

Abstract
Introduction:
Patiromer is a potassium-binding polymer that is not systemically absorbed; however, it may bind coadministered oral drugs in the gastrointestinal tract, potentially reducing their absorption.
Methods:
Twelve randomized, open-label, 3-period, 3-sequence crossover studies were conducted in healthy volunteers to evaluate the effect of patiromer (perpetrator drug) on absorption and single-dose pharmacokinetics (PK) of drugs (victims) that might be commonly used with patiromer. Subjects received victim drug alone, victim drug administered together with patiromer 25.2 g (highest approved dose), and victim drug administered 3 hours before patiromer 25.2 g. The primary PK endpoints were area under the curve (AUC), extrapolated to infinity (AUC0-∞), and maximum concentration (C max). Results were reported as 90% confidence intervals (CIs) about the geometric mean AUC0-∞ and C max ratios with prespecified equivalence limits of 80% to 125%.
Results:
Overall, 370 subjects were enrolled, with 365 receiving ≥1 dose of patiromer; 351 subjects completed the studies and all required treatments. When coadministered with patiromer, the 90% CIs for AUC0-∞ remained within 80% to 125% for 9 drugs (amlodipine, cinacalcet, clopidogrel, furosemide, lithium, metoprolol, trimethoprim, verapamil, and warfarin). The AUC0-∞ point estimate ratios for levothyroxine and metformin with patiromer coadministration were ≥80%, with the lower bounds of the 90% CIs at 76.8% and 72.8%, respectively. For ciprofloxacin, the point estimate for AUC0-∞ was 71.5% (90% CI: 65.3-78.4). For 8 of 12 drugs, point estimates for C max were ≥80% with patiromer coadministration; for ciprofloxacin, clopidogrel, metformin, and metoprolol, the point estimates were <80%. When patiromer was administered 3 hours after each victim drug, the 90% CIs for AUC0-∞ and C max for each drug were within the prespecified 80% to 125% limits.
Conclusion:
For 9 of the 12 drugs coadministered with patiromer, there were no clinically significant drug–drug interactions. For 3 drugs (ciprofloxacin, levothyroxine, and metformin), a 3-hour separation between patiromer and their administration resulted in no clinically significant drug–drug interactions.
Keywords: patiromer, hyperkalemia, potassium-binder, drug–drug interactions, absorption, dose separation
Introduction
Hyperkalemia is common in patients with chronic kidney disease (CKD)1,2 and is associated with increased mortality.3 As the kidneys are the primary organ for eliminating potassium from the body, the risk of hyperkalemia increases as renal function worsens.4,5 Heart failure (HF) and diabetes are more common at higher CKD stages,6 and factors such as hyporeninemic hypoaldosteronism, uncontrolled diabetes, and advanced HF, superimposed on low renal function, likely contribute to hyperkalemia risk.5 In a nested case–control study, the prevalence of hyperkalemia was approximately 60% higher in diabetic versus nondiabetic patients with CKD stage 3.5 A case–control study of ambulatory patients found that congestive HF was independently associated with the risk of developing hyperkalemia even in the presence of angiotensin-converting enzyme inhibitor therapy.7 Renin–angiotensin–aldosterone system inhibitors (RAASi), which are guideline recommended to improve outcomes in HF, proteinuric CKD, and diabetes,8,9 also substantially contribute to hyperkalemia risk.10–12 The association of hyperkalemia with RAASi therapy frequently leads to use of suboptimal doses or even discontinuation of these agents in the same patients who are expected to derive the greatest cardiovascular benefit from them.10,13
Until recently, there were no viable long-term treatment options for the chronic management of patients with hyperkalemia, many of whom have an indication for RAASi medications for cardiorenal protection. A potassium-restricted diet is recommended in patients at risk for hyperkalemia but is often challenging for patients to follow consistently and may impact nutrition in those who otherwise may benefit the most from a heart healthy diet such as the Dietary Approach to Stop Hypertension diet.14,15 Although the potassium binding resin, sodium polystyrene sulfonate (SPS), which exchanges sodium for potassium, was approved more than 50 years ago,16 this agent has not been evaluated in rigorously designed prospective clinical trials. In addition, concerns about the safety of SPS related to reports of colonic necrosis,17 the precaution against its use in patients who cannot tolerate even a small increase in sodium load,16 and tolerability issues related to high rates of gastrointestinal (GI) side effects have limited its use.
Patiromer is a novel, sodium-free, nonabsorbed, potassium-binding polymer that was approved for the treatment of hyperkalemia in the United States in 2015.18 Patiromer acts by exchanging calcium for potassium in the GI tract, primarily in the colon, where the drug was designed to be fully ionized and where the concentration of potassium is high.19 Patiromer’s potassium-binding activity promotes fecal potassium excretion, leading to a decrease in serum potassium.19 In multiple clinical trials, patiromer was generally well tolerated and demonstrated efficacy in both prevention and treatment of hyperkalemia in patients with CKD, HF, and/or diabetes.20–23
Patiromer is not systemically absorbed19; therefore, the potential for drug–drug interactions (DDIs) related to effects on cytochrome P450 isoenzymes or systemic drug transporters is not a clinical concern when patiromer is coadministered with other drugs. However, patiromer has the potential to bind to charged particles in the GI tract, which could lead to reduced absorption of some concomitantly administered oral medications. Previously, during the patiromer development program, 28 orally administered drugs that were likely to be used in patients with CKD having hyperkalemia were tested in vitro. Specifically, the selection of these agents was based on the following criteria: (1) representative drugs from a range of pharmacological drug classes commonly taken by patients with CKD who could be prescribed patiromer, (2) representative narrow therapeutic index drugs, or (3) representative drugs that might be expected to interact based on certain physicochemical characteristics (ie, basic with pKa(s) >9.0, have cationic charges, and/or are hydrophilic). The drugs evaluated in vitro also included examples from all 4 Biopharmaceutics Classification System classes, encompassing a wide range of solubility and permeability, and drugs with known interactions with calcium. The in vitro binding studies were conducted using buffers that represented the physiological pH in 3 different regions of the GI tract: a simulated gastric fluid (pH 1.2), an acetate buffer (pH 4.5), and a simulated intestinal fluid (pH 6.8). All in vitro tests were performed under conditions that reflect the highest proposed clinical dose of patiromer (25.2 g) and the lowest clinical dose of the victim drug and therefore should maximize the possibility of demonstrating an interaction.24
These in vitro binding studies of patiromer and victim drugs served as a screening mechanism and demonstrated that 14 of the 28 victim drugs showed no binding ≥30%,18 which was the threshold considered to indicate the binding of potential clinical relevance,24,25 thereby ruling out the need for additional in vivo studies of these drugs. For 12 of the other 14 drugs that showed in vitro binding ≥30% with patiromer in at least 1 of the 3 matrices tested, it was decided that clinical DDI studies should be conducted, since in vitro studies of other binders (eg, colesevelam25) with 25% binding have had high rates of false-positive findings. The 2 drugs that were not tested were thiamine (commonly available in the diet) and quinidine (a rarely used antiarrhythmic agent). Results of the 12 in vivo studies in healthy volunteers are reported here.
Methods
Study Design
Twelve individual clinical trials were conducted (Celerion, Lincoln, Nebraska, and Tempe, Arizona). Each was a randomized, open-label, 3-period, 3-sequence crossover study. The primary objective of these studies was to evaluate the effect of the perpetrator drug (in this case, patiromer, the drug which might affect the pharmacokinetics [PK] of other drugs) on the single-dose PK of each of the 12 victim drugs (ie, the drugs that might be affected by patiromer) in healthy subjects.
Each study comprised 3 distinct treatment periods defined as the administration of the victim drug with or without patiromer, followed by a washout period. In each treatment period, the victim drug was administered as a single dose alone (treatment A), victim drug administered together with 25.2 g patiromer (treatment B), or victim drug administered at 21 hours after the first patiromer dose and 3 hours before the second patiromer dose (treatment C).
Treatment C was included to establish whether 3-hour separation between administration of the victim drug and patiromer was sufficient to avoid a DDI, if one existed (Figure 1). The order and timing of the victim drug and patiromer administration in treatment C was chosen to replicate a typical administration pattern used in clinical practice (ie, patiromer given daily with the mid-day meal and the victim drug given daily in the morning). The treatment sequences used in all studies were treatments ABC, BCA, and CAB, respectively. Patiromer is recommended to be given with food18; therefore, patiromer and all victim drugs, with the exception of levothyroxine, which is recommended to be given on an empty stomach,26 were administered with food.
Figure 1.

Design of in vivo drug interaction studies: open-label, randomized, 3-way crossover. Treatment A—Each victim drug was administered alone within 30 minutes after the start of a standard breakfast (day 1), except for levothyroxine administered within 40 minutes before breakfast. Treatment B—Victim drugs were administered together with patiromer. Each victim drug was given within 30 minutes after the start of a standard breakfast and patiromer within 10 minutes after the victim drug (day 1), except levothyroxine, administered at 40 minutes before breakfast followed by patiromer administered with breakfast. Treatment C—Victim drugs were administered between 2 patiromer doses. The first dose of patiromer was administered within 30 minutes after the start of a standard lunch (day −1). Each of the victim drugs was administered 21 hours after the first patiromer dose and within 30 minutes of a standard breakfast on day 1 (except levothyroxine, which was administered at 40 minutes prior to standard breakfast). The second patiromer dose was administered 3 hours after the victim drug and within 30 minutes after the start of a standard lunch. aPatiromer and the victim drugs were always administered with meals, except for levothyroxine, which was given on empty stomach, within 40 minutes prior to the meal. bDuration from administration of victim drug to final draw of blood for pharmacokinetics (PK) analysis of drug concentration varied, generally depending on the PK characteristics of the victim drug. Time from the administration of victim drug (day 0) to the beginning of washout (hours): warfarin (168); verapamil (36), lithium (96), trimethoprim (60), amlodipine (144), cinacalcet (144), furosemide (12), metoprolol (36), clopidogrel (32), ciprofloxacin (24), metformin (24), and levothyroxine (48). cDuration of washout before the administration of victim drug after crossover from previous treatment varied, depending on the PK characteristics of the drug. Between-treatment washout periods (in days): warfarin (≥19), verapamil (≥5), lithium (≥10), trimethoprim (≥4), amlodipine (≥14), cinacalcet (≥10), furosemide (≥4), metoprolol (≥5), clopidogrel (≥3), ciprofloxacin (≥3), metformin (≥4), and levothyroxine (≥35).
The study protocols were approved by an independent institutional review board (Chesapeake Research Review, Inc, Columbia, Maryland). The studies were conducted in accordance with the International Conference for Harmonisation Good Clinical Practice guidelines, the principles of the Declaration of Helsinki, and all local and state regulations. All subjects provided written informed consent prior to enrollment.
Study Participants
Inclusion/exclusion criteria were similar across all 12 trials. The trials included healthy, male or female adults, aged 18 to 55 years, with no clinically significant findings in terms of medical history, physical examination, laboratory profiles, vital signs, or electrocardiograms (ECGs) as deemed by the primary investigator. Smoking status was obtained for all the subjects; however, only nonsmokers were enrolled in the cinacalcet, clopidogrel, verapamil, and warfarin trials, due to potential influence of tobacco use on the PK of these drugs.27–30 Smokers (<10 cigarettes/day) could be enrolled in the other trials.
Major exclusion criteria were significant GI disorders, history or presence of hypersensitivity or idiosyncratic reaction to victim drug or related compounds, patiromer or inactive ingredients, or a history of any illness or concomitant medication that, in the opinion of the investigator, might confound the results of the study or pose additional risk. Subjects with a history of or presence of bleeding abnormality or who had increased sensitivity to warfarin based on the genotyping of vitamin K epoxide reductase complex, subunit 1 gene (VKORC1) and cytochrome P450 2C9 gene (CYP2C9) were excluded from the warfarin study. Women of childbearing potential were excluded from the lithium and warfarin trials.
Subjects were screened in the outpatient setting within 28 days prior to the day before patiromer and/or victim drug administration. Safety evaluations included complete physical examination, 12-lead ECG, serum and blood clinical chemistries (including hematology) and urinalysis, drug and alcohol and HIV/hepatitis screen, serum pregnancy test in premenopausal women, and serum follicular stimulating hormone in postmenopausal women. Assessment of inclusion/exclusion criteria and safety evaluations were repeated immediately prior to admission to a clinical research unit (CRU), 2 days before the first dose of the victim drug.
Dosing and Treatments
The oral doses and physicochemical profiles of the victim drugs used in this study are summarized in Table 1. Taking into consideration the dose selection guidelines published by the Food and Drug Administration (FDA)24 and individual bioequivalence recommendations where applicable for an appropriate dose which would be acceptable in healthy volunteers,31 we focused on maximizing the possibility of detecting a DDI, while using a dose level that would be safe for use in healthy subjects. The selected dose of a victim drug was the lowest dose that would provide sufficient concentrations, when given with food, to enable characterization of its PK profile in the event of an interaction with patiromer. In all but 3 cases (levothyroxine, metformin, and warfarin), the dose of the victim drug was consistent with doses used in clinical practice. For these 3 drugs, doses were selected based on the FDA recommendations,32,33 the guidance documents,26,34 or the literature.35,36
Table 1.
Clinical and Physicochemical Profile of Victim Drugs for In Vivo Studies.
| Victim Drug Salt Form | Clinical Dose (mg) | Acid or Base of Salt Form | pKa | MW | BCSa Class | Water Solubility (mg/mL) | GI | |
|---|---|---|---|---|---|---|---|---|
| Influx | Efflux | |||||||
| Amlodipine besylate | 10 | Acid | 9.21 (B) | 567.05 | I | 1 | N | Y |
| Cinacalcet hydrochloride | 90 | Acid | 8.85 (B) | 393.87 | IV | 1.5 | N | N |
| Ciprofloxacin hydrochloride | 500 | Acid | 6.35 (A), 8.34 (B) | 385.82 | IV | 10 | Y | Y |
| Clopidogrel bisulfate | 75 | Acid | 4.66 (B) | 419.9 | II | 100 | N | Y |
| Furosemide | 40 | Base | 3.62 (A), 10.16 (A) | 330.75 | IV | 0.018 | N | Y |
| Levothyroxine sodiumb | 0.6 | Base | 2.00 (A), 6.65 (A), 8.73 (B) | 888.93 | I | 0.15 | Y | N |
| Lithium carbonate | 600 | NA | NA | 73.89 | I | 13.3 | N | N |
| Metformin hydrochloride | 1000 | Acid | 2.94 (B), 13.7 (B) | 165.62 | III | 300 | Y | N |
| Metoprolol tartrate | 100 | Acid | 9.61 (B) | 684.81 | I | 1000 | N | N |
| Trimethoprim | 200 | Base | 7.14 (B) | 290.32 | II | 0.4 | N | Y |
| Verapamil hydrochloride | 120 | Acid | 8.95 (B) | 491.06 | I | 83 | Y | Y |
| Warfarinc sodium | 25 | Base | 4.94 (A) | 308.33 | I | 1000 | N | N |
Abbreviations: A, acid; B, base; BCS, Biopharmaceutics Classification System; GI, gastrointestinal; MW, molecular weight; N, no; NA, not assessed; pKA, acid dissociation constant; Y, yes.
aBCS class I: high permeability, high solubility; class II: high permeability, low solubility; class III: low permeability, high solubility; class IV: low permeability, low solubility. A drug has high permeability when the extent of absorption in humans is determined to be >90% of an administered dose based on mass balance or in comparison with an intravenous reference dose.
bThe 0.6 mg dose of levothyroxine provides adequate exogenous concentrations of thyroid hormone, which can be differentiated from endogenous levels while still considered to be safe to administer as a single dose to healthy subjects.
cThe warfarin dose of 25 mg was chosen to allow sufficient blood levels for PK evaluation but also allowed for pharmacodynamic evaluation of international normalized ratio while not placing subjects at undue risk of bleeding.
The selected patiromer single oral dose of 25.2 g was chosen in these studies as it is the highest approved dose and had been well tolerated in both healthy subjects and patients in phase 1, 2, and 3 clinical trials.18,20,22,37
For each treatment period, all subjects were admitted on day −2. On day −1, subjects were randomized to 1 of 3 treatment sequences according to a randomization scheme. Subjects were required to fast for at least 2 hours prior to a standard lunch on day −1 and overnight for at least 10 hours on day 1. Patiromer was given with meals, and all meals were standardized for similar caloric content (∼2200 total daily calories) and macronutrient composition (∼50% from carbohydrates, ∼20% protein, and ∼30% fat). For subjects assigned to treatment A, the victim drug was administered within 30 minutes after the start of a standard breakfast (day 1), except for levothyroxine, which was given on an empty stomach within 40 minutes before the start of a standard breakfast, as recommended by the prescribing information.26 For subjects assigned to treatment B, victim drugs were administered within 30 minutes after the start of a standard breakfast and patiromer was administered within 10 minutes after the victim drug on day 1 (except levothyroxine, which was administered 40 minutes before breakfast, and patiromer administered with breakfast).
For subjects assigned to treatment C, patiromer was administered within 30 minutes after the start of a standard lunch day −1 (first dose). Victim drug was administered 21 hours after the first patiromer dose and within 30 minutes after the start of a standard breakfast on day 1 (except levothyroxine, which was administered at 40 minutes prior to standard breakfast). The second patiromer dose was administered 3 hours after the victim drug and within 30 minutes after the start of a standard lunch. Depending on the victim drug, subjects were confined to the CRU until after 24- to 72-hour blood draws.
Assessments
Blood was collected from each subject according to a predetermined schedule and was based on the PK characteristics of the victim drug (eg, time to maximum concentration [T max] and apparent elimination half-life [T 1/2]). All studies included a predose blood sample. For levothyroxine, additional predose samples were obtained at 0.25 and 0.5 hours prior to dosing in order to robustly characterize the baseline concentration of endogenous circulating T4 hormone in serum.
Biological matrix (plasma or serum) was analyzed for drug concentration using an appropriate and validated bioanalytical method. Plasma concentrations of the victim drugs were determined using liquid chromatography–tandem mass spectrometry methods validated with respect to accuracy, precision, linearity, sensitivity, and specificity at Celerion, Zurich, Switzerland, Celerion, Lincoln, Nebraska, or inVentiv Health Clinical, Princeton, New Jersey. The analytical range was based on the range needed to adequately characterize the PK of each victim drug.
Pharmacokinetic Analysis and End Points
The key PK parameters describing the rate and extent of systemic exposure of the victim drug with and without patiromer were derived from plasma (or serum) concentration data by noncompartmental methods (Phoenix® WinNonlin® version 6.3; Certara USA, Inc., Princeton, NJ). The area under the plasma concentration–time curve (AUC) from dosing (time 0) until the last measureable time point (AUC0-t), the AUC from dosing, extrapolated to infinity (AUC0-∞), and maximum concentration (C max) constituted the primary end points. Other exploratory observed and estimated PK parameters such as T max and t 1/2 were also determined. For levothyroxine only, due to the presence of endogenous circulating T4 hormone, the PK parameters were adjusted for baseline (endogenous) circulating T4, and the AUC0-48 was employed as the primary AUC end point.
Statistics
Demographic data pooled for all treated subjects are summarized descriptively. Statistical analysis of the derived PK parameters was performed using SAS v9.3 or higher (SAS Institute, Cary, North Carolina). Descriptive statistics included sample size (n); arithmetic mean; standard deviation (SD); and minimum, median, and maximum values, which were calculated for the plasma concentrations and the PK parameters. In addition, geometric means and geometric coefficient of variation percentage were calculated for all PK parameters. Samples from all subjects were assayed, even if the subjects did not complete the study. All subjects who complied with the protocol sufficiently and displayed an evaluable PK profile were included in the statistical analyses.
Sample sizes for each trial were estimated based on the within-subject variability in the primary PK parameters of interest for each victim drug. The 90% confidence interval (CI) for the least squares mean (LSM) ratio for the primary PK end points (AUC0-t or AUC0-48 for levothyroxine), AUC0-∞, and C max were set conservatively at 80% and 125% as default no-effect boundaries, in line with FDA guidance on drug interaction studies,24 with each sample size assuming a type 1 error of 5%. Cross-treatment comparison of the primary PK end points was performed by analysis of variance (ANOVA) with the model including sequence, treatment, and period as fixed effects and subjects nested within sequence as a random effect. Each ANOVA calculated LSM, the difference between treatment LSM, and the standard error associated with this difference. Ratios of LSM were calculated using the exponentiation of the difference in between-treatment LSM derived from the analyses on the in-transformed AUC and C max for plasma victim drug. Consistent with the 2 one-sided tests, a 90% CI for the ratios was derived by exponentiation of the CIs obtained from the difference between treatment LSM resulting from the analyses on the in-transformed AUC and C max. Geometric mean ratios (GMRs) and corresponding 90% CI were calculated as a percentage relative to treatment A.
Safety
Safety during the study and through follow-up was monitored through adverse events (AEs) and vital sign measurements, 12-lead ECGs, and clinical laboratory tests. All subjects who received at least 1 dose of either victim drug or patiromer were prespecified to be included in the safety evaluations.
Results
A total of 370 subjects were enrolled across the 12 studies, of whom 365 subjects received at least 1 dose of patiromer. Among the 5 subjects who did not receive patiromer, 3 were withdrawn due to an AE, 1 was withdrawn due to a positive drug screen, and 1 subject was withdrawn due to protocol violation. Overall, 351 subjects completed their required treatments with both drugs and 19 subjects withdrew from the studies (Figure 2). The most common primary reason for study withdrawal overall was an AE (8 subjects). The mean age across all treatments was 36.1 (18-55) years (Table 2). Most of the subjects were Caucasian (86.3%) and the proportions of men and women were similar (Table 2).
Figure 2.
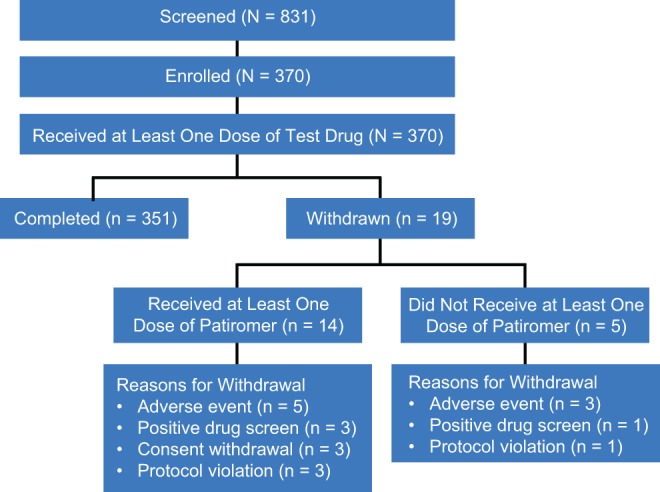
Subject disposition.
Table 2.
Demographic Characteristics of Treated Subjects (N = 370) Across All Studies.
| n (%) | |
|---|---|
| Male | 193 (52.2) |
| Female | 177 (47.8) |
| Age, mean (range), years | 36.1 (18-55) |
| Racea | |
| White | 315 (86.3) |
| Asian | 5 (1.4) |
| African American | 46 (12.6) |
| Native American | 6 (1.6) |
| Other | 1 (0.3) |
| Ethnicity | |
| Hispanic | 210 (56.8) |
| Non-Hispanic | 160 (43.2) |
aThe total percent is >100% as subjects were permitted to check more than 1 race category.
Pharmacokinetic Parameters
The PK profile of each victim drug was adequately characterized in the presence and absence of patiromer. Data are shown in Table 3.
Table 3.
Effect of Patiromer on Pharmacokinetic Parameters of Each Victim Drug.a
| PK Parameter | n | Victim Drug Alone | n | Victim Drug Administered Together With Patiromerb | n | Patiromer Administered 3 Hours After Victim Drugc |
|---|---|---|---|---|---|---|
| Amlodipine, 10 mg | 14 | 13 | 14 | |||
| AUC0-∞, h·ng/mL | 315.7 (34.2) | 253.5 (25.2) | 308.0 (32.4) | |||
| C max, ng/mL | 5.74 (24.6) | 4.78 (20.9) | 5.73 (22.9) | |||
| t max, hours | 9.01 (55.2) | 9.31 (45.0) | 6.29 (35.5) | |||
| t 1/2, hours | 39.1 (39.7) | 36.6 (20.0) | 39.5 (33.6) | |||
| Cinacalcet, 90 mg | 43 | 42 | 40 | |||
| AUC0-∞, h·ng/mL | 528 647 (49.0) | 463 671 (55.6) | 504 614 (47.7) | |||
| C max, ng/mL | 45 788.7 (50.5) | 41 265.4 (61.8) | 46 227.6 (51.5) | |||
| t max, hours | 4.04 (23.9) | 4.53 (55.6) | 3.95 (24.1) | |||
| t 1/2, hours | 70.70 (27.0) | 65.42 (27.2) | 70.30 (24.9) | |||
| Ciprofloxacin, 500 mg | 20 | 18d | 20 | |||
| AUC0-∞, h·ng/mL | 7398.9 (25.6) | 5375.8 (24.6) | 7162.3 (27.7) | |||
| C max, ng/mL | 1565.0 (29.2) | 1035.6 (42.3) | 1635.1 (23.3) | |||
| t max, hours | 1.63 (47.5) | 2.90 (55.6) | 1.50 (48.0) | |||
| t 1/2, hours | 5.36 (17.6) | 5.06 (18.0) | 4.93 (19.6) | |||
| Clopidogrel, 75 mg | 50 | 47e | 50 | |||
| AUC0-∞, h·ng/mL | 10.97 (80.2) | 10.80 (110.7) | 10.74 (86.4) | |||
| C max, ng/mL | 3.83 (92.3) | 2.95 (162.6) | 4.09 (136.7) | |||
| t max, hours | 2.08 (38.0) | 2.98 (44.0) | 2.14 (39.4) | |||
| t 1/2, hours | 6.03 (47.1) | 8.56 (54.0) | 5.16 (47.6) | |||
| Furosemide, 40 mg | 39 | 38f | 38f | |||
| AUC0-∞, h·ng/mL | 1460 (29.4) | 1261 (35.6) | 1376 (31.4) | |||
| C max, ng/mL | 449.6 (63.0) | 357.7 (42.1) | 433.1 (55.1) | |||
| t max, hours | 3.11 (34.7) | 2.62 (54.8) | 3.25 (39.2) | |||
| t 1/2, hours | 3.10 (37.0) | 2.97 (32.0) | 2.82 (24.6) | |||
| Levothyroxine, 0.6 mg | 35 | 34 | 34 | |||
| AUC0-48, h·ng/mLg | 1180.6 (20.9) | 980.21 (25.6) | 1158.9 (20.8) | |||
| C max, ng/mL | 47.96 (23.4) | 45.20 (37.4) | 45.93 (20.1) | |||
| t max, hours | 2.189 (82.0) | 2.163 (66.8) | 2.136 (35.1) | |||
| t 1/2, hours | NA | NA | NA | |||
| Lithium, 600 mg | 16 | 16 | 16 | |||
| AUC0-∞, h·ng/mL | 66 719 (20.8) | 68 462 (21.7) | 64 352 (19.6) | |||
| C max, ng/mL | 3728 (14.6) | 3336 (16.9) | 3513 (13.2) | |||
| t max, hours | 2.275 (26.5) | 3.054 (39.4) | 2.264 (28.3) | |||
| t 1/2, hours | 24.27 (15.4) | 24.87 (17.7) | 24.00 (13.5) | |||
| Metformin, 1000 mg | 17 | 17 | 17 | |||
| AUC0-∞, h·ng/mL | 7954 (18.9) | 6706 (33.9) | 7780 (17.6) | |||
| C max, ng/mL | 1185 (18.5) | 808.2 (28.7) | 1173 (16.9) | |||
| t max, hours | 2.680 (25.4) | 3.390 (41.3) | 2.771 (28.4) | |||
| t 1/2, hours | 4.60 (27.1) | 4.71 (36.2) | 4.20 (22.9) | |||
| Metoprolol, 100 mg | 25 | 25 | 25 | |||
| AUC0-∞, h·ng/mL | 1228 (78.1) | 1085 (87.1) | 1159 (80.6) | |||
| C max, ng/mL | 181.9 (47.1) | 142.9 (54.9) | 189.3 (47.3) | |||
| t max, hours | 2.24 (37.7) | 2.87 (46.5) | 2.02 (43.6) | |||
| t 1/2, hours | 4.468 (37.4) | 4.663 (35.2) | 4.261 (41.1) | |||
| Trimethoprim, 200 mg | 18 | 18 | 18 | |||
| AUC0-∞, h·ng/mL | 25 573 (27.3) | 22 259 (24.1) | 22 380 (26.2) | |||
| C max, ng/mL | 1608.12 (24.3) | 1335.58 (22.3) | 1599.48 (22.4) | |||
| t max, hours | 2.75 (16.8) | 3.53 (40.3) | 2.86 (10.1) | |||
| t 1/2, hours | 9.75 (22.9) | 9.51 (18.5) | 8.79 (22.1) | |||
| Verapamil, 120 mg | 63 | 62 | 62 | |||
| AUC0-∞, h·ng/mL | 865.6 (44.3) | 1210 (29.2) | 875.3 (47.5) | |||
| C max, ng/mL | 165.2 (45.3) | 113.1 (35.7) | 168.8 (61.4) | |||
| t max, hours | 1.948 (45.5) | 2.759 (43.2) | 2.182 (39.9) | |||
| t 1/2, hours | 9.981 (19.8) | 9.219 (15.6) | 9.528 (17.9) | |||
| Warfarin-R, 25 mg | 15 | 14 | 15 | |||
| AUC0-∞, h·ng/mL | 95 350 (24.0) | 95 240 (21.4) | 97 090 (23.0) | |||
| C max, ng/mL | 1416 (14.5) | 1395 (18.6) | 1425 (14.3) | |||
| t max, hours | 4.804 (46.7) | 5.115 (57.0) | 4.805 (50.0) | |||
| t 1/2, hours | 51.94 (19.5) | 51.44 (22.0) | 52.64 (20.5) | |||
| Warfarin-S, 25 mg | 15 | 14 | 15 | |||
| AUC0-∞, h·ng/mL | 53 380 (38.2) | 53 030 (40.2) | 54 240 (39.5) | |||
| C max, ng/mL | 1366 (15.6) | 1354 (23.7) | 1387 (18.3) | |||
| t max, hours | 4.069 (47.8) | 3.468 (63.1) | 3.403 (41.5) | |||
| t 1/2, hours | 37.43 (21.5) | 35.05 (16.3) | 37.77 (18.7) |
Abbreviations: AUC0-∞, area under the plasma concentration time curve from time 0 extrapolated to infinity; C max, maximum observed plasma concentration; CV%, coefficient of variation; LCL, lower confidence limit; NA, not assessed; PK, pharmacokinetic; UCL, upper confidence limit; t max, time to maximum concentration; t 1/2, apparent elimination half-life.
aData presented as arithmetic mean (CV%).
bLevothyroxine which is recommended to be administered from 1/2 hour to 1 hour before meal, and patiromer is recommended to be administered with food, so the 2 drugs were not administered at the same time, and “administered together” represents a 40-minute separation between levothyroxine and patiromer.
cThe first patiromer dose was administered 21 hours before victim drug, and the second patiromer dose was administered 3 hours after victim drug.
dCiprofloxacin C max and t max n = 19.
eClopidogrel C max and t max n = 50.
fFurosemide C max and t max n = 39.
gBaseline-adjusted AUC0-48 is used because extrapolation to infinity is not valid for levothyroxine, because thyroid hormone values do not go to 0 due to endogenous production.
Statistical Comparison of Victim Drugs Administered Together or 3 Hours Before Patiromer
The GMRs and 90% CIs of AUC0-∞ and C max for the 12 victim drugs when administered together with patiromer and when administered 3 hours before patiromer are summarized in Table 4 and Figure 3A and B.
Table 4.
Geometric Mean Ratios.
| Drug | Victim Drug Administered Together With Patiromer,a GMR (90% LCL, UCL) | Victim Drug Administered 3 Hours After Patiromer,b GMR (90% LCL, UCL) | ||
|---|---|---|---|---|
| AUC0-∞ | C max | AUC0-∞ | C max | |
| Amlodipine | 86.3 (82.4, 90.4) | 83.5 (78.4, 88.9) | 98.2 (93.8, 102.8) | 100.2 (94.2, 106.6) |
| Cinacalcet | 86.4 (81.2, 92.0) | 85.5 (75.8, 96.5) | 97.1 (91.1, 103.4) | 99.5 (88.0, 112.5) |
| Ciprofloxacin | 71.5 (65.3, 78.4) | 57.9 (45.4, 73.7) | 95.6 (87.5, 104.4) | 105.4 (82.9, 133.9) |
| Clopidogrel | 90.1 (82.9, 97.8) | 69.1 (62.7, 76.1) | 97.7 (90.2, 105.9) | 102.1 (92.7, 112.4) |
| Furosemide | 84.8 (80.6, 89.2) | 84.2 (73.7, 96.2) | 93.8 (89.2, 98.7) | 95.9 (84.0, 109.6) |
| Levothyroxinec | 81.4 (76.5, 86.7) | 91.6 (84.6, 99.2) | 98.1 (92.1, 104.5) | 95.9 (88.5, 103.8) |
| Lithium | 102.3 (100.2, 104.5) | 89.3 (84.4, 94.5) | 96.1 (94.1, 98.2) | 94.1 (89.0, 99.6) |
| Metformin | 80.6 (72.8, 89.2) | 66.4 (60.7, 72.7) | 98.1 (88.7, 108.6) | 99.2 (90.6, 108.6) |
| Metoprolol | 85.4 (80.8, 90.3) | 76.3 (68.7, 84.6) | 96.3 (91.0, 101.8) | 106.9 (96.3, 118.7) |
| Trimethoprim | 87.8 (84.7, 91.0) | 83.3 (79.8, 86.9) | 87.8 (84.7, 91.0) | 99.9 (95.7, 104.2) |
| Verapamil | 95.9 (92.2, 99.7) | 100.9 (93.5, 108.9) | 100.1 (96.3, 104.0) | 97.7 (90.5, 105.5) |
| Warfarin-R | 99.0 (96.0, 102.2) | 97.8 (93.7, 102.0) | 101.9 (98.9, 105.1) | 100.7 (96.6, 104.9) |
| Warfarin-S | 98.4 (94.8, 102.1) | 98.1 (93.3, 103.1) | 101.1 (97.6, 104.8) | 101.2 (96.3, 106.2) |
Abbreviations: AUC0-∞, area under the plasma concentration time curve from time 0 extrapolated to infinity; C max, maximum observed plasma concentration; GMR, geometric mean ratio; LCL, lower confidence limit; UCL, upper confidence limit.
aLevothyroxine is recommended to be administered from 1/2 hour to 1 hour before meal, and patiromer is recommended to be administered with food, so the 2 drugs were not administered at the same time, and “administered together” represents a 40-minute separation between levothyroxine and patiromer.
bThe first patiromer dose was administered 21 hours before victim drug, and the second patiromer dose was administered 3 hours after victim drug.
cBaseline-adjusted AUC0-48 is used because extrapolation to infinity is not valid for levothyroxine, because thyroid hormone values do not go to 0 due to endogenous production.
Figure 3.

Forest plot of geometric mean ratios (victim/patiromer). A and B, Patiromer administered together with a victim drug: (A) AUC0-∞ and (B) C max. C and D, Victim drugs administered 21 hours after the first patiromer dose and 3 hours before the second patiromer dose: (C) AUC0-∞ and (D) C max. Patiromer and all victim drugs were always administered with food, except for levothyroxine which is recommended to be administered 1/2 to 1 hour before breakfast on an empty stomach. Consequently, patiromer and levothyroxine were not administered at the same time and “administered together” represents a 40-minute separation between levothyroxine and patiromer. aValues adjusted for baseline thyroxine concentration, AUC for 48-hour sampling profile (AUC0-48) is shown because extrapolation to infinity is not valid for levothyroxine due to endogenous thyroxine production. AUC0-48 indicates area under the plasma concentration time curve from time 0 to 48 hours; AUC0-∞, area under the plasma concentration time curve from time 0 extrapolated to infinity; C max, maximum observed plasma concentration; LCL, lower confidence interval limit; N, enrolled subjects; UCL, upper confidence interval limit.
When administered together with patiromer, the 90% CIs for AUC0-∞ remained within the 80% to 125% prespecified bounds for amlodipine, cinacalcet, clopidogrel, furosemide, lithium, metoprolol, trimethoprim, verapamil, and warfarin, indicative of an absence of a PK interaction. The AUC0-∞ point estimates for levothyroxine and metformin were ≥80% of victim drug alone, but the lower bounds of the 90% CIs were 76.5 and 72.8, respectively. For ciprofloxacin, the point estimate and the lower bound of the 90% CI for AUC0-∞ were entirely outside the boundaries from 80% to 125%, suggesting a complex binding interaction (ie, there are likely different but interrelated factors influencing the fraction of victim drug that is bound; Table 4 and Figure 3A).
Of the 9 drugs for which the AUC0-∞ was within the prespecified 90% CIs when administered together with patiromer, the point estimates for C max were ≥80% (range: 83.5%-100.9%) for 7 (amlodipine, cinacalcet, furosemide, lithium, trimethoprim, verapamil, and warfarin; Table 4 and Figure 3A). For 2 of these 9 drugs, the point estimates for C max were <80% (clopidogrel, 69.1% and metoprolol, 76.3%) when administered together with patiromer. For the remaining 3 drugs (ciprofloxacin, levothyroxine, and metformin), the point estimates for C max were 57.9%, 91.6%, and 66.4%, respectively, when administered together with patiromer.
When patiromer was administered 3 hours after each victim drug, the point estimates and 90% CIs for both AUC0-∞ and C max for all 12 victim drugs were well within the 90% CIs of 80% to 125% (Table 4 and Figure 3C, D).
Safety of Patiromer
All subjects who were administered at least 1 dose of patiromer 25.2 g (n = 365) are included in the safety analysis. Across all 12 studies, patiromer was generally well tolerated with safety findings consistent with the approved US prescribing information.18 Overall, 144 (39.5%) subjects experienced at least 1 AE during the studies (Table 5). The most common (>5% of subjects across all drug interaction studies) patiromer-related AEs were GI disorders (all mild, except in 4 subjects with moderate GI AEs), including flatulence (26 [7.1%] subjects), abdominal discomfort/pain (16 [4.4%] subjects), and diarrhea (20 [5.5%] subjects). There were no reported AEs of hypomagnesemia. The AEs leading to study discontinuation were reported in 5 (1.7%) subjects; in 3 subjects, the event was considered by the investigator to be related to patiromer. In 1 subject, the AE leading to discontinuation was GI related (vomiting) but was not considered related to patiromer. One serious AE was reported (supraventricular tachycardia) but was not thought to be related to patiromer. There were no deaths.
Table 5.
Adverse Events in Subjects Administered At Least 1 Dose of Patiromer 25.2 g (n = 365) Across All Studies.
| n (%) | |
|---|---|
| Any adverse event | 144 (39.5) |
| Most common patiromer-related adverse eventsa | |
| Flatulence | 26 (7.1) |
| Diarrhea | 20 (5.5) |
| Abdominal discomfort/pain | 16 (4.4) |
| Adverse events leading to discontinuationb | 5 (1.7) |
| Serious adverse eventc | 1 (0.3) |
aOccurring in >5% of subjects across all drug interaction studies.
bIn 3 subjects, the event was considered by the investigator to be related to patiromer.
cThe serious adverse event was supraventricular tachycardia but was not thought to be related to patiromer.
Discussion
The results of the in vivo studies reported here indicate that 9 of the 12 drugs tested had no PK interaction in terms of extent of absorption (ie, bioavailability) when administered orally together with patiromer as based on the point estimates and 90% CI for AUC. For levothyroxine and metformin, the results showed nominal effects on the extent of absorption (AUC) as evidenced by the lower bounds of the CIs. However, the point estimates for AUC were greater than 80%. In the case of ciprofloxacin, the point estimate of AUC was 71.5% (90% CI, 65.3-78.4), indicating a potential for clinically meaningful DDIs. Of note, the AUC results of the current in vivo drug interaction studies suggest that there was a high rate of false-positive results in the in vitro patiromer binding studies. Of the 14 drugs that were found to bind ≥30% with patiromer in vitro, 12 were assessed in the clinical trials and only 3 showed evidence of potential decreases in the extent of absorption when administered orally together with patiromer (Figure 4).
Figure 4.

Flowchart of drugs tested in and results of in vitro binding and in vivo drug–drug interaction studies.
For 8 of the 12 drugs evaluated in the clinical trials of patiromer, there were no marked changes in the rate of absorption (ie, C max) with point estimates ≥80%. In 4 cases (ciprofloxacin, clopidogrel, metformin, and metoprolol), the point estimate was <80%, with the lowest bound of the 90% CI being 45.4% in the case of ciprofloxacin. Evidently, coadministration of these 4 drugs with patiromer results in a decrease in the rate of absorption, and these findings suggest that complex binding may occur (ie, there are likely many different but interrelated factors influencing the fraction of victim drug that is bound). The clinical consequences of these changes depend on the respective therapeutic range and PK/pharmacodynamic of each drug. However, to put the modest decreases in either AUC or C max into clinical context, in the case of metformin, coadministration with food reduces the AUC and C max similar to patiromer, and the label for metformin recommends administration with or without food,34 confirming that the food effects are not clinically meaningful.
None of the 4 drugs (ciprofloxacin, clopidogrel, metformin, and metoprolol) that showed a reduced C max when administered orally together with patiromer are considered narrow therapeutic range drugs, and all but ciprofloxacin can be titrated to desired clinical effects and are administered chronically unlike acute effect drugs such as pain medications.24,26,34,38 In this respect, AUC is a more important PK parameter than C max from a clinical perspective; therefore, the lower C max for clopidogrel, metformin, and metoprolol are unlikely to bear any clinical significance. The changes in AUC and C max for ciprofloxacin when given with patiromer may be clinically significant and may be an important source of variability when treating patients with this and possibly other quinolones. Decreases in the rate and extent of ciprofloxacin absorption may raise the concern that ciprofloxacin drug concentrations may not exceed the desired minimum inhibitory concentration for commonly encountered bacterial pathogens for most of the recommended dosing interval. Therefore, the PK changes seen with ciprofloxacin when administered together with patiromer suggest that the administration of these 2 medicines should be separated. Evidence of a clinically relevant interaction between patiromer and ciprofloxacin is consistent with the drug interaction profile of fluoroquinolone antibiotics, which includes decreased absorption when administered together with medications that contain multivalent cations and sometimes with food.39–41 Two of the 3 drugs that showed PK drug interactions in this study (levothyroxine26 and ciprofloxacin42) have known interactions with calcium, with prescribing information that recommends dosing separation between these drugs and calcium-containing medications (eg, calcium supplements, antacids). In the current in vivo patiromer studies, a 3-hour separation in the administration of victim drug (given first) and followed by the patiromer dose resulted in an absence of significant changes in either the rate or extent of absorption for all 12 drugs (including the worst case of ciprofloxacin).
We conducted our DDI studies as recommended by the US FDA—in healthy volunteers using the maximal approved dose of patiromer and the lowest possible dose of the victim drug that would allow quantification (ie, worst case scenario) in order to maximize the probability of finding DDIs if they were to occur.24 Additionally, these studies were conducted using a standardized diet (approximately 50% of carbohydrates, 30% of fat, 20% of protein, and 2200 of total daily calories) that reflects a typical diet recommended for adults in the United States.43 The results of these DDI studies are thought to represent those that would be observed in the majority of patients, and there is very little evidence available to refute this assumption. Therefore, we believe the current studies are sufficient to answer the question of potential interactions with these drugs in patients. A limitation is that the studies were conducted with the victim drugs given 3 hours before the second dose of patiromer, and we did not study patiromer given 3 hours before the victim drugs. However, based on the profiles and kinetics of gastric emptying of homogenized solids and liquid nutrients as summarized by Camilleri,44 a 3-hour window between administration of a binder such as patiromer and a victim drug both with particle size <1 mm would be expected to allow a median of 80% of drug to empty from the stomach and thereby avoid potential binding interactions. A 3-hour separation would also be sufficient to avoid potential DDIs from delayed gastric emptying.44
An additional limitation is that levothyroxine in treatment B was not strictly coadministered with patiromer, given the label requirements to dose the former 30 to 60 minutes before breakfast and the latter with meals.18,26
In summary, 12 clinical DDI studies were conducted with patiromer. For 9 drugs, the 90% CIs for the geometric mean AUC ratio remained entirely within 80% to 125% bounds, and for 2 drugs, the point estimate for the geometric mean AUC ratio was ≥80%, indicative of no clinically significant effects, or nominal effects in the case of the 2 drugs, of patiromer on the extent of absorption of coadministered drugs. The exception was ciprofloxacin with a point estimate of 71.5%. Given the uncertainty around the clinical significance of the reduced AUC of ciprofloxacin, it is wise to not coadminister it with patiromer. With regard to the peak oral absorption, patiromer had no significant effect for 8 of the 12 drugs studied with the point estimate of C max ≥80%. Of the remaining 4 drugs, the decrease in C max is likely to be clinically meaningful for only ciprofloxacin. When administration of patiromer is separated from each of the drugs by 3 hours, the results based on 90% CI allow one to conclude that no clinically significant DDIs occur in vivo.
Acknowledgments
Editorial assistance was provided by Julie Obeid of Relypsa, Inc, and Ewa Wandzioch, PhD, of AlphaBioCom, LLC, funded by Relypsa, Inc.
Footnotes
Author Contributions: Lawrence J. Lesko contributed to interpretation and editing the manuscript. Elliot Offman contributed to design and contributed to acquisition, analysis, and interpretation. Christine Taylor Brew contributed to interpretation. Dahlia Garza contributed to acquisition, analysis, and interpretation and drafted the manuscript. Wade Benton contributed to conception and design, contributed to acquisition, analysis, and interpretation, and drafted the manuscript. Martha R. Mayo contributed to conception and design and contributed to interpretation. Alain Romero contributed to design and contributed to analysis and interpretation. Charles Du Mond contributed to conception and design, contributed to acquisition, analysis, and interpretation, and drafted the manuscript. Matthew R. Weir contributed to conception. All authors critically revised the manuscript, gave final approval, and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Lawrence J. Lesko reports fees from Relypsa, Inc, for consulting services. Dr Elliot Offman was an employee of Celerion, Inc. at the time of writing and is currently an employee of Certara. Christine Taylor Brew, Dahlia Garza, Martha Mayo, Alain Romero, and Charles Du Mond are employees of Relypsa, Inc. Wade Benton was an employee of Relypsa when these studies were conducted. Dr Weir reports fees for scientific advisory boards from Relypsa, Inc and from ZS Pharma; personal fees from Akebia, Janssen, AstraZeneca, Amgen, MSD, AbbVie, Novartis, Boston Scientific, and Sandoz outside the submitted work.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was funded by Relypsa, Inc.
References
- 1. Hayes J, Kalantar-Zadeh K, Lu JL, Turban S, Anderson JE, Kovesdy CP. Association of hypo- and hyperkalemia with disease progression and mortality in males with chronic kidney disease: the role of race. Nephron Clin Pract. 2012;120(1):c8–c16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sarafidis PA, Blacklock R, Wood E, et al. Prevalence and factors associated with hyperkalemia in predialysis patients followed in a low-clearance clinic. Clin J Am Soc Nephrol. 2012;7(8):1234–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kovesdy CP. Epidemiology of hyperkalemia: an update. Kidney Int Suppl. 2016;6(1):3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Einhorn LM, Zhan M, Hsu VD, et al. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med. 2009;169(12):1156–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Loutradis C, Tolika P, Skodra A, Avdelidou A, Sarafidis PA. Prevalence of hyperkalemia in diabetic and non-diabetic patients with chronic kidney disease: a nested case-control study. Am J Nephrol. 2015;42(5):351–360. [DOI] [PubMed] [Google Scholar]
- 6. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351(13):1296–1305. [DOI] [PubMed] [Google Scholar]
- 7. Reardon LC, Macpherson DS. Hyperkalemia in outpatients using angiotensin-converting enzyme inhibitors. How much should we worry? Arch Intern Med. 1998;158(1):26–32. [DOI] [PubMed] [Google Scholar]
- 8. O’Gara PT, Kushner FG, Ascheim DD, et al. ; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the American College of Emergency Physicians and Society for Cardiovascular Angiography and Interventions. Catheter Cardiovasc Interv. 2013;82(1):E1–E27. [DOI] [PubMed] [Google Scholar]
- 9. Ponikowski P, Voors AA, Anker SD, et al. ; Authors/Task Force Members. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC): developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129–2200. [DOI] [PubMed] [Google Scholar]
- 10. Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004;351(6):585–592. [DOI] [PubMed] [Google Scholar]
- 11. Bozkurt B, Agoston I, Knowlton AA. Complications of inappropriate use of spironolactone in heart failure: when an old medicine spirals out of new guidelines. J Am Coll Cardiol. 2003;41(2):211–214. [DOI] [PubMed] [Google Scholar]
- 12. Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol. 2005;46(5):845–849. [DOI] [PubMed] [Google Scholar]
- 13. Epstein M, Reaven NL, Funk SE, McGaughey KJ, Oestreicher N, Knispel J. Evaluation of the treatment gap between clinical guidelines and the utilization of renin-angiotensin-aldosterone system inhibitors. Am J Manag Care. 2015;21(11 suppl):S212–S220. [PubMed] [Google Scholar]
- 14. Epstein M, Pitt B. Recent advances in pharmacological treatments of hyperkalemia: focus on patiromer. Expert Opin Pharmacother. 2016;17(10):1435–1448. [DOI] [PubMed] [Google Scholar]
- 15. Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med. 2001;344(1):3–10. [DOI] [PubMed] [Google Scholar]
- 16. Kayexalate® [Sodium Polystyrene Sulfonate]. Prescribing Information. 2014. http://products.sanofi.ca/en/kayexalate.pdf. Accessed May 24, 2016.
- 17. Harel Z, Gilbert C, Wald R, et al. The effect of combination treatment with aliskiren and blockers of the renin-angiotensin system on hyperkalaemia and acute kidney injury: systematic review and meta-analysis. BMJ. 2012;344:e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Veltassa® [patiromer] for oral suspension. Full Prescribing Information. 2016. www.veltassa.com/pi.pdf. Accessed January 20, 2017.
- 19. Li L, Harrison SD, Cope MJ, et al. Mechanism of action and pharmacology of patiromer, a nonabsorbed cross-linked polymer that lowers serum potassium concentration in patients with hyperkalemia. J Cardiovasc Pharmacol Ther. 2016;21(5):456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bakris GL, Pitt B, Weir MR, et al. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: The AMETHYST-DN randomized clinical trial. JAMA. 2015;314(2):151–161. [DOI] [PubMed] [Google Scholar]
- 21. Pitt B, Bakris GL, Bushinsky DA, et al. Effect of patiromer on reducing serum potassium and preventing recurrent hyperkalaemia in patients with heart failure and chronic kidney disease on RAAS inhibitors. Eur J Heart Fail. 2015;17(10):1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weir MR, Bakris GL, Bushinsky DA, et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med. 2015;372(3):211–221. [DOI] [PubMed] [Google Scholar]
- 23. Pitt B, Anker SD, Bushinsky DA, et al. ; PEARL-HF Investigators. Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double-blind, placebo-controlled study in patients with chronic heart failure (the PEARL-HF) trial. Eur Heart J. 2011;32(7):820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guidance for Industry. Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. 2012. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf. Accessed May 24, 2016.
- 25. Walker JR, Brown K, Rohatagi S, et al. Quantitative structure-property relationships modeling to predict in vitro and in vivo binding of drugs to the bile sequestrant, colesevelam (Welchol). J Clin Pharmacol. 2009;49(10):1185–1195. [DOI] [PubMed] [Google Scholar]
- 26. Synthroid® [levothyroxine sodium tablets, USP]. Product Information. 2012. http://www.rxabbvie.com/pdf/synthroid.pdf/. Accessed June 2, 2016.
- 27. Anderson GD, Chan LN. Pharmacokinetic drug interactions with tobacco, cannabinoids and smoking cessation products. Clin Pharmacokinet. 2016;55(11):1353–1368. [DOI] [PubMed] [Google Scholar]
- 28. Fuhr U, Muller-Peltzer H, Kern R, et al. Effects of grapefruit juice and smoking on verapamil concentrations in steady state. Eur J Clin Pharmacol. 2002;58(1):45–53. [DOI] [PubMed] [Google Scholar]
- 29. Gurbel PA, Bliden KP, Logan DK, et al. The influence of smoking status on the pharmacokinetics and pharmacodynamics of clopidogrel and prasugrel: the PARADOX study. J Am Coll Cardiol. 2013;62(6):505–512. [DOI] [PubMed] [Google Scholar]
- 30. Lenzini PA, Grice GR, Milligan PE, et al. Laboratory and clinical outcomes of pharmacogenetic vs. clinical protocols for warfarin initiation in orthopedic patients. J Thromb Haemost. 2008;6(10):1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Food and Drug Administration. Product-specific recommendations for generic drug development. 2013. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm075207.htm. Accessed July 27, 2016.
- 32. Food and Drug Administration. Bioequivalence recommendations for specific products. Draft guidance on metformin hydrochloride. 2008. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm088672.pdf. Accessed June 2, 2016.
- 33. Food and Drug Administration. Bioequivalence recommendations for specific products. Draft guidance on levothyroxine sodium. 2014. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM428208.pdf. Accessed June 2, 2016.
- 34. Glucophage® [metformin hydrochloride] tablets. Product Information. 2009. http://packageinserts.bms.com/pi/pi_glucophage_xr.pdf/. Accessed June 2, 2016.
- 35. Benedek IH, King SY, Powell RJ, Agra AM, Schary WL, Pieniaszek HJ., Jr Effect of moricizine on the pharmacokinetics and pharmacodynamics of warfarin in healthy volunteers. J Clin Pharmacol. 1992;32(6):558–563. [DOI] [PubMed] [Google Scholar]
- 36. Jiang X, Williams KM, Liauw WS, et al. Effect of St John’s wort and ginseng on the pharmacokinetics and pharmacodynamics of warfarin in healthy subjects. Br J Clin Pharmacol. 2004;57(5):592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bushinsky DA, Spiegel DM, Gross C, et al. Effect of patiromer on urinary Ion excretion in healthy adults. Clin J Am Soc Nephrol. 2016;11(10):1769–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rowland M, Tozer TN. Clinical Pharmacokinetics and Pharmacodynamics. Concepts and Applications. 4th ed Philadelphia, PA: Lippincott Williams & Wilkens; 1995. [Google Scholar]
- 39. Radandt JM, Marchbanks CR, Dudley MN. Interactions of fluoroquinolones with other drugs: mechanisms, variability, clinical significance, and management. Clin Infect Dis. 1992;14(1):272–284. [DOI] [PubMed] [Google Scholar]
- 40. Sahai J, Gallicano K, Oliveras L, Khaliq S, Hawley-Foss N, Garber G. Cations in the didanosine tablet reduce ciprofloxacin bioavailability. Clin Pharmacol Ther. 1993;53(3):292–297. [DOI] [PubMed] [Google Scholar]
- 41. Stass H, Kubitza D. Profile of moxifloxacin drug interactions. Clin Infect Dis. 2001;32(suppl 1):S47–S50. [DOI] [PubMed] [Google Scholar]
- 42. Cipro® [ciprofloxacin hydrochloride] tablets, Cipro® [ciprofloxacin] 5% and 10% Oral Suspension. Product Information. 2000. https://www.dartmouth.edu/∼genchem/0102/spring/6winn/pdfs/ciprotab.pdf. Accessed June 14, 2016.
- 43. U.S. Department of Agriculture and U.S. Department of Health and Human Services. Dietary Guidelines for Americans, 2010. 7th ed Washington, DC: U.S. Government Printing Office; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Camilleri M. Drug-resin drug interactions in patients with delayed gastric emptying: what is optimal time window for drug administration? Neurogastroenterol Motil. 2016;28(8):1268–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]