

Abstract
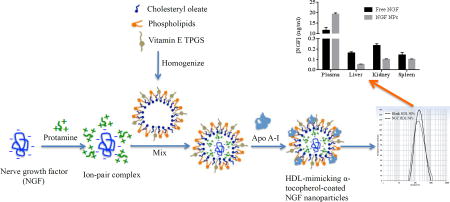
Nerve Growth Factor (NGF) is one of the members of the neurotrophin family with multifaceted functions. However, clinic application of NGF is hurdled by the challenge on formulation development. The objective of this study was to develop novel high-density lipoproteins (HDL)-mimicking nanoparticles (NPs) coated with α-tocopherol to incorporate NGF by a self-assembly approach. The NPs were prepared by an optimized self-assembly method that is simple and scalable. The composition of HDL-mimicking NPs was optimized. The prototype of the HDL-mimicking α-tocopherol-coated NPs contained phosphatidylserine (a negative charged phospholipid) and D- α-Tocopheryl polyethylene glycol succinate (a source of vitamin E) to enhance the entrapment efficiency of apolipoprotein A–I in the NPs. The entrapment efficiency of apolipoprotein A–I was about 30%. The NPs had particle size about 200 nm with a relatively narrow size distribution. Finally, cationic ion-pair agents were optimized to form ion-pairs with NGF to facilitate the incorporation of NGF into the NPs. Protamine sodium salt USP formed an optimal ion-pair complex with NGF. The results showed that the novel HDL-mimicking α-tocopherol-coated NPs successfully encapsulated NGF with over 65% entrapment efficiency by using this ion-pair strategy. In vitro release studies demonstrated a slow release of NGF from NGF NPs in PBS containing 5% BSA at 37°C for 72 hours. Further biodistribution studies showed that intravenously injected NGF NPs significantly increased NGF concentration in plasma and decreased the uptake in liver, spleen and kidney, compared to free NGF in mice.
Keywords: high-density lipoprotein, ion-pair complex, TPGS, apolipoprotein A–I, self-assembly
Graphical abstract
Introduction
Nerve Growth Factor (NGF) is well known for its role in survival, maintenance and differentiating actions on sympathetic and sensory neurons of peripheral nervous system, and maintenance of functional integrity of cholinergic neurons in the central nervous system [1]. Beneficial effects of NGF in peripheral neuropathies, diabetes, skin ulcers, human immune deficiency virus, and ophthalmology make it a potential therapeutic protein [2]. However, NGF administration through various routes like intravenous, subcutaneous or intra-cerebro ventricular infusions caused a variety of undesirable and unwanted effects in patients [3–5]. Other approaches, such as CERE-110 [6] and NsG0202 [7], are invasive requiring brain surgery procedures to incorporate NGF into certain locations of the brain. Therefore, a novel delivery strategy with convenient administration is required for NGF.
The applications of proteins in medications has been limited by their poor stability due to proteolytic and hydrolytic degradation, low permeability across the barriers, and short biologic half-life in the circulatory system [8,9]. Indeed, the half-life of NGF was about 5.4 min by intravenous injection in adult rats [10]. Nanoparticles (NPs) are promising delivery systems for NGF. When encapsulated and delivered by a NP system, the physiochemical properties of the NPs rather than those of NGF will determine the in vivo fate of NGF. Thus, NPs could offer improved transport properties, modified release characteristics and pharmacokinetic profiles after systemic administration [11]. So far, poly(butyl cyanoacrylate) (PBCA) NPs coated with polysorbate 80 [12], a liposome delivery system coated with RMP-7 targeting B2 receptor on brain microvascular endothelial cells [14], and the NGF conjugate with an antibody of the transferrin receptor [15] have been studied to deliver NGF to cross the blood-brain barrier (BBB). However, low NGF loading, low entrapment efficiency (EE) and degradation were significant issues associated with these formulations.
Formulating macromolecules like proteins is challenging because of their hydrophilic properties, charges and big sizes [8]. Ion-pair strategies are promising methods to incorporate macromolecules, such as DNA, siRNA and proteins, into NP systems through charge-charge interaction [16–18]. By neutralizing the charges on proteins and improving lipophilicity, the ion-pair agents can facilitate the encapsulation of the proteins into lipid-based NPs. Protamines and poly-l-lysine (PLL) are the polycationic carriers used to form complex with macromolecules [19–21]. The complexation helped condense macromolecules and protect them from enzymatic degradation. Previous study also demonstrated that the type of protamines affected the formation of the ion-pair complex [20]. Thus, it is important to select an optimal ion-pair agent for protein formulation development.
High-density lipoproteins (HDLs) are natural NPs with the potential to escape the reticuloendothelial system, leading to a long circulation in blood. HDL-mimicking NPs have been developed as drug delivery cargos to target various diseases like atherosclerosis [23], angiogenesis [24], lymphoma [25], Alzheimer’s disease [26], and cancers [27]. HDLs are composed of various components that include triglycerides (3–7%, w/w%) and cholesteryl ester (15–21%, w/w%) in the lipid core, and cholesterol (2–6%, w/w%), phospholipids (20–26%, w/w%), and apolipoproteins (43–58%, w/w%) on the surface [28]. Among phospholipids in HDLs, phosphatidylcholine (PC) is about 70%, sphingomyelin (SM) is about 15% and the rest of phospholipids is about 15%. Apolipoprotein A–I (Apo A–I) (molecular weight 28 kDa) is the major structural HDL apolipoprotein and accounts for about 70% of total HDL proteins. Different approaches were used to prepare HDL-mimicking or recombinant HDL NPs including dialysis method [27], sonication [26], self-assembly [29] and microfluidics [23]. However, all of these methods involved the steps, such as dialysis, gel filtration chromatography and density gradient ultracentrifugation, leading to difficulties in formulation manufacturing and scale-up. Another major limitation of previously methods was very low EE of Apo A–I, resulting in great extra Apo A–I added in NP preparation. These issues limit the use of HDL NPs for pharmaceutical formulations [30]. A new preparation method for HDL-mimicking NPs needs to be developed, which is scalable, reproducible and processable.
Vitamin E is an important nutrient in neurocognitive development [31]. It is not synthesized in vivo, and thus must be consumed through diet and transported to the brain. In vitamin E family, α-tocopherol is the most abundant form found in nature and has the most potent biological activity [32]. The studies showed that α-tocopherol is associated with HDLs in plasma and transported by scavenger receptor class B, type I (SR-BI) across the BBB [33, 34]. D-α-Tocopheryl polyethylene glycol succinate (vitamin E TPGS referred hereon as TPGS) is a water soluble source of vitamin E with extended half-life and enhanced cellular uptake of the drug due to the combination of PEG and vitamin E. Previously, we developed lipid-based NPs using TPGS as a surfactant [35]. In this study, we coated TPGS on the surface of HDL-mimicking NPs to potentially enhance the NPs to cross the BBB. To the best of our knowledge, this is the first report to develop this kind of HDL-mimicking NPs.
The objective of this study is to develop novel HDL-mimicking NPs coated with α-tocopherol to incorporate NGF by a self-assembly approach. Prototype NP composition was optimized. Different ion-pair agents were employed to form an optimal ion-pair with NGF in order to facilitate the encapsulation of NGF. The novel HDL-mimicking α-tocopherol-coated NGF NPs were fully characterized in terms of particle size, EE of Apo A–I and NGF, and loading of Apo A–I and NGF.
Materials and Methods
Materials and Cell culture
Protamine from salmon, protamine grade X, protamine sodium salt USP, poly-lysine and cholesteryl oleate (CO), Sodium chloride, sodium acetate, Triton X-100, bovine serum albumin (BSA), phosphate buffer saline (PBS), phenylmethylsulfonyl fluoride (PMSF) and benzethonium chloride were purchased from Sigma (St. Louis, MO). Sephadex G-50, Sephadex G-100, Sephacryl S-100 and Sepharose CL-4B were also purchased from Sigma-Aldrich (St.Louis, MO). PC, SM, and phosphatidylserine (PS) were purchased from Avanti polar lipids (Alabaster, Alabama). TPGS was provided by BASF as a gift. Apo A–I was purchased from Athens research and technology (Athens, GA). Recombinant human NGF was purchased from Creative Biomart (Shirley, NY). Bradford reagent was obtained from thermo scientific (Rockford, IL). Amicon ultra centrifugal filters (0.5ml) were obtained from Merk Millipore (Germany). Float-A-Lyzer G2 Dialysis device (MWCO 300 kDa) was purchased from Spectrum Laboratories (Rancho Dominguez, CA). Human beta-NGF DuoSet ELISA kit was purchased from R&D Systems (Minneapolis, MN).
Animal
Bcl mice (adult males, 25~30g) were purchased from Charles River Laboratories (Wilmington, MA). All animal experiments were carried out under an approved protocol by the Institutional Animal Care and Use Committee at the University of North Texas Health Science Center.
Optimization of preparation procedure for prototype HDL-mimicking NPs
Blank HDL-mimicking NPs were prepared by a self-assembly method. To maintain NGF bioactivity after NP preparation, we chose low temperature (50°C) or room temperature for preparation. All excipients were dissolved in ethanol to prepare stock solutions. PC (43.1%), SM (8.1%), PS (2.7%), CO (7.7%) and TPGS (38.4%) (percentages based on w/w) were added into a glass vial to form a thin film after removing ethanol by nitrogen. And then 1 ml of milliq water was added into the vial. Five different procedures were evaluated to hydrate the film to form NPs, including: 1) adding water at 50°;C and stirring at 50°;C for 30 min at 600 rpm, 2) adding water at 50°;C and stirring at room temperature (RT) for 30 min at 600 rpm, 3) adding water at RT and stirring at RT for 30 min at 600 rpm, and 4) adding water at 50°;C and homogenizing 5 min using a homogenizer at 8600 rpm, and 5) adding water at RT and homogenizing 5 min using a homogenizer at 8600 rpm. To further evaluate the influence of homogenization time on NP formation, the mixtures were homogenized for 0, 1, 2, 3, 4, 5, and 6 min after adding water at RT. After preparation, particle size and polydispersity index (P.I.) of NPs were measured using a Delsa Nano HC particle analyzer (Beckman Coulter, CA) at 90° light scattering at 25°C.
Development of prototype HDL-mimicking NPs
Nanoparticles without Apo A–I
PC, SM and PS were selected as phospholipid components and CO was selected as the lipid component to develop the HDL-mimicking NPs. To simplify the design and quickly find the optimal compositions, we considered phospholipids as one variable that include PC, SM and PS. The percentage of each phospholipid in the total phospholipids excluding CO was fixed as PC (76%), SM (14%) and PS (10%), which is close to the composition of phospholipids, but doubled the amount of PS, compared to the composition of natural HDLs. To evaluate different ratios of phospholipids and CO, we designed two arrays. In the array #1 (Table 2A and 2B), the ratios of total phospholipids and CO were controlled in a range of 0.6 to 1.6 (total phospholipids/CO, w/w). This array for 3 levels 2 variables (phospholipids and CO) was used to give three different concentrations for each excipient. The array #2 (Table 2C and 2D), an array for 2 levels 2 variables, was used to give the different ratios of total phospholipids and CO in the range of 4.9 to 14 (total phospholipids/CO, w/w). In the array #2, the percentage of each phospholipid in the total phospholipids excluding CO was fixed as PC (80%), SM (15%) and PS (5%). NPs were prepared as described above. After forming the thin film, 1 ml of milliq water at RT was added into the vial and homogenized for 5 min at 8600 rpm to form NPs. To make TPGS-coated NPs, certain amounts of TPGS were added into the compositions in Table 2B and 2D to give a total surfactant (phospholipids + TPGS) in a range of 60 µg/ml to 120 µg/ml. Particle size and P.I. were measured as described above.
Table 2. Development of HDL-mimicking α-tocopherol-coated NPs. Listed are the compositions per 1 ml NPs.
(A) The array #1 with high contents of CO without TPGS, (B) modified 2 A by adding TPGS into the compositions, (C) The array #2 with low contents of CO without TPGS, and (D) modified 2C by adding TPGS into the composition.
| A.
| ||||||
|---|---|---|---|---|---|---|
| Experiment | PC (µg) | SM (µg) | PS (µg) | CO (µg) | Particle size (nm) | P.I. |
| 1–1 | 32 | 6 | 4 | 40 | 275.4 | 0.265 |
| 1–2 | 32 | 6 | 4 | 50 | 383.4 | 0.181 |
| 1–3 | 32 | 6 | 4 | 60 | 242.9 | 0.295 |
| 1–4 | 40 | 7.5 | 5 | 40 | 284.1 | 0.31 |
| 1–5 | 40 | 7.5 | 5 | 50 | 333.6 | 0.301 |
| 1–6 | 40 | 7.5 | 5 | 60 | 404.1 | 0.193 |
| 1–7 | 48 | 9 | 6 | 40 | 386.4 | 0.284 |
| 1–8 | 48 | 9 | 6 | 50 | 282.6 | 0.297 |
| 1–9 | 48 | 9 | 6 | 60 | 255.2 | 0.255 |
| B.
| |||||||
|---|---|---|---|---|---|---|---|
| Experiment | PC (µg) | SM (µg) | PS (µg) | CO (µg) | TPGS (µg) | Particle size (nm) | P.I. |
| 2–1 | 32 | 6 | 4 | 40 | 60 | 173 | 0.261 |
| 2–2 | 32 | 6 | 4 | 50 | 40 | 181.9 | 0.236 |
| 2–3 | 32 | 6 | 4 | 60 | 20 | 198.7 | 0.223 |
| 2–4 | 40 | 7.5 | 5 | 40 | 30 | 173.1 | 0.263 |
| 2–5 | 40 | 7.5 | 5 | 50 | 40 | 190 | 0.239 |
| 2–6 | 40 | 7.5 | 5 | 60 | 20 | 166 | 0.27 |
| 2–7 | 48 | 9 | 6 | 40 | 30 | 173.1 | 0.271 |
| 2–8 | 48 | 9 | 6 | 50 | 10 | 202 | 0.28 |
| 2–9 | 48 | 9 | 6 | 60 | 20 | 211.4 | 0.294 |
| C.
| ||||||
|---|---|---|---|---|---|---|
| Experiment | PC (µg) | SM (µg) | PS (µg) | CO (µg) | Particle size (nm) | P.I. |
| 3–1 | 40 | 7.5 | 2.5 | 5 | 246.6 | 0.312 |
| 3–2 | 40 | 7.5 | 2.5 | 10 | 301.7 | 0.307 |
| 3–3 | 56 | 10.5 | 3.5 | 5 | 269.2 | 0.234 |
| 3–4 | 56 | 10.5 | 3.5 | 10 | 296.1 | 0.332 |
| D.
| |||||||
|---|---|---|---|---|---|---|---|
| Experiment | PC (µg) | SM (µg) | PS (µg) | CO (µg) | TPGS (µg) | Particle size (nm) | P.I. |
| 4–1 | 40 | 7.5 | 2.5 | 5 | 30 | 192.7 | 0.259 |
| 4–2 | 40 | 7.5 | 2.5 | 10 | 30 | 178.9 | 0.283 |
| 4–3 | 56 | 10.5 | 3.5 | 5 | 50 | 171.6 | 0.295 |
| 4–4 | 56 | 10.5 | 3.5 | 10 | 50 | 162 | 0.268 |
Optimization of loading Apo A–I in the prototype HDL-mimicking NPs
Based on the particle size and size distribution, the optimal compositions were selected to load Apo A–I, which are highlighted in Table 2. After homogenization for 5 min as described above, a certain amount of Apo A–I was added into each composition (Table 3). Four different conditions, including 2-hour stirring at RT, 4-hour stirring at RT, 4-hour stirring at RT followed with incubation at 4°C overnight, and 4-hour stirring at RT followed with stirring at 4°C overnight, were evaluated to load Apo A–I. Particle size and size distribution were measured as described above. EE of Apo A–I was analyzed by ultrafiltration. Briefly, 0.2 ml of the NPs were added into Amicon Ultra (Molecular cutoff 100 KDa) and centrifuged at 14000 rpm at 4°C for 3 min. After this, 400 µl water were added into the insert of Amicon to wash the membrane with the same centrifugation condition. Apo A–I was passed through the membrane and washed with the same approach as described above to measure the recovery of Apo A-I in this separation method. The concentration of unloaded (free) Apo A–I in the filtrate was measured by Bradford assay. Loading and EE of Apo A-I were calculated as follows:
| Eq. (1) |
| Eq. (2) |
Table 3. Characterization of the prototype HDL-mimicking α-tocopherol-coated NPs.
Each batch (experiment) contained the same composition as the corresponding batch in Table 2, except for the addition of Apo A–I.
| Experiment | Experiment number in Table 2 |
Apo A–I (µg) |
Particle size (nm) |
P.I. | Theroretical loading of Apo A–I (mole%) |
EE% of Apo A–I |
|---|---|---|---|---|---|---|
| 5–1 | 1–6 | 70 | 194.2 | 0.273 | 1.5 | 8 |
| 5–2 | 2–4 | 80 | 256.7 | 0.264 | 1.9 | 12 |
| 5–3 | 2–6 | 70 | 177.8 | 0.291 | 1.4 | 16 |
| 5–4 | 2–7 | 70 | 152 | 0.253 | 1.5 | 18 |
| 5–5 | 3–3 | 80 | 251.7 | 0.33 | 2.8 | 5 |
| 5–6 | 4–2 | 70 | 148.5 | 0.26 | 2.43 | 26 |
| 5–7 | 4–3 | 80 | 173.8 | 0.305 | 2.12 | 20 |
Further optimization on Apo A–I loading was studied based on the composition of the batch 4-2. To optimize Apo A–I loading, different amounts of Apo A–I were added into the NPs (Table 4) by changing the amount of PC, but keeping the same amounts of SM, PS, CO and TPGS in the batch 4-2. Loading and EE of Apo A–I were measured and calculated as described above.
Table 4. Influence of Apo A–I loading on the prototype HDL-mimicking α-tocopherol-coated NPs (n=3).
Batch 5–6 in Table 3 was modified by changing the contents of PC and Apo A–I to obtain batch 5–8 and 5–9.
| Experiment | PC (µg) |
Apo A–I (µg) |
Particle size (nm) |
P.I. | Loading of Apo A–I (%, w/w) |
EE% of Apo A–I |
|---|---|---|---|---|---|---|
| 5–6 | 40 | 70 | 145 ± 5 | 0.289 ± 0.012 | 43.8 | 31 ± 5.4 |
| 5–8 | 39 | 106 | 152 ± 5 | 0.265 ± 0.012 | 54.3 | 31 ± 3.6 |
| 5–9 | 38 | 140 | 156 ± 11 | 0.273 ± 0.001 | 61.4 | 26 ± 2.5 |
Particle size stability of prototype HDL-mimicking NPs at 4°C
The physical stability of the prototype HDL-mimicking NPs was assessed over time at 4°C. Prior to particle size measurement, NPs were allowed to equilibrate to RT. One milliliter of NPs was used to measure the particle size and P.I as described above.
Development of NGF-loaded HDL-mimicking NPs
Optimization of ion-pair complex for NGF
To efficiently load NGF into the NPs, poly-lysine and three types of protamines were tested to form an ion-pair complex with NGF. Protamines included protamine from salmon, protamine grade X and protamine sodium salt USP. Poly-lysine, protamines and NGF were dissolved in water at the concentration of 1 mg/ml. NGF was added into poly-lysine or protamine solutions at 0.8:1, 1:1, and 1:1.2 ratios (NGF : polymer, w/w). The complex was allowed to stand at RT for 10 min, and then diluted with 1 ml of water or PBS to measure particle size as described above and also to measure zeta potential using the particle analyzer. The optimal ratio of the complex was determined according to particle size and zeta potential.
Preparation of NGF-loaded HDL-mimicking NPs
Poly-lysine and protamine USP were selected to prepare NGF-loaded NPs. Briefly, 10 µg of NGF was mixed with 10 µg poly-lysine or protamine USP (1:1, NGF : polymer, w/w) and kept for 10 min at RT to form the complex. PC, SM, PS, CO and TPGS ethanol solutions (Table 6) were mixed and then ethanol was removed by nitrogen to form the thin film as described above. Two procedures were tested to add the NGF complex into NPs. In the first procedure, the NGF complex was added into the thin film, and then 1 ml of water at RT was added and homogenized for 5 min to incorporate NGF. In the second procedure, 1 ml of water at RT was first added into the thin film and homogenized for 5 min, and then the NGF complex was added into the solution. After the addition of NGF complex, the solution was incubated at 37°C for 30 min, and then stirred at RT for 30 min until cooling in order to incorporate NGF. The defined amount of Apo A–I was added into each solution and stirred at RT overnight to form the final NGF-loaded HDL-mimicking α-tocopherol-coated NPs. Particle size and zeta potential were measured as described above.
Table 6. The composition of the final HDL-mimicking α-tocopherol-coated NGF NPs.
| Unit | PC | SM | PS | CO | TPGS | Apo A–I | Cationic polymer | NGF |
|---|---|---|---|---|---|---|---|---|
| µg | 59 | 11 | 4 | 15 | 45 | 159 | 10 | 10 |
| w/w% | 19 | 4 | 1 | 5 | 14 | 51 | 3 | 3 |
Determination of NGF entrapment efficiency in NGF-loaded HDL-mimicking NPs
Gel filtration chromatography was used to separate unloaded NGF from NGF NPs. To determine the fractions containing NGF, 200 µl of NGF solution (10 µg/ml) were added on a Sepharose 4B–CL column and eluted with PBS. Twelve fractions (about 1 ml for each) were collected and measured for the concentrations of NGF using a Sandwich ELISA method developed based on a Sandwich ELISA kit for NGF. In a separate experiment, 200 µl of NGF HDL-mimicking NPs were eluted from the same column. The intensity in each fraction was measured using the particle analyzer to determine fractions containing NPs. The concentrations of NGF in fraction 5 to fraction 10 were measured and added together to calculate the amount of unloaded NGF. Loading and EE of NGF were calculated using equation (1) and (2) as described above.
In vitro release study
The release of NGF from NGF NPs (n=4) was studied using a dialysis method. The release medium was PBS (pH 7) containing 5% BSA to mimic the physiological condition in blood. Briefly, 200 µl NGF NPs and 400 µl release medium were loaded into the dialysis tube (MWCO 300 kDa). Then the dialysis tube was placed into a 30 ml release medium and shaken at a 37°C at 135 rpm. At the time intervals (1, 2, 4, 6, 8, 24, 48 and 72 hours), 100 µl of the release medium were withdrawn and replaced with an equal volume of fresh medium. The amounts of released NGF in the medium were analyzed by a NGF Sandwich ELISA kit. As a control, free NGF (n=4) was studied in parallel.
Tissue distribution of NGF NPs
Mice were randomly divided to three groups (n=3). Saline, free NGF and NGF NPs were injected, respectively, through tail vein at a dose of 40 µg/kg for each group. After injection, mice were sacrificed at 30 min, and blood, brain, liver, spleen and kidney were collected. Blood samples were centrifuged at 3400 rpm at 4°C for 5 min to obtain plasma. Plasma and tissues were stored at −80°C until analyzed. For tissue samples, 100 mg of tissues were suspended in a 10-times volume of extraction buffer (0.05M sodium acetate, 1.0 M sodium chloride, 1% Triton X-100, 1% BSA, 0.2 mM PMSF, and 0.2 mM benzethonium chloride) and homogenized at 4°C as previously reported [36]. The concentrations of NGF in plasma and tissues were measured by the Sandwich ELISA kit.
Statistical Analysis
Statistical analysis of the data including ANOVA and t-test, wherever needed, was performed using Graph Pad Prism software. Results were considered significant if p < 0.05.
Results and discussion
Optimal procedure for NP preparation
Significant efforts have been devoted to the use of recombinant lipoprotein-like NPs as drug delivery vehicles and diagnostic agents, because most of these particles resemble natural lipoprotein structures and are considered highly biocompatible and safe. Given the limitations of currently available preparation methods for scale-up, we tested five different procedures to prepare the HDL-mimicking NPs by self-assembly. Table 1 shows the results for four procedures and Figure 1 shows the effect of different homogenization times on particle size at RT. Efficient mixing is the key to prepare the NPs less than 200 nm. Increase of temperature did not help in decreasing the particle size. Since homogenization is a common technique used to prepare liquid formulations at industrial scales, we enhanced the mixing efficiency by homogenization. With short-time homogenization, we can produce particle size at 183.9 nm with a narrow size distribution (P.I. < 0.3). To further evaluate the influence of homogenization time on particle size, different homogenization times were studied. As shown in Figure 1, there were no significant differences in particle size among 3-min, 4-min, 5-min and 6-min homogenization (p > 0.05). Thus, 5-min homogenization was selected to prepare NPs. The new preparation method developed here is easy to be scaled up with appropriate reproducibility.
Table 1. Evaluation of preparation procedures for blank HDL-mimicking nanoparticle formation.
| Preparation conditions | Particle size (nm) | P.I. |
|---|---|---|
| 50°C water + 30 min stirring at 50°C | 347.7 ± 19.4 | 0.322 ± 0.0075 |
| 50°C water + 30 min stirring at RT | 297.7 ± 21.5 | 0.296 ± 0.0118 |
| RT water + 30 min stirring at RT | 335.4 ± 18.7 | 0.320 ± 0.0125 |
| 50°C water + 5 min homogenization | 183.9 ± 7.0 | 0.276 ± 0.030 |
The data are presented as the mean of the mean particle size of NPs in different batches ± standard deviation (SD) (n=3).
P.I. means polydispersity index that indicates size distribution of NPs. When P.I. < 0.35, NPs present as one single peak in the measurement (n=3).
Figure 1.

Influence of homogenization times on nanoparticle preparation. Excipients were homogenized for 0, 1, 2, 3, 4, 5 or 6 min to form the nanoparticles. Data are presented as the mean of particle size ± SD (n=3). # p > 0.05.
Prototype HDL-mimicking NPs
Accurate amounts of excipients in the NPs are keys to prepare self-assembled NPs. As mentioned above, natural HDLs are composed of multiple components. Given the complexity of compositions in nature HDL particles, it is challenging to mimic and artificially synthesize HDL NPs. An effective experimental design assists designing and optimizing the study that includes multiple variables and parameters. In this study, we designed the compositions of HDL-mimicking NPs based on the natural composition of HDL. Moreover, we optimized the compositions by changing the amounts of total phospholipids, CO and TPGS. According to the results of two arrays, we selected the optimal compositions of PC, SM, PS and CO and then add TPGS to further develop our desired NPs. The detailed rationale to design the arrays is described in the Methods section. The results are presented in Table 2A and 2B. Without TPGS, particle size was > 250 nm (Table 2A and 2C). The addition of TPGS decreased particle size (< 200 nm) and also narrowed size distribution (Table 2B and 2D). However, a high concentration of TPGS (batch 2-1) did not influence particle size compared to other batches. The ratio of phospholipids and CO did not influence to particle size as small particle size (< 200 nm) was obtained in both arrays (Table 2B and 2D). As shown in Table 2B, batch 2-1, 2–4, 2–6 and 2–7 gave smaller particle size compared to other batches. However, the total amount of the surfactants in batch 2-1 was very high, potentially leading to instability of NPs; thus, batch 2–4, 2–6 and 2–7 were selected to load Apo A–I. In Table 2D, all four batches produced similar NPs. We chose batch 4-2 and 4-3 to represent batches with different amounts of CO to load Apo A–I.
Apo A–I entrapment efficiency
HDL NPs have been studied for drug delivery for 25 years. However, there are presently no lipoprotein-based formulations in clinical use or in clinical trials. The major obstacle in the development of lipoprotein-based NPs is difficulty in obtaining the apolipoprotein starting material. Most synthetic HDL NPs reported very low EE and loading for Apo A–I because of high molecular weight of Apo A–I. In order to solve the problem, some studies tried to use peptide fragments to replace Apo A–I in synthetic HDL NPs; however, the EE% of Apo A–I remained low. For example, EE% of Apo A–I mimetic 4F peptide in HDL-mimicking NPs prepared via self-assembly was less than 10% and the real loading was about 2% [29]. As a result, a great amount of Apo A–I mimetic peptide (1 mg/ml) was added into the HDL preparation. With high cost of this starting material, it is very challenging to translate HDL NPs towards pharmaceutical applications.
The study showed that Apo A–I formed stable complex with zwitterionic phospholipids (e.g. PC) only at temperatures near liquid crystalline transition temperature (Tc) [37]. With small change of Tc, Apo A–I dissociated from PC. In contrast, complexes of Apo A–I with acidic phospholipid (e.g. PS) is thermodynamically stable over a wide temperature range > Tc. The data suggest that the involvement of acidic phospholipids in accelerating the rate of formation of HDL in vivo at 37°C. Indeed, another study demonstrated that the contents of negatively charged PS and phosphatidic acid increased progressively with increase in hydrated density of HDL [38]. Specifically, the content of PS revealed positive correlations with all metrics of HDL functionality. These results proved the important role of negatively charged phospholipids, especially PS, in nature HDLs. However, PS has not been used as a component in previously reported HDL NPs. We hypothesized that including PS in NP compositions will increase Apo A–I loading and EE%. According to this hypothesis, we designed the initial arrays (Table 2A and 2C).
We used membrane separation to measure EE% of Apo A–I. Proteins tend to bind with separation membranes. Thus, we measured the recovery of Apo A–I from Amicon Ultra. The result revealed that about 50% Apo A–I were detected in the filtrate after the initial centrifugation. After washing the membrane with 400 µl of water, the recovery of Apo A–I in the filtrate was 84.3% ± 4.5, demonstrating that the method was sufficient to collect free Apo A–I in the filtrate. We loaded Apo A–I into the batches highlighted in Table 2B and 2D in order to prepare HDL-mimicking NPs. The results from four different preparation conditions showed that the initial 4-hour stirring at RT was crucial to get homogenous NPs, and incubation overnight was important to get appropriate EE% of Apo A–I. Thus, we selected 4-hour stirring at RT followed with incubation at 4°C overnight to load Apo A–I. It was observed that drug formulations with TPGS resulted in high drug encapsulation efficiency along with high cellular uptake and therapeutic effects in in vitro and in vivo respectively [39]. To understand the influence of TPGS on EE% of Apo A–I, we also selected batch 1–6 from Table 2A and batch 3–3 from Table 2C as representative batches to load Apo A–I. As shown in Table 3, all of the batches (batch 5-2, 5-3, 5-4, 5–6, and 5–7) that contained TPGS in the compositions had higher EE% of Apo A–I, compared to the batches without TPGS (batch 5-1 and 5–5). These results suggested that the addition of TPGS improved EE% of Apo A–I. The highest EE% of Apo A–I was provided by the batch 5–6. To clearly know the influence of Apo A–I loading on its EE%, we designed another two batches by only replacing the amount of PC with Apo A–I while keeping the same amounts of other excipients in the batch 5–6 (Table 4). By this design, we can minimize the influence from the change of NP composition. The profiles show that increasing Apo A–I loading did not change EE% of Apo A–I (Figure 2). The EE% of Apo A–I was over 26% - about 3-fold higher than those reported in literatures [29]. Consequently, the real content of Apo A–I in our NPs was over 16% close to the Apo A–I content in natural HDLs. Thus, we chose the composition of the batch 5–8 to prepare NGF-loaded NPs. The optimal blank HDL-mimicking NPs had particle size around 150 nm with a narrow and monodispersed size distribution (Figure 3). In our preparation, we added about 0.14 mg/ml of Apo A–I to achieve a sufficient Apo A–I content in the NPs, which dramatically decreased the use of Apo A–I compared to previously reported NPs.
Figure 2.

Relationship of Apo A–I loading and entrapment efficiency %. # p > 0.05 for the entrapment efficiency %.
Figure 3.

Particle size and size distribution of blank and NGF HDL-mimicking nanoparticles.
Ion-pair complex for NGF
NGF is a 120-amino acid polypeptide homodimer. It exists as monomer with 13 kDa and forms dimer by a disulfide bond in aqueous condition. Positively charged amino acids dominate in the NGF monomer chain; however, after folding, the surface potential of the NGF dimer is negative as positively charged basic groups forms a positive groove at one end of the dimer that is responsible for the binding affinity of NGF to its receptor [40]. Therefore, we hypothesized that a cationic polymer will be a suitable complex agent to form an ion-pair complex with NGF to facilitate encapsulation of NGF into the NPs. First, we tested if cationic polymers could form complexes with NGF. After mixing protamine with NGF at 1:1 ratio (w/w), we easily visualized formation of white precipitates, directly indicating the formation of the complex. Next, we measured particle size and zeta potential of the complexes that were formed by mixing each cationic polymer with NGF at different ratios. As we expected, zeta potential changed from positive to negative while decreasing the concentrations of protamine, protamine sulfate USP and poly-D-lysine (Table 5). The results confirmed that NGF has negative charge on the surface and using cationic polymers for complexation is appropriate for NGF. PC and SM are neutral phospholipids and TPGS is a non-ionic surfactant. PS is negative-charged phospholipids. Thus, our HDL-mimicking NPs are negatively charged. A desirable complex should not only contain a minimal amount of the cationic polymer to produce sufficient complexation but also keep the complex slightly positively charged to be entrapped into the negatively charged HDL-mimicking NPs. As shown in Table 5, large aggregation was shown at the ratio of 1:1 of NGF to protamine, suggesting that a complex formed and tended to aggregate. Importantly, at the ratio of 1:1, the complex had slightly positive charge, which was what we preferred as described above. Compared with other tested protamines, protamine sulfate USP showed more favorite properties in terms of particle size and zeta potential. Moreover, protamine sulfate USP is approved by the Food and Drug Administration for injection. Therefore, we chose protamine sulfate USP as the ion-pair agent to prepare NGF HDL-mimicking NPs. In contrast to protamine, we did not observe the same trend on particle size for poly-D-lysine. The ratio of NGF to poly-D-lysine at 1:1 and 1.2:1 produced similar particle size; however, the zeta potential was more sensitive for the ratio change compared to protamines. These results suggested that protamines were superior to poly-D-lysine since the complexation of NGF by using poly-D-lysine could more difficult to be qualified and controlled than those by using protamines. We included poly-D-lysine in the following study as a comparison. One concern while using charge-charge interaction for formulations is instability of the ion-pair complex because of the competition from other ions in physiological fluid. To verify the stability of the NGF complexes, we mixed the NGF/protamine USP or NGF/poly-D-lysine complexes with PBS and then measured particle size. In PBS, particle size of the NGF/protamine complex and the NGF/poly-D-lysine complex was 725.3 nm and 957.6 nm, respectively, indicating both complexes were stable.
Table 5. Ion-pair complexes of protamines or poly-D-Lysine with NGF at different ratios.
| NGF : Polycation | Protamine free base | Protamine salt from salmon | Protamine sulfate USP | Poly-D-Lysine | ||||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| (w/w) in water | Size (nm) | Potential (mV) | Size (nm) | Potential (mV) | Size (nm) | Potential (mV) | Size (nm) | Potential (mV) |
| 0.8 : 1 | 554.3 | 12.22 | 278.5 | 0.34 | 562.6 | 0.86 | 529.3 | 0.54 |
| 1 : 1 | 863.6 | 0.58 | 589.4 | 0.22 | 802.6 | 0.30 | 805.5 | −0.82 |
| 1.2 : 1 | 596.4 | −0.71 | 543.7 | 0.59 | 356.0 | −0.32 | 830.0 | −4.53 |
NGF-loaded HDL-mimicking nanoparticles
To load 10 µg/ml of NGF, we modified the composition of the batch 5–8 (Table 4) by increasing each excipient for 1.5 times. The final composition of the NGF HDL-mimicking NPs is shown in Table 6. Theoretically, the NGF loading was 3.2% and the Apo A–I loading was 50.8%. We evaluated two procedures to add the NGF complex into the NPs. Both procedures, adding NGF complex before and after homogenization, did not show difference on particle size and size distribution. To preserve the bioactivity of NGF in the NPs, we decided to add NGF complex after homogenization. Also, after the addition of Apo A–I, stirring the NPs at RT overnight provided higher EE% of NGF compared to incubation at 4°C overnight. As a consequence, NGF HDL-mimicking NPs were prepared by stirring at RT overnight after adding Apo A–I. Incorporation of NGF into the NPs did not influence particle size and size distribution compared to the blank HDL-mimicking NPs (Figure 3).
To measure the EE% of NGF, we first tried to use Amicon Ultra (molecule cutoff 100 kDa) to separate free NGF and NGF-loaded NPs. However, free NGF did not pass the membrane, probably due to the 3-D structure of the NGF dimer (26 kDa) in the aqueous solution. Thus, we tested several gel filtration columns including Sephadex G-50, Sephadex G-100, Sephacryl S-100 and Sepharose CL-4B. Only Sepharose CL-4B completely separated NGF NPs and free NGF. As shown in Figure 4, fractions of 2 to 4 contained NGF NPs. We calculated the EE% of NGF based on the concentrations of free NGF from fraction 6 to fraction 10 after the column separation. Different ELISA methods were evaluated to quantitatively measure the concentration of NGF. A direct ELISA method worked very well for NGF standard solution that was in PBS. However, cationic polymers, protamine sulfate USP and poly-D-lysine, increased the NGF absorbance in the direct ELISA method. Next, we evaluated a commercial NGF ELISA kit. Protamine sulfate USP and poly-D-lysine did not interfere with NGF measurement using the sandwich ELISA method. Characterization of NGF HDL-mimicking NPs is shown in Table 7. Both NGF HDL-mimicking NPs had relatively narrow size distribution. D90, the size which 90% of the distribution lies below, was smaller than 550 nm and D10, the size which 10% of the distribution lies below, was bigger than 75 nm. As we expected, NGF/protamine sulfate USP NPs had higher NGF EE% than NGF/poly-D-lysine NPs. The variation of zeta potential on NGF/protamine sulfate USP NPs also was smaller than that of NGF/poly-D-lysine NPs. It could be because the charge density of poly-D-lysine is relatively high compared to protamine sulfate USP; thus, small change on poly-D-lysine amounts significantly influenced complex formation and zeta potential. Also, the NGF/poly-D-lysine complex had negative zeta potential, which may be not preferable for the negatively charged NPs. To the best of our knowledge, our study is the first study to encapsulate NGF into HDL-mimicking α-tocopherol-coated NPs. We did not use hazard organic solvents as previously reported methods (e.g. chloroform), and all excipients in our NPs are naturally present, minimizing the toxicity of the NPs. In our study, we used PBS to wash the gel filtration column to separate free NGF and NGF NPs for the measurement of EE%. Unloaded NGF and loosely bound NGF (on the NP surface) were separated and washed out as free NGF from fraction 6 to 10. Therefore, the 65.9% of NGF we measured for the EE% should be entrapped in the core of the NPs so that they did not dissociate from the NPs during the column separation and elution by PBS. This result suggests that the HDL-mimicking α-tocopherol-coated NPs could protect NGF from degradation and systemically deliver NGF to treat diseases.
Figure 4.

Separation of the NGF NPs from free NGF by a gel filtration Sepharose CL-4B column. The NGF NPs were measured based on particle intensity. Free NGF was measured by a Sandwich ELISA method.
Table 7. Characterization of the HDL-mimicking α-tocopherol-coated NGF NPs using protamine sulfate USP and Poly-D-Lysine as ion-pair agents, respectively (n=3).
| NGF HDL-mimicking NPs | Particle size (nm) | P.I. | EE% of NGF | Zeta potential (mV) |
|---|---|---|---|---|
| Protamine sulfate USP | 171.4 ± 6.6 | 0.239 ± 0.01 | 65.9 ± 1.4 | −12.5 ± 1.9 |
| Poly-D-lysine | 160.1 ± 11.7 | 0.282 ± 0.005 | 49.1 ± 1.7 | −24.9 ± 8.1 |
Physical stability studies of nanoparticles
Stability measurement for optimized HDL-mimicking NPs was performed on basis of particle size and P.I. The batch 4-2 in Table 2D was stable over six months at 4°C (Figure 5). Batch 2–4, 2–6 and 2–7 in Table 2B were stable over three months at 4°C (Figure 6). The prototype HDL-mimicking α-tocopherol-coated NPs (batch 5–8 in Table 4) were stable over two months at 4°C, and the NGF HDL-mimicking NPs were stable over one month at 4°C. However, considering degradation potentials of both Apo A–I and NGF during long-term storage in aqueous solutions, we are studying the lyophilization of the NGF NPs to make them as powders for long-term storage. The stability results demonstrated that the NPs developed in this study were stable with or without Apo A–I. Thus, we developed not only the NGF HDL-mimicking α-tocopherol-coated NPs but also the stable lipid NPs that did not contain Apo A–I. We will further characterize these lipid NPs and evaluate their potential applications for drug delivery.
Figure 5.

Long-term stability of the batch 4-2 that did not contain Apo A–I. The batch was monitored for particle size and P.I. over six months. Data are presented as the mean particle size.
Figure 6.

Long-term stability of the prototype HDL-mimicking α-tocopherol-coated NPs. Batch 2–4, 2–6, and 2–7 in Table 2B consist of three different compositions. Each batch was prepared in triplicate and monitored for particle size and P.I. over three months. For all tested NPs, P. I. < 0.3. Data are presented as the mean particle size of three batches at the certain composition. # p > 0.05. within the group.
In vitro release study
The release profiles of free NGF and NGF NPs are shown in Figure 7. Free NGF passed through the membrane readily and reached 83% in the first hour. We observed the tendency of NGF to bind with the membrane when we tested the entrapment efficiency. In the release studies, we added 5% BSA to reduce the binding of NGF as well as matching the BSA concentration in blood. The result indicated that 5% BSA efficiently prevented the binding of NGF to the membrane. With this advance, we can accurately measure the released NGF from NGF NPs. NGF NPs showed a slow release without a burst release. Only 5.5% of NGF was released within 1 hour. The release of NGF reached a plateau at 8 hours (9.9%) and kept over 72 hours. The release results demonstrated that NGF was entrapped in the core of the NPs, which aligns with the result of the entrapment efficiency.
Figure 7.

In vitro release profiles of free NGF and NGF NPs in 5% BSA-PBS solution (pH 7). Data are presented as the mean ± SD (n=4).
Biodistribution
One of our hypotheses was that NPs can protect NGF from degradation and control NGF release in order to improve the half-life of NGF after intravenous injection. Hence, we measured the biodistribution of NGF NPs in mice. As shown in Figure 8, NGF NPs increased the plasma concentration of NGF by 1.7 fold compared to free NGF. For tissues, NGF NPs decreased the tissue uptake by 3 fold in liver, 2.3 fold in kidney and 1.4 fold in spleen. The results demonstrated that the NPs prolonged the circulation of NGF in blood. As shown in the release studies (Figure 7), NGF was entrapped inside the NPs and slowly released from the NPs. Thus, the NPs protected NGF from degradation in vivo, leading to a long circulation in blood and reduced uptake in tissues (Figure 8). When the NPs are used to deliver NGF to brain, the prolonged circulation would provide more opportunity for the brain uptake compared to free NGF. Therefore, the novel HDL-mimicking NPs are very promising to deliver NGF through intravenous injection.
Figure 8.

Biodistribution of NGF NPs after mice were intravenously injected 40 µg/kg of NGF for 30 min (n=3). NGF NPs resulted in significantly higher NGF concentration in plasma compared to free NGF (p < 0.05). For other tissues, NGF NPs led to lower NGF concentrations compared to free NGF (p < 0.05).
Conclusions
The results of this present study demonstrate that appropriate design of NP compositions efficiently facilitated novel HDL-mimicking α-tocopherol-coated NP development. The novel NPs prepared by self-assembly are scalable. The usage of Apo A–I in NP preparation dramatically decreased because of the remarkable increase of Apo A–I EE%. These improvements encourage the potential pharmaceutical applications of HDL-mimicking NPs at an industrial level. The HDL-mimicking α-tocopherol-coated NPs are able to sufficiently encapsulate proteins and prolong their circulation in blood after intravenous injection, in particular NGF. The favorable features of NGF HDL-mimicking α-tocopherol-coated NPs warrant the further evaluation of their biological activities in vitro and in vivo.
Acknowledgments
This work was supported by NIH R03 NS087322-01 to Dong, X.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aloe L, Rocco ML, Bianchi P, Manni L. Nerve growth factor: from the early discoveries to the potential clinical use. J. Transl. Med. 2012;10 doi: 10.1186/1479-5876-10-239. 239-5876-10-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu. Rev. Neurosci. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- 3.Apfel SC. Nerve growth factor for the treatment of diabetic neuropathy: what went wrong what went right and what does the future hold? Int. Rev. Neurobiol. 2002;50:393–413. doi: 10.1016/s0074-7742(02)50083-0. [DOI] [PubMed] [Google Scholar]
- 4.McArthur JC, Yiannoutsos C, Simpson DM, Adornato BT, Singer EJ, Hollander H, Marra C, Rubin M, Cohen BA, Tucker T, Navia BA, Schifitto G, Katzenstein D, Rask C, Zaborski L, Smith ME, Shriver S, Millar L, Clifford DB, Karalnik IJ. A phase II trial of nerve growth factor for sensory neuropathy associated with HIV infection. AIDS Clinical Trials Group Team 291. Neurology. 2000;54:1080–1088. doi: 10.1212/wnl.54.5.1080. [DOI] [PubMed] [Google Scholar]
- 5.Eriksdotter Jonhagen M, Nordberg A, Amberla K, Backman L, Ebendal T, Meyerson B, Olson L, Seiger, Shigeta M, Theodorsson E, Viitanen M, Winblad B, Wahlund LO. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 1998;9:246–257. doi: 10.1159/000017069. [DOI] [PubMed] [Google Scholar]
- 6.Mandel RJ. CERE-110, an adeno-associated virus-based gene delivery vector expressing human nerve growth factor for the treatment of Alzheimer’s disease. Curr. Opin. Mol. Ther. 2010;12:240–247. [PubMed] [Google Scholar]
- 7.Wahlberg LU, Lind G, Almqvist PM, Kusk P, Tornoe J, Juliusson B, Soderman M, Sellden E, Seiger A, Eriksdotter-Jonhagen M, Linderoth B. Targeted delivery of nerve growth factor via encapsulated cell biodelivery in Alzheimer disease: a technology platform for restorative neurosurgery. J. Neurosurg. 2012;117:340–347. doi: 10.3171/2012.2.JNS11714. [DOI] [PubMed] [Google Scholar]
- 8.Pinto Reis C, Neufeld RJ, Ribeiro AJ, Veiga F. Nanoencapsulation II. Biomedical applications and current status of peptide and protein nanoparticulate delivery systems. Nanomedicine. 2006;2:53–65. doi: 10.1016/j.nano.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Vaishya R, Khurana V, Patel S, Mitra AK. Long-term delivery of protein therapeutics. Expert Opin. Drug Deliv. 2015;12:415–440. doi: 10.1517/17425247.2015.961420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tria MA, Fusco M, Vantini G, Mariot R. Pharmacokinetics of nerve growth factor (NGF) following different routes of administration to adult rats. Exp. Neurol. 1994;127:178–183. doi: 10.1006/exnr.1994.1093. [DOI] [PubMed] [Google Scholar]
- 11.Zhang S, Uludag H. Nanoparticulate systems for growth factor delivery. Pharm. Res. 2009;26:1561–1580. doi: 10.1007/s11095-009-9897-z. [DOI] [PubMed] [Google Scholar]
- 12.Kurakhmaeva KB, Djindjikhashvili IA, Petrov VE, Balabanyan VU, Voronina TA, Trofimov SS, Kreuter J, Gelperina S, Begley D, Alyautdin RN. Brain targeting of nerve growth factor using poly(butyl cyanoacrylate) nanoparticles. J. Drug Target. 2009;17:564–574. doi: 10.1080/10611860903112842. [DOI] [PubMed] [Google Scholar]
- 13.Olivier JC. Drug transport to brain with targeted nanoparticles. NeuroRx. 2005;2:108–119. doi: 10.1602/neurorx.2.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Y, Ye L, Zhang X, Cui W, Lou J, Nagai T, Hou X. Transport of nerve growth factor encapsulated into liposomes across the blood-brain barrier: in vitro and in vivo studies. J. Control. Release. 2005;105:106–119. doi: 10.1016/j.jconrel.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Granholm AC, Backman C, Bloom F, Ebendal T, Gerhardt GA, Hoffer B, Mackerlova L, Olson L, Soderstrom S, Walus LR. NGF and anti-transferrin receptor antibody conjugate: short and long-term effects on survival of cholinergic neurons in intraocul ar septal transplants. J. Pharmacol. Exp. Ther. 1994;268:448–459. [PubMed] [Google Scholar]
- 16.Dai WG, Dong LC. Characterization of physiochemical and biological properties of an insulin/lauryl sulfate complex formed by hydrophobic ion pairing. Int. J. Pharm. 2007;336:58–66. doi: 10.1016/j.ijpharm.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 17.Meyer JD, Manning MC. Hydrophobic ion pairing: altering the solubility properties of biomolecules. Pharm. Res. 1998;15:188–193. doi: 10.1023/a:1011998014474. [DOI] [PubMed] [Google Scholar]
- 18.Yuan H, Jiang SP, Du YZ, Miao J, Zhang XG, Hu FQ. Strategic approaches for improving entrapment of hydrophilic peptide drugs by lipid nanoparticles. Colloids Surf. B Biointerfaces. 2009;70:248–253. doi: 10.1016/j.colsurfb.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez Ferreiro M, Tillman L, Hardee G, Bodmeier R. Characterization of complexes of an antisense oligonucleotide with protamine and poly-L-lysine salts. J. Control. Release. 2001;73:381–390. doi: 10.1016/s0168-3659(01)00296-6. [DOI] [PubMed] [Google Scholar]
- 20.Sorgi FL, Bhattacharya S, Huang L. Protamine sulfate enhances lipid-mediated gene transfer. Gene Ther. 1997;4:961–968. doi: 10.1038/sj.gt.3300484. [DOI] [PubMed] [Google Scholar]
- 21.Owens DR. Insulin preparations with prolonged effect. Diabetes Technol. Ther. 2011;1(13 Suppl):S5–14. doi: 10.1089/dia.2011.0068. [DOI] [PubMed] [Google Scholar]
- 22.Camont L, Chapman MJ, Kontush A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol. Med. 2011;17:594–603. doi: 10.1016/j.molmed.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez-Gaytan BL, Fay F, Lobatto ME, Tang J, Ouimet M, Kim Y, van der Staay SE, van Rijs SM, Priem B, Zhang L, Fisher EA, Moore KJ, Langer R, Fayad ZA, Mulder WJ. HDL-mimetic PLGA nanoparticle to target atherosclerosis plaque macrophages. Bioconjug. Chem. 2015;26:443–451. doi: 10.1021/bc500517k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tripathy S, Vinokour E, McMahon KM, Volpert OV, Thaxton CS. High Density Lipoprotein Nanoparticles Deliver RNAi to Endothelial Cells to Inhibit Angiogenesis. Part Part Syst. Charact. 2014;31:1141–1150. doi: 10.1002/ppsc.201400036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang S, Damiano MG, Zhang H, Tripathy S, Luthi AJ, Rink JS, Ugolkov AV, Singh AT, Dave SS, Gordon LI, Thaxton CS. Biomimetic, synthetic HDL nanostructures for lymphoma. Proc. Natl. Acad. Sci. U. S. A. 2013;110:2511–2516. doi: 10.1073/pnas.1213657110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song Q, Huang M, Yao L, Wang X, Gu X, Chen J, Chen J, Huang J, Hu Q, Kang T, Rong Z, Qi H, Zheng G, Chen H, Gao X. Lipoprotein-based nanoparticles rescue the memory loss of mice with Alzheimer’s disease by accelerating the clearance of amyloid-beta. ACS Nano. 2014;8:2345–2359. doi: 10.1021/nn4058215. [DOI] [PubMed] [Google Scholar]
- 27.Rui M, Tang H, Li Y, Wei X, Xu Y. Recombinant high density lipoprotein nanoparticles for target-specific delivery of siRNA. Pharm. Res. 2013;30:1203–1214. doi: 10.1007/s11095-012-0957-4. [DOI] [PubMed] [Google Scholar]
- 28.Shuhei N, Soderlund S, Jauhiainen M, Taskinen MR. Effect of HDL composition and particle size on the resistance of HDL to the oxidation. Lipids Health. Dis. 2010;9 doi: 10.1186/1476-511X-9-104. 104-511X-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marrache S, Dhar S. Biodegradable synthetic high-density lipoprotein nanoparticles for atherosclerosis. Proc. Natl. Acad. Sci. U. S. A. 2013;110:9445–9450. doi: 10.1073/pnas.1301929110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacko AG, Nair M, Prokai L, McConathy WJ. Prospects and challenges of the development of lipoprotein-based formulations for anti-cancer drugs. Expert Opin. Drug Deliv. 2007;4:665–675. doi: 10.1517/17425247.4.6.665. [DOI] [PubMed] [Google Scholar]
- 31.Morris MC. Nutritional determinants of cognitive aging and dementia. Proc. Nutr. Soc. 2012;71:1–13. doi: 10.1017/S0029665111003296. [DOI] [PubMed] [Google Scholar]
- 32.Joshi YB, Pratico D. Vitamin Ein aging dementia and Alzheimer’s disease. Biofactors. 2012;38:90–97. doi: 10.1002/biof.195. [DOI] [PubMed] [Google Scholar]
- 33.Balazs Z, Panzenboeck U, Hammer A, Sovic A, Quehenberger O, Malle E, Sattler W. Uptake and transport of high-density lipoprotein (HDL) and HDL-associated alpha-tocopherol by an in vitro blood-brain barrier model. J. Neurochem. 2004;89:939–950. doi: 10.1111/j.1471-4159.2004.02373.x. [DOI] [PubMed] [Google Scholar]
- 34.Goti D, Hrzenjak A, Levak-Frank S, Frank S, van der Westhuyzen DR, Malle E, Sattler W. Scavenger receptor class B type I is expressed in porcine brain capillary endothelial cells and contributes to selective uptake of HDL-associated vitamin E. J. Neurochem. 2001;76:498–508. doi: 10.1046/j.1471-4159.2001.00100.x. [DOI] [PubMed] [Google Scholar]
- 35.Dong X, Mattingly CA, Tseng M, Cho M, Adams VR, Mumper RJ. Development of new lipid-based paclitaxel nanoparticles using sequential simplex optimization. Eur. J. Pharm. Biopharm. 2009;72:9–17. doi: 10.1016/j.ejpb.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolbeck R, Bartke I, Eberle W, Barde YA. Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J. Neurochem. 1999;72:1930–1938. doi: 10.1046/j.1471-4159.1999.0721930.x. [DOI] [PubMed] [Google Scholar]
- 37.Surewicz WK, Epand RM, Pownall HJ, Hui SW. Human apolipoprotein A–I forms thermally stable complexes with anionic but not with zwitterionic phospholipids. J. Biol. Chem. 1986;261:16191–16197. [PubMed] [Google Scholar]
- 38.Camont L, Lhomme M, Rached F, Le Goff W, Negre-Salvayre A, Salvayre R, Calzada C, Lagarde M, Chapman MJ, Kontush A. Small dense high-density lipoprotein-3 particles are enriched in negatively charged phospholipids: relevance to cellular cholesterol efflux, antioxidative, antithrombotic, anti-inflammatory, and antiapoptotic functionalities. Arterioscler. Thromb. Vasc. Biol. 2013;33:2715–2723. doi: 10.1161/ATVBAHA.113.301468. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33:4889–4906. doi: 10.1016/j.biomaterials.2012.03.046. [DOI] [PubMed] [Google Scholar]
- 40.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]