

Summary
Skin microbiota can impact allergic and autoimmune responses, wound healing and anti-microbial defense. We investigated the role of skin microbiota in cutaneous leishmaniasis and found that human patients infected with Leishmania braziliensis develop dysbiotic skin microbiota, characterized by increases in the abundance of Staphylococcus and/or Streptococcus. Mice infected with L. major exhibit similar changes depending upon disease severity. Importantly, this dysbiosis is not limited to the lesion site, but is transmissible to normal skin distant from the infection site, and to skin from co-housed naïve mice. This observation allowed us to test whether a preexisting dysbiotic skin microbiota influences disease, and we found that challenging dysbiotic naïve mice with L. major or testing for contact hypersensitivity results in exacerbated skin inflammatory responses. These findings demonstrate that a dysbiotic skin microbiota is not only a consequence of tissue stress, but also enhances inflammation, which has implications for many inflammatory cutaneous diseases.
Keywords: Leishmania, microbiota, cutaneous inflammation, dysbiosis
eTOC Blurb
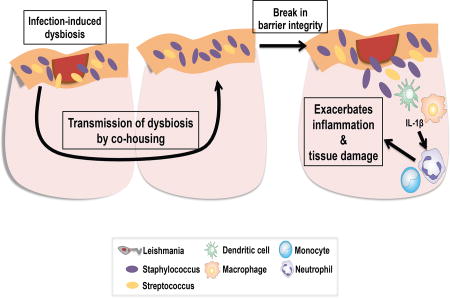
The role of skin commensal microbes in dermal cellular responses is largely unknown. Gimblet et al. investigate the role of the skin microbiota during cutaneous leishmaniasis. Leishmania infection creates a dysbiotic skin microbiota that is transmissible to naïve skin. Additionally, dysbiosis prior to infection or injury exacerbates skin inflammation.
Introduction
The skin is a barrier and the body’s first line of defense against injury and infection. It also hosts commensal populations of bacteria, fungi and viruses that may influence wound healing, the immune response to infection, and inflammatory responses that occur in chronic diseases (Canesso et al., 2014; Grice et al., 2010; Naik et al., 2012). Though there are strong associations between certain human diseases and changes in the skin microbiota (Kong et al., 2012; Loesche et al., 2016; Oh et al., 2013), the consequences of such changes are unclear, including the role of skin commensal microbes in modulating dermal cellular responses. Animal models in which microbial communities can be manipulated are essential to determine whether these changes influence the outcome of disease.
Cutaneous leishmaniasis is caused by intracellular protozoan parasites and is characterized by a spectrum of clinical manifestations, ranging from self-healing single lesions to chronic, and in some cases metastatic, lesions (Scott and Novais, 2016). The factors responsible for chronic disease in leishmaniasis are still being defined, although it is clear that some of the most severe forms of the disease are not caused by uncontrolled parasite replication, but rather an exaggerated immune response leading to excessive inflammation (Antonelli et al., 2005; Lopez Kostka et al., 2009; Santos Cda et al., 2013; Gonzalez-Lombana et al., 2013; Novais et al., 2013; Crosby et al., 2014). Unfortunately, there is no vaccine for leishmaniasis and drug treatment is often ineffective, which provides the impetus for better understanding the factors that drive the destructive inflammatory responses. Some of these severe forms of disease can be mimicked in mice, which can develop healing or non-healing disease following L. major infection depending upon whether a dominant Th1 or Th2 response develops (Scott and Novais, 2016). Less well understood is the role the skin microbiota plays in cutaneous leishmaniasis. Although it has been reported that the course of infection in germ free mice differs from conventional mice (de Oliveira et al., 1999; Naik et al., 2012; Oliveira et al., 2005), how the skin microbiota changes in patients and conventional mice, and whether such changes influence disease is less clear.
In this study, we found that infection with leishmania parasites causes a decrease in bacterial diversity in the skin that is characterized by communities dominated by Staphylococcus spp. and/or Streptococcus spp in both humans and mice. We hypothesized that disease-associated shifts in the skin microbiota (“dysbiosis”) contribute to lesion pathology and dermal cellular responses, including immune and inflammatory responses in L. major infection. To test this we utilized a mouse model of cutaneous leishmaniasis, and found that infection with L. major changed the skin microbiota in a manner dependent on disease severity. Leishmania-induced dysbiosis was not confined to the site of infection, but occurred globally on the skin of infected mice, and moreover, was transferred to uninfected co-housed mice. Colonization of skin with Staphylococcus xylosus isolated from the dysbiotic mice increased inflammatory responses in a contact hypersensitivity model, although not in normal skin, indicating that dysbiosis might exacerbate disease. Dysbiotic microbiota, when transferred to naïve mice prior to leishmania infection, increased disease pathology compared to control animals. Taken together these results indicate that the skin microbiota influences the inflammatory response in leishmaniasis and other inflammatory skin conditions. This work has significant implications for the treatment of cutaneous leishmaniasis and other skin diseases, and highlights the potential of the skin microbiota as a therapeutic target.
Results
Characterization of microbiota colonizing human leishmaniasis lesions and skin
Dysbiosis in skin microbiota is often associated with inflammation and disease (Grice et al., 2010; Kobayashi et al., 2015; Kong et al., 2012; Oh et al., 2013), suggesting that cutaneous lesions in leishmaniasis might also exhibit changes in the skin-residing bacterial communities. To test this, we analyzed the microbiota of 44 patients infected with L. braziliensis (72.7% male, 27.3% female, median age, 27 years old), with lesions present at various body sites (Supplemental Table 1). We collected 2–3 skin swabs for each patient including the lesion, adjacent skin near the lesion, and unaffected contralateral skin of the same body site as the lesion (Figure 1A). Taxanomic composition of unaffected contralateral skin fell within the normal range of what has been previously observed of the healthy skin microbiome and colonizing microbiota was assessed with respect to gender and no significant differences were found (Meisel et al., 2016; Supplemental Figure 1A). Bacterial diversity was significantly lower in lesions compared to unaffected contralateral skin and adjacent skin sites, as measured by the observed species-level operational taxonomic units (OTUs) and Shannon Diversity indices (Figure 1B).
Figure 1. Lesions from cutaneous leishmaniasis patients also have a dysbiotic skin microbiota.

(A) Swabs were collected from the lesion, nearby adjacent skin, and contralateral skin sites for 16S rRNA analysis. (B) Bacterial diversity was assessed by the number of observed species-level OTUs and Shannon Index. (C) Bar charts represent intragroup mean Bray-Curtis dissimilarity between each skin site. (D) PCoA values for weighted UniFrac analysis were plotted and colored based on the Dirichilet multinomial cluster assignment. (E) Stacked bar charts represent the proportion of the top 10 taxa present in each Dirichilet cluster. Swabs were collected from an n = 44 patients. **, p < 0.01; ***, p < 0.001; ****, p , 0.0001. See also Figure S1, Table S1, and Table S2.
Interestingly, the skin microbiota on the adjacent skin sites appeared more similar in composition to the lesions than to the contralateral skin (Supplemental Figure 1A). To quantify the similarity between each site where specimens were collected, we used the Bray Curtis dissimilarity metric of shared microbial community structure. We observed that lesion and adjacent skin shared greater microbial community structure compared to contralateral and adjacent skin (Figure 1C). This data suggests that microbiota colonizing the lesion is shared with adjacent skin sites, which may have implications in the immune responses at those sites.
We then applied a Dirichlet multinomial mixture model-based approach to assign the lesions to different community types (CTs) based on their taxonomic composition. Lesions clustered into 3 CTs (Figure 1D and Supplemental Figure 1B) with distinct bacterial compositions. The top discriminating taxa in CT1 was Staphylococcus aureus, CT2 displayed a heterogeneous composition with no dominating taxa, and CT3 was dominated by an unclassified species of Streptococcus (Figure 1E and Supplemental Table 2). These results suggest that cutaneous leishmaniasis lesions are colonized with microbiota similar to other cutaneous ulcers (Kong et al., 2012; Oh et al., 2013; Loesche et al., 2016), but display less heterogeneity of the colonizing microbiota, which is driven primarily by proportions of Staphylococcus aureus and Streptococcus spp. in this cohort. Interestingly, neither bacteria were associated with larger lesion sizes (Supplementary Figure 1C), but lesion size may not be a good predictor of disease severity or outcome. Additional epidemiologic studies may be needed to further evaluate the influence of the skin microbiota in cutaneous leishmaniasis, yet these results clearly demonstrate that infection with leishmania alters the skin microbiota, creating several types of dysbiosis.
L. major infection induces changes to the skin microbiota in mouse models
Since the influence on disease of a dysbiosis is difficult to evaluate in humans, we employed a mouse model of leishmaniasis to assess the role dysbiosis might play in cutaneous leishmaniasis. C57BL/6 mice were infected in the ear with L. major parasites, which led to the development of a lesion that resolved by 12 weeks post-infection (Figure 2A–B). Prior to infection, and at 6 and 12 weeks post-infection, swabs were collected from the ventral and dorsal ear skin and sequencing of the 16S ribosomal RNA gene was employed to assess skin microbial diversity and composition. Alpha diversity, as measured by the number of observed species-level OTUs and Shannon Diversity indices, decreased at 6 weeks post-infection, but returned to pre-infection levels upon lesion resolution (Figure 2C–D). This shift in alpha diversity was paralleled by a significant increase in the relative abundance of Staphylococcus spp. after lesion development that returned to pre-infection levels once the lesions resolved (Figure 2E). Speciation of the Staphylococcus species associated with L. major infection was performed by biochemical typing in an automated system (Microscan, Beckman Coulter, Irvine, CA, USA) and isolates were identified as S. xylosus, a common commensal bacteria found on mouse skin (Nagase et al., 2002). Isolates were then confirmed as S. xylosus by MALDI-TOF mass spectrometry using the Bruker MALDI Biotyper System (Beckman Coulter, Irvine, CA, USA). The MALDI-TOF identification score for all isolates tested was >2.299 which indicates a secure genus identification and a highly probable species identification. Since infections can often lead to changes in the intestinal microbiota (Kamdar et al., 2016; Lozupone et al., 2013), we also analyzed the fecal microbiota of infected mice, but found no significant changes in the fecal bacterial populations throughout the course of infection with L. major (Figure 2F), demonstrating that dysbiosis caused by infection is localized to the skin.
Figure 2. L. major infection alters the skin microbiota.

C57BL/6 mice were intradermally infected in the ear with 2 × 106 L. major parasites. (A) Lesion size and (B) pathology were assessed over 12 weeks of infection. Swabs were collected from the ear at 0, 6, and 12 weeks post-infection and bacterial diversity was assessed by (C) number of observed species-level OTUs and (D) Shannon Index. Stacked bar charts represent the proportion of the top 10 taxa present (E) from ear swabs and (F) from fecal pellets at 0, 6, and 12 weeks post-infection. Each column represents the proportion of taxa for an individual mouse. Data represent two independent experiments (n = 1 skin swab each from 15 mice and n = 1 fecal pellet each from 10 mice). *, p < 0.05; ***, p < 0.001.
L. major induced dysbiosis differs depending on the severity of the disease
Inflammatory responses induced by a variety of skin insults lead to changes in the skin microbiota (Grice et al., 2010; Gontcharova et al., 2010; Kong et al., 2012; Oh et al., 2013; Loesche et al., 2016), but whether the magnitude of the insult alters the nature or degree of the dysbiosis is not known. To address this we compared the microbiota from L. major infected C57BL/6 mice that resolve their infection and BALB/c mice that develop severely ulcerated non-healing lesions (Figure 3A–B) (Scott and Novais, 2016). Similar to C57BL/6 mice, BALB/c mice had significantly lower alpha diversity at 6 weeks post-infection (Figure 3C). However, in contrast to the dominance of Staphylococcus spp. found on lesions of C57BL/6 mice, BALB/c mice had a dominance of Streptococcus spp. at 6 weeks post-infection, (Figure 3D). To rule out the possibility that the increase in Streptococcus in non-healing BALB/c mice was due to differences in the mouse strain, we depleted IL-12 in C57BL/6 mice, which leads to non-healing lesions similar to those seen in BALB/c mice (Heinzel et al., 1989; Scharton-Kersten et al., 1995). As expected, anti-IL-12 mAb treated mice developed large non-healing lesions (Figure 3E–F). We first analyzed the skin microbiota of naïve mice treated with anti-IL-12 prior to infection and found that treatment does not significantly alter the proportions of Staphylococcus spp. or Streptococcus spp. (Supplemental Figure 2A). However after 2 weeks of L. major infection, Staphylococcus spp. made up a high proportion of the skin microbiota in the anti-IL-12 treated mice, while remaining low in the isotype treated mice until 4 weeks post-infection (Figure 3G and Supplemental Figure 2B). At 6 weeks post-infection, the relative abundance of Streptococcus spp. remained less than 1% of the total population in control mice, but it increased significantly in anti-IL-12 treated mice to >50% relative abundance (Figure 3G and Supplemental Figure 2B), further demonstrating that Streptococcus spp. are associated with more severely ulcerated lesions. Taken together, our data suggest that L. major infection elicits severity-dependent changes in the skin microbiota.
Figure 3. Skin microbiota alterations in L. major infection are dependent on disease severity.

C57BL/6 and BALB/c mice were intradermally infected with L. major parasites. Lesional severity was assessed by (A) ear thickness and (B) a pathology score over the course of infection. Swabs for sequencing of 16S rRNA genes were collected from the lesions at 0 and 6 weeks post-infection. (C) Alpha diversity was assessed by Shannon Index. (D) Stacked bar charts represent the proportion of the top 10 taxa present in each sample. Data are representative of two independent experiments (n = 1 skin swab each from 10 mice in each group). C57BL/6 mice were treated with an isotype or anti-IL-12 mAb and intradermally infected in the ear with L. major parasites. Lesional severity was assessed by (E) ear thickness and (F) a pathology score over the course of infection. Anti-IL-12 mAb treated mice were euthanized at 6 weeks post-infection due to severe disease. (G) Swabs were collected from the lesions at 2, 4 and 6 weeks post-infection and proportions of Staphylococcus and Streptococcus were assessed. Data are representative of two independent experiments (n = 1 skin swab each from 10 mice in each group). *, p < 0.05; **, p < 0.01. See also Figure S2.
S. xylosus mediated inflammation is dependent on tissue damage
To determine if the dysbiosis caused by L. major infection would influence skin inflammatory responses, we topically associated naïve mice with S. xylosus (Figure 4A). One week following colonization with S. xylosus mice exhibited a significantly higher relative and absolute abundance of Staphylococcus spp. compared with naïve mice by culture-independent (Figure 4B) and culture-dependent assays (Figure 4C).
Figure 4. Staphylococcus xylosus isolated from L. major lesions causes inflammation only when injected intradermally.

(A) C57BL/6 mice were topically colonized with 108-109 S. xylosus every other day for a total of 4 applications; naïve mice were unassociated. (B) Prior to and 14 days post colonization, swabs were collected to analyze the proportion of Staphylococcus. (C) Ear lysates from naïve and S. xylosus colonized mice were cultured on mannitol salt agar plates and colony forming units were counted after overnight incubation at 37°C. (D) Ear thickness was assessed in naïve and colonized mice. (E) Flow cytometry analysis was performed for the frequency of CD4+, CD8+, and CD11b+, IL-1β+, and Ly6G+ cells in the ears of naïve or colonized mice 14 days post-association. Cells were pregated on live, singlet, CD45+ cells. Data are representative of two independent experiments (n = 1 ear tissue each from 4 mice in each group). C57BL/6 mice were topically colonized or intradermally infected in the ear with S. xylosus. Fourteen days later, skin was harvested and mRNA expression was assessed for (E) cytokine and (F) chemokine genes. Data are representative of one experiment (n = 1 ear tissue each from 5 mice in each group). ns = not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p , 0.0001. See also Figure S3.
Interestingly, S. xylosus colonization did not influence ear thickness (Figure 4D), the frequency or total cell numbers of CD4+ T cells, CD8+ T cells, CD11b+ myeloid cells, IL-1β production, Ly6G+ neutrophils (Figure 4E and Supplemental Figure 3A–E), or T cell cytokine production (Supplemental Figure 3F–G). To determine if S. xylosus incites inflammation upon breach of the skin barrier, we injected mice intradermally with S. xylosus and analyzed the inflammatory response in the skin. These mice had significantly higher expression of Il17, Tnfa, Il1b, Cxcl1, and Ccl2 compared with either naïve or colonized mice (Figure 4F–G), suggesting that S. xylosus might contribute to skin inflammation when the skin barrier is compromised.
While skin colonized with S. xylosus appeared immunologically normal, based on the results above we hypothesized that the response to damage might differ between normal and dysbiotic skin. We tested this idea using a model of contact hypersensitivity in which sensitizing and challenging the skin with a known skin irritant, dinitrofluorobenzene (DNFB), increases transepidermal water loss, an indication of skin barrier dysfunction and inflammation (Figure 5A). Naïve C57BL/6 mice were colonized with S. xylosus prior to sensitization with DNFB (Figure 5B). DNFB challenge resulted in a significant increase in neutrophils (CD11b+ Ly6G+) and expression of pro-IL-1β from myeloid cells (Figure 5C–D). A similar immune response was observed when mice were colonized with S. xylosus during the challenge phase of DNFB treatment (Supplemental Figure 4A). Since IL-17 and IL-1 can both lead to an increase in neutrophil recruitment, we investigated whether these cytokines played a role in the increase of neutrophils in S. xylosus treated mice. Mice colonized with S. xylosus were treated with an isotype control mAb, anti-IL-17A mAb, or anti-IL-1R mAb prior to DNFB challenge (Figure 5E), and neutralizing IL-17 or IL-1 decreased neutrophil recruitment (Figure 5F). Thus, it appears that a commensal such as S. xylosus can induce IL-17 and IL-1 expression in conditions of tissue stress and damage, leading to increased inflammation.
Figure 5. S. xylosus colonization exacerbates skin inflammation during contact hypersensitivity.

(A) C57BL/6 mice were sensitized with DNFB or vehicle control on the belly and challenged with DNFB or vehicle 5 days later. Transepidermal water loss was measured on ear skin of vehicle control and DNFB treated mice. (B) C57BL/6 mice were topically associated with 108-109 S. xylosus every other day for a total of 4 applications and control C57BL/6 mice were left unassociated. The next day, control and S. xylosus associated mice were sensitized on the belly with DNFB. 5 days later, control and S. xylosus associated mice were challenged with DNFB. Representative flow cytometry plots and graphs depict the expression of (C) CD11b+ Ly6G+ cells and (D) CD11b+ IL-1β+ cells. (E) C57BL/6 mice were topically associated with 108-109 S. xylosus every other day for a total of 4 applications and then treated with isotype, anti-IL-17, or anti-IL-1R mAbs prior to sensitization and challenge with DNFB. (F) Graphs depict the expression of CD11b+ Ly6G+ cells in the skin of treated mice. All data are representative of two independent experiments (n = 1 ear tissue each from 5 mice in each group). *, p < 0.05; **, p < 0.01; ***, p < 0.001. See also Figure S4.
To determine if colonization with Streptococcus spp. might have a similar effect, we isolated Streptococcus from L. major infected mice that had been treated with anti-IL-12 mAb. The immune responses in mice sensitized and challenged with DNFB colonized with Streptococcus was unchanged (Supplemental Figure 4B). However, we were unable to achieve stable colonization with the streptococcal isolate (Supplemental Figure 4C), suggesting that this particular lesion-associated Streptococcus isolate requires additional as yet undefined nutrients or other conditions to colonize normal skin. We hypothesized that the Streptococcus isolate would establish a better colonization on lesional skin, thus we infected mice with L. major, allowed the lesion to develop, and then colonized with the Streptococcus isolate. Indeed, Streptococcus colonized the lesion better than naïve skin (Supplemental Figure 4D). Interestingly, colonization with the Streptococcus isolate did not exacerbate lesion development or the inflammatory response in the skin (Supplemental Figure 4E–F).
L. major-induced dysbiosis is transmissible to uninfected skin
The observation that the lesional microbiota of human cutaneous leishmaniasis extends to adjacent seemingly normal skin sites prompted us to ask if the same was true in the mouse model of L. major infection. To answer this question, we compared the bacterial composition at the lesion site (infected ear) and the contralateral ear of infected mice. As expected, the infected ear was dominated by Staphylococcus spp. at the peak of infection. Interestingly, the contralateral ear also had a high proportion of Staphylococcus spp., despite the absence of infection (Figure 6A). We also observed higher bacterial loads on the infected and contralateral ears when compared to naïve skin (Figure 6B). These data demonstrate that in the mouse model, the dysbiotic microbiota caused by L. major infection is transmissible to the non-inflamed, contralateral ear.
Figure 6. L. major induced dysbiosis is transmissible to uninfected skin.

(A) C57BL/6 mice were intradermally infected with L. major and swabs were collected from the infected and contralateral ears at 6 weeks post-infected for 16S rRNA gene analysis. Stacked bar charts represent the proportion of the top 10 taxa present in each sample. Data are representative of three independent experiments (n = 1 swab of each ear from 15 mice). (B) Swabs from naïve or L. major infected C57BL/6 mice were cultured on mannitol salt agar plates and CFUs were counted to determine bacteria burden. Data are representative of 1 experiment (For naïve group, n = 1 swab from the ear of 10 mice; for infected and contralateral ears, n = 1 swab of each ear from 12 mice). (C) Naïve C57BL/6 mice were co-housed with L. major infected mice for 6 weeks, while control naïve mice were housed separately. Swabs were collected from co-housed naïve and control naïve mice. Stacked bar charts represent the proportion of taxa present in each sample. Data are representative of two independent experiments (For infected group, n = 1 swab of each ear from 15 mice; for co-housed naïve, n = 1 swab of one ear from 10 mice; for control naïve, n = 1 swab of one ear from 5 mice). (D) Bar graphs depict ear thickness of control and co-housed naïve mice. (E) Cells were isolated from the ears of co-housed naïve mice and control naïve mice to assess for CD4+, CD8+, and CD11b+, IL-1β+, and Ly6G+ cells by flow cytometry. Data are representative of one experiment (Co-housed naïve, n = 1 ear tissue each from 4 mice; control naïve, n = 1 ear tissue each from 5 mice). ns = not significant; **, p < 0.01; ***, p < 0.001. See also Figure S5.
A dysbiotic intestinal microbiota is often transmissible by simply co-housing mice (Elinav et al., 2011; Zenewicz et al., 2013). Whether transmission of the skin microbiota also occurs is less clear, although co-habiting families may share their skin microbiota (Song et al., 2013). To directly address this issue we tested if naïve mice co-housed with L. major infected mice might acquire their dysbiotic microbiota. C57BL/6 mice were infected with L. major and co-housed with naïve mice for 6 weeks, while a group of control naïve mice were housed separately. Similar to the infected and contralateral ears, the skin of the co-housed naïve mice also acquired a high abundance of Staphylococcus spp., while the control naïve mice maintained a diverse population of bacteria (Figure 6C). Similar to what we observed with S. xylosus colonized mice, the ear thickness and immune response in co-housed naïve mice were not altered by dysbiosis (Figure 6D–E and Supplemental Figure 5). Our data demonstrate that the dysbiotic skin microbiota caused by L. major infection is transmissible to naïve mice without causing inflammation. This model allows us to assess the consequences of dysbiosis in inflammatory responses occurring in the skin.
L. major-induced dysbiosis exacerbates disease during inflammation and infection
While we and others have shown that colonizing mice with a single organism at high levels can alter immune responses (Figure 5) (Naik et al., 2012; Naik et al., 2015), whether a naturally transmitted dysbiosis would alter immune skin immune responses has not been tested. To assess this, we co-housed naïve mice with L. major infected mice for 6 weeks to create “naïve” dysbiotic mice. Control mice were housed separately and never exposed to L. major infected mice. We then compared the contact hypersensitivity responses of both groups of mice to DNFB. Dysbiotic co-housed mice had significantly more neutrophils and pro-IL-1β production in the skin than control mice (Figure 7A–B), similar to mice colonized with high numbers of bacteria.
Figure 7. Dysbiosis exacerbates inflammation during DNFB treatment and L. major infection.

Naïve C57BL/6 mice acquired dysbiotic microbiota after co-housing with L. major infected mice for 6 weeks. Control and dysbiotic mice were then sensitized and challenged with DNFB. Representative flow cytometry plots and graphs of skin cells depict the expression of (A) CD11b+ Ly6G+ cells and (B) CD11b+ IL-1β+ cells. Control and dysbiotic mice were intradermally infected with L. major parasites and the cells from the lesions were collected at 5 weeks post-infection. Representative flow cytometry plots and graphs of skin cells depict the expression of (C) CD11b+ Ly6G+ cells and (D) CD11b+ IL-1β+ cells. (E) A pathology score was used to assess disease severity over 5 weeks post-infection. (F) Representative ear skin sections stained with hemotoxylin and eosin of L. major infected control and dysbiotic mice. (G) Parasite burdens were assessed using a limiting dilution assay after 5 weeks post-infection. Data are representative of two independent experiments (For dysbiotic group, n = 1 ear tissue each from 4 mice; for control group, n = 1 ear tissue each from 5 mice). ns = not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****.
Taken together, our results suggested that mice with dysbiotic skin might respond differently to infection with L. major when compared with normal mice. To determine if this was the case, naïve mice were co-housed with L. major infected mice for 6 weeks and then infected with L. major. At 5 weeks post-infection, we analyzed the inflammatory cells and cytokines in the lesions of control and dysbiotic mice. Similar to DNFB challenge, L. major infected skin had significantly more neutrophils and IL-1β in dysbiotic mice compared to control mice (Figure 7C–D). Furthermore, the dysbiotic mice had significantly greater lesion severity, characterized by increased skin ulceration, than control mice (Figure 7E–F) despite similar parasite burdens (Figure 7G). These findings demonstrate that the skin microbiota influences the magnitude of lesion severity following infection with L. major.
Discussion
Interactions between the immune system and the microbiota can be either beneficial or harmful, depending on the context (Gaboriau-Routhiau et al., 2009; Naik et al., 2012; Atarashi et al., 2013; Naik et al., 2015; Kobayashi et al., 2015). In our studies, we found that leishmania infections in humans and mice change the composition of the skin microbiota. The nature of the changes in mice differed depending on the severity of inflammation, with Staphylococcus spp. dominant in moderate lesions and Streptococcus spp. increasing in more severe lesions in mice infected with L. major. In humans, we found individuals with a dominance of Staphylococcus aureus, Streptococcus spp., or a mixture of both, although whether these distinct skin microbiota influences the outcome of disease is yet unknown. However, our studies in mice clearly suggest that further studies in patients are warranted.
Why dysbiosis occurs during cutaneous leishmaniasis, or in other inflammatory conditions, is unknown. Innate defenses, such as antimicrobial peptides (AMPs), can target certain bacteria and play a role in disrupting the microbiota in the intestine and in the skin (Cogen et al., 2010; Dorschner et al., 2001; Natsuga et al., 2016; Nizet et al., 2001; Salzman et al., 2010), and may also in part contribute to the dysbiosis caused by L. major infection. We found that infection with L. major causes changes in AMP expression in the skin (Supplemental Figure 6), and mice deficient in a cathelicidin-type antimicrobial peptide (CAMP) appear more susceptible to infection with L. amazonensis (Kulkarni et al., 2006). Whether this deficiency in CAMP causes changes in the skin microbiota remains to be determined, but these results in addition to our own findings suggest that AMPs in cutaneous leishmaniasis warrant further investigation. How AMPs might promote these changes is unclear, but virulence factors can make bacteria resistant to AMPs and both Staphylococcus spp. and Streptococcus spp. express genes that protect them from AMP killing (Kristian et al., 2005; Peschel et al., 1999; Peschel et al., 2001), potentially providing them with a survival advantage during L. major infection.
One difficulty in studying the microbiota is assessing how changes in the skin microbiota influence disease, since skin dysbiosis is the consequence of the inflammatory response in the skin. While transmissibility of dysbiotic microbiota has been demonstrated in the intestinal tract (Elinav et al., 2011; Zenewicz et al., 2013), our data demonstrates transmissibility of the skin microbiota in a murine model. In this study and previous studies, colonization with a single bacterial species enhanced skin inflammation (Naik et al., 2012). In our studies, enhanced inflammation was only observed when there was pre-existing tissue damage and inflammation. The differences between these studies is likely due to differences in bacterial species. Although mono-colonization will be essential for dissecting how particular bacteria alter immune responses, it will not replicate the complex changes that might be associated with a naturally occurring dysbiosis. Our ability to generate a mouse with dysbiotic skin microbiota overcomes this issue, and has allowed us to demonstrate that a naturally acquired dysbiosis promotes increased inflammatory responses, and in the case of cutaneous leishmaniasis increased disease. It is not clear how this transmission occurs, although consistent with our results, evidence from human studies indicates that the environment influences the skin microbiota (Song et al., 2013), and L. major infections in mice may provide a model to study the mechanisms involved.
The findings from our mouse model of cutaneous leishmaniasis are similar to the dysbiosis that occurs during human cutaneous leishmaniasis. Interestingly, the different topological sites of our samples did not show any differences in the skin microbiota, although we only had a few samples from moist and sebaceous sites. Yet comparable to what has been reported by culture dependent and independent methods (Isaac-Marquez and Lezama-Davila, 2003; Sadeghian et al., 2011; Layegh et al., 2015; Salgado et al., 2016), our results demonstrated that Staphylococcus aureus and an unclassified species of Streptococcus are highly abundant on lesional skin. This dysbiosis was also present on skin sites adjacent to the lesion. However unlike our mouse model, the dysbiotic skin microbiota did not appear to be transmissible to contralateral skin sites. It is not yet clear why the dysbiosis is confined to the lesional and adjacent skin sites in human cutaneous leishmaniasis but it is likely to be due to differences in grooming and environmental conditions between mice and humans. However, the similarities in the dysbiotic microbiota between the mouse model and human cutaneous leishmaniasis demonstrate the utility of our model system to study the role of skin microbiota during leishmania infections.
One of our findings was that skin dysbiosis does not cause immunologic changes in the skin or disease by itself, nor did topical colonization with S. xylosus. However, in mice where tissue stress is induced by contact hypersensitivity to DNFB, S. xylosus exacerbated the inflammatory response, assessed by increased recruitment of neutrophils and upregulated expression of IL-1β. These results are consistent with other studies showing that mice with barrier defects allow Staphylococcus to penetrate the epidermal barrier and subsequently increase cytokine expression in the skin (Nakatsuji et al., 2016). In some situations the cytokine production may be protective, such as during a fungal infection (Naik et al., 2015). However, in cutaneous leishmaniasis, neutrophils and IL-1β are associated with increased pathology rather than the restriction of parasites (Charmoy et al., 2016; Fernandez-Figueroa et al., 2012; Gimblet et al., 2015; Gonzalez-Lombana et al., 2013; Novais et al., 2014; Voronov et al., 2010). Thus, we hypothesize that L. major infection disturbs skin barrier integrity while simultaneously inducing a dysbiosis in the skin microbiota, which taken together leads to the increased recruitment of neutrophils and IL-1β recruiting cells to the skin, and causes increased lesion severity.
These results raise the obvious question of what role systemic or topical antibiotics might play in moderating inflammatory responses associated with leishmaniasis (Grice, 2014). As previous studies with germ-free mice indicate that commensal bacteria may contribute to lesion severity in cutaneous leishmaniasis (de Oliveira et al., 1999; Naik et al., 2012; Oliveira et al., 2005), and our studies demonstrate that dysbiosis exacerbates disease, it is reasonable to predict that antibiotic treatment might be beneficial in leishmaniaisis. While we have been unsuccessful in moderating disease in mice by antibiotic treatment, there are examples of antibiotic therapy being protective in some cutaneous leishmaniasis patients (Aguiar et al., 2010; Ben Salah et al., 2013; Kim et al., 2009; Krolewiecki et al., 2002). However, there are other studies that find no effect of antibiotic treatment (Iraji and Sadeghinia, 2005; Neva et al., 1997), and moreover when such treatment shows a positive outcome the mechanism involved is not clear. Given the different outcomes of studies looking at antibiotic treatment, and taken together with our results, it appears that the role of antibiotics in treatment needs further investigation.
In summary, our findings indicate that the skin microbiota not only changes during leishmania infection, but when transmitted to naïve mice can enhance disease to leishmania. These findings have obvious consequences when considering how to limit disease severity in cutaneous leishmaniasis. Moreover, since we find that the dominant bacteria associated with a leishmania-induced dysbiosis differs depending upon the severity of disease in mice, further epidemiologic studies with patients to determine the consequences of differing types of dysbiosis are warranted. Finally, we found that dysbiotic skin microbiota can be transmitted to conventional naïve mice, which provides a model to define how and when dysbiosis might influence control of other infections, autoimmune diseases and wound healing.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elizabeth Grice (egrice@upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Female C57BL/6 and BALB/c mice 6–8 weeks old were purchased from the Charles River Laboratories (Durham, NC). All mice were maintained in specific pathogen-free facilities at the University of Pennsylvania. The mice were under the care of a veterinarian and were healthy, immunocompetent, and required no drugs or treatment prior to experimentation. Cages were changed twice per week with glove changes between handling each cage. Unless stated otherwise, a minimum of 5 mice were used based on variability observed in previous experiments with L. major. Mice were randomly assigned to experimental groups by investigators. Investigators were not blinded in this study. Prior to infection, mice were anesthetized using a ketamine and xylazine mixture and monitored until the mice were fully awake. At the end of the experiments, mice were humanely euthanized using carbon dioxide inhalation. All procedures involving mice were performed in accordance with the guidelines of the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC).
Human Cutaneous Leishmaniasis Subjects
All cutaneous leishmaniasis patients were seen at the health post in Corte de Pedra, Bahia, Brazil, which is a well-known area of L. braziliensis transmission. The criteria for diagnosis were a clinical picture characteristic of cutaneous leishmaniasis in conjunction with documentation of DNA of L. braziliensis by PCR, or parasite isolation or documentation of amastigotes in lesion biopsies by histopathology. In all cases, swabs were collected before therapy. There were 44 patients, both male (72.7%) and female (27.3%), with a median age of 27 years. This study was conducted according to the principles specified in the Declaration of Helsinki and under local ethical guidelines (Ethical Committee of the Maternidade Climerio de Oliveira, Salvador, Bahia, Brazil; and the University of Pennsylvania Institutional Review Board). This study was approved by the Ethical Committee of the Federal University of Bahia (Salvador, Bahia, Brazil)(010/10) and the University of Pennsylvania IRB (Philadelphia, PA)(813390). All patients provided written informed consent for the collection of samples and subsequent analysis.
Parasite and Bacterial Cultures
L. major (WHO/MHOM/IL/80/Friedlin wild-type L. major) promastigotes were grown to the stationary phase in Schneider’s Drosophila medium (GIBCO BRL, Grand Island, NY, USA) supplemented with 20% heat-inactivated fetal bovine serum (FBS, Invitrogen USA), 2 mM L-glutamine, 100 U of penicillin and 100 mg of streptomycin per mL. Infective-stage promastigotes (metacyclics) were isolated from 4–5 day old (L. major) stationary culture by density gradient separation by Ficoll (Sigma) (Spath and Beverley, 2001). An isolate of S. xylosus and alpha-hemolytic Streptococcus was cultured from the ears of L. major infected mice. For topical associations and infections, the bacteria was cultured in Brain heart infusion (BHI) media (Remel, Lenexa, KS, USA) shaking for 12 hours at 37°C.
METHOD DETAILS
Leishmania infection and in vivo antibody depletions
Mice were inoculated intradermally in the ear with 10 µL of PBS containing 2 × 106 L. major metacyclic promastigotes. Lesion development was measured weekly by ear thickness with a digital caliper (Fisher Scientific). Mice were also assessed for pathology, using the following score system: no lesion (0), swelling/redness (1), deformation of the ear pinna (2), ulceration (3), partial tissue loss (4), and total tissue loss (5). Parasite burden in lesion tissues was assessed using a limiting dilution assay as previously described (Zaph et al., 2004). In specified experiments, mice were treated with 500µg of anti-IL-12 mAb (BioXcell, clone R1‒5D9, RRID:AB_1107700) one day prior to infection and then twice per week for the duration of the experiment. Equal amounts of an isotype control, Rat IgG2a (BioXcell, clone 2A3, RRID:AB_1107769) was given in all experiments using in vivo antibody treatments.
Bacterial topical associations, intradermal infections, and CFU quantification
For topical associations,108-109 CFUs of bacteria were applied to the entire mouse body using sterile cotton swabs, every other day for a total of 4 times. For intradermal infections, mice were inoculated with 10µL of 108-109 CFU bacteria/mL culture. For CFU quantification, the dermal sheets of the mouse ears were homogenized in 1mL of PBS using a tissue homogenizer (FastPrep-24, MP Biomedical) and plated on tryptic soy blood agar (Remel) or mannitol salt agar (Acumedia) in serial dilutions. Plates were incubated overnight at 37°C and CFUs were counted t he next day.
Contact hypersensitivity and antibody treatments
For sensitization, 1-fluoro-2,4-dinitrobenzene (DNFB) (Sigma-Aldrich) was added to a 3:1 acetone:olive oil dissolvent to get a final concentration of 0.5%. Mice were treated on the belly with 30µL of the mixture. During the challenge phase, mice were treated with 20µL of 0.3% DNFB (in 3:1 acetone:olive oil) on the ear once a day, for a total of 3 days. The mice were euthanized 24 hours after the last challenge. In some experiments, mice were treated with 500µg of a Rat IgG2a isotype monoclonal antibody (BioXcell, clone 2A3, RRID:AB_1107769), an anti-mouse IL-17A monoclonal antibody (BioXcell, clone 17F3, RRID:AB_10950102), or an anti-mouse IL-1R monoclonal antibody (BioXcell, clone JAMA-147, RRID: AB_2661843), one day prior and one day after the first challenge with DNFB.
Preparation of dermal sheets
The dorsal and ventral sides of the mouse ear were split mechanically and placed dermis side down in a 24 wells plate in RPMI 1640 containing 0.25 mg/mL of Liberase TL (Roche, Diagnostics Corp.) and 10 µg/mL DNase I (Sigma-Aldrich). Ears were incubated for 90 min at 37° C in a 24-well plate. D ermal cell suspensions were prepared by dissociation on 40 µm cell strainer (Falcon) in PBS containing 0.05% BSA and 20 µM EDTA.
Antibodies and flow cytometry
Single cell suspensions from the ear were obtained as described above. For analysis of surface markers and intracellular cytokines, some cells were incubated for 4 h with 10 µg/mL of brefeldin A, 50 ng/mL of PMA and 500 ng/mL ionomycin (Sigma-Aldrich). Before staining, cells were incubated with anti-mouse CD16/CD32 mouse Fc block (Thermo Fisher Scientific, RRID:AB_467135) and 10% rat-IgG in PBS containing 0.1% BSA. Cells were stained for dead cells with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Molecular Probes) and surface markers (CD4 [BioLegend, clone RM4-5, RRID:AB_11219790], CD8β [BioLegend, clone YTS156.7.7, RRID:AB_2260149], TCRγδ [BD Biosciences, clone GL3, RRID: AB_2661844], CD45 [Thermo Fisher Scientific, clone 30-F11, RRID:AB_493714], Ly6G [Thermo Fisher Scientific, clone 1A8-Ly6g, RRID:AB_2637123], CD11b [BioLegend, clone M1/70, RRID:AB_11125575]) followed by fixation with 2% of formaldehyde and permeablization with 0.2% saponin/PBS. Intracellular cytokine staining was performed for pro-IL-1β (Thermo Fisher Science, clone NJTEN3, RRID:AB_10670739), IL-17 (Thermo Fisher Scientific, clone eBio17B7, RRID:AB_763579), and IFN-γ (Thermo Fisher Scientific, clone XMG1.2, RRID:AB_1257211). The data were collected using LSRII flow cytometer (BD) and analyzed using FlowJo software (Tree Star).
RNA isolation, purification, and quantitative real-time PCR
Total RNA was extracted from ear tissue samples in 500µL of RLT lysis buffer (QIAgen). The sample was homogenized using a tissue homogenizer (FastPrep-24, MP Biomedical), and total RNA was extracted according to the recommendations of the manufacturer and further purified using the RNeasy Mini kit (QIAgen). RNA was reverse transcribed using high capacity RNA-to-cDNA Kit (Applied Biosystems). Real-time RT-PCR was performed on a ViiA™ 7 Real-Time PCR System (Applied Biosystems). Relative quantities of mRNA for several genes were determined using SYBR Green PCR Master Mix (Applied Biosystems) and by the comparative threshold cycle method, as described by the manufacturer. mRNA levels for each sample were normalized to the ribosomal protein S11 gene (RPS11). The primer sequences are reported in Supplemental Table 3.
Microbiota collection, sequencing, and analysis
Microbiota samples were collected from the ear of mice using a swab (Catch-all Sample Collection Swab, Epicentre) moistened in Yeast Cell Lysis Buffer (from MasterPure Yeast DNA Purification Kit; Epicentre). DNA was isolated from swab specimens using the PureLink Genomic DNA Mini Kit (Invitrogen) and amplification of the 16S–V4 region for the murine samples, and 16S-V1-V3 region for the human samples, was performed as previously described (Hannigan et al., 2014; Meisel et al., 2016). Sequencing of 16S rRNA amplicons was performed at the Penn Next Generation Sequencing Core using the Illumina MiSeq platform with 150 bp paired-end ‘V4’ chemistry for murine samples and with 300 bp paired-end ‘V1-V3’ chemistry for the human samples. For the fecal samples, DNA was isolated using the PowerSoil DNA Isolation Kit (Mo Bio) and sequencing of the 16S rRNA amplicons was conducted using 250bp paired-end ‘V4’ chemistry with dual index primers (Kozich et al., 2013).
QUANTIFICATION AND STATISTICAL ANALYSIS
Pre-processing and community characterization of 16S rRNA sequence data
Sequence pre-processing followed methods previously described (Hannigan et al., 2014), but modified by subsampling at 5000 sequences per sample for murine samples, and at 1000 sequences per sample for human samples. QIIME 1.8.0 (Caporaso et al., 2010) was used for initial stages of sequence analysis. Sequences were clustered into OTUs (operational taxonomic units, a proxy for ‘species’) using UCLUST (Edgar, 2010) at 97% sequence similarity. Bacterial diversity was calculated using the following alpha diversity indices: Shannon diversity index and the number of observed OTUs. Relative abundance of bacteria was calculated based on taxonomic classification of sequences using the RDP classifier (Wang et al., 2007) at a confidence threshold of 0.8. Microbiota data was analyzed with the R statistical software environment (ww.r-project.org). Statistical significance was determined using two-sample Wilcoxon tests and corrected for multiple comparisons by FDR where appropriate. Dirichlet multinomial mixture modeling was performed using the R package Dirichlet Multinomial and calculated as previously reported (Loesche et al., 2016).
Sample sizes
n represents the number of patients, mice, or swabs collected from each mouse as described in legends of each figure.
Statistical analysis
Results represent means ± SEM. Data were analyzed using Prism 7.0 (GraphPad Software, San Diego, CA). Statistical significance was determined by one-way ANOVA when comparing more than two groups and by an unpaired two-tailed Student’s t test to compare means of lesion sizes, parasite burdens, and cytokine production from different groups of mice. Variances were equal between experimental groups. Statistically significant differences were defined as * when p values were <0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
DATA AND SOFTWARE AVAILABILITY
All 16S rRNA gene sequence data will be publicly available on the NCBI Sequence Read Archive with accession number PRJNA389688.
Supplementary Material
Highlights.
Leishmania infection alters the skin microbiota of both humans and mice
Dysbiosis is characterized by a dominance of Staphylococcus and/or Streptococcus
Naïve mice acquire dysbiosis when co-housed with leishmania-infected Mice
Acquiring a dysbiotic microbiota prior to infection exacerbates skin inflammation
Acknowledgments
Research was funded by grants from the National Institutes of Health, F31 AI 114227 to CG; RO1 AI 1068432 and U01 AI088650 to PS; R00AR060873 and R01AR066663 to EAG; P50 AI030639 to E.M.C. Funding was also provided by the PennChop Microbiome Program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to thank Ba Nguyen for technical assistance with experiments, as well as the laboratory of Dr. Yasmine Belkaid (NIAID, LPD, NIH) for bacterial isolate sequencing and identification.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, C.G., P.S., E.M.C. and E.A.G.; Methodology, C.G., P.S., and E.A.G.; Software, C.G., J.S.M, and M.A.L.; Validation, C.G.; Formal Analysis, C.G., J.S.M., M.A.L.; Investigation, C.G., S.D.C., J.H., F.O.N., C.W.B..A.M.M.; Resources, P.S., E.A.G., S.C.R., L.P.C., E.M.C., D.P.B., and C.W.B.; Data Curation, C.G., J.S.M., F.O.N., L.P.C., and E.M.C.; Writing – Original Draft, C.G., P.S., and E.A.G.; Writing – Review & Editing, C.G., J.S.M., M.A.L., S.D.C., J.H., F.O.N., A.M.M., C.W.B., D.P.B., S.C.R., L.P.C., E.M.C., P.S., and E.A.G.; Visualization, C.G. and J.S.M.; Supervision, P.S., E.A.G.; Funding Acquisition, P.S.,E.A.G and E.M.C..
The authors list no conflicts of interest.
References
- Aguiar MG, Pereira AM, Fernandes AP, Ferreira LA. Reductions in skin and systemic parasite burdens as a combined effect of topical paromomycin and oral miltefosine treatment of mice experimentally infected with leishmania (leishmania) amazonensis. Antimicrob Agents Chemother. 2010;54:4699–4704. doi: 10.1128/AAC.00809-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli LR, Dutra WO, Almeida RP, Bacellar O, Carvalho EM, Gollob KJ. Activated inflammatory T cells correlate with lesion size in human cutaneous leishmaniasis. Immunol. Lett. 2005;101:226–230. doi: 10.1016/j.imlet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- Ben Salah A, Ben Messaoud N, Guedri E, Zaatour A, Ben Alaya N, Bettaieb J, Gharbi A, Belhadj Hamida N, Boukthir A, Chlif S, et al. Topical paromomycin with or without gentamicin for cutaneous leishmaniasis. N. Engl. J. Med. 2013;368:524–532. doi: 10.1056/NEJMoa1202657. [DOI] [PubMed] [Google Scholar]
- Canesso MC, Vieira AT, Castro TB, Schirmer BG, Cisalpino D, Martins FS, Rachid MA, Nicoli JR, Teixeira MM, Barcelos LS. Skin wound healing is accelerated and scarless in the absence of commensal microbiota. J. Immunol. 2014;193:5171–5180. doi: 10.4049/jimmunol.1400625. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charmoy M, Hurrell BP, Romano A, Lee SH, Ribeiro-Gomes F, Riteau N, Mayer-Barber K, Tacchini-Cottier F, Sacks DL. The Nlrp3 inflammasome, IL-1beta, and neutrophil recruitment are required for susceptibility to a nonhealing strain of leishmania major in C57BL/6 mice. Eur. J. Immunol. 2016;46:897–911. doi: 10.1002/eji.201546015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogen AL, Yamasaki K, Sanchez KM, Dorschner RA, Lai Y, MacLeod DT, Torpey JW, Otto M, Nizet V, Kim JE, et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from staphylococcus epidermidis, a normal resident of the skin. J. Invest. Dermatol. 2010;130:192–200. doi: 10.1038/jid.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby EJ, Goldschmidt MH, Wherry EJ, Scott P. Engagement of NKG2D on bystander memory CD8 T cells promotes increased immunopathology following leishmania major infection. PLoS Pathog. 2014;10:e1003970. doi: 10.1371/journal.ppat.1003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira MR, Tafuri WL, Nicoli JR, Vieira EC, Melo MN, Vieira LQ. Influence of microbiota in experimental cutaneous leishmaniasis in swiss mice. Rev. Inst. Med. Trop. Sao Paulo. 1999;41:87–94. doi: 10.1590/s0036-46651999000200005. [DOI] [PubMed] [Google Scholar]
- Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A streptococcus. J. Invest. Dermatol. 2001;117:91–97. doi: 10.1046/j.1523-1747.2001.01340.x. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Figueroa EA, Rangel-Escareno C, Espinosa-Mateos V, Carrillo-Sanchez K, Salaiza-Suazo N, Carrada-Figueroa G, March-Mifsut S, Becker I. Disease severity in patients infected with leishmania mexicana relates to IL-1 beta. PLoS Negl Trop. Dis. 2012;6:e1533. doi: 10.1371/journal.pntd.0001533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–689. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- Gimblet C, Loesche MA, Carvalho L, Carvalho EM, Grice EA, Artis D, Scott P. IL-22 protects against tissue damage during cutaneous leishmaniasis. PLoS One. 2015;10:e0134698. doi: 10.1371/journal.pone.0134698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gontcharova V, Youn E, Sun Y, Wolcott RD, Dowd SE. A comparison of bacterial composition in diabetic ulcers and contralateral intact skin. Open Microbiol. J. 2010;4:8–19. doi: 10.2174/1874285801004010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Lombana C, Gimblet C, Bacellar O, Oliveira WW, Passos S, Carvalho LP, Goldschmidt M, Carvalho EM, Scott P. IL-17 mediates immunopathology in the absence of IL-10 following leishmania major infection. PLoS Pathog. 2013;9:e1003243. doi: 10.1371/journal.ppat.1003243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA. The skin microbiome: Potential for novel diagnostic and therapeutic approaches to cutaneous disease. Semin. Cutan. Med. Surg. 2014;33:98–103. doi: 10.12788/j.sder.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Snitkin ES, Yockey LJ, Bermudez DM, NISC Comparative Sequencing Program. Liechty KW, Segre JA. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc. Natl. Acad. Sci. U. S. A. 2010;107:14799–14804. doi: 10.1073/pnas.1004204107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan GD, Hodkinson BP, McGinnis K, Tyldsley AS, Anari JB, Horan AD, Grice EA, Mehta S. Culture-independent pilot study of microbiota colonizing open fractures and association with severity, mechanism, location, and complication from presentation to early outpatient follow-up. J. Orthop. Res. 2014;32:597–605. doi: 10.1002/jor.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel FP, Sadick MD, Holaday BJ, Coffman RL, Locksley RM. Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine leishmaniasis. evidence for expansion of distinct helper T cell subsets. J. Exp. Med. 1989;169:59–72. doi: 10.1084/jem.169.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iraji F, Sadeghinia A. Efficacy of paromomycin ointment in the treatment of cutaneous leishmaniasis: Results of a double-blind, randomized trial in isfahan, iran. Ann. Trop. Med. Parasitol. 2005;99:3–9. doi: 10.1179/136485905X16372. [DOI] [PubMed] [Google Scholar]
- Isaac-Marquez AP, Lezama-Davila CM. Detection of pathogenic bacteria in skin lesions of patients with chiclero's ulcer. reluctant response to antimonial treatment. Mem. Inst. Oswaldo Cruz. 2003;98:1093–1095. doi: 10.1590/s0074-02762003000800021. [DOI] [PubMed] [Google Scholar]
- Kamdar K, Khakpour S, Chen J, Leone V, Brulc J, Mangatu T, Antonopoulos DA, Chang EB, Kahn SA, Kirschner BS, et al. Genetic and metabolic signals during acute enteric bacterial infection alter the microbiota and drive progression to chronic inflammatory disease. Cell. Host Microbe. 2016;19:21–31. doi: 10.1016/j.chom.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Chung HJ, Bleys J, Ghohestani RF. Is paromomycin an effective and safe treatment against cutaneous leishmaniasis? A meta-analysis of 14 randomized controlled trials. PLoS Negl Trop. Dis. 2009;3:e381. doi: 10.1371/journal.pntd.0000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, Kong HH, Amagai M, Nagao K. Dysbiosis and staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity. 2015;42:756–766. doi: 10.1016/j.immuni.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, NISC Comparative Sequence Program, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq illumina sequencing platform. Appl. Environ. Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, Gallo RL, Nizet V. D-alanylation of teichoic acids promotes group a streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J. Bacteriol. 2005;187:6719–6725. doi: 10.1128/JB.187.19.6719-6725.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krolewiecki A, Leon S, Scott P, Abraham D. Activity of azithromycin against leishmania major in vitro and in vivo. Am. J. Trop. Med. Hyg. 2002;67:273–277. doi: 10.4269/ajtmh.2002.67.273. [DOI] [PubMed] [Google Scholar]
- Kulkarni MM, McMaster WR, Kamysz E, Kamysz W, Engman DM, McGwire BS. The major surface-metalloprotease of the parasitic protozoan, leishmania, protects against antimicrobial peptide-induced apoptotic killing. Mol. Microbiol. 2006;62:1484–1497. doi: 10.1111/j.1365-2958.2006.05459.x. [DOI] [PubMed] [Google Scholar]
- Layegh P, Ghazvini K, Moghiman T, Hadian F, Zabolinejad N, Pezeshkpour F. Bacterial contamination in cutaneous leishmaniasis: Its effect on the lesions' healing course. Indian J. Dermatol. 2015;60 doi: 10.4103/0019-5154.152560. 211-5154.152560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesche M, Gardner SE, Kalan L, Horwinski J, Zheng Q, Hodkinson BP, Tyldsley AS, Franciscus CL, Hillis SL, Mehta S, et al. Temporal stability in chronic wound microbiota is associated with poor healing. J. Invest. Dermatol. 2016 doi: 10.1016/j.jid.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Kostka S, Dinges S, Griewank K, Iwakura Y, Udey MC, von Stebut E. IL-17 promotes progression of cutaneous leishmaniasis in susceptible mice. J. Immunol. 2009;182:3039–3046. doi: 10.4049/jimmunol.0713598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Li M, Campbell TB, Flores SC, Linderman D, Gebert MJ, Knight R, Fontenot AP, Palmer BE. Alterations in the gut microbiota associated with HIV-1 infection. Cell. Host Microbe. 2013;14:329–339. doi: 10.1016/j.chom.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel JS, Hannigan GD, Tyldsley AS, SanMiguel AJ, Hodkinson BP, Zheng Q, Grice EA. Skin microbiome surveys are strongly influenced by experimental design. J. Invest. Dermatol. 2016;136:947–956. doi: 10.1016/j.jid.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase N, Sasaki A, Yamashita K, Shimizu A, Wakita Y, Kitai S, Kawano J. Isolation and species distribution of staphylococci from animal and human skin. J. Vet. Med. Sci. 2002;64:245–250. doi: 10.1292/jvms.64.245. [DOI] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, Conlan S, Himmelfarb S, Byrd AL, Deming C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. 2015;520:104–108. doi: 10.1038/nature14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, Quinones M, Koo L, Conlan S, et al. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337:1115–1119. doi: 10.1126/science.1225152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chen TH, Two AM, Chun KA, Narala S, Geha RS, Hata TR, Gallo RL. Staphylococcus aureus exploits epidermal barrier defects in atopic dermatitis to trigger cytokine expression. J. Invest. Dermatol. 2016 doi: 10.1016/j.jid.2016.05.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuga K, Cipolat S, Watt FM. Increased bacterial load and expression of antimicrobial peptides in skin of barrier-deficient mice with reduced cancer susceptibility. J. Invest. Dermatol. 2016;136:99–106. doi: 10.1038/jid.2015.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neva FA, Ponce C, Ponce E, Kreutzer R, Modabber F, Olliaro P. Non-ulcerative cutaneous leishmaniasis in honduras fails to respond to topical paromomycin. Trans. R. Soc. Trop. Med. Hyg. 1997;91:473–475. doi: 10.1016/s0035-9203(97)90290-x. [DOI] [PubMed] [Google Scholar]
- Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414:454–457. doi: 10.1038/35106587. [DOI] [PubMed] [Google Scholar]
- Novais FO, Carvalho LP, Graff JW, Beiting DP, Ruthel G, Roos DS, Betts MR, Goldschmidt MH, Wilson ME, de Oliveira CI, et al. Cytotoxic T cells mediate pathology and metastasis in cutaneous leishmaniasis. PLoS Pathog. 2013;9:e1003504. doi: 10.1371/journal.ppat.1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novais FO, Carvalho LP, Passos S, Roos DS, Carvalho EM, Scott P, Beiting DP. Genomic profiling of human leishmania braziliensis lesions identifies transcriptional modules associated with cutaneous immunopathology. J. Invest. Dermatol. 2014 doi: 10.1038/jid.2014.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Freeman AF, NISC Comparative Sequencing Program. Park M, Sokolic R, Candotti F, Holland SM, Segre JA, Kong HH. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013;23:2103–2114. doi: 10.1101/gr.159467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira MR, Tafuri WL, Afonso LC, Oliveira MA, Nicoli JR, Vieira EC, Scott P, Melo MN, Vieira LQ. Germ-free mice produce high levels of interferon-gamma in response to infection with leishmania major but fail to heal lesions. Parasitology. 2005;131:477–488. doi: 10.1017/S0031182005008073. [DOI] [PubMed] [Google Scholar]
- Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J. Exp. Med. 2001;193:1067–1076. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F. Inactivation of the dlt operon in staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 1999;274:8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- Sadeghian G, Ziaei H, Bidabadi LS, Baghbaderani AZ. Decreased effect of glucantime in cutaneous leishmaniasis complicated with secondary bacterial infection. Indian J. Dermatol. 2011;56:37–39. doi: 10.4103/0019-5154.77549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado VR, Queiroz AT, Sanabani SS, Oliveira CI, Carvalho EM, Costa JM, Barral-Netto M, Barral A. The microbiological signature of human cutaneous leishmaniasis lesions exhibits restricted bacterial diversity compared to healthy skin. Mem. Inst. Oswaldo Cruz. 2016;111:241–251. doi: 10.1590/0074-02760150436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos Cda S, Boaventura V, Ribeiro Cardoso C, Tavares N, Lordelo MJ, Noronha A, Costa J, Borges VM, de Oliveira CI, Van Weyenbergh J, et al. CD8(+) granzyme B(+)-mediated tissue injury vs. CD4(+)IFNgamma(+)-mediated parasite killing in human cutaneous leishmaniasis. J. Invest. Dermatol. 2013;133:1533–1540. doi: 10.1038/jid.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharton-Kersten T, Afonso LC, Wysocka M, Trinchieri G, Scott P. IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J. Immunol. 1995;154:5320–5330. [PubMed] [Google Scholar]
- Scott P, Novais FO. Cutaneous leishmaniasis: Immune responses in protection and pathogenesis. Nat. Rev. Immunol. 2016;16:581–592. doi: 10.1038/nri.2016.72. [DOI] [PubMed] [Google Scholar]
- Song SJ, Lauber C, Costello EK, Lozupone CA, Humphrey G, Berg-Lyons D, Caporaso JG, Knights D, Clemente JC, Nakielny S, et al. Cohabiting family members share microbiota with one another and with their dogs. Elife. 2013;2:e00458. doi: 10.7554/eLife.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spath GF, Beverley SM. A lipophosphoglycan-independent method for isolation of infective leishmania metacyclic promastigotes by density gradient centrifugation. Exp. Parasitol. 2001;99:97–103. doi: 10.1006/expr.2001.4656. [DOI] [PubMed] [Google Scholar]
- Voronov E, Dotan S, Gayvoronsky L, White RM, Cohen I, Krelin Y, Benchetrit F, Elkabets M, Huszar M, El-On J, et al. IL-1-induced inflammation promotes development of leishmaniasis in susceptible BALB/c mice. Int. Immunol. 2010;22:245–257. doi: 10.1093/intimm/dxq006. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaph C, Uzonna J, Beverley SM, Scott P. Central memory T cells mediate long-term immunity to leishmania major in the absence of persistent parasites. Nat. Med. 2004;10:1104–1110. doi: 10.1038/nm1108. [DOI] [PubMed] [Google Scholar]
- Zenewicz LA, Yin X, Wang G, Elinav E, Hao L, Zhao L, Flavell RA. IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J. Immunol. 2013;190:5306–5312. doi: 10.4049/jimmunol.1300016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.