that loss of PKCζ upregulates MAP3K1 (Fig 4). In addition, TGF-β and EGF can activate MAP3K1 [80]. Nonetheless, Pard3 is the unique component in this polarity complex and the effect of the reduction of Pard3 is more consistent and significant (Fig 2-3). Thus, loss of Pard3 may serve as a switch to turn on MAP3K1 after TGF-β stimulation.
The importance of the coordinated regulation of the three polarity complex components is also evident in seemingly discrepant results. Although PKCζ knockdown decreases Pard3 to a similar extent as in A549-sh-Pard3 cells, there is no increase in tumors in A549-sh-PKCζ (Fig 3). This may be due to the decreased cell proliferation of A549-sh-PKCζ cells (Fig 3C). Even though they can invade (Fig 2E) but are not able to grow to a palpable tumor (Fig 3D-E). This is presumably caused by the different levels of PKCζ in these two cell lines: there is complete loss of PKCζ in A549-sh-PKCzeta, while about 30% of PKCζ remains in A549-sh-Pard3 (Fig 3B). Although we don't know whether PKCζ activity is required for this effect, in an experiment with PKCζ inhibitor, inhibition of PKCζ has no effect on A549 cell invasion in normal condition, while prevents invasion in hypoxia, in which PKCζ is activated and degraded (supplemental Fig SF1). Therefore, we reason that it is likely that activation and subsequent loss of PKCζ/Pard3/Pard6 complex, but not the PKCζ activity per se, are key to lung cancer cell invasion. Another example is that siRNA-Par6b leads to marginal levels of Pard3, but not enhanced invasion as seen in siRNA-Pard3 (Fig 2). This may be explained by the compensation effect of Pard6b as we show that in siRNA-Pard3 cells, both Pard6a and Pard6b are down, whereas in siRNA-pard6b cells, Pard6a remains (Fig 2A and Fig 2D). Similarly, siRNA-Pard6a also does not change Pard6b levels or increase invasion (Fig 2C). Another explanation may be the compartmentalization of Pard3, as there is a compartment-specific function of Pard3 [81]. Thus, despite their unique functions of each component, these components act together to fully achieve the function of a polarity complex.
Another upregulated molecule after Pard3 suppression is fibronectin (FN1), a well-known downstream target of TGF-β1. As a major component of extracellular matrix, fibronectin is well known to regulate cancer cell proliferation, migration, and invasion. During development, perturbation of the polarity complex disrupts polarized fibronectin fibril assembly on mesodermal tissue surfaces [82]. Intriguingly, Pard6 appears to not be involved in the regulation of fibronectin organization [83]. Activation of PKCζ is required for arachidonic acid (AA) signaling. AA stimulates induction of membrane type 3-matrix metalloproteinase (MT3-MMP) and fibronectin degradation in human umbilical cord blood-derived mesenchymal stem cells [84]. In rat mesangial cells (RMCs), BMP-7 simultaneously decreases PKCζ and fibronectin [85]. However, in the microarray assay, we did not find any changes in the expression levels of fibronectin receptor integrins, suggesting that Pard3 mainly regulates the abundance of fibronectin to control ECM assembly during EMT.
The alteration of the PKCζ/Pard3/Pard6 levels between human lung cancer tissues and normal tissues varies, depending on the methods of measurement (Fig 5). For example, in the mRNA levels, PKCZ (the gene that encodes PKCζ) and Pard3 are significantly down in human adenocarcinoma, PARD6A mRNA remains unchanged. In immunohistochemical staining, only Pard6b exhibits distinguishable downregulation, whereas Western blot analysis show both PKCζ [30] and Pard6b are reduced. One possible explanation for these discrepancies is the sensitivity and specificity of the antibodies we used in this study. However, we are able to show that Pard3, PKCζ, and E-cadherin levels are reduced in the KRas-driven mouse lung cancer model by Western blot analysis, suggesting an overall downregulation of PKCζ/Pard3/Pard6 in human and mouse lung adenocarcinam.
We observe that the expression of PKCζ/Pard3/Pard6b may also be regulated by epigenetic regulation. In lymphoblastoid cell lines (LCLs), we find that a methylation locus in the gene body of Pard3 is positively associated with the expression of Pard3 (Fig 6B) and that methylation of Pard3 increases cellular sensitivity to carboplatin (Fig 6A). Suppression of Pard3 also displays a differential resistance to chemotherapy drugs carboplatin and cisplatin (Fig 6C-F). Consistently, Fischer and colleagues demonstrate that EMT contributes to chemoresistance [86].
In summary, we show that PKCζ/Pard3/Pard6b is downregulated in lung cancer by hypoxia or likely hypomethylation, leading to EMT, invasion, and chemoresistance (Fig 6G). Although in current study we did not pursue the molecular mechanism underlying downregulation of PKCζ/Pard3/Pard6 during hypoxia, one mechanism is likely the ubiquitination and degradation of these proteins as we have shown the ubiquitination and degradation of PKCζ during hypoxia [30]. Another mechanism could be hypomethylation of the Pard3 gene (Fig 6). Thus, the loss of Pard3 may represent a novel biomarker for NSCLC, and Pard3 status may help determine whether cisplatin or carboplatin is a better therapeutic drug for a specific subset of NSCLC patients. Further studies are warranted to determine whether Pard3 is hypomethylated in lung cancer and whether targeting the downregulation of PKCζ/Pard3/Pard6b may be a novel approach to treat invasive lung cancer in the future.
Supplementary Material
Highlights.
PKCζ/Pard3/Pard6b is downregulated in lung cancer by hypoxia or hypomethylation.
Suppression of the PKCζ/Pard3/Pard6 causes EMT, invasion, and lung colonization.
Suppression of the PKCζ/Pard3/Pard6 leads to chemoresistance.
Acknowledgments
This work was supported in part by the University of Illinois Cancer Center Pilot Subsidy Program (GZ), NIH R01HL123804 (GZ), the Robert H. Lurie Comprehensive Cancer Center-Developmental Funds P30CA060553 and R21HG006367 (WZ), supplemental funding from the UIC Center for Clinical and Translational Science (UL1RR029879), and HL-048129 and HL-071643 (JIS). We would also like to thank Dr. Lawrence M. Knab and Lynn C. Welch for her assistance with human lung cancer samples and Miranda Sun for her assistance on the proofreading of our manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. CA: a cancer journal for clinicians. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Eramo A, Haas TL, De Maria R. Oncogene. 2010;29:4625–4635. doi: 10.1038/onc.2010.207. [DOI] [PubMed] [Google Scholar]
- 3.Sun S, Schiller JH, Gazdar AF. Nat Rev Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 4.Herbst RS, Heymach JV, Lippman SM. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piperdi B, Walsh WV, Bradley K, Zhou Z, Bathini V, Hanrahan-Boshes M, Hutchinson L, Perez-Soler R. J Thorac Oncol. 2012;7:1032–1040. doi: 10.1097/JTO.0b013e31824de2fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta GP, Massague J. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Chambers AF, Groom AC, MacDonald IC. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 8.MacDonald IC, Groom AC, Chambers AF. BioEssays : news and reviews in molecular, cellular and developmental biology. 2002;24:885–893. doi: 10.1002/bies.10156. [DOI] [PubMed] [Google Scholar]
- 9.Zavadil J, Bottinger EP. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 10.Thiery JP, Sleeman JP. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 11.Peinado H, Olmeda D, Cano A. Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 12.Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 13.Tse JC, Kalluri R. J Cell Biochem. 2007;101:816–829. doi: 10.1002/jcb.21215. [DOI] [PubMed] [Google Scholar]
- 14.Lara-Guerra HCC, Schwock J, Pintilie M, Hwang DM, Leighl NB, Waddell TK, Tsao MS. Lung Cancer. 2011 doi: 10.1016/j.lungcan.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 15.Shintani Y, Okimura A, Sato K, Nakagiri T, Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, Matsumoto T, Matsuura N, Ohta M, Okumura M. Ann Thorac Surg. 2011;92:1794–1804. doi: 10.1016/j.athoracsur.2011.07.032. [DOI] [PubMed] [Google Scholar]
- 16.Tischler V, Pfeifer M, Hausladen S, Schirmer U, Bonde AK, Kristiansen G, Sos ML, Weder W, Moch H, Altevogt P, Soltermann A. Mol Cancer. 2011;10:127. doi: 10.1186/1476-4598-10-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryant JLBJ, Balko JM, Willian M, Timmons R, Frolov A, Black EP. Br J Cancer. 2011 doi: 10.1038/bjc.2011.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 19.Martin-Belmonte F, Mostov K. Curr Opin Cell Biol. 2008;20:227–234. doi: 10.1016/j.ceb.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki A, Ohno S. Journal of Cell Science. 2006;119:979–987. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- 21.Ohno S. Curr Opin Cell Biol. 2001;13:641–648. doi: 10.1016/s0955-0674(00)00264-7. [DOI] [PubMed] [Google Scholar]
- 22.Aranda V, Nolan ME, Muthuswamy SK. Oncogene. 2008;27:6878–6887. doi: 10.1038/onc.2008.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soriano EV, Ivanova ME, Fletcher G, Riou P, Knowles PP, Barnouin K, Purkiss A, Kostelecky B, Saiu P, Linch M, Elbediwy A, Kjar S, O'Reilly N, Snijders AP, Parker PJ, Thompson BJ, McDonald NQ. Dev Cell. 2016;38:384–398. doi: 10.1016/j.devcel.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bose R, Wrana JL. Curr Opin Cell Biol. 2006;18:206–212. doi: 10.1016/j.ceb.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Smith L, Chen L, Reyland ME, DeVries TA, Talanian RV, Omura S, Smith JB. J Biol Chem. 2000;275:40620–40627. doi: 10.1074/jbc.M908517199. [DOI] [PubMed] [Google Scholar]
- 26.Etienne-Manneville S. Oncogene. 2008;27:6970–6980. doi: 10.1038/onc.2008.347. [DOI] [PubMed] [Google Scholar]
- 27.Moscat J, Diaz-Meco MT, Wooten MW. Cell Death Differ. 2009;16:1426–1437. doi: 10.1038/cdd.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fields AP, Murray NR. Adv Enzyme Regul. 2008;48:166–178. doi: 10.1016/j.advenzreg.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JKV, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PVK, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 30.Queisser MA, Dada LA, Deiss-Yehiely N, Angulo M, Zhou G, Kouri FM, Knab LM, Liu J, Stegh AH, DeCamp MM, Budinger GRS, Chandel NS, Ciechanover A, Iwai K, Sznajder JI. Am J Respir Crit Care Med. 2014;190:688–698. doi: 10.1164/rccm.201403-0463OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galvez AS, Duran A, Linares JF, Pathrose P, Castilla EA, Abu-Baker S, Leitges M, Diaz-Meco MT, Moscat J. Mol Cell Biol. 2009;29:104–115. doi: 10.1128/MCB.01294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Oncogene. 2014;33:5225–5237. doi: 10.1038/onc.2013.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, Hunter T, Brognard J. Newton AC, Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kitaichi T, Yasui K, Gen Y, Dohi O, Iwai N, Tomie A, Yamada N, Terasaki K, Umemura A, Nishikawa T, Yamaguchi K, Moriguchi M, Sumida Y, Mitsuyoshi H, Naito Y, Zen Y, Itoh Y. Hum Pathol. 2017;62:134–140. doi: 10.1016/j.humpath.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Bonastre E, Verdura S, Zondervan I, Facchinetti F, Lantuejoul S, Chiara MD, Rodrigo JP, Carretero J, Condom E, Vidal A, Sidransky D, Villanueva A, Roz L, Brambilla E, Savola S, Sanchez-Cespedes M. Cancer Res. 2015;75:1287–1297. doi: 10.1158/0008-5472.CAN-14-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Saad S, Al-Shibli K, Donnem T, Persson M, Bremnes RM, Busund LT. Br J Cancer. 2008;99:1476–1483. doi: 10.1038/sj.bjc.6604713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaikh D, Zhou Q, Chen T, Ibe JC, Raj JU, Zhou G. Cell Signal. 2012;24:2396–2406. doi: 10.1016/j.cellsig.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 38.Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI. American Journal of Physiology - Lung Cellular & Molecular Physiology. 2009;297:L1120–1130. doi: 10.1152/ajplung.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Q, Pardo A, Konigshoff M, Eickelberg O, Budinger GRS, Thavarajah K, Gottardi CJ, Jones J, Varga J, Selman M, Sznajder JI, Raj JU, Zhou G, Faseb J. 2011;25:3032–3044. doi: 10.1096/fj.10-177824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen T, Zhou G, Zhou Q, Tang H, Ibe JCF, Cheng H, Gou D, Chen J, Yuan JXJ, Raj JU. Am J Respir Crit Care Med. 2015;191:678–692. doi: 10.1164/rccm.201405-0941OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou G, Dada LA, Chandel NS, Iwai K, Lecuona E, Ciechanover A, Sznajder JI, Faseb J. 2008;22:1335–1342. doi: 10.1096/fj.07-8369com. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Q, Chen T, Ibe JCF, Raj JU, Zhou G. Cell Cycle. 2011;10:2233–2234. doi: 10.4161/cc.10.13.16307. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Q, Chen T, Ibe JC, Raj JU, Zhou G. FEBS Lett. 2012;586:1510–1515. doi: 10.1016/j.febslet.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 45.Kanehisa M, Goto S. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benjamini YH. Yosef Journal of the Royal Statistical Society, Series B. 1995;57:289–300. [Google Scholar]
- 48.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 49.Ibe JCF, Zhou Q, Chen T, Tang H, Yuan JXJ, Raj JU, Zhou G. Am J Respir Cell Mol Biol. 2013;49:609–618. doi: 10.1165/rcmb.2012-0446OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moen EL, Zhang X, Mu W, Delaney SM, Wing C, McQuade J, Myers J, Godley LA, Dolan ME, Zhang W. Genetics. 2013;194:987–996. doi: 10.1534/genetics.113.151381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang W, Duan S, Kistner EO, Bleibel WK, Huang RS, Clark TA, Chen TX, Schweitzer AC, Blume JE, Cox NJ, Dolan ME. Am J Hum Genet. 2008;82:631–640. doi: 10.1016/j.ajhg.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang RS, Kistner EO, Bleibel WK, Shukla SJ, Dolan ME. Mol Cancer Ther. 2007;6:31–36. doi: 10.1158/1535-7163.MCT-06-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X, Moen EL, Liu C, Mu W, Gamazon ER, Delaney SM, Wing C, Godley LA, Dolan ME, Zhang W. Hum Mol Genet. 2014;23:5893–5905. doi: 10.1093/hmg/ddu313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z, Zheng Y, Zhang X, Liu C, Joyce BT, Kibbe WA, Hou L, Zhang W. Hum Genet. 2016;135:223–232. doi: 10.1007/s00439-015-1628-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang XZW. Genetics. 2016 [Google Scholar]
- 56.Eadon MT, Wheeler HE, Stark AL, Zhang X, Moen EL, Delaney SM, Im HK, Cunningham PN, Zhang W, Dolan ME. Hum Mol Genet. 2013;22:4007–4020. doi: 10.1093/hmg/ddt240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Erler JT, Giaccia AJ. Cancer Res. 2006;66:10238–10241. doi: 10.1158/0008-5472.CAN-06-3197. [DOI] [PubMed] [Google Scholar]
- 58.Harris AL. Nature Reviews Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 59.Peinado H, Cano A. Nat Cell Biol. 2008;10:253–254. doi: 10.1038/ncb0308-253. [DOI] [PubMed] [Google Scholar]
- 60.Gusarova GA, Dada LA, Kelly AM, Brodie C, Witters LA, Chandel NS, Sznajder JI. Molecular & Cellular Biology. 2009;29:3455–3464. doi: 10.1128/MCB.00054-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singer BB, Scheffrahn I, Kammerer R, Suttorp N, Ergun S, Slevogt H. PLoS One. 2010;5:e8747. doi: 10.1371/journal.pone.0008747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hong KP, Shin MH, Yoon S, Ji GY, Moon YR, Lee OJ, Choi SY, Lee YM, Koo JH, Lee HC, Lee GK, Kim SR, Lee KH, Han HS, Choe KH, Lee KM, Hong JM, Kim SW, Yi JH, Ji HJ, Kim YB, Song HG. Biomaterials. 2015;67:32–41. doi: 10.1016/j.biomaterials.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 63.Oida Y, Gopalan B, Miyahara R, Branch CD, Chiao P, Chada S, Ramesh R. Mol Cancer Ther. 2007;6:1440–1449. doi: 10.1158/1535-7163.MCT-06-0374. [DOI] [PubMed] [Google Scholar]
- 64.Parker BC, Engels M, Annala M, Zhang W. J Pathol. 2014;232:4–15. doi: 10.1002/path.4297. [DOI] [PubMed] [Google Scholar]
- 65.Fidler MJ, Shersher DD, Borgia JA, Bonomi P. Ther Adv Med Oncol. 2012;4:51–60. doi: 10.1177/1758834011427576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M. Proc Natl Acad Sci U S A. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, Grosveld F, Philipsen S. PLoS One. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Su LJ, Chang CW, Wu YC, Chen KC, Lin CJ, Liang SC, Lin CH, Whang-Peng J, Hsu SL, Chen CH, Huang CYF. BMC Genomics. 2007;8:140. doi: 10.1186/1471-2164-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stearman RS, Dwyer-Nield L, Zerbe L, Blaine SA, Chan Z, Bunn PA, Jr, Johnson GL, Hirsch FR, Merrick DT, Franklin WA, Baron AE, Keith RL, Nemenoff RA, Malkinson AM, Geraci MW. Am J Pathol. 2005;167:1763–1775. doi: 10.1016/S0002-9440(10)61257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, Murphy SE, Yang P, Pesatori AC, Consonni D, Bertazzi PA, Wacholder S, Shih JH, Caporaso NE. Jen J, PLoS One. 2008;3:e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beer DG, Kardia SLR, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JMG, Iannettoni MD, Orringer MB, Hanash S. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 72.Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I. Proc Natl Acad Sci U S A. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamagata N, Shyr Y, Yanagisawa K, Edgerton M, Dang TP, Gonzalez A, Nadaf S, Larsen P, Roberts JR, Nesbitt JC, Jensen R, Levy S, Moore JH, Minna JD, Carbone DP. Clin Cancer Res. 2003;9:4695–4704. [PubMed] [Google Scholar]
- 74.Franchini DM, Schmitz KM, Petersen-Mahrt SK. Annu Rev Genet. 2012;46:419–441. doi: 10.1146/annurev-genet-110711-155451. [DOI] [PubMed] [Google Scholar]
- 75.International HapMap C. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 76.International HapMap C. Nature. 2005;437:1299–1320. [Google Scholar]
- 77.Marques E, Englund JI, Tervonen TA, Virkunen E, Laakso M, Myllynen M, Makela A, Ahvenainen M, Lepikhova T, Monni O, Hautaniemi S, Klefstrom J. Oncogene. 2016;35:1386–1398. doi: 10.1038/onc.2015.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ali SA, Justilien V, Jamieson L, Murray NR, Fields AP. Cancer Cell. 2016;29:367–378. doi: 10.1016/j.ccell.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fields AP, Frederick LA, Regala RP. Biochem Soc Trans. 2007;35:996–1000. doi: 10.1042/BST0350996. [DOI] [PubMed] [Google Scholar]
- 80.Suddason T, Gallagher E. Cell Death Differ. 2015;22:540–548. doi: 10.1038/cdd.2014.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lv XB, Liu CY, Wang Z, Sun YP, Xiong Y, Lei QY, Guan KL. EMBO Rep. 2015;16:975–985. doi: 10.15252/embr.201439951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goto T, Davidson L, Asashima M, Keller R. Curr Biol. 2005;15:787–793. doi: 10.1016/j.cub.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 83.Sipes NS, Feng Y, Guo F, Lee HO, Chou FS, Cheng J, Mulloy J, Zheng Y. J Biol Chem. 2011;286:36469–36477. doi: 10.1074/jbc.M111.283176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oh SY, Lee SJ, Jung YH, Lee HJ, Han HJ. Cell Death Dis. 2015;6:e1750. doi: 10.1038/cddis.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yeh CH, Chang CK, Cheng MF, Lin HJ, Cheng JT. Biochem Biophys Res Commun. 2009;382:292–297. doi: 10.1016/j.bbrc.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 86.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, Choi H, El Rayes T, Ryu S, Troeger J, Schwabe RF, Vahdat LT, Altorki NK, Mittal V, Gao D. Nature. 2015;527:472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.