

Supplemental Digital Content is available in the text.
Keywords: arrhythmia, blebbistatin, electrophysiology, heart, mechanoreceptors
Abstract
Background—
External chest impacts (commotio cordis) can cause mechanically induced premature ventricular excitation (PVEM) and, rarely, ventricular fibrillation (VF). Because block of stretch-sensitive ATP-inactivated potassium channels curtailed VF occurrence in a porcine model of commotio cordis, VF has been suggested to arise from abnormal repolarization caused by stretch activation of potassium channels. Alternatively, VF could result from abnormal excitation by PVEM, overlapping with normal repolarization-related electric heterogeneity. Here, we investigate mechanisms and determinants of PVEM induction and its potential role in commotio cordis–induced VF.
Methods and Results—
Subcontusional mechanical stimuli were applied to isolated rabbit hearts during optical voltage mapping, combined with pharmacological block of ATP-inactivated potassium or stretch-activated cation-nonselective channels. We demonstrate that local mechanical stimulation reliably triggers PVEM at the contact site, with inducibility predicted by local tissue indentation. PVEM induction is diminished by pharmacological block of stretch-activated cation-nonselective channels. In hearts where electrocardiogram T waves involve a well-defined repolarization edge traversing the epicardium, PVEM can reliably provoke VF if, and only if, the mechanical stimulation site overlaps the repolarization wave edge. In contrast, application of short-lived intraventricular pressure surges neither triggers PVEM nor changes repolarization. ATP-inactivated potassium channel block has no effect on PVEM inducibility per se, but shifts it to later time points by delaying repolarization and prolonging refractoriness.
Conclusions—
Local mechanical tissue deformation determines PVEM induction via stretch-activation of cation-nonselective channels, with VF induction requiring PVEM overlap with the trailing edge of a normal repolarization wave. This defines a narrow, subject-specific vulnerable window for commotio cordis–induced VF that exists both in time and in space.
WHAT IS KNOWN
Mechanical impact-induced changes in cardiac electrophysiology (commotio cordis) can result in ventricular fibrillation, with a generally low incidence attributed to a short vulnerable period during the ECG T wave.
Possible mechanisms include abnormal repolarization caused by intraventricular pressure surge–mediated opening of stretch-activated potassium channels or premature excitation caused by local tissue deformation–mediated opening of stretch-activated cation-nonselective channels.
WHAT THE STUDY ADDS
T-wave impact–associated pressure surges have no electrophysiological effect, local deformation induces ectopic excitation that can be prevented by pharmacological block of stretch-activated cation-nonselective channels, and block of stretch-activated potassium channels plays no relevant role except shifting the vulnerable period.
Ventricular fibrillation occurs if, and only if, mechanically induced premature excitation occurs right on the trailing edge of the preceding normal repolarization wave as it traverses the ventricles.
This means that the vulnerable window for mechanical induction of ventricular fibrillation is short (in time) and narrow (in space), explaining why mechanically induced ventricular fibrillation is rare.
For over a century, it has been known that the heart is an exquisitely mechanosensitive organ.1 Subcontusional mechanical stimulation of the heart (commotio cordis [CC]), whether by intracardiac device/tissue interactions or by chest impacts, can result in a variety of heart rhythm changes, including ectopy (eg, mechanically induced premature ventricular excitation [PVEM]) and sustained rhythm disturbances (eg, ventricular fibrillation [VF]).2
PVEM is common in patients with central monitoring catheters3,4 or intracardiac pacing wires.5–7 It is also presumed to be common in CC-prone sports, such as baseball or ice hockey, and CC-induced VF is one of the most common causes of death in youth athletes in the United States.8 However, mechanisms of PVEM genesis and determinants for VF induction are ill-understood.
What is known is that stretch can trigger PVEM in myocardial cells,9 tissue,10 and whole heart.11 This mechanoelectric feedback12–14 can be explained by stretch-activated cation-nonselective channels (SACNS),15 which depolarize diastolic transmembrane potential (Vm) in cardiomyocytes16 and whole hearts.17 During the action potential (AP) plateau, stretch accelerates Vm repolarization by activating SACNS17,18 (their reversal potential is approximately half-way between peak and resting Vm levels19) or by opening stretch-sensitive potassium-selective currents,20 causing early AP shortening. In CC, the mechanosensitive ATP-inactivated potassium (KATP) current21 has been implicated in VF induction, as administration of glibenclamide (a nonspecific KATP blocker) reduced mechanical VF induction in a porcine model of CC.22
Electrophysiological outcomes of CC are modulated by mechanical stimulus characteristics, such as location, area, and duration.23 Studies in the porcine model have characterized mechanical inducibility of VF as inversely related to impact area and duration, rising with projectile stiffness and, most strikingly, occurring only during a narrow vulnerable period: 15 to 30 ms before the ECG T-wave peak.8,24,25
Generally, VF vulnerability during the T wave is related to repolarization dynamics. In the case of locally acting mechanical stimuli, as ventricular repolarization is spatially nonuniform, timing relative to the ECG will not translate into a similarly defined timing relative to the Vm of affected cardiomyocytes.26 Computational simulations have suggested that mechanically induced VF can occur only when a suprathreshold mechanical stimulus occurs at the trailing edge of the preceding normal repolarization wave.27,28 In this situation, PVEM in excitable tissue arises directly adjacent to refractory tissue, resulting in a region of functional conduction block around which reentry can ensue. As a wave of repolarization travels across the ventricles, the condition for such overlap will be met in different locations at different times, and for very brief periods only. From this site dependence of critical mechanical stimulation timing follows the hypothesis that the vulnerable window for CC-induced VF exists both in time and in space. Experimental assessment of this computationally predicted mechanism is still outstanding.
An interesting additional question concerns the role of the large, but short-lived, intraventricular pressure surge that accompanies precordial impacts. In a porcine CC model, VF-inducing impacts cause pressure surges whose amplitude correlates with both impact severity and probability of VF induction.29 Yet similarly pronounced intraventricular pressure surges are seen with coughing,30 where they are not normally associated with induction of sustained arrhythmias. Therefore, the question as to whether the intraventricular pressure surge is causal for CC-induced VF, or is a covariate of those impact properties that determine arrhythmogenicity, requires experimental verification.
Thus, the goals of our study were to assess, in the Langendorff-perfused rabbit heart (in which mechanoelectric feedback responses are well preserved31), mechanisms of PVEM induction, and their potential role in triggering VF. Our specific aims were to determine whether (1) PVEM induction depends on the degree of local tissue deformation, deformation rate, applied force, stress, or intraventricular pressure surges; (2) CC effects on cardiac electrophysiology are caused by SACNS or mechanosensitive KATP; and (3) spatiotemporal overlap of mechanical stimulation with the trailing edge of the repolarization wave is critical for VF induction. These aims were addressed by controlled application of subcontusional local epicardial mechanical stimuli, or intraventricular volume bursts, during optical Vm mapping in the absence or presence of suitable pharmacological blockers of SACNS or KATP.
Methods
Ethical Approval
This study was performed, under local ethical approval, in strict accordance with the UK Home Office Animals (Scientific Procedures) Act of 1986. Details of experimental protocols have been reported following the Minimum Information About a Cardiac Electrophysiology Experiment reporting standard,32 repository (https://www.micee.org/?q=node/00001374). Detailed methods are given in the Data Supplement.
Heart Preparation
Langendorff-perfused hearts from female New Zealand White rabbits were instrumented with custom-made polyethylene balloons in the left ventricle (LV) connected to a transducer for the measurement of intraventricular pressure and a servomotor-controlled syringe for rapid bidirectional volume alteration. Surface ECG was recorded using 2 spring-loaded monopolar Ag/AgCl pellet electrodes. The experimental setup can be seen in Figure I in the Data Supplement.
Voltage Optical Mapping
Hearts were loaded with a voltage-sensitive dye (di-4-ANBDQPQ) and an electromechanical uncoupler (blebbistatin). Optical mapping was performed with a previously described system using light-emitting diodes for illumination and a 511 frames/s camera for observation.33
Mechanical Stimulation
Local mechanical stimuli were applied to the ventricular epicardium using the Soft Tissue Impact Characterisation Kit,34 with the area of indentation determined by impact probe surface and the magnitude of indentation monitored at ≈35 μm resolution using an optical grating. Images and characteristics of a typical stimulation are shown in Figure 1 and Movie I in the Data Supplement. For all stimuli, local excitation was assessed by optical Vm mapping. Intraventricular pressure surges were applied by brief bursts in intraventricular balloon volume (active inflation and deflation). Local mechanical stimulation and pressure surge timing relative to the ECG were controlled. Tissue integrity was assessed by analysis of creatine kinase activity in coronary effluent.34
Figure 1.
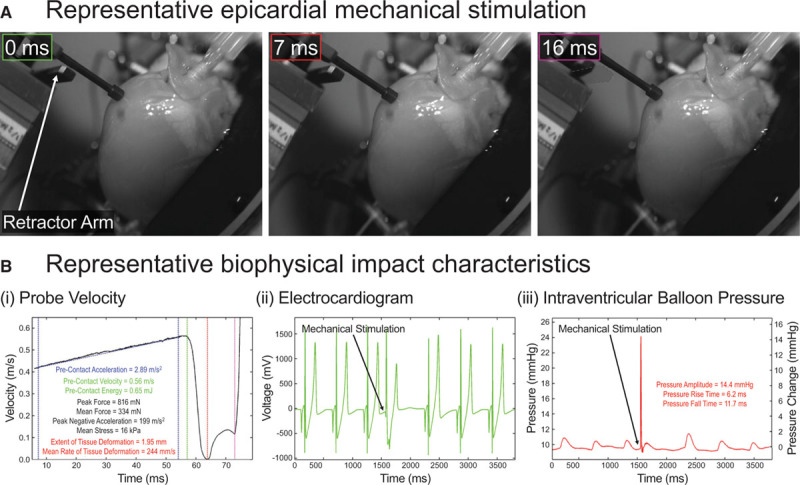
Subcontusional local epicardial mechanical stimulation. A, Images of a typical epicardial mechanical stimulus (from Movie I in the Data Supplement). B, Mechanical stimulation characteristics, showing: (i) probe velocity (blue lines indicate period of probe acceleration; green, red, and purple lines show time of initial tissue contact [0 ms in A], full probe deceleration [7 ms in A] followed by reversal of probe direction initially by tissue recoil, and subsequently by retractor arm activation [16 ms in A, dashed outline showing resting arm position]), along with calculated precontact probe acceleration, velocity, and kinetic energy; peak and mean force; peak negative acceleration; mean stress; and extent and mean rate of tissue deformation (Methods in the Data Supplement for calculations); (ii) surface ECG showing mechanically induced premature ventricular excitation; and (iii) intraventricular balloon pressure, along with impact-induced pressure surge amplitude and rise/fall times.
Experimental Protocols
Four experimental series were performed.
Series 1 tested inducibility of PVEM and VF by local mechanical stimulation (n=32; n=number of heart preparations). Subcontusional mechanical stimuli of ≈0.5 mJ were applied to 3.1 mm2 contact areas across the LV freewall, and additional locations in some of these hearts: LV apex (n=4); LV base (n=4); LV/right ventricular (RV) border (n=4); and RV freewall (with an RV intraventricular balloon, n=11). Coupling interval to the preceding sinus beat was shortened (5 ms steps) from late diastole until either VF was induced or PVEM ceased to be elicited (entering the electric refractory period).
Series 2 investigated determinants of PVEM threshold and compared properties of mechanically and electrically induced ventricular excitation (n=7). In all hearts, local mechanical stimuli were applied to various locations of the LV and RV freewall using 3.1 or 28.3 mm2 probes in randomized order during late diastole (at ≈75% of the cycle length). Stimulation energy was reduced from ≈0.5 mJ until PVEM ceased to occur (ie, when below threshold). At each of the mechanically targeted LV locations, hearts were stimulated electrically, using a concentric bipolar stimulation electrode.
Series 3 assessed the roles of SACNS and KATP channels in mechanically induced responses using pharmacological blockers of SACNS (50, 250, and 500 μmol/L streptomycin, n=6; 500 nmol/L Grammostola spatulata MechanoToxin-4, n=5) and KATP channels (5 and 10 μmol/L glibenclamide, n=6).
Series 4 examined electrophysiological effects of intraventricular pressure surges (n=6). A rapid change in LV balloon volume (20 μL), producing pressure amplitudes that mimicked those seen during ≈0.5 mJ epicardial mechanical stimulation, was applied by active balloon inflation and deflation during diastole. This was repeated with volumes raised by 22 μL increments, normally up to a maximum of 130 μL.
Statistics
Data analysis was performed using custom programs in Matlab. Values are presented as mean±SD. For statistical tests, P<0.05 was taken to signify statistically significant differences between means. Comparison of local mechanically and electrically induced excitation and the effects of glibenclamide application were assessed by Wilcoxon signed-rank test (as distribution normality cannot be assumed). Mechanical stimulation characteristics at PVEM threshold were compared by 1-way ANOVA with Tukey–Kramer post hoc test. Linear correlation between the change in intraventricular balloon volume and pressure surge amplitude, rise time, or fall time was assessed by Pearson correlation.
Results
PVEM and VF Induction
All local mechanical stimuli applied to the ventricular epicardium with an amplitude of ≈0.5 mJ and a coupling interval outside the refractory period (n=32 hearts; m=409 stimuli analyzed, all with confirmed lack of elevated creatine kinase activity), irrespective of stimulation location, resulted in PVEM. This was associated with a change in activation pattern compared with sinus rhythm (compare Figure 2A and 2B) which, in terms of epicardially mapped Vm dynamics, generally propagated in an apicobasal direction, frequently involving multiple sites of epicardial breakthrough (Figure 2A; Movie II in the Data Supplement). In 6 of these hearts, the inducibility of PVEM was also tested before the application of blebbistatin (ie, while still beating), which similarly showed 100% incidence (m=60), demonstrating that PVEM induction is not conditional on electromechanical uncoupling.
Figure 2.
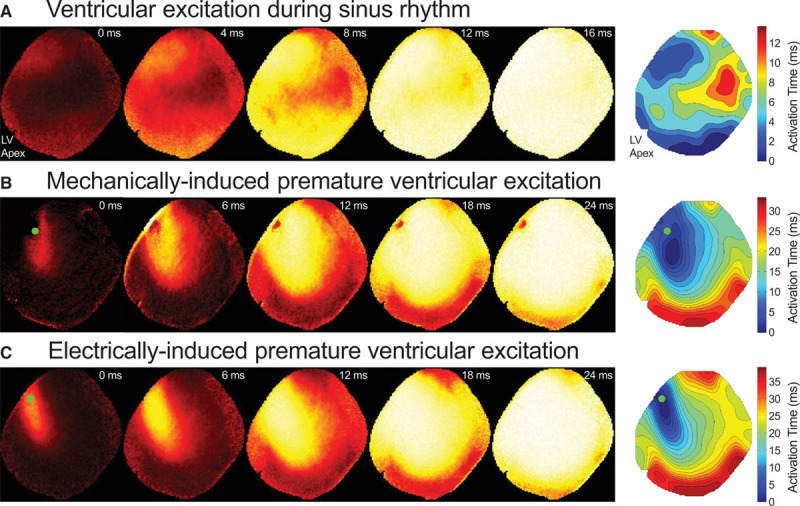
Left ventricular excitation visualized by epicardial optical mapping. Representative recordings during sinus rhythm (A) and mechanically (B) or electrically induced (C) excitation (from Movies II through IV in the Data Supplement) and maps of activation time (0 ms represents earliest epicardial activation in the associated map). Mechanical and electric stimulations were triggered at the same midlevel left ventricular (mid-LV) freewall location (green dots). Isochrones represent 2 ms steps. Note that the dark region at the mechanical stimulation location is an artifact caused by the probe entering the field of view, and the notch at the LV apex is caused by the surface ECG electrode.
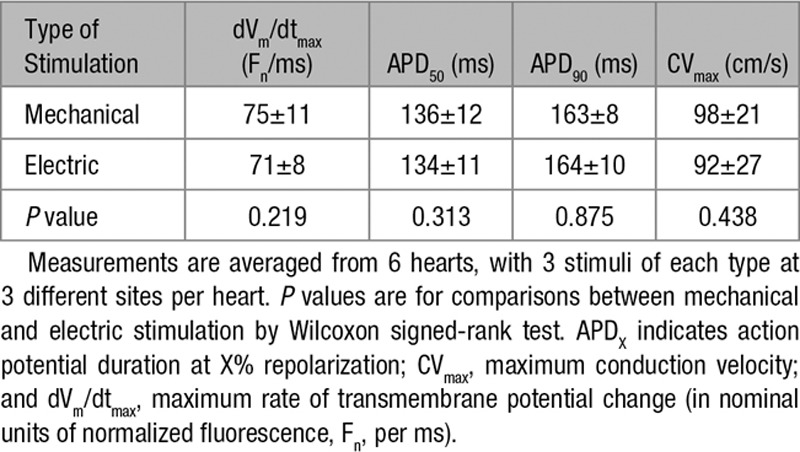
All PVEM, including those in the absence of an electromechanical uncoupler, originated focally from the stimulation site, confirmed by optical mapping (Figure 2B; Movie III in the Data Supplement). Mechanically and electrically induced ventricular excitation, triggered at the same location, resulted in downstream patterns of electric activation (compare Figure 2B and 2C; Movies III and IV in the Data Supplement) more similar to one another than to sinus activation (dVm/dtmax, AP duration, and conduction velocity are given in the Table; n=6, m=18).
Table.
Comparison of Excitation Induced Mechanically or Electrically at the Same Site (Midlevel Left Ventricular Freewall)
Most PVEM-inducing stimuli resulted in a single ventricular activation. In 3 of 32 hearts, local mechanical stimulation during the T wave gave rise to instantaneous induction of VF (Figure 3A; Movie V in the Data Supplement). VF episodes lasted 30 to 60 s before spontaneously converting to sinus rhythm. Where observed, mechanically induced VF was repeatable (m=13), when stimulation site and coupling interval were kept constant (including attempts with reduced mechanical stimulation energy, as long as suprathreshold for PVEM, m=5).
Figure 3.
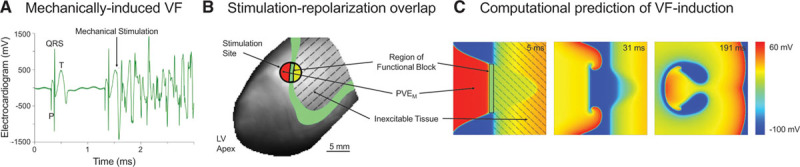
Mechanically induced ventricular fibrillation (VF). A, Surface ECG showing mechanical stimulation in early T wave, resulting in instantaneous VF (Movie V in the Data Supplement). B, Spatial interrelation of stimulation site and 50% repolarization isochrone (green). C, Comparison with previously published computational 2-dimensional modeling predictions,27 showing mechanically induced premature ventricular excitation (PVEM; red) arising directly adjacent to inexcitable tissue (yellow), forming a region of functional block (black rectangle) around which sustained reentry occurs. Adapted from Garny and Kohl27 with permission of the publisher. Copyright © 2004, John Wiley and Sons. Authorization for this adaptation has been obtained both from the owner of the copyright in the original work and from the owner of copyright in the translation or adaptation.
In all cases of VF induction, optical mapping showed a well-defined trailing edge of the preceding depolarization wave, traversing the epicardial surface at the time of mechanical stimulation (Movie V in the Data Supplement). VF-inducing PVEM induction sites corresponded to the 50% repolarization isochrone (Figure 3B), supporting previous 2-dimensional (Figure 3C)27 and 3-dimensional28 computational model predictions. A different repolarization pattern, near-uniform repolarization of the epicardial surface (Movie II in the Data Supplement), was seen in the other ≈90% of hearts. In these preparations, PVEM did not cause VF at any coupling interval or impact location.
Determinants of PVEM Threshold
For mechanical stimuli, applied during late diastole to either the LV or RV freewall using different probe contact areas, PVEM threshold corresponded to significantly different values of mean rate of local tissue deformation, peak force, mean stress, and intraventricular pressure surge amplitude (n=7, m=405), suggesting that none of these parameters scale well with arrhythmogenicity. However, local tissue indentation, required for PVEM induction, was similar across all groups (Figure 4).
Figure 4.
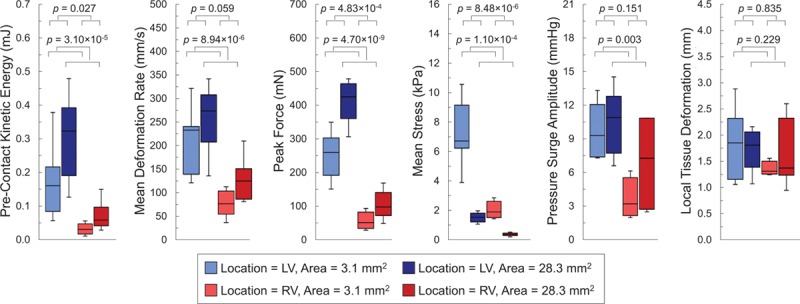
Mechanically induced premature ventricular excitation (PVEM) thresholds. Box-and-whisker plots of mechanical stimulation characteristics (precontact kinetic energy, mean deformation rate, peak force, mean stress, intraventricular pressure surge amplitude, and local tissue deformation) at PVEM threshold with varying combinations of freewall contact location (midleft ventricular [mid-LV; blues boxes] and midright ventricular freewall [mid-RV; red boxes]) and contact area (3.1 mm2 [lighter boxes] and 28.3 mm2 [darker boxes]). Only local tissue indentation (far right) forms a uniform predictor of PVEM threshold across the various stimulation protocols. Measurements from 7 hearts. P values are for comparison of contact location and area by 2-way ANOVA with Tukey–Kramer post hoc tests.
Role of SACNS and KATP Channels
Application of Grammostola spatulata MechanoToxin-4 (500 nmol/L) reduced PVEM inducibility in all preparations, on average by 62% (n=5, m=48/77 PVEM inductions prevented). In contrast, streptomycin (≤500 μmol/L) had no effect on PVEM inducibility (n=6, m=0/270 PVEM prevented).
The KATP channel blocker glibenclamide (≤10 μmol/L) also had no effect on PVEM inducibility (n=6, m=0/78 PVEM prevented), but resulted in a ≈10% decrease in heart rate (from 138±10 to 125±13 beats per minute; P=0.031). This was associated with slowed repolarization (Figure II in the Data Supplement) and a 12% increase in refractory period for PVEM (from 218±32 to 244±41 ms; P=0.031).
Electrophysiological Effects of Intraventricular Volume Pulses
Pressure surges, observed during epicardial mechanical stimulation (Figure 1B), were mimicked by the application of intraventricular volume bursts. Pressure surge amplitude (r=0.904, P=1.07×10−7; Figure 5A) and pressure rise time (r=0.723, P=0.008) increased linearly with the size of rapid intraventricular balloon volume change, while pressure fall time was not correlated with the change in balloon volume (r=0.428, P=0.166).
Figure 5.
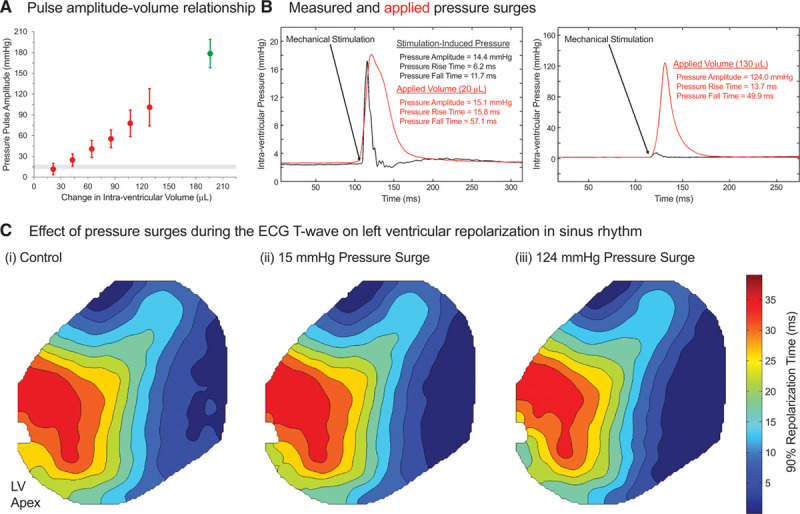
Intraventricular volume pulses. A, Relationship between intraventricular balloon volume pulse and peak pressure, averaged from 6 hearts. Red points indicate volumes assessed in the standard experiments (which were not associated with elevated creatine kinase release; Figure III in the Data Supplement), whereas a mechanically induced premature ventricular excitation (PVEM)–inducing excessive volume pulse (200 μL) is shown in green. The gray horizontal band indicates the range of pressure amplitudes measured during PVEM-inducing epicardial mechanical stimulation. B, Representative pressure surges measured during epicardial mechanical stimulation (black) and with active injection/retraction of 20 μL (left) or 130 μL (right) intraventricular volume pulses (red). C, Representative optical mapping data for 90% repolarization time (0 ms represents earliest repolarization in the associated map) in sinus rhythm during (i) control and application of (ii) 15 mm Hg or (iii) 124 mm Hg pressure surges (using stimuli shown in B) applied at the peak of the T wave. Isochrones represent 5 ms steps. Note that the notch at the left ventricular (LV) apex is caused by the surface ECG electrode in the field of view.
Diastolic pressure surges (all with confirmed lack of elevated creatine kinase activity), even if about an order of magnitude larger than those measured during PVEM-inducing epicardial stimuli (101±27 versus 15±3 mm Hg, induced by volume bursts of 130 versus 20 µL, respectively), did not trigger a single PVEM (Figure 5B; n=6, m=108). If applied during the ECG T wave, no change in repolarization pattern was observed (Figure 5C; n=6, m=108). In contrast, pressure surges of 178±21 mm Hg (induced by volume bursts of 200 µL) were needed to trigger excitation (Figure 5A).
Discussion
Summary of Principal Findings
PVEM is induced reliably in diastole, using subcontusional local stimuli (≈0.5 mJ in isolated rabbit heart). While accompanied by intraventricular pressure surges (≈15 mm Hg), the origin of electric activation always coincides with the mechanical contact site (similar to repetitive local mechanical stimulation35). PVEM induction is correlated with the degree of local tissue indentation, but not with indentation rate, force, stress, or intraventricular pressure surge amplitude, in keeping with earlier suggestions that mechanical deformation at the contact site is a key determinant for mechanoelectric signal transduction.34 Underlying mechanisms involve SACNS, as PVEM induction is reliably attenuated by the specific36 SACNS blocker Grammostola spatulata MechanoToxin-4. The lack of streptomycin effects, which is an efficient SACNS blocker in vitro,37 confirms results from a porcine model of CC38 and is in keeping with earlier reports on the limited efficacy of streptomycin for acute SACNS block in native myocardium.39 The mechanosensitive KATP channel blocker glibenclamide has no effect on PVEM inducibility per se, but shifts PVEM to later time points as a result of reduced sinus rate, delayed repolarization, and prolonged electric refractoriness. As a result, impacts applied early after the original refractory period, identified before drug application, can become ineffective.
With reduction of the coupling interval between preceding sinus activation and mechanical stimulation, PVEM continues until stimulation timing enters the refractory period. If, and only if, repolarization is associated with a well-defined wave edge, PVEM reliably and repeatably triggers VF. This occurs only if stimulation site and timing are such that PVEM induction overlaps with the 50% repolarization isochrone of the preceding sinus beat.
In contrast, intraventricular pressure surges mimicking those seen during PVEM-inducing epicardial stimuli do not result in a single occurrence of PVEM, nor in noticeable changes in repolarization, even if increased in amplitude by an order of magnitude.
Taken together, these results suggest that ventricular excitation upon local mechanical stimulation is a result of SACNS activation, caused by local tissue deformation. The presence of a well-delineated trailing repolarization wave, and spatiotemporal colocalization of mechanically affected tissue with the trailing edge of the preceding wave, are preconditions for VF induction. The vulnerable window for CC-induced VF is thus defined in both time and space, explaining the short period during which mechanical stimuli can induce reentry for any given contact location, and why mechanically induced VF is so rare in real life. Our findings may have future implications for predictive assessment of individual athletes’ CC risk (a target which until now has been lacking40), through measurement of ventricular repolarization patterns using noninvasive imaging techniques, such as electrocardiographic imaging41 or overall dispersion of repolarization by the T-peak to T-end interval42 (whose prolongation has been shown to be an independent risk marker for sudden cardiac death43).
Mechanisms of PVEM Induction
Previously reported PVEM induction by transiently increased intraventricular volume in isolated hearts11,44–46 occurred in different studies at remarkably similar pulse volumes (which define tissue stretch; eg, 750 μL pulses resulted in near 100% PVEM occurrence in hearts isolated from ≈2 kg rabbits11). However, associated changes in intraventricular pressure (a function of the interaction between volume pulse dynamics and tissue viscoelastic properties47) show high variability.45 In addition, where seen, ventricular excitation on volume loading is focal, often originating in the posterolateral LV,11 the most compliant region where tissue distension is likely to be largest.
This suggests that stretch of (part of the) myocardium is a primary determinant of electrophysiological responses to mechanical stimulation. Our results support this view, as the threshold for PVEM, regardless of contact location (LV, RV) and area (3.1 and 28.3 mm2), correlates with local tissue deformation only (indentation depth). In our hands, independently applied intraventricular pressure surges up to an order of magnitude larger than those seen with epicardial stimulation do not result in PVEM. Note that pressure surges in our study were achieved by rapid application of comparatively small volume pulses (20–130 μL), using active balloon inflation and deflation to mimic more closely the CC setting, where pressure surges (1) are exceedingly short lived and (2) arise without intraventricular volume increase.
Our results also identify SACNS as a key contributor to PVEM, as application of Grammostola spatulata MechanoToxin-4 reduces its inducibility. This role of SACNS in depolarizing cells to threshold during mechanical stimulation is further reflected by the lack of an additional delay between mechanical stimulation and the onset of electric excitation when the coupling interval is reduced (as SACNS are not inactivated by normal AP dynamics).
Although mechanisms by which external energy delivery give rise to ectopy are clearly different for mechanical and electric stimulation, our results confirm that, once excitation and ventricular activation occur, their downstream characteristics are similar, as previously shown in open-chest, anesthetized dogs48 and rat isolated hearts (in which similar downstream calcium dynamics were also seen).33 Electric stimulation initially excites a narrower tissue region than mechanical stimulation (elongated in the locally prevailing cell direction; compare early activation isochrones in Figure 2B and 2C and Movies III and IV in the Data Supplement), as local electric stimulation affects a smaller tissue region (contact area of the stimulation electrode was one-hundredth that of the mechanical probe, 0.031 versus 3.1 mm2).
Mechanisms of VF Induction
In the porcine model of CC, a critical determinant of electrophysiological outcome is precordial impact timing relative to the ECG.8,24,25 Impacts just before the T-wave peak can induce VF, whereas impacts at other stages of the cardiac cycle result in different transient rhythm disturbances.49 The vulnerable time window for precordial impact-induced VF in pig is short: ≈15 ms,49 compared to ≥100 ms for extracorporeal electric stimulation in other large animals (dogs).50 In smaller hearts, vulnerable windows are narrower: we could not trigger VF if impact timing varied by ±5 ms, whereas the electric vulnerable window in rabbit is ≈20 to 25 ms.51
A pig model of CC also highlighted a correlation of VF inducibility with the amplitude of impact-induced intraventricular pressure surges, suggesting that the rapid pressure increase may be causal for electric effects.29 Furthermore, block of mechanosensitive KATP channels by glibenclamide reduced VF occurrence.22 This observation motivated the hypothesis that CC-induced VF represents an acquired form of abnormal repolarization.8,24
The role of intraventricular pressure surges is supported by experiments with acute LV balloon inflation in rabbit isolated hearts.44 Pressure surges of >200 mm Hg, caused by volume injections of 800 to 1600 μL (a ≈80%–160% increase in intraventricular volume; no assessment of tissue integrity reported) result in increased dispersion of repolarization and, occasionally, VF (11% of volume pulses), in an amplitude/timing-dependent manner. In our hands, using hearts of similar size, intraventricular volume increases >500 μL cause structural damage (assessed by creatine kinase release; Figure III in the Data Supplement). In contrast, the exceedingly short-lived pressure surges during CC occur in the absence of a rise in intraventricular volume. Given that tissue viscoelasticity dampens the translation of mechanical stress into stretch, it is likely that CC-induced pressure surges cause little, if any, tissue distension beyond what is induced locally by precordial impact. In keeping with this suggestion, excitation in the in situ pig heart during CC-inducing impacts is focal and originates from tissue immediately underneath the extracorporeal impact site.52
In the present study, rapid infusion and active retraction of 20 μL into the LV balloon result in pressure surges similar in amplitude to those seen during PVEM, albeit with slightly slower dynamics (as expected from an intervention that is not isovolumic per se). Increasing volume pulses up to 130 μL (≈10%–15% of LV volume) gives rise to pressure surges up to an order of magnitude larger than those during PVEM-inducing stimulation, but still does not result in excitation or marked changes in repolarization (volumes exceeding ≈200 μL are needed to trigger PVEM in diastole; Figure 5A). This suggests that precordial impact-induced pressure surges are not the main driver of mechanical VF induction, but a covariate of impact severity.
Of course, it is possible that pressure surges have effects on repolarization in whole animals that are not preserved in isolated hearts, in particular if they involve nervous system responses. Given that CC-induced VF is near instantaneous and that mechanically induced changes in cardiac electrophysiology persist in situ after surgical and pharmacological denervation,53 it remains unlikely that these would be major determinants of electrophysiological outcomes.
In terms of possible roles of KATP channels in CC-induced VF, it is noteworthy that under conditions of normal oxygen supply, KATP channels are inactivated54 and not responsive to mechanical stimulation.19,21,55 In our experiments, application of the KATP channel blocker glibenclamide has no effect on PVEM inducibility, which is in agreement with previous reports from anesthetized pigs.22 We observe a glibenclamide-induced slowing of sinus rate, delayed repolarization, and an increase (by ≈25 ms) in refractory period for PVEM. This is in keeping with nonspecific effects reported for glibenclamide.56,57 The observed changes in repolarization timing and refractoriness exceed the narrow vulnerable time window for VF inducibility. Therefore, glibenclamide application may shift vulnerability for mechanical VF induction past critical timings, identified in control conditions. This could explain the previously reported reduction in VF occurrence during precordial impact with glibenclamide in the pig, where impact timings that previously induced VF failed after drug application.22
In contrast to the view that mechanically induced VF arises primarily out of repolarization abnormalities, 2-dimensional27 and 3-dimensional28 computational modeling, including representations of SACNS, suggests that CC-induced reentry is a consequence primarily of abnormal excitation. In particular, if a mechanical stimulus overlaps the trailing edge of the normal repolarization wave, it can induce VF by causing PVEM in tissue that has regained excitability (ie, where Vm levels are below SACNS reversal potential), while at the same time not only increasing electrophysiological heterogeneity by regional AP shortening in tissue that is still excited (ie, where Vm levels are above SACNS reversal potential) but, crucially, by forming a region of functional conduction block at the intersection of the normal repolarization wave edge and the PVEM-induced excitation. Our results support this prediction. In all cases where epicardial stimulation resulted in VF, the mechanically affected tissue overlapped with the 50% repolarization isochrone, traversing the epicardial surface. The vulnerable time window for CC-induced VF is therefore location specific, existing both in time and in space.
Limitations
The key limitation is the low incidence of local mechanical stimulation-induced VF (n=3/32 animals, 9%), which prevented a more systematic assessment of the vulnerable window. This incidence is in keeping, however, with the exquisite dependence of VF induction on repolarization dynamics, stimulation site, and stimulation timing, whose conditions will be met only rarely, in hearts that display a specifically well-delineated repolarization wave. The necessary conditions occurred in a small subset of hearts only, because of intersubject variability, but in hearts that fulfilled the preconditions, VF was reliably induced. The low incidence of VF in our study is in line with the rarity of CC-induced VF in humans8 and the individual susceptibility in the present gold standard in vivo model of CC.58 In addition, in physiological conditions, Langendorff-perfused rabbit hearts show an exceedingly low probability of VF induction also using electric stimulation.59 This could be further compounded by a reduction in arrhythmia susceptibility by blebbistatin,60 suggesting that our observations form a conservative estimate.
Conclusions
Our findings demonstrate that local subcontusional mechanical stimuli can reliably trigger PVEM, originating at the probe–tissue contact site, and require SACNS, whose activation scales with the degree of local tissue deformation. PVEM cause VF if, and only if, there is overlap of mechanical stimulation with the trailing edge of the preceding repolarization wave. As a result, there is a subject-specific set of vulnerable windows for CC-induced VF both in time and in space.
Acknowledgments
We are indebted to the late Christian Boulin (European Molecular Biology Laboratory, Heidelberg) for his help with instrumentation development. We thank William Stevenson (Cardiovascular Division, Brigham and Women’s Hospital) and Denis Loiselle (Department of Physiology, University of Aukland) for their helpful comments on early stages of the work; Avi Epstein (European Molecular Biology Laboratory, Heidelberg) for his technical support; Jan-Christoph Edelmann, Callum Johnston, Elizabeth Jones, Urszula Siedlecka, and Eva Rog-Zielinska (National Heart and Lung Institute, Imperial College London) for their experimental assistance; Leslie Loew (Richard D. Berlin Center for Cell Analysis and Modeling, University of Connecticut Health Center) for providing access to di-4-ANBDQPQ; Fred Sachs, Tom Suchyna, Philip Gottlieb (Department of Physiology and Biophysics, State University of New York at Buffalo), and Tonus Therapeutics for providing access to Grammostola spatulata MechanoToxin-4 (GsMTx-4); and Stefan Luther and Johannes Schröder-Schetelig (Max Planck Institute for Dynamics and Self-Organisation, Göttingen) for providing the MultiRecorder software (http://www.bmp.ds.mpg.de/multirecorder.html).
Sources of Funding
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC, EP/F042868/1 to Dr Quinn), the British Heart Foundation (BHF, PG/09/066 to Dr Quinn), the European Research Council (ERC, Advanced Grant CardioNECT to Dr Kohl), and the Magdi Yacoub Institute. Dr Quinn was an EPSRC Postdoctoral Fellow; Dr Kohl was a BHF Senior Fellow.
Disclosures
None.
Supplementary Material
Footnotes
The Data Supplement is available at http://circep.ahajournals.org/lookup/suppl/doi:10.1161/CIRCEP.116.004777/-/DC1.
References
- 1.Nesbitt AD, Cooper PJ, Kohl P. Rediscovering commotio cordis. Lancet. 2001;357:1195–1197. doi: 10.1016/S0140-6736(00)04338-5. doi: 10.1016/S0140-6736(00)04338-5. [DOI] [PubMed] [Google Scholar]
- 2.Kohl P, Nesbitt AD, Cooper PJ, Lei M. Sudden cardiac death by Commotio cordis: role of mechano-electric feedback. Cardiovasc Res. 2001;50:280–289. doi: 10.1016/s0008-6363(01)00194-8. [DOI] [PubMed] [Google Scholar]
- 3.Fiaccadori E, Gonzi G, Zambrelli P, Tortorella G. Cardiac arrhythmias during central venous catheter procedures in acute renal failure: a prospective study. J Am Soc Nephrol. 1996;7:1079–1084. doi: 10.1681/ASN.V771079. [DOI] [PubMed] [Google Scholar]
- 4.Kusminsky RE. Complications of central venous catheterization. J Am Coll Surg. 2007;204:681–696. doi: 10.1016/j.jamcollsurg.2007.01.039. doi: 10.1016/j.jamcollsurg.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 5.Bõhm A, Pintér A, Préda I. Ventricular tachycardia induced by a pacemaker lead. Acta Cardiol. 2002;57:23–24. doi: 10.2143/AC.57.1.2005375. doi: 10.2143/AC.57.1.2005375. [DOI] [PubMed] [Google Scholar]
- 6.Lee JC, Epstein LM, Huffer LL, Stevenson WG, Koplan BA, Tedrow UB. ICD lead proarrhythmia cured by lead extraction. Heart Rhythm. 2009;6:613–618. doi: 10.1016/j.hrthm.2009.01.039. doi: 10.1016/j.hrthm.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 7.Lindsay AC, Wong T, Segal O, Peters NS. An unusual twist: ventricular tachycardia induced by a loop in a right ventricular pacing wire. QJM. 2006;99:347–348. doi: 10.1093/qjmed/hcl043. doi: 10.1093/qjmed/hcl043. [DOI] [PubMed] [Google Scholar]
- 8.Maron BJ, Estes NA., III Commotio cordis. N Engl J Med. 2010;362:917–927. doi: 10.1056/NEJMra0910111. doi: 10.1056/NEJMra0910111. [DOI] [PubMed] [Google Scholar]
- 9.White E, Boyett MR, Orchard CH. The effects of mechanical loading and changes of length on single guinea-pig ventricular myocytes. J Physiol. 1995;482(pt 1):93–107. doi: 10.1113/jphysiol.1995.sp020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lab MJ. Depolarization produced by mechanical changes in normal and abnormal myocardium. J Physiol. 1978;284:143P–144P. [PubMed] [Google Scholar]
- 11.Franz MR, Cima R, Wang D, Profitt D, Kurz R. Electrophysiological effects of myocardial stretch and mechanical determinants of stretch-activated arrhythmias. Circulation. 1992;86:968–978. doi: 10.1161/01.cir.86.3.968. [DOI] [PubMed] [Google Scholar]
- 12.Quinn TA, Kohl P, Ravens U. Cardiac mechano-electric coupling research: fifty years of progress and scientific innovation. Prog Biophys Mol Biol. 2014;115:71–75. doi: 10.1016/j.pbiomolbio.2014.06.007. doi: 10.1016/j.pbiomolbio.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Quinn TA. The importance of non-uniformities in mechano-electric coupling for ventricular arrhythmias. J Interv Card Electrophysiol. 2014;39:25–35. doi: 10.1007/s10840-013-9852-0. doi: 10.1007/s10840-013-9852-0. [DOI] [PubMed] [Google Scholar]
- 14.Quinn TA. Cardiac mechano-electric coupling: a role in regulating normal function of the heart? Cardiovasc Res. 2015;108:1–3. doi: 10.1093/cvr/cvv203. doi: 10.1093/cvr/cvv203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peyronnet R, Nerbonne JM, Kohl P. Cardiac mechano-gated ion channels and arrhythmias. Circ Res. 2016;118:311–329. doi: 10.1161/CIRCRESAHA.115.305043. doi: 10.1161/CIRCRESAHA.115.305043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Craelius W. Stretch-activation of rat cardiac myocytes. Exp Physiol. 1993;78:411–423. doi: 10.1113/expphysiol.1993.sp003695. [DOI] [PubMed] [Google Scholar]
- 17.Zabel M, Koller BS, Sachs F, Franz MR. Stretch-induced voltage changes in the isolated beating heart: importance of the timing of stretch and implications for stretch-activated ion channels. Cardiovasc Res. 1996;32:120–130. [PubMed] [Google Scholar]
- 18.White E, Le Guennec JY, Nigretto JM, Gannier F, Argibay JA, Garnier D. The effects of increasing cell length on auxotonic contractions; membrane potential and intracellular calcium transients in single guinea-pig ventricular myocytes. Exp Physiol. 1993;78:65–78. doi: 10.1113/expphysiol.1993.sp003671. [DOI] [PubMed] [Google Scholar]
- 19.Kohl P, Bollensdorff C, Garny A. Effects of mechanosensitive ion channels on ventricular electrophysiology: experimental and theoretical models. Exp Physiol. 2006;91:307–321. doi: 10.1113/expphysiol.2005.031062. doi: 10.1113/expphysiol.2005.031062. [DOI] [PubMed] [Google Scholar]
- 20.Kelly D, Mackenzie L, Hunter P, Smaill B, Saint DA. Gene expression of stretch-activated channels and mechanoelectric feedback in the heart. Clin Exp Pharmacol Physiol. 2006;33:642–648. doi: 10.1111/j.1440-1681.2006.04392.x. doi: 10.1111/j.1440-1681.2006.04392.x. [DOI] [PubMed] [Google Scholar]
- 21.Van Wagoner DR. Mechanosensitive gating of atrial ATP-sensitive potassium channels. Circ Res. 1993;72:973–983. doi: 10.1161/01.res.72.5.973. [DOI] [PubMed] [Google Scholar]
- 22.Link MS, Wang PJ, VanderBrink BA, Avelar E, Pandian NG, Maron BJ, Estes NA., III Selective activation of the K(+)(ATP) channel is a mechanism by which sudden death is produced by low-energy chest-wall impact (commotio cordis). Circulation. 1999;100:413–418. doi: 10.1161/01.cir.100.4.413. [DOI] [PubMed] [Google Scholar]
- 23.Schlomka G. Commotio cordis und ihre folgen. Die einwirkung stumpfer brustwandtraumen auf das herz. Ergebn Inn Med Kinderheilkd. 1934;47:1–91. [Google Scholar]
- 24.Link MS. Commotio cordis: ventricular fibrillation triggered by chest impact-induced abnormalities in repolarization. Circ Arrhythm Electrophysiol. 2012;5:425–432. doi: 10.1161/CIRCEP.111.962712. doi: 10.1161/CIRCEP.111.962712. [DOI] [PubMed] [Google Scholar]
- 25.Link MS. Pathophysiology, prevention, and treatment of commotio cordis. Curr Cardiol Rep. 2014;16:495. doi: 10.1007/s11886-014-0495-2. doi: 10.1007/s11886-014-0495-2. [DOI] [PubMed] [Google Scholar]
- 26.Quinn TA, Kohl P. Combining wet and dry research: experience with model development for cardiac mechano-electric structure-function studies. Cardiovasc Res. 2013;97:601–611. doi: 10.1093/cvr/cvt003. doi: 10.1093/cvr/cvt003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garny A, Kohl P. Mechanical induction of arrhythmias during ventricular repolarization: modeling cellular mechanisms and their interaction in two dimensions. Ann N Y Acad Sci. 2004;1015:133–143. doi: 10.1196/annals.1302.011. doi: 10.1196/annals.1302.011. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Kohl P, Trayanova N. Induction of ventricular arrhythmias following mechanical impact: a simulation study in 3D. J Mol Histol. 2004;35:679–686. doi: 10.1007/s10735-004-2666-8. doi: 10.1007/s10735-004-2666-8. [DOI] [PubMed] [Google Scholar]
- 29.Link MS, Maron BJ, Wang PJ, VanderBrink BA, Zhu W, Estes NA., III Upper and lower limits of vulnerability to sudden arrhythmic death with chest-wall impact (commotio cordis). J Am Coll Cardiol. 2003;41:99–104. doi: 10.1016/s0735-1097(02)02669-4. [DOI] [PubMed] [Google Scholar]
- 30.Dubiel JP, Pyzik Z. [Pressure increases in the left ventricle and aorta during an involuntarily induced cough reflex]. Acta Physiol Pol. 1972;23:587–596. [PubMed] [Google Scholar]
- 31.Quinn TA, Kohl P. Rabbit models of cardiac mechano-electric and mechano-mechanical coupling. Prog Biophys Mol Biol. 2016;121:110–122. doi: 10.1016/j.pbiomolbio.2016.05.003. doi: 10.1016/j.pbiomolbio.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quinn TA, Granite S, Allessie MA, Antzelevitch C, Bollensdorff C, Bub G, Burton RA, Cerbai E, Chen PS, Delmar M, Difrancesco D, Earm YE, Efimov IR, Egger M, Entcheva E, Fink M, Fischmeister R, Franz MR, Garny A, Giles WR, Hannes T, Harding SE, Hunter PJ, Iribe G, Jalife J, Johnson CR, Kass RS, Kodama I, Koren G, Lord P, Markhasin VS, Matsuoka S, McCulloch AD, Mirams GR, Morley GE, Nattel S, Noble D, Olesen SP, Panfilov AV, Trayanova NA, Ravens U, Richard S, Rosenbaum DS, Rudy Y, Sachs F, Sachse FB, Saint DA, Schotten U, Solovyova O, Taggart P, Tung L, Varró A, Volders PG, Wang K, Weiss JN, Wettwer E, White E, Wilders R, Winslow RL, Kohl P. Minimum Information about a Cardiac Electrophysiology Experiment (MICEE): standardised reporting for model reproducibility, interoperability, and data sharing. Prog Biophys Mol Biol. 2011;107:4–10. doi: 10.1016/j.pbiomolbio.2011.07.001. doi: 10.1016/j.pbiomolbio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee P, Bollensdorff C, Quinn TA, Wuskell JP, Loew LM, Kohl P. Single-sensor system for spatially resolved, continuous, and multiparametric optical mapping of cardiac tissue. Heart Rhythm. 2011;8:1482–1491. doi: 10.1016/j.hrthm.2011.03.061. doi: 10.1016/j.hrthm.2011.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooper PJ, Epstein A, Macleod IA, Schaaf ST, Sheldon J, Boulin C, Kohl P. Soft tissue impact characterisation kit (STICK) for ex situ investigation of heart rhythm responses to acute mechanical stimulation. Prog Biophys Mol Biol. 2006;90:444–468. doi: 10.1016/j.pbiomolbio.2005.07.004. doi: 10.1016/j.pbiomolbio.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Quinn TA, Kohl P. Comparing maximum rate and sustainability of pacing by mechanical vs. electrical stimulation in the Langendorff-perfused rabbit heart. Europace. 2016;18(suppl 4):iv85–iv93. doi: 10.1093/europace/euw354. doi: 10.1093/europace/euw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suchyna TM, Johnson JH, Hamer K, Leykam JF, Gage DA, Clemo HF, Baumgarten CM, Sachs F. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J Gen Physiol. 2000;115:583–598. doi: 10.1085/jgp.115.5.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belus A, White E. Streptomycin and intracellular calcium modulate the response of single guinea-pig ventricular myocytes to axial stretch. J Physiol. 2003;546(pt 2):501–509. doi: 10.1113/jphysiol.2002.027573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garan AR, Maron BJ, Wang PJ, Estes NA, III, Link MS. Role of streptomycin-sensitive stretch-activated channel in chest wall impact induced sudden death (commotio cordis). J Cardiovasc Electrophysiol. 2005;16:433–438. doi: 10.1046/j.1540-8167.2005.40664.x. doi: 10.1046/j.1540-8167.2005.40664.x. [DOI] [PubMed] [Google Scholar]
- 39.Cooper PJ, Kohl P. Species- and preparation-dependence of stretch effects on sino-atrial node pacemaking. Ann N Y Acad Sci. 2005;1047:324–335. doi: 10.1196/annals.1341.029. doi: 10.1196/annals.1341.029. [DOI] [PubMed] [Google Scholar]
- 40.Link MS, Estes NA, III, Maron BJ American Heart Association Electrocardiography and Arrhythmias Committee of Council on Clinical Cardiology, Council on Cardiovascular Disease in Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and American College of Cardiology. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 13: Commotio Cordis: A Scientific Statement From the American Heart Association and American College of Cardiology. Circulation. 2015;132:e339–e342. doi: 10.1161/CIR.0000000000000249. doi: 10.1161/CIR.0000000000000249. [DOI] [PubMed] [Google Scholar]
- 41.Rudy Y. Noninvasive electrocardiographic imaging of arrhythmogenic substrates in humans. Circ Res. 2013;112:863–874. doi: 10.1161/CIRCRESAHA.112.279315. doi: 10.1161/CIRCRESAHA.112.279315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Opthof T, Coronel R, Wilms-Schopman FJ, Plotnikov AN, Shlapakova IN, Danilo P, Jr, Rosen MR, Janse MJ. Dispersion of repolarization in canine ventricle and the electrocardiographic T wave: Tp-e interval does not reflect transmural dispersion. Heart Rhythm. 2007;4:341–348. doi: 10.1016/j.hrthm.2006.11.022. doi: 10.1016/j.hrthm.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 43.Panikkath R, Reinier K, Uy-Evanado A, Teodorescu C, Hattenhauer J, Mariani R, Gunson K, Jui J, Chugh SS. Prolonged Tpeak-to-tend interval on the resting ECG is associated with increased risk of sudden cardiac death. Circ Arrhythm Electrophysiol. 2011;4:441–447. doi: 10.1161/CIRCEP.110.960658. doi: 10.1161/CIRCEP.110.960658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bode F, Franz MR, Wilke I, Bonnemeier H, Schunkert H, Wiegand UK. Ventricular fibrillation induced by stretch pulse: implications for sudden death due to commotio cordis. J Cardiovasc Electrophysiol. 2006;17:1011–1017. doi: 10.1111/j.1540-8167.2006.00547.x. doi: 10.1111/j.1540-8167.2006.00547.x. [DOI] [PubMed] [Google Scholar]
- 45.Hansen DE, Craig CS, Hondeghem LM. Stretch-induced arrhythmias in the isolated canine ventricle. Evidence for the importance of mechanoelectrical feedback. Circulation. 1990;81:1094–1105. doi: 10.1161/01.cir.81.3.1094. [DOI] [PubMed] [Google Scholar]
- 46.Hansen DE, Borganelli M, Stacy GP, Jr, Taylor LK. Dose-dependent inhibition of stretch-induced arrhythmias by gadolinium in isolated canine ventricles. Evidence for a unique mode of antiarrhythmic action. Circ Res. 1991;69:820–831. doi: 10.1161/01.res.69.3.820. [DOI] [PubMed] [Google Scholar]
- 47.Kohl P, Hunter P, Noble D. Stretch-induced changes in heart rate and rhythm: clinical observations, experiments and mathematical models. Prog Biophys Mol Biol. 1999;71:91–138. doi: 10.1016/s0079-6107(98)00038-8. [DOI] [PubMed] [Google Scholar]
- 48.Brooks CM, Gilbert JL, Suckling EE. Excitable cycle of the heart as determined by mechanical stimuli. Proc Soc Exp Biol Med. 1964;117:634–637. doi: 10.3181/00379727-117-29656. [DOI] [PubMed] [Google Scholar]
- 49.Link MS, Wang PJ, Pandian NG, Bharati S, Udelson JE, Lee MY, Vecchiotti MA, VanderBrink BA, Mirra G, Maron BJ, Estes NA., III An experimental model of sudden death due to low-energy chest-wall impact (commotio cordis) N Engl J Med. 1998;338:1805–1811. doi: 10.1056/NEJM199806183382504. doi: 10.1056/NEJM199806183382504. [DOI] [PubMed] [Google Scholar]
- 50.Shibata N, Chen PS, Dixon EG, Wolf PD, Danieley ND, Smith WM, Ideker RE. Influence of shock strength and timing on induction of ventricular arrhythmias in dogs. Am J Physiol. 1988;255(4)(pt 2):H891–H901. doi: 10.1152/ajpheart.1988.255.4.H891. [DOI] [PubMed] [Google Scholar]
- 51.Behrens S, Li C, Kirchhof P, Fabritz FL, Franz MR. Reduced arrhythmogenicity of biphasic versus monophasic T-wave shocks. Implications for defibrillation efficacy. Circulation. 1996;94:1974–1980. doi: 10.1161/01.cir.94.8.1974. [DOI] [PubMed] [Google Scholar]
- 52.Alsheikh-Ali AA, Akelman C, Madias C, Link MS. Endocardial mapping of ventricular fibrillation in commotio cordis. Heart Rhythm. 2008;5:1355–1356. doi: 10.1016/j.hrthm.2008.03.009. doi: 10.1016/j.hrthm.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 53.Quinn TA, Kohl P. Mechano-sensitivity of cardiac pacemaker function: pathophysiological relevance, experimental implications, and conceptual integration with other mechanisms of rhythmicity. Prog Biophys Mol Biol. 2012;110:257–268. doi: 10.1016/j.pbiomolbio.2012.08.008. doi: 10.1016/j.pbiomolbio.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 55.Van Wagoner DR, Lamorgese M. Ischemia potentiates the mechanosensitive modulation of atrial ATP-sensitive potassium channels. Ann N Y Acad Sci. 1994;723:392–395. [PubMed] [Google Scholar]
- 56.Babes A, Fischer MJ, Filipovic M, Engel MA, Flonta ML, Reeh PW. The anti-diabetic drug glibenclamide is an agonist of the transient receptor potential Ankyrin 1 (TRPA1) ion channel. Eur J Pharmacol. 2013;704:15–22. doi: 10.1016/j.ejphar.2013.02.018. doi: 10.1016/j.ejphar.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 57.Rabe A, Disser J, Frömter E. Cl- channel inhibition by glibenclamide is not specific for the CFTR-type Cl- channel. Pflugers Arch. 1995;429:659–662. doi: 10.1007/BF00373986. [DOI] [PubMed] [Google Scholar]
- 58.Alsheikh-Ali AA, Madias C, Supran S, Link MS. Marked variability in susceptibility to ventricular fibrillation in an experimental commotio cordis model. Circulation. 2010;122:2499–2504. doi: 10.1161/CIRCULATIONAHA.110.955336. doi: 10.1161/CIRCULATIONAHA.110.955336. [DOI] [PubMed] [Google Scholar]
- 59.Kurz RW, Xiao-Lin R, Franz MR. Increased dispersion of ventricular repolarization and ventricular tachyarrhythmias in the globally ischaemic rabbit heart. Eur Heart J. 1993;14:1561–1571. doi: 10.1093/eurheartj/14.11.1561. [DOI] [PubMed] [Google Scholar]
- 60.Lou Q, Li W, Efimov IR. The role of dynamic instability and wavelength in arrhythmia maintenance as revealed by panoramic imaging with blebbistatin vs. 2,3-butanedione monoxime. Am J Physiol Heart Circ Physiol. 2012;302:H262–H269. doi: 10.1152/ajpheart.00711.2011. doi: 10.1152/ajpheart.00711.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]