

Abstract
There are currently no FDA-approved antidotes for H2S/sulfide intoxication. Sodium nitrite, if given prophylactically to Swiss Webster mice, was shown to be highly protective against the acute toxic effects of sodium hydrosulfide (~LD40 dose) with both agents administered by intraperitoneal injections. However, sodium nitrite administered after the toxicant dose did not detectably ameliorate sulfide toxicity in this fast-delivery, single-shot experimental paradigm. Nitrite anion was shown to rapidly produce NO in the bloodstream, as judged by the appearance of EPR signals attributable to nitrosylhemoglobin and methemoglobin, together amounting to less than 5% of the total hemoglobin present. Sulfide-intoxicated mice were neither helped by the supplemental administration of 100% oxygen nor were there any detrimental effects. Compared to cyanide-intoxicated mice, animals surviving sulfide intoxication exhibited very short knockdown times (if any) and full recovery was extremely fast (~15 min) irrespective of whether sodium nitrite was administered. Behavioral experiments testing the ability of mice to maintain balance on a rotating cylinder showed no motor impairment up to 24 h post sulfide exposure. It is argued that antagonism of sulfide inhibition of cytochrome c oxidase by NO is the crucial antidotal activity of nitrite rather than formation of methemoglobin.
Graphical Abstract
INTRODUCTION
Methods for generating hydrogen sulfide gas (H2S) from household chemical products obtainable at multiple retail outlets have been publicized through the Internet, and suicide by H2S inhalation is an emerging trend in some countries.1,2 Significantly, it appears that neighbors not in the immediate vicinity of the release site, or emergency responders, were sometimes affected/injured,3,4 leading to a growing realization that H2S might find application as a weapon.1 Additionally, there are approximately 102 known commercial sources of H2S, resulting in thousands of occupational exposures per year in the US alone, including individuals engaged in paper pulping, tanning, vulcanizing, management of animal waste, sewer maintenance, heavy water production,5,6 and, especially, natural gas mining.7 Preventable deaths of inadequately protected would-be rescuers, both co-workers and emergency personnel, have been reported.8 It is, therefore, of concern that no FDA-approved antidote, or reliable protocol, for treating acute H2S intoxication is currently available.9 Emergency medicine bulletins/pamphlets issued by several international, federal, and state authorities10–16 suggest the use of cyanide antidote kits containing nitrite-thiosulfate, or cobalamin, but the basic science that would justify this approach is lacking. Moreover, there are conflicting anecdotal case reports attesting to both the success and failure of cyanide therapeutics administered in situations where H2S was known or suspected to be the toxic agent.17 More seriously, we have recently shown that the production of hydrosulfide (HS−) in the blood from thiosulfate administered as a cyanide antidote is measurably toxic.18 Therefore, the suggested use of the nitrite-thiosulfate combination as a therapy for H2S poisoning is alarming. Certainly the toxicology of H2S shares features in common with that of cyanide; for instance, both toxins are highly efficient disruptors of mitochondrial electron-transport chain function19–21 with approximately identical inhibition constants (Ki) for cytochrome c oxidase.18,22 It follows that in developing potential therapies for treating acute H2S intoxication initial efforts should be directed toward overcoming inhibition of cytochrome c oxidase and the associated rapid cardiopulmonary collapse.9
The first pKa of H2S is 6.9–7.0 (37–25 °C), with the second pKa being inaccessible in water,23,24 which results in aqueous solutions of approximately 30–25% H2S and 70–75% HS− at pH 7.4, irrespective of whether it was initially introduced as the molecular gas or as a salt (e.g., NaHS). In keeping with what seems to be a common convention, we refer to this mixture at physiological pH as sulfide (and do not mean to imply S2− at any time herein). Haouzi et al.25 have recently shown that, at sublethal levels, intravenously administered aqueous sulfide becomes converted to other forms in the bloodstream of sheep and rats within 1 min of infusion. This seems to be incompatible with the findings of Truong et al.,26 who reported that cobalamin given intraperitoneally greatly increases the survival rate of sulfide toxicity in mice when administered 2 min after the toxicant. Curiously, these latter authors report death following sulfide intoxication in a 0–24 h window with no mention of the rapid effects that we describe below. To further confound matters, suspected human victims of H2S inhalation reaching the clinic have been reported to succumb to the poisoning hours after the exposure,20,27 possibly indicating additional slower mechanisms of toxicity subsidiary to cytochrome c oxidase inhibition. In this study, we attempt to resolve these puzzling observations with regard to the acute toxicity of sulfide.
Sodium nitrite has periodically been mentioned as a possible antidote for H2S for over 30 years,20,28–30 on the basis of the assumption that detoxification would result from nitrite-induced generation of methemoglobin (metHb) followed by binding of sulfide to form sulfidomethemoglobin (metHbSH).9 However, we propose this widely held mechanistic belief to be erroneous. The matter is a critical issue to overcome because concerns that increased metHb levels (methemoglobinemia) would further challenge the oxygen utilization capabilities of already sulfide-compromised individuals have almost certainly hindered the rational investigation of nitrite as a possible frontline antidote. In the related case of acute cyanide intoxication, we have shown that there need be no significant methemoglobinemia following administration of sodium nitrite (<2% metHb18), as the principle antidotal action is to generate nitric oxide (NO) that ameliorates inhibition of cytochrome c oxidase function.31,32 Herein, we have examined the antidotal activity of sodium nitrite toward sulfide toxicity and the potential ameliorating effects of supplemental oxygen, employing a combination of behavioral assessments on mice and spectroscopic (EPR) measurements on drawn blood. A variety of sequelae, secondary to sulfide poisoning, have been anecdotally reported in humans, including neurological defects. Accordingly, we have performed some neuromuscular assessments on mice following recovery from sulfide intoxication to determine if any improved survivability observed with nitrite administration might also be associated with undesirable neurological consequences.
EXPERIMENTAL METHODS
Chemicals
All reagents were ACS grade, or better, used without further purification, and, unless stated to the contrary, were purchased from Fisher or Sigma-Aldrich. Argon, carbon dioxide, nitric oxide, nitrogen, and oxygen gases were purchased from Matheson Incorporated and, with the exception of nitric oxide (see Enzyme Preparation and Cell Culture), used without further purification. Sodium hydrosulfide or sodium cyanide solutions were prepared in septa-sealed vials with minimized headspaces immediately prior to use, and volumetric transfers were made with gastight syringes. Sodium hydrosulfide was obtained as NaHS·xH2O (Sigma), where x was determined to be 2.5 essentially following procedures described by Koltoff et al.33 Briefly, concentrations of HS− were determined by quantitative reaction with excess iodine (2HS− + I2 → S + 2H+ + 2I−) followed by titration of the liberated iodide (as I− + I3−) with silver nitrate (precipitating AgI + AgI3) using an Ag+-sensitive ion-selective potentiometric electrode (Accumet Silver/Sulfide Combination ISE 13-620-551) to detect the end point.
Animals, Exposure, and Blood Collection
All animal procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (protocol no. 13092637). Veterinary care was provided by the Division of Laboratory Animal Research of the University of Pittsburgh. Male Swiss Webster (CFW) mice weighing 40–50 g were purchased from Taconic (Hudson, NY). Adult animals were 16–18 weeks old and were housed four per cage. The mice were allowed access to food and water ad libitum. Experiments commenced after the animals were allowed to adapt to their new environment for 1 week. A series of experiments testing the efficacy of sodium nitrite as a sulfide antidote were performed. All solutions were prepared by dilutions into sterilized saline in septa-sealed vials using gastight syringes for all transfers and administered through ~0.1 mL intraperitoneal (ip) injections. In general, a group of at least 6 mice were tested for each experimental point. At the end of exposures and tests, mice were euthanized with CO2. Juvenile male Swiss Webster mice 6–8 weeks old, 25–35 g, supplied by Taconic, were treated in the same fashion.
For collection of blood samples, mice were first euthanized in an atmosphere of carbon dioxide and blood was drawn by cardiac puncture into a 1 mL syringe containing 25 μL of 500 mM EDTA. The blood was expelled into the bottom of a quartz EPR tube containing an argon atmosphere and then frozen by immersion in liquid nitrogen. This entire process could comfortably be completed in 2 min. The cryogenically preserved sample was stored in liquid nitrogen and subsequently transferred to the EPR spectrometer without ever having been thawed. Samples of heart tissue were also collected. Euthanized animals were perfused with 5 mL of PBS by cardiac puncture, and then the hearts were removed and immediately frozen in liquid nitrogen. After storage at −80 °C, individual hearts were thawed and homogenized in 1 mL of PBS, and a 200 μL aliquot of this homogenate was introduced into an EPR tube and frozen in liquid nitrogen before subsequent transfer to the spectrometer.
Righting-Recovery Testing
Lengths of time required for recovery of righting ability in mice were determined based on some of the recommendations of Crankshaw et al.34 regarding their measurement of the righting reflex but adopting a simpler procedure. Following ip administration of toxicant (±NaNO2), mice were placed in a transparent but dark green-colored plastic tube (Kaytee CritterTrail, available from pet stores) in a supine position. The time duration from the toxicant injection until the mouse flipped from the supine to a prone position in the plastic tube was taken as the end point.
RotaRod Testing
To assess motor skill learning and recovery following intoxication, we used the accelerating RotaRod (Coulbourn Instruments, Whitehall, PA), a rotating cylindrical apparatus (4 to 40 rpm) on which the mice were placed. The animals were evaluated for three trials per time point on three consecutive days, with a resting time of 30 s between each trial. An individual trial was considered to have ended when the mouse either fell off or had remained on the rotating cylinder for 60 s. Latency to fall and highest speed reached were recorded for each trial. Mice were trained for eight trials on the first day by placing them on an accelerating RotaRod for 60 s, during which time the rotation rate was varied linearly from 4 to 11.2 rpm. On the day of the toxicity experiments (day 2), animals were tested for a single set of three trials, with the same parameters as those used during training to establish a baseline performance, before injection. Trained animals were tested at 15 min intervals after toxicant administration for 2 h and an additional time 24 h after injection, by placing them on an accelerating RotaRod for 60 s, accelerating from 4 to 22 rpm. Motor performance was determined to be the highest rotation speed reached before the animal fell off the apparatus, determined from the mean rpm in three trials for each mouse at each experimental time point. For comparison between groups, the mean performance in preinjection testing for a group was used to normalize all of the other experimental points of that group.
Enzyme Preparation and Cell Culture
Bovine pulmonary artery endothelial cells (BPAEC) were purchased from Lonza and used at passages 4–8. Cells were grown in Opti-MEM media supplemented with 10% fetal bovine serum, 5 mM glutamate, penicillin, and streptomycin under 5% CO2. Cytochrome c oxidase was prepared as previously described31 from intact bovine heart mitochondria using a modified Harzell–Beinert procedure (without the preparation of Keilin–Hartree particles). The enzyme was determined to be spectroscopically pure if the 444 to 424 nm absorbance ratio for the reduced enzyme was 2.2 or higher. Derivatives were prepared in 50 mM potassium phosphate, 1 mM in sodium EDTA, and 0.1% in lauryl maltoside, pH 7.4–7.8, to concentrations of 10–80 μM (in enzyme). Enzyme concentrations were determined as total heme a using the differential extinction coefficient of Δε604 = 12 mM−1 cm−1 for the reduced minus oxidized electronic absorption spectra.35 Concentrations throughout are given on a per enzyme concentration basis (NOT per [heme a]). Ferrocytochrome c:O2 oxidoreductase activity was determined spectrophotometrically employing the high ionic strength method of Sinjorgo et al.36 Electronic absorption spectra were measured and photometric determinations were made using Shimadzu UV-1650PC and UV-2501PC spectrophotometers. Nitric oxide (for reactions with cytochrome c oxidase) was scrubbed with water and KOH pellets prior to use, bubbled through anaerobic buffer (prepared by bubbling argon through the solution), and added to enzyme samples volumetrically with gastight syringes. Buffered solutions never exhibited any significant change of pH (i.e., < 0.05 pH units) following NO additions.
Electron Paramagnetic Resonance (EPR)
X-band (9 GHz) EPR spectra were recorded on a Bruker ESP 300 spectrometer equipped with an Oxford Instruments ESR 910 flow cryostat for ultra-low-temperature measurements. Access to this instrument and the software (SpinCount) used to analyze the EPR spectra were provided by Professor Michael Hendrich, Carnegie Mellon University. Quantification of EPR signals was performed by simulating the spectra using known (or determined) parameters for each sample in question. Simulations employed a least-squares fitting method to match the line shape and signal intensity of a selected spectrum. Simulated spectra were expressed in terms of an absolute intensity scale, which could then be related to sample concentration through comparison with a CuII(EDTA) spin standard of known concentration.
Data Analysis
Statistical data was analyzed using GraphPad Prism 6 software by t-test. A p-value ≤ 0.05 was considered to be statistically significant.
RESULTS
Comparison of Sulfide and Cyanide Toxicities
It is frequently noted that there are parallels between the acute toxicities of H2S versus HCN, and it will be instructive to explore this comparison further (e.g., Table 1). In our procedures, where the toxicants are given to mice as ip injections of sodium salts in saline solutions at approximately LD50 doses, those animals that succumb typically do so within 2–4 min, in keeping with the well-documented rapid action of these poisons. In the particular case of sulfide, we have to date observed deaths in excess of 200 animals, with more than 98% of these occurring within 5 min of the toxicant dose, 2 individuals died in the 5–10 min period, and only 1 individual in the 10 min to 24 h window. Consequently, we are very surprised by the methodology and observations of Truong et al.,26 who report returning mice to their cages after giving similar ip doses of aqueous NaHS to record deaths 24 h later, with no mention of any fatalities in the first few minutes following administration of the toxicant. The antidotal regimen adopted by these same authors involved ip administration of hydroxycobalamin solutions at 2 min following the toxicant dose and not at any later times in the 24 h experimental window before deaths were recorded. A time delay of only 2 min between the toxicant and antidote doses for efficacy suggests to us that these authors were probably dealing with the same kind of acute response as we now report (i.e., the majority of deaths within a few minutes of the toxicant dose) but for some reason left this unclear.
Table 1.
Summary of HCN and H2S Toxicological Observations in Mice
| NaCN/HCN18 (~99% HCN at pH 7.4, 37 °C) | NaHS/H2S (~25% H2S at pH 7.4, 37 °C) | |
|---|---|---|
| strain/sex | Swiss Webster/males | Swiss Webster/males |
| supplier | Charles River | Taconic |
| age | 17 weeks | 17 weeks |
| median weight | 40 g | 43 g |
| mean weight | 39.8 ± 6.6 ga | 43.0 ± 4.0 ga |
| toxicant dose | 5.0 mg/kg (LD33 − LD50) ip | 16 mg/kg (LD40) ip |
| total knockdowns | 100% | 71% (20 of 28) |
| knockdown duration | 30.5 ± 8 mina | 3.5 ± 1.4 mina |
| survival with knockdown | 50−67% | 40% (8 of 20) |
| mode of death | respiratory paralysis | respiratory paralysis |
| time to full recovery | <2 h | ~15 min |
The quoted uncertainties are standard deviations.
A period of unconsciousness, or knockdown, is a common acute symptom of exposure to both sulfide9 and cyanide.37 With a simplified modification of a procedure developed by Crankshaw et al.,34 we have previously assessed sublethal cyanide intoxication in mice and its antagonism using the observed duration of knockdown to indicate extent of incapacitation and recovery.18,38,39 Injection of mice with 0.1 mmol/kg (5 mg/kg, ip) NaCN in saline results in loss of consciousness, with clear indication of the onset of narcosis (animals stagger or are motionless) beginning at around 1 min following administration of the toxicant. Shortly thereafter, the animals may be placed on their backs (supine position) and observed until they regain consciousness, at which time they turn themselves to an upright (prone) position. The observed righting-recovery time generally lasts around 30 min, with about 60% survival in the case of cyanide intoxication at the stated dose. The method has proven to be suitable for demonstrating the efficacy of putative antidotes given before or up to 20 min after the cyanide.18 Unfortunately, however, the same method proved to be impractical for use with respect to sulfide as the toxicant. Mice that experienced sulfide-induced knockdowns were more likely to die than to survive, and, in fact, only about one-quarter of all surviving animals experienced knockdown (Table 1). Furthermore, using 0.29 mmol/kg (16 mg/kg, ip) NaHS in saline (57% survival) caused a knockdown duration of only 3.5 ± 1.4 min (n = 28). Increasing the dose incrementally did not lead to any significant lengthening of knockdown times observed, and at 20 mg/kg, all of the mice injected (n = 6) died within 5 min. That is, in the case of sulfide intoxication, knockdown/righting-recovery was the atypical response, variable and short, necessitating that an unreasonably large number of animals would have been needed to demonstrate any beneficial effects of putative antidotes by this method. Therefore, we reluctantly investigated the efficacy of NaNO2 toward antagonism of NaHS toxicity using death or survival (observed at 15 min and 24 h) as the end point.
In addition to the clear differences in frequency of response and knockdown duration noted above with sulfide- and cyanide-dosed mice, the recovery times were also found to be quite distinct. Following a toxicant dose (~LD40) with no antidote given, sulfide-intoxicated mice took about 15 min to recover essentially normal behavior, whereas almost 2 h had elapsed before cyanide-intoxicated mice exhibited similar recovery (Table 1 and RotaRod experiments described below). It appears from these observations that the part(s) of the central nervous system dealing with consciousness is (are) significantly more deeply affected by sublethal cyanide intoxication than is the case with sulfide. The experimentally established LD40 doses of NaHS and NaCN, 16 and 5.0 mg/kg, respectively, represent a molar ratio of ~3:1 (total sulfide/total cyanide). As the relevant pKa’s are 6.9 and 9.2, respectively,23,24 this implies [H2S] ≅ [HCN] circulating in the bloodstreams at pH 7.4; that is, the toxicant species expected to be most readily membrane-permeable were administered at the same level. This makes sense, given that we can anticipate the rate of diffusion of the toxicants from the bloodstream to depend upon mass action and the principle target molecule, cytochrome c oxidase, is similarly inhibited by sulfide and cyanide.22 In summary, while the pattern of sublethal narcosis clearly differs between the two toxicants, our present data are fully consistent with the notion that the two mechanisms of lethality are essentially analogous, with pulmonary function being principally affected.
Potential Antidotes to Sulfide Poisoning
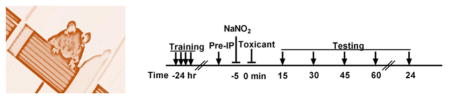
Doubts and conflicting reports have continued to persist regarding the usefulness of sodium nitrite as a sulfide antidote for almost 40 years.9,28,40–42 On the basis of our recent experience with regard to cyanide and nitrite, we suspected that some of the confusion in the literature might stem from an incorrect understanding of how the antidotal action of nitrite might best be mechanistically explained. The general consensus is that cyanide exerts its acute toxicity primarily on the central nervous system through inhibition of cytochrome c oxidase, with death primarily the result of interruption to the pulmonary nervous supply.37,43 We have shown that nitric oxide (NO) relieves the inhibition of cytochrome c oxidase by cyanide,31,32 leading to the suggestion that the antidotal action of nitrite toward cyanide poisoning involves the anion functioning as an NO donor to alleviate electron-transport chain inhibition by cyanide.18,38 Consequently, we set out to investigate whether sodium nitrite might also be antidotal toward sulfide poisoning through a mechanism in which the anion acts as an NO donor. We began with a prophylactic paradigm (Figure 1A). Previously, 12 mg/kg NaNO2 was established as the optimal antidotal dose in the case of acute cyanide toxicity.18,38 This same dose, however, resulted in only a modest improvement in survival of 79% (Figure 1B) for sulfide toxicity. When 24 mg/kg NaNO2 was administered 5 min before a 16 mg/kg NaHS dose, survival was significantly increased to 93% (Figure 1B), compared to 57% in the case of controls given no nitrite (Figure 1B). Several groups of authors have suggested that delivery of supplemental oxygen during treatment for sulfide poisoning may be beneficial,44–47 and, indeed, while the supporting evidence is anecdotal, the idea can be rationalized on the basis that the known detoxification pathway of sulfide uses oxygen.9,17 We investigated the matter experimentally. Following prophylactic doses of NaNO2, mice were maintained under normoxic conditions for 5 min and then placed in a 100% oxygen environment immediately after injections of NaHS. Control animals were given just NaHS and placed in the 100% oxygen chamber. Exposures to 100% oxygen were discontinued after 15 min or at time of death (<15 min). There was no detectable improvement in survival of mice provided with the supplemental oxygen following the toxicant dose (Figure 1B) compared to those maintained under normoxic conditions throughout (Figure 1B). While this result could be considered negative, it is, nevertheless, important. Faced with patients in respiratory distress, it is quite normal practice for emergency responders to provide supplemental oxygen if available. Therefore, it is comforting that our data show no effect of supplemental oxygen, neither good nor bad, suggesting that the protocol should at least do no harm in cases of sulfide intoxication.
Figure 1.

Prophylactically administered NaNO2 ameliorates NaHS toxicity in mature and juvenile mice. (A) Injection paradigm. (B) Mice (Swiss Webster males, 16–18 weeks of age) were given NaHS in saline (16 mg/kg, ip), and times until death were recorded. The duration of survival (breathing cessation) was measured from the time of the sulfide injection (t = 0). Survival quotients are shown with surviving mice/total mice written above the bar. NaNO2 (12 or 24 mg/kg, ip) was given 5 min prior to NaHS injection. Supplemental oxygen (100% O2) was administered for either 15 min or until death immediately after NaHS injections (* p ≤ 0.05 vs NaHS injection alone). (C) Juvenile mice (Swiss Webster males, 6–8 weeks old) were injected (ip) with either 16, 18, or 20 mg/kg NaHS, and survival was recorded as for adults. In addition, 24 mg/kg NaNO2 was given 5 min before 18 mg/kg NaHS (* p ≤ 0.05 vs 18 mg/kg NaHS injection).
Unfortunately, in experiments where NaNO2 (24 mg/kg) was administered 1–2 min after the toxicant (NaHS, 16 mg/kg), there was no significant improvement in survival observed (data not shown). While NaNO2 was not beneficial when administered after sulfide doses, we were able to confirm the results of Truong et al.26 in which administration of hydroxycobalamin acetate (ip) at 2 min after the NaHS dose (ip) increased survival to 80% (data not shown). We interpret these observations to reinforce the idea that cobalamin binds sulfide circulating in the bloodstream, slowing the passage of the toxicant to the tissues in the critical 2–4 min period during which the majority of deaths were observed. Nitrite, on the other hand, does not exert its primary action in the bloodstream (see below), and the NO released must reach inhibited mitochondria within tissues to reverse the effect of the toxicant. As the maximal release of NO occurs about 5 min after ip injection,18 this method of NaNO2 delivery is too slow for post-NaHS use in the current scenario, and, ultimately, alternate methods (e.g., aerosol inhalation) will probably have to be considered.
Sulfide Toxicity in Juvenile Mice
It is of interest to determine if juveniles are more susceptible to the effects of sulfide. To this end, we carried out a series of experiments examining the dose response to sulfide and the efficacy of nitrite as an antidote in 6–8 week old mice. As shown in Figure 1C, juveniles proved to be slightly more resistant to sulfide, as a dose of 18 mg/kg (Figure 1C) was needed to obtain a similar level of survival compared to adult mice exposed to 16 mg/kg. It is of interest to note that at 2 mg/kg more (20 mg/kg) survival dropped precipitously from 67 to 14%. This kind of steep relationship was also observed in preliminary testing with adults (data not shown). Survival was improved significantly upon the prophylactic administration NaNO2 (Figure 1C) at the same dose (24 mg/kg) as that in the adults. Also like the adults, those sublethally intoxicated juvenile subjects recovered quickly (~15 min), and all deaths occurred within 5 min of receiving the toxicant dose; none were observed in the 5 min to 24 h time window.
Recovery of Neuromuscular Function Following Acute Sulfide Intoxication
There is anecdotal evidence in the occupational medicine literature for neurological dysfunction in humans following acute exposures to H2S.9,27 To address this possibility experimentally, we employed an approach based on the RotaRod device, primarily a determinant of neuromuscular coordination, but there is also assessment of muscle strength, with a more limited cognitive component. The mice were trained 24 h before the intoxication procedures, and baseline peformance was established at 1 h prior to administration of toxicant. Following toxicant injections, mice were then tested every 15 min, up to 1 h, and subsequently at 24 h (Figure 2A). There was no significant difference in RotaRod performance of mice (Figure 2B) given 16 mg/kg NaHS (●), 24 mg/kg NaNO2 (□), or NaNO2 + NaHS (▲). In general, the animals improve with practice, so the increased performance between 15 and 45 min post-intoxication should be taken to indicate a learning curve, as it has a slope similar to that observed during the training period (Figure 2B). These data show that all of the animals have essentially recovered neuromuscular coordination at 15 min after administration of NaHS, irrespective of whether nitrite was given. A comparison (Figure 2C) of the RotaRod performace of mice given 16 mg/kg NaHS (●), 5.0 mg/kg NaCN (◆), or NaNO2 + NaCN (□) showed that the cyanide-intoxicated mice had a longer recovery time (~2 h) compared to sulfide (15 min). Furthermore, while administering nitrite shortened the recovery time, the performance of these nitrite-treated and cyanide-intoxicated mice still clearly lagged behind that of the sulfide-intoxicated animals (Figure 2C). Nevertheless, it is to be noted that, in the case of both toxicants, there was no indication of persistent impairment of neuromuscular coordination or readily apparent cognitive (learning/memory) issues as reported for humans; all of the surviving animals rapidly made full recoveries, as judged by the RotaRod testing, and did not develop any latent problems at 24 h.
Figure 2.

RotaRod testing of neuromuscular coordination following NaHS/NaCN/NaNO2 exposures in adult Swiss Webster mice. (A) RotaRod testing paradigm: arrows indicate RotaRod testing times; lines with bars indicate injection times (all ip). Mice were trained on the RotaRod 24 h before injection, and a baseline performance was obtained 1 h before injection (Preip). Mice were tested every 15 min after injections for 1 h to assess recovery. (B) Comparison of performance for injections of 16 mg/kg NaHS (●), 24 mg/kg, NaNO2 (□) and 24 mg/kg NaNO2 injected 5 min prior to 16 mg/kg NaHS dose (▲). (C) Comparison of performance for injections of 16 mg/kg NaHS (●), 6.4 mg/kg NaCN (◆), or 24 mg/kg NaNO2 injected 5 min prior to 6.4 mg/kg NaCN (□). Numbers of animals (in parentheses) used in each set of experiments: NaHS (6), NaNO2 (8), NaNO2 + NaHS, (8), NaCN (9), NaNO2 + NaCN (10). (*, p ≤ 0.05 vs controls.)
Nitrite-Dependent Release of NO in Blood and Heart Muscle
We have previously shown that minority hemoglobin species in blood samples (HbNO, metHb, and metHbS) can be quite accurately determined using EPR spectroscopy.18,38 The addition of sodium nitrite to mice followed by euthanasia with CO2 and sample preparation (see below) led to dose-dependent (0–24 mg/kg) EPR signals, identified as HbNO and metHb. In general, the amount of HbNO observed in the blood (a maximum of 0.5 mM, or ~6% of the total hemoglobin) was roughly 2 to 3 times the amount of metHb (0.15 mM, or ~2% of the total hemoglobin). Followed over time, the signal intensities peaked at 10–15 min and measurably persisted up to 1 h after administration of the nitrite dose.18,38 The presence of both signals could be construed as evidence for the presence of NO in the blood rather than nitrite (HbO2 + NO → metHb + nitrate; Hb + NO → HbNO), but as the signals were not present in the spectra of blood samples taken from control animals, they clearly arose in a nitrite-dependent manner. In the present study, mice were given NaNO2 (12–24 mg/kg in saline, ip) and then later euthanized in an atmosphere of CO2 starting at 7 min after the nitrite dose. We chose this delay because previously the maximal level of NO-dependent EPR signals was found between 5 and 15 min following NaNO2 administration. Within 2 min of starting euthanasia, blood had been withdrawn by cardiac puncture, 250 μL was dispensed into an EPR tube, and the sample cryogenically preserved by immersion in liquid nitrogen. When required, NaHS (16 mg/kg in saline, ip) was administered either alone or 5 min following the nitrite dose (i.e., 2 min before commencing euthanasia).
The EPR spectra of control blood samples, from animals given neither sulfide nor nitrite, exhibit only weak signals arising from transferrin at ~1600 gauss (not shown). In samples drawn from mice given NaNO2, both metHb (~1100 gauss) and HbNO (~3400 gauss) EPR signals were readily observed (Figure 3A). The HbNO exhibits a three-line hyperfine due to the interaction of the nuclear spin of the nitrogen atom in NO with the electron spin.48,49 Most of the HbNO probably accumulates during euthanasia, as the animal will rapidly become systemically anaerobic before the blood sample can be drawn and cryogenically preserved. Consequently, while the HbNO level contributes to the quantitative estimation of effective NO concentration at time of sacrifice, no other useful information can be deduced from these particular signals. The blood of animals treated with NaHS alone yielded EPR spectra that were essentially the same as those of controls, containing only very weak signals and certainly nothing that could be associated with the toxicant dose (Figure 3B). On the other hand, the blood of mice administered sulfide 5 min after the nitrite exhibited EPR spectra that were interesting in a couple of respects. First, the known EPR signal associated with metHbSH (rhombic in nature with features at 2736, 3057 (crossover) and 3715 gauss18) was routinely observed (Figure 3C). There are, however, three overlapping sets of EPR signals present: metHb (~1100 gauss), HbNO (~3400 gauss), and the metHbSH signal. To quantitate these signals, we simulated the spectra using the program SpinCount (M. T. Hendrich, Carnegie Mellon University; see Experimental Methods), and the resulting signal intensities are presented in Table 2. The concentration of metHbS detected was 0.13 mM, or <2% of the total hemoglobin, which equates to 0.5 μmol in a mouse of total blood volume ~4 mL (0.13 mM × 4 mL = 0.52 μmol). Now, a 16 mg/kg dose of NaHS in a 45 g mouse equates to 12.9 μmol (16/56 × 40/1000 = 12.9 μmol). Thus, only 4% of the total sulfide dose given was scavenged by the blood as metHbSH. Interestingly, if nitrite was given to mice 2 min after administration of the sulfide, with both reagents provided at the same doses as above, any EPR signals associated with the binding of HS− to metHb were weaker and in the majority of cases none were detected (e.g., Figure 3D), seemingly consistent with the rapid elimination of HS− from the bloodstream before nitrite can release NO and generate metHb. Examination of samples of juvenile mouse blood drawn from animals subjected to the same nitrite/sulfide treatments as adults revealed no significant differences in the EPR signals observed (not shown).
Figure 3.

EPR spectra (X-band, 10 K) of whole mouse blood. (A) EPR spectrum of drawn blood following dose of 24 mg/kg NaNO2 administered ip 5–10 min prior to sacrifice showing clear evidence for the generation of nitric oxide. Signal (1) at ~1100 gauss: metHb; signal (2) at ~3300 gauss: HbNO. The combined intensities of the metHb plus HbNO signals represents <5% of the total heme present in the blood (~9 mM). (B) Spectrum of blood following dose of 16 mg/kg NaHS. This dose of NaHS roughly amounts to a maximal concentration of ~2.8 mM in the blood. (C) Spectrum of blood following dose of 24 mg/kg of NaNO2 (t = 0) and 16 mg/kg of NaHS at 2 min. The signals at ~2700, 3060, and 3700 gauss all arise from metHbSH (designated signal 3) and represent <3% (~0.2 mM) of the total Hb (~200 mM). (D) Spectrum of blood following NaHS injection 2 min prior to NaNO2. (metHbSH signals not detected.)
Table 2.
Quantitation of EPR Signals (X-band, 10 K) Observed in Mouse Tissuea
| whole blood | minced heart | ||||
|---|---|---|---|---|---|
|
|
|
||||
| metHb (μM) | HbNO (μM) | metHbSH (μM) | metMb (μM) | MbNO (μM) | |
| control | <5 | 0 | 0 | <2 | 0 |
| NaHS (16 mg/kg) | <5 | 0 | 0 | <2 | 0 |
| NaNO2 (24 mg/kg) | 90 (30) | 190 (30) | 0 | 15 (3) | 50 (5) |
| NaNO2 + NaHSb | 60 (30) | 190 (40) | 130 (30) | 7 (2) | 54 (3) |
EPR samples were made 7 min after injections (ip) to the mice (Swiss Webster, 16–18 week old males) by either cardiac puncture (2 min after euthanasia) or by mincing the heart tissue and quickly freezing samples in EPR tubes for later analysis. Values are means (3–6 samples) of signal concentrations determined either by integration or by signal simulation, with standard errors quoted in parentheses (see Experimental Methods).
5 min delay. Heart tissue samples are diluted ~4 fold.
Mouse hearts were removed from animals exposed to either 24 mg/kg NaNO2 alone, 16 mg/kg NaHS alone, or the nitrite/sulfide combinations immediately after euthanasia, flash frozen in liquid nitrogen, and stored at −80 °C prior to preparation of EPR samples as described in the Experimental Methods. EPR signals consistent with metmyoglobin (metMb) and nitrosylmyoglobin (MbNO) formation were observed in animals treated with nitrite (Figure 4A). EPR signals of MbNO lack the three-line hyperfine pattern of HbNO and, thus, the two spectra are readily distinguishable (see Figures 3A and 4A). There was very little sulfidometmyoglobin (metMbSH) signal present in any of the spectra obtained from the hearts of animals given NaHS (Figures 4B–D). The level of metMb was diminished in samples where NaNO2 had been given before the NaHS (Figure 3C and Table 2), but any explanation of this could only be speculative in the absence of detectable metMbSH formation. For practical purposes, the detection limit of single-scan EPR measurements is a few micromolar, so it is certainly the case that biologically significant levels of potentially EPR detectable species can arise but remain below detection. Thus, these data are not very informative regarding the movement of sulfide species into cells, but they do confirm significant trafficking of NO/nitrite from the bloodstream into the heart muscle. Presumably, this also applies to other soft vascularized tissues, but since most do not contain high levels of constitutive traps (like myoglobin), the assertion cannot be verified by our EPR methodology in such cases. While tissues of the central nervous system are of prime importance in the present context, our EPR findings certainly do not exclude their efficient uptake of NO/nitrite, and at least NO is known to cross the blood–brain barrier.50
Figure 4.

EPR spectra (X-band, 10 K) of mouse heart tissue. (A) EPR spectrum of minced heart tissue following dose of 24 mg/kg NaNO2 administered ip 5–10 min prior to sacrifice showing clear evidence for the generation of nitric oxide. Signal (4) at ~1100 gauss: metMb; signal (5) at ~3300 gauss: MbNO. The combined intensities of the metMb plus MbNO signals approach 100% of the total heme (~260 μM) present in the heart. (B) Spectrum of heart tissue following dose of 16 mg/kg NaHS. (C) Spectrum of heart tissue following dose of 24 mg/kg of NaNO2 (t = 0) and 16 mg/kg of NaHS at 2 min. No signals due to metMbSH were observed. (D) Spectrum of heart tissue following NaHS injection 2 min prior to NaNO2. (metHbSH signals not detected.)
Reactions with the Crucial Target of Sulfide Toxicity, Cytochrome c Oxidase
In many cases, changes in oxidation state and substitution of ligands at the oxygen-binding (active site) heme a3 in cytochrome c oxidase can be conveniently followed by electronic absorption spectroscopy (e.g., Figure 5). As prepared, the fully oxidized (resting) enzyme exhibits an absorption spectrum with two distinct features: a Soret band at 420–422 nm (Figure 5A, dot-dash trace) and a visible-region band at ~600 nm (Figure 5B, dot-dash trace). Upon the addition of sulfide, using a 5-fold excess of NaHS over enzyme, the spectrum changes to yield a more prominent Soret band at 428 nm (Figure 5A, dotted trace) and an increased intensity of the ~600 nm band (Figure 5B, dotted trace). It should be remembered that these absorption spectral envelopes (Figure 5A,B) result from two sets of overlapping signals arising from heme a and heme a3 (any CuA and CuB contributions are minimal). This lack of resolution in the spectra means that while such measurements are very good indeed at revealing whether or not something has happened at either heme a or heme a3 (oxidaton–reduction and/or ligand substitution) they do not necessarily reveal exactly which heme was involved or exactly what happened chemically; this usually has to be inferred from other information. It is quite clear from the absorption spectra that, following addition of sulfide, a new derivative of the enzyme has been formed, probably HS− (rather than S2− at pH 7.4) has bound to heme a3 of the enzyme, but the oxidations states of the metal cofactors remain unspecified. On the basis of earlier studies,19 it is most likely that HS− became bound to ferric heme a3 with reduction of CuB, and, indeed, our own EPR measurements with samples prepared in parallel proved to be in keeping with this assertion (not shown). Following exposure of the tentatively identified hydrosulfide adduct of the enzyme to excess NO gas, the 428 nm Soret band sharpened and increased in intensity (Figure 5A, dashed trace), whereas the visible-region band hardly changed (Figure 5B, dashed trace). The new spectrum obtained after the NO addition is reminiscent of that of a partially reduced NO adduct of the enzyme, where NO is bound to ferrous heme a3 while heme a remains in the ferric form.31 Interestingly, upon the addition of a strong reductant (sodium dithionite) to the partially reduced sulfide-inhibited NO adduct, a more complicated envelope was obtained, with two maxima at 428 and 442 nm (Figure 5A, solid trace) together with a 2-fold stronger band at 603 nm in the visible region (Figure 5B, solid trace). Absorption maxima at 442–444 and 603–605 nm are invariably associated with fully reduced derivatives of cytochrome c oxidase. The lack of any great shift in the 428 nm band of the NO adduct upon the addition of dithionite confirms that NO was bound to a ferrous heme in both cases. Also, the 428 and 442 nm features were slightly variable in relative intensity between samples (not shown), indicating that they represent two distinct chemical species (one associated with heme a and the other, with heme a3) rather than being two bands in the spectrum of a single chromophore. The present data do not definitively identify to which heme NO binds, but we assume this to be the oxygen binding site of heme a3, which, in turn, suggests that the 442 nm band arises from reduced heme a. More importantly, the absorption spectra do unambiguously show that NO is able to displace the inhibitory ligand from the sulfide-inhibited enzyme. Moreover, when oxygen was admitted to the closed vessel containing the NO adduct, the absorption spectrum reverted to that of the starting, fully oxidized enzyme (not shown), confirming that any NO inhibition is transient and providing a mechanistic basis for nitrite-derived NO antagonizing sulfide inhibition of cytochrome c oxidase.
Figure 5.

Electronic absorption spectra of cytochrome c oxidase derivatives showing displacement of HS− by NO. Samples were prepared in 100 mM aqueous potassium phosphate buffer, pH 7.4, 0.05% lauryl maltoside, 25 °C, 1.00 cm pathlength. (Dash-dot trace) Cytochrome c oxidase as isolated (oxidized, resting), 5 μM in enzyme; (dotted trace) partially reduced sulfide adduct 5 μM in enzyme, 0.2 mM in NaHS; (dashed trace) partially reduced sulfide adduct plus NO, 5 μM in enzyme, 0.2 mM in NaHS, 1.9 mM (1.0 atm) NO; (solid trace) partially reduced sulfide adduct plus NO, 5 mM in enzyme, 0.2 mM in NaHS, 1.9 mM (1.0 atm) NO, plus ~1 mM in Na2S2O4. (A) Soret region 380–470 nm. (B) Q (or α) band region 570–650 nm.
Amelioration of Sulfide Toxicity by Nitrite in Cultured Cells
We have previously shown that proliferating (subconfluent) bovine pulmonary artery endothelial cells (BPAEC) can be used to investigate changes in mitochondrial function within the cellular environment.51 Furthermore, as BPAEC do not contain hemoglobin/myoglobin, it was possible to show with this cell line that NO antagonizes cell death due to cyanide in a manner that does not depend upon methemoglobin, or metmyoglobin, formation.32 Leavesley et al.52 have reported a similar effect using NO donors in a neuronal line of cultured cells inhibited with KCN. In the previous studies with BPAEC, we successfully used the metabolic indicator dye alamarBlue to monitor cell proliferation. However, sulfide was found to interact directly with alamarBlue, reducing the dye and preventing its implementation in the current studies. Instead, we used propidium iodide staining as a marker of cell death, having first established that propidium iodide did not react with sulfide. Recent studies have shown that nitrite can be easily converted to NO in various cells/tissues.53–55 Plated cells were covered with Parafilm, and inoculations of reagents were made by injections through the cover to slow the loss rates of gaseous H2S and NO. Compared to controls, application of NaNO2 did result in increased cell death after 1 h (Figure 6), which is not surprising, as the NO released will inhibit the mitochondrial electron-transport chain to some extent (i.e., transiently). Addition of NaHS (alone) to the media resulted in considerably more cell death 1 h later (Figure 6), showing that the sulfide dose is considerably more inhibitory/toxic than the nitrite-derived NO dose. Most importantly, however, when the same doses of nitrite and sulfide were given together, their toxic effects did not combine additively; rather, the level of cell death observed corresponded to that seen with nitrite alone (Figure 6). In other words, nitrite is observed to ameliorate the sulfide toxicity. These findings strongly support the proposition that methemoglobin formation by nitrite is not required for significant antidotal activity, but, instead, NO generated from nitrite displaces bound sulfide from cytochrome c oxidase.
Figure 6.

Resistance of bovine pulmonary artery endothelial cells (BPAEC) to sulfide toxicity is increased in the presence of sodium nitrite. Effect of NaNO2 on BPAEC and BPAEC treated with NaHS. BPAEC were plated and then covered with Parafilm just prior to experiments. Aqueous solutions (in media) of both NaNO2 (1 mM) and NaHS (5 mM) were injected through the Parafilm into the cell media of wells while shaking the plates gently. Plates were incubated at 37 °C for 1 h and then treated with both SYBR Green and propidium iodide dyes. Cell counts were taken using a Zeiss IM 35 fluorescent microscope and an Infinity 2 camera with the Infinity Analyze software.
DISCUSSION
It is widely accepted that the lethal acute toxicities of HCN and H2S are similar, cytochrome c oxidase within the central nervous system is the primary target for inhibition, and death results from respiratory paralysis.9,37 We have found nothing, to date, that would lead us to suspect that any of these assertions might be incorrect. It has also, however, previously been widely accepted that the antidotal action of sodium nitrite toward cyanide intoxication involves oxidation of hemoglobin to metHb, which then detoxifies cyanide through formation of cyanomethemoglobin (metHbCN). This, in turn, frequently led to the tautological opinion that sodium nitrite was an undesirable choice for a cyanide antidote because it would result in methemoglobinemia in patients with already challenged respiratory function.56–58 Contrary to these hypotheses, our laboratory has shown in a series of studies that the nitrite anion probably functions as an NO donor able to reverse cyanide inhibition at cytochrome c oxidase and, at efficacious antidotal levels, metHb formation is minimal (<2% total hemoglobin).18,31,32,38 In support of this position, Lavon has very recently shown that the administration of isosorbide dinitrate in rabbits ameliorates cyanide toxicity without any metHb formation.59 Now, in the present study of sulfide toxicity and its amelioration by nitrite, we have demonstrated an analogy. Sodium nitrite can clearly be protective against acute sulfide toxicity in mice (Figures 1 and 2), whereas metHb accumulation in the blood is negligible (Figure 3). Quantitation of the EPR data revealed that only 4% of the NaHS dose given to mice became trapped as metHbSH. This is less than the experimental variability of the toxicant delivery, since our dose error was about ±1 mg/kg (of 16 mg/kg total) or ca. ± 6%, not enough to explain the observed level of protection. Using an approach suggested independently by Chen60 and Way,61 who, before our group, were the most vocal critics of the nitrite-metHb hypothesis, we attempted to further suppress nitrite-dependent metHb formation with methylene blue. Certainly, sodium nitrite was still fully protective against acute sulfide toxicity in mice also treated with methylene blue (data not shown), but we found only a modest (<20%) reduction in levels of metHb by EPR, in keeping with our position regarding the insignificance of metHb but not adding much weight to the argument. The current rather ambiguous results of the methylene blue experiment could be due to the relatively low levels of metHb formation we now obtain. The present data obtained with isolated enzyme (Figure 5) and cultured cells (Figure 6) support the notion that nitrite-derived NO reversing the sulfide inhibition at cytochrome c oxidase is likely to be the principle antidotal mechanism.
Our findings are that experimental animals acutely poisoned with sulfide (LD40 given intraperitoneally as an aqueous NaHS solution) either succumb within minutes (typically, <5 min) or recover fully (by RotaRod assay) within about 15 min (Figure 2) irrespective of whether antidote was given. These observations seem to be in broad agreement with the recent report of Haouzi et al.,25 who also found, following administration directly into the vasculature of sheep and rats at sublethal levels, free sulfide species reacted and disappeared from the bloodstream within 1 min. In short, effects that can truly be described as acute are very rapid. On the other hand, there are numerous accounts of suspected victims of H2S gas inhalation reaching the clinic exhibiting acute symptoms such as unconsciousness and respiratory difficulty.9,27 Typically, these patients will have presented at the clinic approximately half an hour or more after collapse, with many having received ventillatory support, and some succumb to the poisoning hours after the exposure. This chain of events is quite unlike observations made with laboratory animals, suggesting additional and slower mechanisms of toxicity are important in real-world exposures. While inhibition of cytochrome c oxidase may represent the key acute toxic action of sulfide, it has been suggested that sulfide may engage a broad range of other biological targets such as carbonic anhydrase,62 monoamine oxidase,63 cholinesterase, and Na+/K+-ATPase.64 Alternately, sulfide inhibition of mitochondrial electron-transport chains in various parts of the brain, which is probably not relieved by ventillatory support, may lead to damagingly low ATP levels; sometimes this is misleadingly referred to as brain/tissue hypoxia in relation to cyanide/sulfide toxicity, whereas bioenergetic hypoxia might be more appropriate, as there will actually be net hyperoxic conditions due to suppressed oxidative phosphorylation. Distinguishing between these possibilities will have to await further studies, but the current findings do reinforce the view from the experimental toxicologist’s perspective that clinical presentations of sulfide poisoning can never be accurately described as acute (the patient would either be dead or require no treatment); instead, they represent a range of more complicated post-acute conditions.
The current results (Figures 1 and 2) suggest that sodium nitrite may be useful only prophylactically and cannot be administered quickly enough to be of any antidotal value when given after toxicant dose. However, the lengthy survival time of sulfide-poisoned human victims compared to the laboratory animals indicates that this need not be so. There may be around half an hour or so, especially in the ambulance and before arrival at the clinic, during which time a beneficial intervention could be made with sodium nitrite or other antidotes like decorporating agents. NO crosses the blood–brain barrier, and, consequently, nitrite administration may be a practical way to address the potential brain bioenergetic hypoxia during ventillatory support. At this time, what is lacking is a good experimental paradigm for studying real-world exposures, which are almost all occupational accidents involving H2S gas inhalation for a duration of at least several minutes. This is the first obvious limitation of the simple ip injection approach we have adopted so far, which has such a short period of effect that meaningful testing of putative antidotes post-toxicant delivery is impractical, if at all possible. At some point, inhalation exposures of animals will be required to address some particular pulmonary issues, such as the lung edema commonly found at autopsy in humans,65 and which, so far, has not been evident in our animals. Nevertheless, there is some value in pursuing noninhalation methods, as some of the pulmonary issues are likely due to H2S acting as an irritant, and it will be important to disentangle these from systemic toxic effects and their amelioration. An equally important limitation of the present study is the use of a mouse model. Humans and other larger mammals exposed to H2S tend to exhibit coma and death without the transient and reversible torpor, associated with bradycardia and hypopnea, observed for small animals like mice. Perhaps this pattern of protective response helps the mice to avoid neurological sequelae in a manner not available to larger mammals.
Acknowledgments
Funding
(1) CounterACT Program, National Institutes of Health Office of the Director (NIH OD), and the National Institute of Neurological Disorders and Stroke (NINDS), grant no. NS089896 to J.P. and L.L.P.; (2) NSF grant no. CHE1126268 to M.P. Hendrich (toward purchase of EPR equipment).
ABBREVIATIONS
- EDTA
ethylenediaminetetraacetate
- EPR
electron paramagnetic resonance
- Hb
hemoglobin
- Mb
myoglobin
- ip
intraperitoneal
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Adkins J. Hydrogen Sulfide Suicide: Latest Technique Hazardous to First Responders and the Public. Regional Organized Crime Information Center Special Research Report, Bureau of Justice Assistance, U.S. Department of Justice; Washington, DC: 2010. [Google Scholar]
- 2.Morii D, Miyagatani Y, Nakamae N, Murao M, Taniyama K. Japanese experience of hydrogen sulfide: the suicide craze in 2008. J Occup Med Toxicol. 2010;5:28. doi: 10.1186/1745-6673-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reedy SJ, Schwartz MD, Morgan BW. Suicide fads: frequency and characteristics of hydrogen sulfide suicides in the United States. West J Emerg Med. 2011;12:300–304. [PMC free article] [PubMed] [Google Scholar]
- 4.Truscott A. Suicide fad threatens neighbours, rescuers. Can Med Assoc J. 2008;179:312–313. doi: 10.1503/cmaj.080878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellenhorn MJ, Barceloux DG. Medical Toxicology: Diagnosis and Treatment of Human Poisoning. Elsevier; New York: 1988. [Google Scholar]
- 6.Sood A. Toxic gases. In: Rosenstock L, Cullen MR, Brodkin CA, Redlich CA, editors. Textbook of Clinical Occupational and Environmental Medicine. Elsevier Saunders; Philadelphia, PA: 2005. pp. 1087–1098. [Google Scholar]
- 7.Prior MG, Roth SH, Green FHY, Hulbert WC, Reiffenstein RJ. Executive Summary. In: Prior MG, Roth SH, Green FHY, Hulbert WC, Reiffenstein RJ, editors. International Conference on Hydrogen Sulphide Toxicity. University of Alberta; Banff, Alberta, Canada: 1989. pp. v–vi. [Google Scholar]
- 8.Snyder JW, Safir EF, Summerville GP, Middleberg RA. Occupational fatality and persistent neurological sequelae after mass exposure to hydrogen sulfide. Am J Emerg Med. 1995;13:199–203. doi: 10.1016/0735-6757(95)90094-2. [DOI] [PubMed] [Google Scholar]
- 9.Toxicological Profile for Hydrogen Sulfide. Agency for Toxic Substances and Disease Registry, Division of Toxicology; Atlanta, GA: 2006. [PubMed] [Google Scholar]
- 10.Hydrogen Sulfide, Medical Management Guidelines. Agency for Toxic Substances and Disease Registry; Atlanta, GA: 2014. [Google Scholar]
- 11.Treatment and Management of H2S Poisoning: First Aid and CPR Training, Courses and Re-Certifications in Calgary. First Aid Calgary, Calgary. 2015 http://www.firstaidcalgary.ca.
- 12.High Chemicals: Hydrogen Sulfide, Poison Facts. KUMED; Kansas City, KS: 2015. http://www.kumed.com. [Google Scholar]
- 13.Protocol No. 5: Hydrogen Sulfide, Poison Facts. Florida State Emergency Response Commission; Tallahasse, FL: 2015. http://www.floridadisaster.org. [Google Scholar]
- 14.Fujita Y, Fujino Y, Onodera M, Kikuchi S, Kikkawa T, Inoue Y, Niitsu H, Takahashi K, Endo S. A fatal case of acute hydrogen sulfide poisoning caused by hydrogen sulfide: hydroxocobalamin therapy for acute hydrogen sulfide poisoning. J Anal Toxicol. 2011;35:119–123. doi: 10.1093/anatox/35.2.119. [DOI] [PubMed] [Google Scholar]
- 15.Hydrogen Sulfide, Sulfides and Mercaptans. System Protocols: Hazardous Materials Medical Response Team, Washtenaw/Livingston MCA; Ann Arbor, MI: 2013. http://www.ewashtenaw.org. [Google Scholar]
- 16.Protocol R-8: Cyanide/Hydrogen Sulfide Poisoning. Ada County Paramedics; Boise, ID: 2010. http://www.adaweb.net. [Google Scholar]
- 17.Mihajlovic A. MS Thesis. University of Toronto; Toronto, Canada: 1999. Antidotal Mechanisms for Hydrogen Sulfide Toxicity. [Google Scholar]
- 18.Cambal LK, Swanson MR, Yuan Q, Weitz AC, Li HH, Pitt BR, Pearce LL, Peterson J. Acute, sublethal cyanide poisoning in mice is ameliorated by nitrite alone: complications arising from concomitant administration of nitrite and thiosulfate as an antidotal combination. Chem Res Toxicol. 2011;24:1104–1112. doi: 10.1021/tx2001042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill BC, Woon TC, Nicholls P, Peterson J, Greenwood C, Thomson AJ. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem J. 1984;224:591–600. doi: 10.1042/bj2240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guidotti TL. Hydrogen sulphide. Occup Med. 1996;46:367–371. doi: 10.1093/occmed/46.5.367. [DOI] [PubMed] [Google Scholar]
- 21.Dorman DC, Moulin FJ, McManus BE, Mahle KC, James RA, Struve MF. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: correlation with tissue sulfide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicol Sci. 2002;65:18–25. doi: 10.1093/toxsci/65.1.18. [DOI] [PubMed] [Google Scholar]
- 22.Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 23.Kabil O, Banerjee R. Redox biochemistry of hydrogen sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Housecroft CE, Sharpe AG. Inorganic Chemistry. 2. Pearson Education Ltd; Harlow, UK: 2005. [Google Scholar]
- 25.Haouzi P, Sonobe T, Torsell-Tubbs N, Prokopczyk B, Chennuel B, Klingerman CM. In vivo interactions between cobalt or ferric compounds and the pools of sulphide in the blood during and after H2S poisoning. Toxicol Sci. 2014;141:493–504. doi: 10.1093/toxsci/kfu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Truong DH, Mihajlovic A, Gunness P, Hindmarsh W, O’Brien PJ. Prevention of hydrogen sulfide (H2S)-induced mouse lethality and cytotoxicity by hydroxocobalamin (vitamin B(12a)) Toxicology. 2007;242:16–22. doi: 10.1016/j.tox.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 27.Burnett WW, King EG, Grace M, Hall WF. Hydrogen sulfide poisoning: review of 5 years’ experience. Can Med Assoc J. 1977;117:1277–1280. [PMC free article] [PubMed] [Google Scholar]
- 28.Hall AH, Rumack BH. Hydrogen sulfide poisoning: an antidotal role for sodium nitrite? Vet Hum Toxicol. 1997;39:152–154. [PubMed] [Google Scholar]
- 29.Osbern LN, Crapo RO. Dung lung: a report of toxic exposure to liquid manure. Ann Int Med. 1981;95:312–314. doi: 10.7326/0003-4819-95-3-312. [DOI] [PubMed] [Google Scholar]
- 30.Hoidal CR, Hall AH, Robinson MD, Kulig K, Rumack BH. Hydrogen sulfide poisoning from toxic inhalations of roofing asphalt fumes. Ann Emerg Med. 1986;15:826–830. doi: 10.1016/s0196-0644(86)80383-3. [DOI] [PubMed] [Google Scholar]
- 31.Pearce LL, Bominaar EL, Hill BC, Peterson J. Reversal of cyanide inhibition of cytochrome c oxidase by the auxiliary substrate nitric oxide: an endogenous antidote to cyanide poisoning? J Biol Chem. 2003;278:52139–52145. doi: 10.1074/jbc.M310359200. [DOI] [PubMed] [Google Scholar]
- 32.Pearce LL, Lopez Manzano E, Martinez-Bosch S, Peterson J. Antagonism of nitric oxide toward the inhibition of cytochrome c oxidase by carbon monoxide and cyanide. Chem Res Toxicol. 2008;21:2073–2081. doi: 10.1021/tx800140y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koltoff IM, Sandell EB, Meehan EJ, Bruckenstein S. Quantitative Chemical Analysis. 4. Macmillan; New York: 1969. [Google Scholar]
- 34.Crankshaw DL, Goon DJ, Briggs JE, Delong D, Kuskowski M, Patterson SE, Nagasawa HT. A novel paradigm for assessing efficacies of potential antidotes against neurotoxins in mice. Toxicol Lett. 2007;175:111–117. doi: 10.1016/j.toxlet.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Gelder BF. On cytochrome c oxidase. I The extinction coefficients of cytochrome a and cytochrome a3. Biochim Biophys Acta. 1966;118:36–46. doi: 10.1016/s0926-6593(66)80142-x. [DOI] [PubMed] [Google Scholar]
- 36.Sinjorgo KM, Hakvoort TB, Durak I, Draijer JW, Post JK, Muijsers AO. Human cytochrome c oxidase isoenzymes from heart and skeletal muscle; purification and properties. Biochim Biophys Acta. 1987;890:144–150. doi: 10.1016/0005-2728(87)90015-6. [DOI] [PubMed] [Google Scholar]
- 37.Toxicological Profile for Cyanide. Agency for Toxic Substances and Disease Registry, Division of Toxicology; Atlanta, GA: 2006. [PubMed] [Google Scholar]
- 38.Cambal LK, Weitz AC, Li HH, Zhang Y, Zheng X, Pearce LL, Peterson J. Comparison of the relative propensities of isoamyl nitrite and sodium nitrite to ameliorate acute cyanide poisoning in mice and a novel antidotal effect arising from anesthetics. Chem Res Toxicol. 2013;26:828–836. doi: 10.1021/tx400103k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benz OS, Yuan Q, Amoscato AA, Pearce LL, Peterson J. Metalloporphyrin Co(III)TMPyP ameliorates acute, sublethal cyanide toxicity in mice. Chem Res Toxicol. 2012;25:2678–2686. doi: 10.1021/tx300327v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beck JF, Bradbury CM, Connors AJ, Donini JC. Nitrite as antidote for acute hydrogen sulfide intoxication? Am Ind Hyg Assoc J. 1981;42:805–809. doi: 10.1080/15298668191420738. [DOI] [PubMed] [Google Scholar]
- 41.Huang CC, Chu NS. A case of acute hydrogen sulfide (H2S) intoxication successfully treated with nitrites. J Formosan Med Assoc. 1987;86:1018–1020. [PubMed] [Google Scholar]
- 42.Smith RP, Kruszyna R, Kruszyna H. Management of acute sulfide poisoning. Effects of oxygen, thiosulfate, and nitrite. Arch Environ Health. 1976;31:166–169. doi: 10.1080/00039896.1976.10667212. [DOI] [PubMed] [Google Scholar]
- 43.Ballantyne B, Salem H. Experimental, clinical, occupational toxicological, and forensic aspects of hydrogen cyanide with particular reference to vapor exposure. In: Salem H, Katz SA, editors. Inhalation Toxicology. CRC Taylor & Fancis; Boca Raton, FL: 2006. pp. 717–802. [Google Scholar]
- 44.Whitcraft DD, III, Bailey TD, Hart GB. Hydrogen sulfide poisoning treated with hyperbaric oxygen. J Emerg Med. 1985;3:23–25. doi: 10.1016/0736-4679(85)90215-x. [DOI] [PubMed] [Google Scholar]
- 45.Smilkstein MJ, Bronstein AC, Pickett HM, Rumack BH. Hyperbaric oxygen therapy for severe hydrogen sulfide poisoning. J Emerg Med. 1985;3:27–30. doi: 10.1016/0736-4679(85)90216-1. [DOI] [PubMed] [Google Scholar]
- 46.Goldenberg I, Shoshani O, Mushkat Y, Bentur Y, Melamed Y, Shupak A. Hyperbaric oxygen for hydrogen sulfide poisoning. Harefuah. 1994;127:300–302. [PubMed] [Google Scholar]
- 47.Gunn B, Wong R. Noxious gas exposure in the outback: two cases of hydrogen sulfide toxicity. Emerg Med. 2001;13:240–246. doi: 10.1046/j.1442-2026.2001.00220.x. [DOI] [PubMed] [Google Scholar]
- 48.Fago A, Crumbliss AL, Peterson J, Pearce LL, Bonaventura C. The case of the missing NO-hemoglobin: spectral changes suggestive of heme redox reactions reflect changes in NO-heme geometry. Proc Natl Acad Sci USA. 2003;100:12087–12092. doi: 10.1073/pnas.2032603100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fago A, Crumbliss AL, Hendrich MP, Pearce LL, Peterson J, Henkens R, Bonaventura C. Oxygen binding to partially nitrosylated hemoglobin. Biochim Biophys Acta. 2013;1834:1894–1900. doi: 10.1016/j.bbapap.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lawther BK, Kumar S, Krowidi H. Blood–brain barrier. Contin Educ Anaesth, Crit Care Pain. 2011;11:128–132. [Google Scholar]
- 51.Stitt-Fischer MS, Ungerman RK, Wilen DS, Wasserloos K, Renz LM, Raub SE, Peterson J, Pearce LL. Manganese superoxide dismutase is not protective in bovine pulmonary artery endothelial cells at systemic oxygen levels. Radiat Res. 2010;174:679–690. doi: 10.1667/RR2062.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leavesley HB, Li L, Prabhakaran K, Borowitz JL, Isom GE. Interaction of cyanide and nitric oxide with cytochrome c oxidase: implications for acute cyanide toxicity. Toxicol Sci. 2008;101:101–111. doi: 10.1093/toxsci/kfm254. [DOI] [PubMed] [Google Scholar]
- 53.Bueno M, Wang J, Mora AL, Gladwin MT. Nitrite signaling in pulmonary hypertension: mechanisms of bioactivation, signaling, and therapeutics. Antioxid Redox Signaling. 2013;18:1797–1809. doi: 10.1089/ars.2012.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feelisch M, Fernandez BO, Bryan NS, Garcia-Saura MF, Bauer S, Whitlock DR, Ford PC, Janero DR, Rodriguez J, Ashrafian H. Tissue processing of nitrite in hypoxia: an intricate interplay of nitric oxide-generating and -scavenging systems. J Biol Chem. 2008;283:33927–33934. doi: 10.1074/jbc.M806654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lundberg JO, Weitzberg E. NO generation from inorganic nitrate and nitrite: role in physiology, nutrition and therapeutics. Arch Pharm Res. 2009;32:1119–1126. doi: 10.1007/s12272-009-1803-z. [DOI] [PubMed] [Google Scholar]
- 56.Geller RJ, Barthold C, Saiers JA, Hall AH. Pediatric cyanide poisoning: causes, manifestations, management, and unmet needs. Pediatrics. 2006;118:2146–2158. doi: 10.1542/peds.2006-1251. [DOI] [PubMed] [Google Scholar]
- 57.Hamel J. A review of acute cyanide poisoning with a treatment update. Crit Care Nurse. 2011;31:72–81. doi: 10.4037/ccn2011799. quiz 82. [DOI] [PubMed] [Google Scholar]
- 58.Borron SW. Recognition and treatment of acute cyanide poisoning. J Emerg Nurs. 2006;32:S12–18. doi: 10.1016/j.jen.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 59.Lavon O. Early administration of isosorbide dinitrate improves survival of cyanide-poisoned rabbits. Clin Toxicol. 2015;53:22–27. doi: 10.3109/15563650.2014.990564. [DOI] [PubMed] [Google Scholar]
- 60.Chen KK, Rose CL, Clowes GHA. Methylene blue, nitrites and sodium thiosulfate against cyanide poisoning. Proc Soc Exp Biol Med. 1933;31:250–251. [Google Scholar]
- 61.Holmes RK, Way JL. Mechanism of cyanide antagonism by sodium nitrite. Pharmacologist. 1982;24:182. [Google Scholar]
- 62.Nicholson RA, Roth SH, Zhang A, Zheng J, Brookes J, Skrajny B, Bennington R. Inhibition of respiratory and bioenergetic mechanisms by hydrogen sulfide in mammalian brain. J Toxicol Environ Health, Part A. 1998;54:491–507. doi: 10.1080/009841098158773. [DOI] [PubMed] [Google Scholar]
- 63.Warenycia MW, Smith KA, Blashko CS, Kombian SB, Reiffenstein RJ. Monoamine oxidase inhibition as a sequel of hydrogen sulfide intoxication: increases in brain catecholamine and 5-hydroxytryptamine levels. Arch Toxicol. 1989;63:131–136. doi: 10.1007/BF00316435. [DOI] [PubMed] [Google Scholar]
- 64.Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol. 1992;32:109–134. doi: 10.1146/annurev.pa.32.040192.000545. [DOI] [PubMed] [Google Scholar]
- 65.Knight LD, Presnell SE. Death by sewer gas: case report of a double fatality and review of the literature. Am J Forensic Med Pathol. 2005;26:181–185. [PubMed] [Google Scholar]