

Abstract
Thyroid neoplasms encompass a variety of lesions that range from benign adenomas to malignancies. These latter can be well-differentiated, poorly differentiated or undifferentiated (anaplastic) carcinomas. More than 95% of thyroid cancers are derived from thyroid follicular cells, while 2-3% (medullary thyroid cancers, MTC) originate from calcitonin producing C-cells. Over the last decade, investigators have developed a clearer understanding of genetic alterations underlying thyroid carcinogenesis. A number of point mutations and translocations are involved, not only in its tumorigenesis, but also as have potential use as diagnostic and prognostic indicators and therapeutic targets. Many occur in genes for several important signaling pathways, in particular the mitogen-activated protein kinase (MAPK) pathway. Sporadic (isolated) lesions account for 75% of MTC cases, while inherited MTC, often in association with multiple endocrine neoplasia (MEN) type 2A and 2B syndromes, constitute the remainder. However, non-MEN familial MTC may also occur. Advances in genetic testing have revolutionized the management of MTC, with prospects of genetic screening, testing and early prophylactic thyroidectomy. Ethical concerns of these advances are addressed.
Keywords: Thyroid cancer, mutations, undifferentiated thyroid cancer, differentiated thyroid cancer, medullary thyroid
Introduction
Thyroid cancer is the most common type of endocrine malignancy with an incidence that has steadily increased for the past three decades (Davies and Welch, 2006; Pellegriti et al.,2013). Deaths from thyroid cancers alone account for more deaths than all of the other endocrine malignancies combined in the U.S.A(Davies and Welch 2006), with an estimate of 62,450 new cases and 1,950 deaths for 2014 (Pellegriti et al., 2013). The increased incidence of thyroid cancer diagnoses has been attributed, in part, to improved detection of small or subclinical thyroid nodules by thyroid ultrasonography and other imaging techniques; however, increased incidence of thyroid tumors of all sizes has also been reported (Albores-Saavedra et al., 2007).
Thyroid cancer typically presents as a thyroid nodule. However, thyroid nodules are commonly and incidentally found and may be seen in up to 50% of patients older than 60 years of age. Only about 5% of thyroid nodules are malignant (Brito et al., 2013). Epithelial malignant cancers of the thyroid arise from two different types of parenchymal cells, follicular and parafollicular. Follicular cells line the colloid follicles, concentrate iodine and are predominantly involved in production of thyroid hormones. These cells give rise to well differentiated and anaplastic thyroid cancers. The parafollicular or C cells, which are spread among the thyroid follicles, are responsible for the production of calcitonin and give rise to medullary thyroid cancer (MTC). (Burgess andTucker, 2006).
While well differentiated thyroid carcinomas (DTC) account for 90% of all thyroid cancers, medullary thyroid carcinomas account for 2-3%, and anaplastic carcinomas and poorly differentiated carcinomas for the remaining 7 to 8%. The DTCs are further divided histologically to papillary thyroid cancer (80–85%), follicular thyroid cancer (10–15%) and Hurtle cell carcinoma (3–5%). Overall, DTCs have a very good prognosis with long term disease free survival close to 95% for papillary thyroid cancers (PTC) and 80% for follicular thyroid cancers (FTC). The MTCs are clinically classified as sporadic or familial cancers. Sporadic MTCs occur as localized cancers with infrequent lymph node involvement (unifocal) and correspond to 70% of all cases, while familial cancers are typically diagnosed as advanced disease (multifocal) in the remaining 30% of the cases (Kloos et al., 2009). Familial MTCs have been described as part of the multiple endocrine neoplasia (MEN 2a) syndrome which includes the presence of pheochromocytoma and parathyroid hyperplasia, and of the MEN 2b syndrome that also include pheochromocytoma and mucosal neuromas and/or gastrointestinal ganglioneuromas.
Most thyroid cancers are well-differentiated papillary carcinomas or follicular carcinomas and are associated with a low mortality rate, particularly in patients with stage I or II disease (survival rate more than 98%). However, a subset of these patients will have recurrent disease. In addition, patients who present with a higher stage disease or distant metastases and patients with poorly differentiated or anaplastic thyroid cancer have high mortality rates (Hansford and Mulligan, 2000). Accurate identification of subsets of patients with risk factors for aggressive disease and higher mortality rates can help to guide management and prevent overtreatment of patients with low-risk disease.
In addition to environmental factors, genetic factors are involved in thyroid cancer predisposition. Aside from the well-characterized familial forms of medullary thyroid cancer, an individual whose first degree relative is diagnosed with non-medullary thyroid cancer has a fourfold to tenfold higher risk than those in the general population (Moses et al., 2011; Jung et al., 2014). The last several years have seen dramatic advances in our understanding of the genomics of thyroid cancer. Major technological advances have allowed scientists to interrogate DNA and RNA changes at a depth and speed that were previously impossible. Several genetic abnormalities have been involved in the pathogenesis of thyroid cancers, which induce dysregulation of the MAPK and phosphatidylinositol-3 Kinase (P13K)/AKT signaling pathways (Frich et al., 2001). (fig.3)
Figure 1.
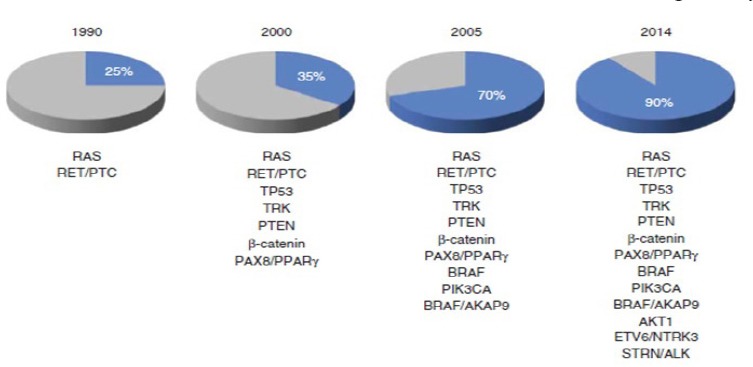
Progress in Identifying Mutational Markers in Thyroid Cancer (Hsiao And Nikiforov, 2014).
Figure 2.
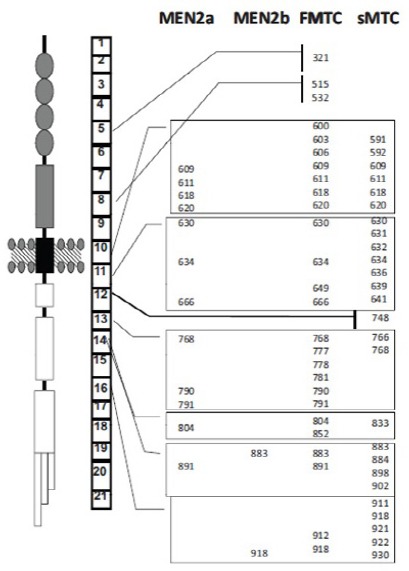
It Shows the RET Receptor, the Gene with Its Exons, and the Most Common Mutations in Hereditary and Sporadic MTC (Taccaliti, 2011).
Figure 3.

Molecular Pathway in Thyroid Cancer. The molecular pathogenesis of thyroid cancer includes dysregulation of the MAPK, phosphatidylinistol-3kinase (PI3KYAKT, and the TSHR cAMP signaling pathway. The MAPK pathway is frequently activated in thyroid cancer through point mutation of the BRAF and RAS and RET/PTC (Hsiao and Nikiforov, 2014).
Mutations
RET/PTC rearrangements
The RET proto-oncogene is a 20-exon gene located on chromosome 10q11.2 and encodes a membrane tyrosine kinase receptor. It is expressed in thyroid C cells, but not in normal follicular cells (Fenton et al., 2000). The RET can be activated by fusion with various partners, in which the 3’ or tyrosine kinase domain of the RET gene is fused with the 5’ domain of a constitutively expressed foreign gene. This fusion results in a permanent expression of the rearranged RET. These rearranged genes have coiled-coil domains that activate ret kinase through permanent dimerization. Because these rearrangements were originally found only in papillary thyroid cancers they were called RET/PTC (de Groot et al., 2006 Santoro et al., 1994). Point mutation of RET proto-oncogene result in an unregulated dominant activation of receptor.
RET mutation and medullary thyroid carcinoma
Somatic and germline mutations of RET play an important role in the development of sporadic and familial forms of MTC, respectively. Genetic diagnosis has an important role in differentiating sporadic from familial MTC. Furthermore, depending on the location of the mutation, patients can be classified into risk classes. Therefore, genetic testing of RET plays a critical role not only in diagnosis but also in assessing the prognosis and course of MTC (Agrawal et al., 2013).
MTCs are characterized by activating mutations of RET proto-oncogene which occur prevalently as point alteration of codons. In 98% of MEN 2a families, a germ-line activating RET mutation can be detected. Germline RET mutations indicate hereditary MTC and determine the lifetime risk for developing MTC, which is nearly 100% for RET mutation carriers. A high prevalence of de novo RET mutations (over 50%) has been identified in MEN 2b patients, and to a lesser extent in MEN 2a/FMTC patients (Taccaliti et al., 2011).
Also, germ-line RET mutations are frequently detected in apparently sporadic MTC patients, indicating the importance of genetic testing in all MTC patients, even without a clear indication for hereditary disease. Somatic RET mutations can be detected in tumor tissue of 40-60% of sporadic MTC patients. The contribution to tumor development of somatic RET mutations in MTC pathogenesis is unclear. Somatic RET mutations are not consistently distributed within primary tumors and metastases, indicating that the mutation can occur during progression of the tumor or that MTC is a disease of polyclonal origin (Taccaliti et al., 2011). Probably in these cases, somatic RET mutations merely contribute to the disease phenotype instead of causing it (Figure 2).
RAS mutations
The RAS genes (HRAS, KRAS and NRAS) encode a 21 kD protein (p21) involved in signal transmission from cell membrane receptors to growth factors to the nucleus to both the MAPK and PI3K/AKT pathways (Figure 3). The KRAS is activated by point mutations mostly in codon 12 or 61 and sometimes in codon 13 or 59. In thyroid cancers, point mutations are found in the three RAS genes, with NRAS being the most frequently involved. RAS mutations are found in 10-20% of papillary thyroid carcinoma and in 25% of poorly differentiated thyroid cancers. The RAS-mutated papillary thyroid carcinomas are typically of the follicular variant. RAS mutations are also seen in 20-40% of follicular adenomas (Wells et al., 2013). RAS mutation mostly HRAS,KRAS and NRAS have been found in 68% of sporadic MTC without RET mutation (Marques et al., 2002).
Mutations in the P13K/AKT pathway
P13K/AKT pathway may be driven by activating RAS point mutation. Inactivating mutations or deletions of the tumor suppressor gene PTEN activate the P13K/AKT pathway and constitute the genetic basis for follicular thyroid cell tumor genesis in Cowden syndrome (Dahia et al., 1997). Activating mutations of PIK3CA are common in follicular thyroid carcinoma, poorly differentiated thyroid carcinoma and anaplastic thyroid carcinoma. AKT1 mutations were only found in metastatic thyroid cancers. Promoter methylation of PTEN, which is consistent with the loss of its expression is associated with genetic alterations of P13K-AKT pathway in follicular thyroid carcinoma and anaplastic thyroid carcinoma, including mutations of RAS, PIK3CA and PTEN (Garcia-Rostan et al., 2005; Houet al., 2007). Over-expression of proteins involved in this pathway is frequently found in aggressive thyroid cancers.
BRAF
The BRAF provides instructions for making a protein that helps the transmission of chemical signals from outside the cell to the nucleus. This protein is part of a signaling pathway known as the RAS/MAPK pathway, which controls several important cell functions. Specifically, the RAS/MAPK pathway regulates the growth and division (proliferation) of cells, the process by which cells mature to carry out specific functions (differentiation), cell movement (migration), and the programmed self-destruction of cells (apoptosis). Chemical signaling through this pathway is essential for normal development before birth (Chiosea et al., 2009; Ciampi and Nikiforov, 2005).
Point mutation in BRAF is found in approximately 45% of papillary thyroid cancers (PTC) and less frequently in poorly differentiated and in anaplastic thyroid cancers. In most cases, the p.V600E mutation causes constitutive activation of this serine/threonine kinase. In few cases other BRAF mutations such as the p.K601E mutation, small inframe insertions or deletions, or BRAF rearrangement can occur (Kim TY et al., 2005; Adeniran et al., 2006). BRAF mutations are identified in 60% of classic PTC, 80% of tall-cell variant PTC, and only 10% of follicular variant PTC.
Many studies demonstrated a significant association of BRAF p.V600E with poor clinicopathological outcomes of PTC, including aggressive pathological features, increased recurrence risk, loss of radioiodine avidity, treatment failures and mortality. However, other small number of studies have not confirmed the prognostic impact of BRAF mutations (Ito et al., 2009; Liu et al., 2005). In addition to serving as a driver mutation in papillary thyroid cancer, activation of the MAPK pathway via BRAF mutations results in decreased expression of sodium iondine symporter, TSH receptor, and thyroglobulin, resulting in a relative iodine refractory state (Ricarte-Filho et al., 2009).
Other gene abnormalities
Mutations in hTERT have been found in follicular cell derived thyroid cancers, but were absent in benign lesions and in medullary thyroid cancers (Landa et al., 2013). These hTERT mutations have a significantly higher prevalence in aggressive thyroid tumors including widely invasive oncocytic carcinoma and anaplastic thyroid carcinoma. In papillary thyroid cancer, a hTERT mutation has been more frequently found in advanced tumors and in tumors with a BRAF p.V600E mutation, and the rate of recurrence is 8 times greater in tumors with both mutations, as compared to patients who lack both mutations (Liu et al., 2013).
Other important genes that are mutated in thyroid tumorigenesis include CTNNB1 (β-catenin) that is involved in Wnt signaling, and TP53 a tumor suppressor. These mutations have been mostly found in poorly differentiated and anaplastic thyroid cancer, in particular TP53 mutations that may participate in de-differentiation of these tumors (Garcia-Rostan et al., 2001; Dobashi et al., 1994)
Mutations in TSHR (thyroid stimulating hormone receptor) are found in 40% to 60% of benign hyperfunctioning adenomas. Activating mutations of TSHR have also been found in the rare hyperfunctioning follicular carcinomas with high radioiodine uptake and thyrotoxicosis. The role of TSHR in the tumorigenesis of some hypofunctioning thyroid tumors is unclear (Donghi et al., 1993).
The gene for the alpha polypeptide chain (αs) of the heterotrimeric G protein Gs can be activated to the putative oncogene gsp by specific point mutations at codons 201 and 227. Such mutations have been reported in 40% of human growth hormone-secreting pituitary adenomas and in a single autonomously functioning thyroid adenoma. However the role of this mutation in thyroid tumors is unclear
In Hűrthle-cell thyroid carcinoma, mutations of NDUFA13 (NADH:ubiquinone oxidoreductase subunit A13 are fairly common but classical genetic alterations frequently found in other thyroid cancer subtypes, such as RET/PTC, RAS or BRAF mutations are not found (Nishihara et al., 2009).
Oncogenic gene amplification or copy-number gains are more prevalent in poorly differentiated and anaplastic than in differentiated thyroid carcinomas, suggesting that these genetic alterations are important for the progression and aggressiveness of thyroid cancer. This is particularly the case for genes encoding receptor tyrosine kinases (RTKs) and for the genes encoding P13K-AKT pathway members. Many of the genes with copy-number gains are proto-oncogenes, and an increased protein expression will induce activation of the signaling pathways in which they are involved (Liu et al., 2008). TRK rearrangements are found in 1-5% of papillary thyroid carcinomas and at higher frequencies in patients with a history of radiation exposure (Leeman-Neill et al., 2014). Anaplastic lymphoma kinase gene, (ALK) was found in 9% of poorly differentiated thyroid cancers, 4% of anaplastic thyroid cancers, and 1% of papillary thyroid cancers (Leeman-Neill et al., 2014). The PAX8/PPARɤ rearrangement is found in 30-40% of follicular carcinomas. It is also found, at lower prevalence, in the follicular variant of papillary thyroid carcinoma and in follicular adenomas (Leeman-Neill et al., 2013; Xing et al., 2005).
Altered Signaling Pathways in Thyroid Cancer
The MAPK and P13-1AKT signaling pathways
The importance of the MAPK pathway has been well established in the tumorigenesis of papillary thyroid cancer. The MAPK pathway is driven by activating mutations, including BRAF and RAS mutations, by RET/PTC, TRK or ALK rearrangements. One of these mutations is found in 70-80% of papillary thyroid cancers and genetic abnormalities are found in over 95% of cases by using massively parallel next generation sequencing (NGS). These driver mutations are mutually exclusive, indicating that this mutation may be responsible for the occurrence of the papillary thyroid cancer. Activation of this pathway induces the activation of phosphatases, resulting in feedback mechanisms that will limit the activation of the pathway (Bongarzone et al., 1998). MAPK-mediated thyroid tumorigenesis involves a wide range of secondary molecular alterations that synergize and amplify the oncogenic activity of this pathway, such as genome-wide hypermethylation and hypomethylation and altered expression of miRNAs. Upregulation of various oncogenic proteins can occur that drive cancer cell proliferation, growth, migration and survival, as well as tumor angiogenesis, invasion and metastasis. These include chemokines, vascular endothelial growth factor A (VEGFA), C-MET, nuclear factor κB (NFκB), matrix metalloproteinases (MMPs), hypoxia-inducible factor 1α (HIF1α), transforming growth factor β1 (TGFβ1). Many of these proteins are key constituents of the extracellular matrix microenvironment (Kimura et al., 2003; Liu et al., 2010). All these alterations permit the distinction of two main groups of papillary thyroid cancers: one group includes tumors that harbor a RAS mutation and another group includes tumors that harbor a BRAF mutation. This second group is heterogeneous but in general tumors have a more aggressive and less differentiated phenotype.
The MAPK and P13K-AKT pathways are primarily involved in differentiated papillary and follicular thyroid carcinoma, respectively. As genetic alterations accumulate and both pathways become activated, the tumor progresses into poorly differentiated and anaplastic thyroid carcinoma (Musholt et al.,2000). The coexistence of multiple genetic alterations to members of the MAPK pathway also occurs, as exemplified by the simultaneous presence of BRAF p.V600E mutation, RAS mutations and RET-PTC in some aggressive recurrent papillary and anaplastic thyroid cancers. In metastatic tissues, the dominant genetic abnormalities present in the primary tumor are usually found with additional abnormalities in some.
Impairment of the Iodide-Handling Machinery
Aberrant activation of the MAPK pathway has a crucial role in the impairment of the iodide uptake and metabolism. In addition to serving as a driver mutation in papillary thyroid cancer, activation of the MAPK pathway via BRAF mutation results in decreased expression of NIS, TPO, TSH-R, and Tg resulting in a relative iodine refractory state. Inhibition of the activated MAPK pathway with a BRAF inhibitor or a MEK inhibitor was able to restore the effectiveness of RAI therapy in mouse thyroid cancers with BRAF activation, and in small cohorts of RAI refractory differentiated thyroid cancer patients (Xing, 2009). Activation of the PI3K-AKT pathway was also shown to down regulate the iodide uptake and metabolism in thyroid cells both in vitro and in vivo (Xing, 2009).
Clinical implications
Remarkable advances in the translation of molecular findings in thyroid cancer to the clinic have occurred recently.
Diagnosis of thyroid cancer
The diagnostic accuracy for thyroid nodules that are otherwise diagnostically indeterminate by conventional cytology assessment is improved by the search for genetic markers in Spell it out Fine needle aspiration biopsy (FNAB) samples, including BRAF mutation, RAS mutations, RET-PTC and PAX8-PPARᶌ rearrangements. More recently the diagnostic performance of this strategy was further improved by the use of Next-generation sequencing (NGS). Also, the use of a gene-expression classifier permits to rule out malignancy in half of the nodules with indeterminate cytology results (Baloch et al., 2008;Bartolazzi et al., 2008). The differentiation of radiation induced thyroid tumors from thyroid tumors occurring in the absence of radiation exposure may benefit from mutation analysis. Rearrangements are more frequently and point mutations less frequently found after radiation exposure. Also the study of the expression of a panel of genes helps in this differentiation.
Prognosis
The prognostic application of BRAF p.V600E mutation has also been the subject of many clinical studies. It appeared that BRAF p.V600E is associated with poorer clinicopathological outcomes even in conventionally low-risk patients. However, the use of mutation status in clinical practice is unclear and most patients are treated according to their risk of death or recurrence; furthermore in patients with refractory advanced disease, the RAS or BRAF mutation status did not appear to be an independent factor of survival or a predictive factor of response to anti-angiogenic therapy (Musholt et al., 2000; Verrienti et al., 2015).
Environmental factors and thyroid oncogenesis
Several will known environmental risk factors affect the risk of thyroid cancer through different mechanisms:
Radiation exposure
The most well-accepted environmental risk factor for thyroid cancer especially DTC is exposure to ionizing radiation, which increases the risk of thyroid malignancy from 5 to 50% (Robbins et al., 1991). The first association between DTC and radiation was observed in 1950, in children who received X-ray therapy in the thymus (Duffy and Fitzgerald, 1950). The atomic bomb explosions in Japan in 1945 (Nagataki et al., 1994), the radioactive contamination of the Marshall Islands in 1954 (Cronkite et al., 1995) and Chernobyl radioactive fallout accident in 1986 (Tuttle and Becker,2000; Williams,2002). The effects of environmental radiation exposure include multiple DNA injuries due to concentration of radioactive iodine in the thyroid gland, leading to the deregulation of cell growth and proliferation coupled with an impaired ability of T-cells to fight cancer cells, allowing them to multiply (Yarilin et al., 1993). Sporadic PTC developed post-Chernobyl are characterized by constitutive activation of effectors along the RAS–RAF–MAPK signaling pathway, and the most frequent genetic alterations are rearrangements of the RET/PTC gene. (Fugazzola et al., 1995; Soares et al., 2003, Lima et al., 2004).
Iodine intake and eating habits
Iodine intake may influence the incidence and prevalence of thyroid disease in general and of thyroid cancer in particular (Wartofsky, 2010). In fact, iodine deficiency is associated with an increased risk of FTC, whereas chronically high iodine intake may increase the risk of PTC (Knobel and Medeiros-Neto, 2007). The prevalence of BRAF mutations was significantly higher in high-iodine content regions when compared with normal-iodine-content regions. In some studies iodine exerts protective effects on thyroid cancer cell lines by attenuating acute BRAF oncogene-mediated microRNA deregulation (Fuziwara and Kimura, 2013).
Excess carbohydrate and protein diet and high body weight (BMI > 25 kh/m2) increase the risk of thyroid cancer (Marcello et al., 2012). The accumulation of carbohydrate and the impairment of insulin regulation might lead to a deregulation of the PI3K/AKT pathway, disorganization of cell growth and proliferation which has been strongly related to DTC development and progression (Gomez Saez, 2011; Bartholomeusz and Gonzalez-Angulo, 2012).
Smoking
Studies showed that smoking reduces thyroid cell proliferation by exerting effects on TSH, estrogen, or other mechanisms (Rossing et al., 2000). TSH concentration was significantly lower in smokers than in ex-smokers and nonsmokers, but not triiodothyronine (T3) concentrations (Ericsson and Lindgarde, 1991). DTC risk was reduced by 40% among smokers with PTC and FTC, indicating that cigarette smoking exerts a protective effect against the development of DTC (Mack et al., 2003). Other studies on Japanese, Swedish, and Norwegian populations showed a reduced risk of DTC in smoking patients (Galanti et al., 1996;Nagano et al., 2007; Suzuki et al., 2007;Meinhold et al., 2010;Kabat et al., 2012).
Volcanic areas
Some of the highest DTC incidences worldwide were observed among people living in volcanic areas such as Iceland (Arnbjornsson et al., 1986), Hawaii (Kolonel et al., 1990), French Polynesia (Curado et al., 2007), New Caledonia (Truong et al., 2007), and Sicily (Pellegriti et al., 2009). Environmental carcinogens of volcanic origin could be responsible for gene mutations favoring thyroid carcinogenesis, patients with DTC living in volcanic areas have a higher rate of BRAF (V600E) mutation. (Frasca et al., 2008).
Xenobiotics
These substances may originate from natural sources (plants and bacteria), but most of them are derived from human activities because of their application, such as flame retardants, pesticides, repellents, or thermal insulators. Xenobiotics act mainly as disrupting chemicals (endocrine-disrupting chemical (EDC)) that influence the physiological functions and the hormone production of several endocrine glands (Marcello et al, 2014). Possible cause of thyroid oncogenesis is BRAF and / or RAS mutations, BRAFV600E point mutation these mutations were markedly increased in DTC cases with high exposure to chemicals, (Jung et al., 2014).
Medullary thyroid carcinoma
MTC accounts for approximately 5-8% of all thyroid cancer. Clinically, MTC is mainly sporadic (70-80%), but hereditary pattern is present in 20-30% of cases, transmitted as an autosomal dominant trait (de Groot et al., 2006). The sporadic form of MTC is observed in 60-70 years old patients with a palpable thyroid nodule indistinguishable from any other thyroid nodule. Neck lymph node metastases are detected in at least 50% of patients and may reveal the disease. Metastases outside the neck, in liver, lungs or bones, are initially present in 10 -20% of the cases. Hereditary MTC may be part of “multiple endocrine neoplasia type 2” (MEN2) and is divided into three clinical forms:
A) MEN2a is characterized by the presence of MTC in combination with pheochromocytoma and/or hyperparathyroidism. Cutaneous lichen amyloidosis has been observed in some families (Santoro et al.,1994) and Hirschsprung’s disease has been observed in a few families with MEN2a (Kluijfhout et al.,2015). The typical onset of this condition is in the third or fourth decade of life. Almost all gene carriers will develop MTC, but this depends on the mutation (Agrawal et al., 2013). The risk of developing unilateral or bilateral pheochromocytoma is as high as 57% (both for germline mutations of codon 918 and 634), and 15– 30% of codon 634 mutations carriers will develop hyperparathyroidism.
B) MEN2b in which MTC is accompanied by pheochromocytoma, multiple mucosal neuromas and marfanoid habitus. MEN2b is the rarest and most aggressive form of MEN2 based on its development of MTC earlier in life, usually before the age of 5-10 years. It is frequently associated with extension beyond the thyroid capsule, with lymph node and distant metastases at the time of diagnosis. Patients also have chronic constipation and colonic cramping due to the presence of Hisrchsprung disease. More than 50% of cases are de novo germline mutations. The higher mortality rate of MEN2B reflects its more advanced stage at presentation, rather than the tumour behavior once established (Verrienti et al., 2015; Kluijfhout et al., 2015).
C) FMTC (Familial Medullary Thyroid Carcinoma) occurs when MTC is the only clinical feature. Clinical presentation of cancer is at a later age and a relatively more favorable prognosis. The most rigid definition is multigenerational transmission of MTC in which no family member has pheochromocytoma or hyperparathyroidism; a less rigid definition is the presence of MTC in four affected family members without other manifestations of MEN2A. Histology is peculiar in hereditary MTC; C-cell hyperplasia is always associated with hereditary MTC with bilaterality and multicentricity as a consequence when the patients over 5-years old, carry the codon 918 or codon 634 mutation. Whereas, sporadic MTC generally presents as a single tumor confined to one thyroid lobe, except for 5-9% of patients. Tumor metastasizes early to paratracheal and lateral cervical lymph nodes; lymph nodes metastases are found in 20-30% of patients with MTC smaller than 1 cm in diameter, in 50% with tumor between 2 and 4 cm and in up to 90% of the patients with tumors that are more than 4 cm in diameter or infiltrating surrounding tissues. The prognosis of MTC is intermediate between well differentiated and anaplastic thyroid cancer. Its prognosis is worse compared to papillary and follicular thyroid cancer and better than the prognosis in anaplastic thyroid cancer. Therefore, an early diagnosis is fundamental for a good prognosis in these patients (Taccaliti et al., 2011). Genetic abnormalities are present in MTC. Hereditary forms are characterized by germline mutations while sporadic MTC showed somatic alterations in 40-60% of patients (Lallier et al., 1998; Machens et al., 2013).
Recently, a French study has demonstrated that the prognostic factor of disease free survival after surgery in young patients with RET germline mutation is best predicted by TNM staging and preoperative basal calcitonin(CT) level below 30 pg./ml. Basal CT, class D genotype, and age constitute key determinants to preoperatively decide timely surgery (Abdelhakim et al., 2009). Moreover, genetic screening of germline RET mutations permits the identification of unsuspected FMTC in apparently sporadic MTC patients. Therefore, RET genetic testing of patients with apparently sporadic MTC represents an important tool for the preclinical diagnosis and early treatment of unsuspected affected family members and allows the identification of a relevant percentage of hidden FMTC (Abdelhakim et al., 2009; Lallier et al., 1998). Given the high chance of a RET mutation carrier to develop MTC at some point during life, these patients should be offered prophylactic thyroidectomy. In cases of MEN2a/FMTC mutations of ATA level C risk, prophylactic total thyroidectomy should be carried out before 5 years of age. In patients with RET mutations of ATA level A and B risk, prophylactic thyroidectomy may be delayed beyond the first 5 years in the setting of a normal basal and/or stimulated serum CT and normal annual cervical ultrasound starting at 5 years of age. Prophylactic level VI central compartment neck dissection may not be necessary in patients with MEN 2a/FMTC who undergo prophylactic thyroidectomy within their first 3-5 years of life unless there is clinical or radiological evidence of lymph node metastases, thyroid nodules more than 5 mm in size or a serum basal CT >40 pg/ml in a child >6 months old. Children with MEN 2b (ATA level D risk) should have thyroidectomy as soon as possible, preferably within the first year of life. Prophylactic level VI neck dissection may not be necessary in patients with MEN 2b, unless there is clinical or radiological evidence of lymph node metastases. In RET mutation-positive patients, screening for pheochromocytoma, including annual plasma metanephrines and normetanephrines, or 24-h urine collection for metanephrines and normetanephrines, begins by 8 years of age in carriers of RET mutation associated with MEN 2b and codons 630 and 634, and by 20 years of age in carriers of other MEN 2a RET mutations. Patients with RET mutation associated with FMTC alone should be screened periodically from 20 years of age. Abdominal imaging is not indicated in the absence of symptoms or biochemical data. Screening for hyperparathyroidism should be carried out with the same interval by measuring serum calcium and parathyroid hormone.
Molecular targeted therapy
The MAPK and P13-AKT pathways, from tyrosine kinase receptors in the cell membrane to the various downstream signaling relay molecules, such as BRAF, MEK, AKT and mTOR, are therapeutic targets that are being actively tested for treatment of RAI refractory thyroid cancer with novel small-molecule protein-kinase inhibitors. A promising therapeutic strategy is genetic-based targeting of thyroid cancer, as supported by many preclinical studies demonstrating the selective inhibition of BRAF p.V600E-mutant thyroid cancer cells targeting of the P13K-AKT pathway may also be genetically guided, as genetic alterations that activate this pathway confer thyroid cancer cells with remarkable increased sensitivities to AKT and mTOR inhibitors (Solomon and Rischin, 2011). The involvement of multiple signaling pathways in aggressive thyroid cancer suggests that it may be necessary to target them simultaneously for effective treatment. Synergistic effects of simultaneously targeting the MAPK and P13K-AKT pathways were even more pronounced in cells that harbored genetic alterations in both pathways (Tufano et al., 2012).
Summary and Conclusions
From the 1990s, when pathogenesis of only approximately 25% of thyroid cancers was understood, to the present, when genes involved in the pathogenesis of over 90% of thyroid cancers have been described, much progress has been made in elucidating the molecular mechanisms underlying thyroid cancer. This progress provides the basis upon which new diagnostic and prognostic markers, as well as new targeted therapies, have been developed.
The molecular pathogenesis of differentiated thyroid cancer, with specific signaling pathways and activating point mutations, has been successfully elucidated. Some of the specific genes tend to be mutated in more aggressive and advanced thyroid cancer. Other discoveries include mutations in thyroid stimulating hormone receptor (TSHR) that were shown to play a role in thyroid tumorigenesis.
Some environmental carcinogens in the industrialized lifestyle may have specifically affected the thyroid. Among potential carcinogens, the increased exposure to medical radiations is the most likely risk factor. Other factors specific for the thyroid like increased iodine intake, increasing prevalence of obesity and a large class of xenobiotics to which we are exposed.
Medullary thyroid carcinoma (MTC), whether sporadic or hereditary, with or without other combinations, have specific genetic abnormalities in the RET proto-oncogene system. The issue of prophylactic thyroidectomy, and its timing, have been outlined and debated. Ethical and psychosocial considerations of predictive testing for mutations have been discussed and debated.
Hereditary MTC, as part of MEN2, is one of the first hereditary syndromes to be described (Sakorafas et al.,2008). The syndrome has been utilized as an example for understanding and analyzing the prediction of testing for gene carriers and timing of prophylactic thyroidectomy at the proper age in childhood.
Related ethical issues, including the possibility of performing pre-implantation genetic diagnosis (PGD) to identify genetic mutations in fertilized ova, the issue of the ethics and legality of genetic testing prior to employment and insurance, and involving other family members in hereditary cancer syndrome screening and management have been debated.
References
- Abdelhakim A, Barlier A, Kebbou M, et al. RET genetic screening in patients with medullary thyroid cancer:The Moroccan experience. J Can Res Ther. 2009;5:198–202. doi: 10.4103/0973-1482.57126. [DOI] [PubMed] [Google Scholar]
- Abdelhakim A, Anne B, Mohamed K, et al. Progression of medullary thyroid cancer in RET carriers of ATA class A and C mutations. J Clin Endocrinol Metab. 2014;99:286–92. doi: 10.1210/jc.2013-3343. [DOI] [PubMed] [Google Scholar]
- Adeniran AJ, Zhu Z, Gandhi M, et al. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am J Surg Pathol. 2006;30:216–22. doi: 10.1097/01.pas.0000176432.73455.1b. [DOI] [PubMed] [Google Scholar]
- Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab. 2013;98:364–9. doi: 10.1210/jc.2012-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albores-Saavedra J, Henson DE, Glazer E, Schwartz AM. Changing patterns in the incidence and survival of thyroid cancer with follicular phenotype –papillary, follicular, and anaplastic:a morphological and epidemiological study. Endocr Pathol. 2007;18:1, 7. doi: 10.1007/s12022-007-0002-z. [DOI] [PubMed] [Google Scholar]
- Arnbjornsson E, Arnbjornsson A, Olafsson A. Thyroid cancer incidence in relation to volcanic activity. Arch Environ Health. 1986;41:36, 40. doi: 10.1080/00039896.1986.9935763. [DOI] [PubMed] [Google Scholar]
- Baloch ZW, LiVolsi VA, Asa SL, et al. Diagnostic terminology and morphologic criteria for cytologic diagnosis of thyroid lesions:a synopsis of the national cancer institute thyroid fine-needle aspiration state of the science conference. Diagn Cytopathol. 2008;36:425, 37. doi: 10.1002/dc.20830. [DOI] [PubMed] [Google Scholar]
- Bartolazzi A, Orlandi F, Saggiorato E, et al. Galectin-3-expression analysis in the surgical selection of follicular thyroid nodules with indeterminate fine-needle aspiration cytology:a prospective multicentre study. Lancet Oncol. 2008;9:543, 9. doi: 10.1016/S1470-2045(08)70132-3. [DOI] [PubMed] [Google Scholar]
- Bartholomeusz C, Gonzalez-Angulo AM. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:121, 30. doi: 10.1517/14728222.2011.644788. [DOI] [PubMed] [Google Scholar]
- Bongarzone I, Vigneri P, Mariani L, et al. RET/NTRK1 rearrangements in thyroid gland tumors of the papillary carcinoma family:correlation with clinicopathological features. Clin Cancer Res. 1998;4:223–8. [PubMed] [Google Scholar]
- Brito JP, Yarur AJ, Prokop LJ. Prevalence of thyroid cancer in multinodular goiter vs. single nodule:a systematic review and meta-analysis. Thyroid. 2013;23:449, 55. doi: 10.1089/thy.2012.0156. [DOI] [PubMed] [Google Scholar]
- Burgess JR, Tucker P. Incidence trends for papillary thyroid carcinoma and their correlation with thyroid surgery and thyroid fine-needle aspirate cytology. Thyroid. 2006;16:47, 53. doi: 10.1089/thy.2006.16.47. [DOI] [PubMed] [Google Scholar]
- Chiosea S, Nikiforova M, Zuo H, Ogilvie J. A novel complex BRAF mutation detected in a solid variant of papillary thyroid carcinoma. Endocr Pathol. 2009;20:122. doi: 10.1007/s12022-009-9073-3. [DOI] [PubMed] [Google Scholar]
- Ciampi R, Nikiforov YE. Alterations of the BRAF gene in thyroid tumors. Clin Endocrinol. 2005;63:588–93. doi: 10.1385/ep:16:3:163. [DOI] [PubMed] [Google Scholar]
- Cronkite EP, Bond VP, Conard RA. Medical effects of exposure of human beings to fallout radiation from a thermonuclear explosion. Stem Cells. 1995;13:49, 57. [PubMed] [Google Scholar]
- Curado MP, Edwards B, Shin HR, et al. Cancer incidence in five continents, IARC Scientific Publications, IX No. 160. 2007 [Google Scholar]
- Dahia PL, Marsh DJ, Zheng Z, Zedenius J. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 1997;57:4710–13. [PubMed] [Google Scholar]
- Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States 1973–2002. JAMA. 2006;295:2164–7. doi: 10.1001/jama.295.18.2164. [DOI] [PubMed] [Google Scholar]
- de Groot JW, Links TP, Plukker JT. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr Rev. 2006;27:535, 60. doi: 10.1210/er.2006-0017. [DOI] [PubMed] [Google Scholar]
- Dobashi Y, Sugimura H, Sakamoto A, et al. Stepwise participation of p53 gene mutation during dedifferentiation of human thyroid carcinomas. Diagn Mol Pathol. 1994;3:9–14. doi: 10.1097/00019606-199403010-00003. [DOI] [PubMed] [Google Scholar]
- Donghi R, Longoni A, Pilotti S, et al. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J Clin Invest. 1993;91:1753, 60. doi: 10.1172/JCI116385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy BJ, Jr, Fitzgerald PJ. Cancer of the thyroid in children:a report of 28 cases. J Clin Endocrinol Metab. 1950;10:1296, 08. doi: 10.1210/jcem-10-10-1296. [DOI] [PubMed] [Google Scholar]
- Ericsson UB, Lindgarde F. Effects of cigarette smoking on thyroid function and the prevalence of goitre, thyrotoxicosis and autoimmune thyroiditis. J Intern Med. 1991;229:67–71. doi: 10.1111/j.1365-2796.1991.tb00308.x. [DOI] [PubMed] [Google Scholar]
- Fenton CL, Lukes Y, Nicholson The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J Clin endocrinol Metab. 2000;85:1170, 5. doi: 10.1210/jcem.85.3.6472. [DOI] [PubMed] [Google Scholar]
- Frasca F, Nucera C, Pellegriti G, et al. BRAFV600E mutation and the biology of papillary thyroid cancer. Endocrine-Related Cancer. 2008;15:191, 205. doi: 10.1677/ERC-07-0212. [DOI] [PubMed] [Google Scholar]
- Frich L, Glattre E, Akslen LA. Familial occurrence of nonmedullary thyroid cancer:a population-based study of 5673 first-degree relatives of thyroid cancer patients from Norway. Cancer Epidem Biomar. 2001;10:113, 7. [PubMed] [Google Scholar]
- Fugazzola L, Pilotti S, Pinchera A, et al. Oncogenic rearrangements of the RET proto-oncogene in papillary thyroid carcinomas from children exposed to the Chernobyl nuclear accident. Cancer Res. 1995;55:5617–20. [PubMed] [Google Scholar]
- Fuziwara CS, Kimura ET. High iodine blocks a Notch/miR-19 loop activated by the BRAFV600E oncoprotein and restores the response to TGFb in thyroid follicular cells. Thyroid. 2013;24:453–62. doi: 10.1089/thy.2013.0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanti MR, Hansson L, Lund E, et al. Reproductive history and cigarette smoking as risk factors for thyroid cancer in women:a population-based case–control study. Cancer Epidemiol Biomarkers Prev. 1996;5:425–31. [PubMed] [Google Scholar]
- Garcia-Rostan G, Camp RL, et al. b-Catenin dysregulation in thyroid neoplasms:down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am J Pathol. 2001;158:987–96. doi: 10.1016/s0002-9440(10)64045-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rostan G, Costa AM, Pereira-Castro I, Salvatore G, Hernandez R. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005;65:10199–207. doi: 10.1158/0008-5472.CAN-04-4259. [DOI] [PubMed] [Google Scholar]
- Gomez Saez JM. Diagnostic and prognostic markers in differentiated thyroid cancer. Current Genomics. 2011;12:597, 608. doi: 10.2174/138920211798120826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford JR, Mulligan LM. Multiple endocrine neoplasia type 2 and RET:from neoplasia to neurogenesis. J Med Genet. 2000;37:817, 27. doi: 10.1136/jmg.37.11.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou P, Liu D, Shan Y, Hu S, Studeman K. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res. 2007;13:1161–70. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- Hsiao SJ, Nikiforov YE. Molecular approaches to thyroid cancer diagnosis endocr relat cancer. Endocr Relat Cancer. 2014;21:301–13. doi: 10.1530/ERC-14-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Yoshida H, Maruo R, Morita S, Takano T. BRAF mutation in papillary thyroid carcinoma in a Japanese population:its lack of correlation with high-risk clinicopathological features and disease-free survival of patients. Endocr J. 2009;56:89–97. doi: 10.1507/endocrj.k08e-208. [DOI] [PubMed] [Google Scholar]
- Jung CK, Little MP, Lubin JH. The increase in thyroid cancer incidence during the last four decades is accompanied by a high frequency of BRAF mutations and a sharp increase in RAS mutations. J Clin Endocrinol Metab. 2014;99:276–85. doi: 10.1210/jc.2013-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat GC, Kim MY, Wactawski-Wende J, Rohan TE. Smoking and alcohol consumption in relation to risk of thyroid cancer in postmenopausal women. Cancer Epidemiol. 2012;36:335, 40. doi: 10.1016/j.canep.2012.03.013. [DOI] [PubMed] [Google Scholar]
- Kelly LM, Barila G, Liu P, et al. Identification of the transforming STRN–ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci U S A. 2014;111:4233, 8. doi: 10.1073/pnas.1321937111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TY, Kim WB, Song JY. The BRAFV600E mutation is not associated with poor prognostic factors in Korean patients with conventional papillary thyroid microcarcinoma. Clin Endocrinol. 2005;63:588–93. doi: 10.1111/j.1365-2265.2005.02389.x. [DOI] [PubMed] [Google Scholar]
- Kimura ET, Nikiforova MN, ZZhu Z, et al. High prevalence of BRAF mutations in thyroid cancer:genetic evidence for constitutive activation of the RET/PTC–RAS–BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454, 7. [PubMed] [Google Scholar]
- Kloos RT, Eng C, Evans DB, Francis F. Medullary thyroid cancer:management guidelines of the American thyroid association. Thyroid. 2009;19:565, 612. doi: 10.1089/thy.2008.0403. [DOI] [PubMed] [Google Scholar]
- Kluijfhout WP, van Beek DJ, Verrijn Stuart AA, et al. Postoperative complications after prophylactic thyroidectomy for very young patients with multiple endocrine neoplasia Type 2:Retrospective cohort analysis. Medicine (Baltimore) 2015;94:e1108. doi: 10.1097/MD.0000000000001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobel M, Medeiros-Neto G. Relevance of iodine intake as a reputed predisposing factor for thyroid cancer. Arq Bras Endocrinol Metab. 2007;51:701–12. doi: 10.1590/s0004-27302007000500007. [DOI] [PubMed] [Google Scholar]
- Kolonel LN, Hankin JH, Wilkens LR, Fukunaga FH, Hinds MW. An epidemiologic study of thyroid cancer in Hawaii. Cancer Causes Control. 1990;1:223, 34. doi: 10.1007/BF00117474. [DOI] [PubMed] [Google Scholar]
- Lallier D, St-Vil D, Giroux M, et al. Prophylactic thyroidectomy for medullary thyroid carcinoma in gene carriers of MEN2 syndrome. J Pediatr Surg. 1998;33:846–8. doi: 10.1016/s0022-3468(98)90656-x. [DOI] [PubMed] [Google Scholar]
- Landa I, Ganly I, Chan TA, Mitsutake N. Frequent somatic TERT promoter mutations in thyroid cancer:higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab. 2013;98:1562–6. doi: 10.1210/jc.2013-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeman-Neill RJ, Brenner AV, Little MP, et al. RET/PTC and PAX8/PPARg chromosomal rearrangements in post-Chernobyl thyroid cancer and their association with iodine-131 radiation dose and other characteristics. Cancer. 2013;119:1792–9. doi: 10.1002/cncr.27893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeman-Neill RJ, Kelly LM, Liu P, et al. ETV6–NTRK3 is a common chromosomal rearrangement in radiationassociated thyroid cancer. Cancer. 2014;120:799–807. doi: 10.1002/cncr.28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima J, Trovisco V, Soares P, et al. BRAF mutations are not a major event in post-Chernobyl childhood thyroid carcinomas. J Clin Endocrinol Metab. 2004;89:4267, 71. doi: 10.1210/jc.2003-032224. [DOI] [PubMed] [Google Scholar]
- Liu RT, Chen YJ, Chou FF. No correlation between BRAFV600E mutation and clinicopathological features of papillary thyroid carcinomas in Taiwan. Clin Endocrinol (Oxf) 2005;63:461–6. doi: 10.1111/j.1365-2265.2005.02367.x. [DOI] [PubMed] [Google Scholar]
- Liu T, Wang N, Cao J, Sofiadis A, Dinets A. The age-and shorter telomere-dependent TERT promoter mutation in follicular thyroid cell-derived carcinomas. Oncogene. 2014;33:4978, 84. doi: 10.1038/onc.2013.446. [DOI] [PubMed] [Google Scholar]
- Liu X, Bishop J, Shan Y, et al. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer. 2010;20:603–10. doi: 10.1530/ERC-13-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Hou P, Ji M, et al. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. Clin Endocrinol Metab. 2008;93:3106–16. doi: 10.1210/jc.2008-0273. [DOI] [PubMed] [Google Scholar]
- Mack WJ, Preston-Martin S, Dal Maso L, et al. A pooled analysis of case–control studies of thyroid cancer:cigarette smoking and consumption of alcohol, coffee, and tea. Cancer Causes Control. 2003;14:773–85. doi: 10.1023/a:1026349702909. [DOI] [PubMed] [Google Scholar]
- Marcello MA, Sampaio AC, Geloneze B, et al. Obesity and excess protein and carbohydrate consumption are risk factors for thyroid cancer. Nutr Cancer. 2012;64:1190, 5. doi: 10.1080/01635581.2012.721154. [DOI] [PubMed] [Google Scholar]
- Marcello MA, Malandrino P, Almeida J, et al. The influence of the environment on the development of thyroid tumors:a new appraisal. Endocr Relat Cancer. 2014;21:235, 54. doi: 10.1530/ERC-14-0131. [DOI] [PubMed] [Google Scholar]
- Marques AR, Espadinha C, Catarino AL. Expression of PAX8–PPARg1 rearrangements in both follicular thyroid carcinomas and adenomas. J Clin Endocrinol Metab. 2002;87:3947, 52. doi: 10.1210/jcem.87.8.8756. [DOI] [PubMed] [Google Scholar]
- Meinhold CL, Ron E, Schonfeld SJ, et al. Nonradiation risk factors for thyroid cancer in the US Radiologic Technologists Study. Am J Epidemio. 2010;171:242, 52. doi: 10.1093/aje/kwp354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses W, Weng J, Kebebew E. Prevalence, clinicopathologic features, and somatic genetic mutation profile in familial versus sporadic nonmedullary thyroid cancer. Thyroid. 2011;21:367, 71. doi: 10.1089/thy.2010.0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musholt TJ, Musholt PB, Khaladj N, et al. Prognostic significance of RET and NTRK1 rearrangements in sporadic papillary thyroid carcinoma. Surgery. 2000;128:984–93. doi: 10.1067/msy.2000.110845. [DOI] [PubMed] [Google Scholar]
- Nagano J, Mabuchi K, Yoshimoto Y, et al. A case–control study in Hiroshima and Nagasaki examining nonradiation risk factors for thyroid cancer. J Epidemiol. 2007;17:76, 85. doi: 10.2188/jea.17.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagataki S, Shibata Y, Inoue S, et al. Thyroid diseases among atomic bomb survivors in Nagasaki. JAMA. 1994;272:364, 70. [PubMed] [Google Scholar]
- Nishihara E, Amino N, Maekawa K, et al. Prevalence of TSH receptor and Gsa mutations in 45 autonomously functioning thyroid nodules in Japan. Endocr J. 2009;56:791–8. doi: 10.1507/endocrj.k09e-073. [DOI] [PubMed] [Google Scholar]
- Pellegriti G, De Vathaire F, Scollo C, et al. Papillary thyroid cancer incidence in the volcanic area of Sicily. J Natl Cancer Inst. 2009;101:1575, 83. doi: 10.1093/jnci/djp354. [DOI] [PubMed] [Google Scholar]
- Pellegriti G, Frasca F, Regalbuto C, Squatrito S, Vigneri R. Worldwide increasing incidence of thyroid cancer:Update on epidemiology and risk factors. J Cancer Epidemiol. 2013;2013:965212. doi: 10.1155/2013/965212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricarte-Filho JC, Ryder M, Chitale DA. Mutational profile of advanced primary and metastatic radioactive iodinerefractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69:4885–9. doi: 10.1158/0008-5472.CAN-09-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J, Merino MJ, Boice JD, Jr, et al. Thyroid cancer:a lethal endocrine neoplasm. Ann Intern Med. 1991;115:133, 47. doi: 10.7326/0003-4819-115-2-133. [DOI] [PubMed] [Google Scholar]
- Rossing MA, Cushing KL, Voigt LF, Wicklund KG, Daling JR. Risk of papillary thyroid cancer in women in relation to smoking and alcohol consumption. Epidemiology. 2000;11:49, 54. doi: 10.1097/00001648-200001000-00011. [DOI] [PubMed] [Google Scholar]
- Sakorafas GH, Friess H, Peros G. The genetic basis of hereditary medullary thyroid cancer:clinical implications for the surgeon, with a particular emphasis on the role of prophylactic thyroidectomy. Endocr Relat Cancer. 2008;15:871, 84. doi: 10.1677/ERC-08-0098. [DOI] [PubMed] [Google Scholar]
- Santoro M, Dathan NA, Berlingieri MT. Molecular characterization of RET/PTC3;a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene. 1994;9:509, 16. [PubMed] [Google Scholar]
- Solomon B, Rischin D. Progress in molecular targeted therapy for thyroid cancer:Vandetanib in medullary thyroid cancer. J Clin Oncol. 2011;30:119–33. doi: 10.1200/JCO.2011.37.8638. [DOI] [PubMed] [Google Scholar]
- Soares P, Trovisco V, Rocha AS, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene. 2003;22:4578–80. doi: 10.1038/sj.onc.1206706. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Matsuo K, Wakai K, et al. Effect of familial history and smoking on common cancer risks in Japan. Cancer. 2007;109:2116, 23. doi: 10.1002/cncr.22685. [DOI] [PubMed] [Google Scholar]
- Taccaliti A, Silvetti F, Palmonella G, Boscaro M. Genetic alterations in medullary thyroid cancer. Curr Genomics. 2011;12:618, 25. doi: 10.2174/138920211798120835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong T, Rougier Y, Dubourdieu D, et al. Time trends and geographic variations for thyroid cancer in New Caledonia, a very high incidence area (1985–1999) Eur J Cancer Prev. 2007;16:62–70. doi: 10.1097/01.cej.0000236244.32995.e1. [DOI] [PubMed] [Google Scholar]
- Tufano RP, Teixeira GV, Bishop J, Carson KA, Xing M. BRAF mutation in papillary thyroid cancer and its value in tailoring initial treatment:a systematic review and meta-analysis. Medicine. 2012;19:274, 86. doi: 10.1097/MD.0b013e31826a9c71. [DOI] [PubMed] [Google Scholar]
- Tuttle RM, Becker DV. The Chernobyl accident and its consequences:update at the millennium. Semin Nucl Med. 2000;30:133, 40. doi: 10.1053/nm.2000.5412. [DOI] [PubMed] [Google Scholar]
- Verrienti A, Carbone A, Bellitti P, et al. Anovelduoble mutation VAL648ILE and VAL 804 LEU of RET protooncogene in multiple endocrine neoplasia type 2. Endocr Prac. 2015;21:1248–54. doi: 10.4158/EP15838.OR. [DOI] [PubMed] [Google Scholar]
- Wartofsky L. Increasing world incidence of thyroid cancer:increased detection or higher radiation exposure? Hormones. 2010;9:103, 8. doi: 10.14310/horm.2002.1260. [DOI] [PubMed] [Google Scholar]
- Wells SA, Robinson BG, Pacini F, et al. Hereditary medullary thyroid carcinoma. J Clin Endocrinol Metab. 2013;98:3148–64. doi: 10.1210/jc.2013-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. Cancer after nuclear fallout:lessons from the Chernobyl accident. Nat Rev Cancer. 2002;2:543, 9. doi: 10.1038/nrc845. [DOI] [PubMed] [Google Scholar]
- Xing M, Westra WH, Tufano RP, et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab. 2005;90:6373–9. doi: 10.1210/jc.2005-0987. [DOI] [PubMed] [Google Scholar]
- Xing M. BRAF mutation in papillary thyroid microcarcinoma:the promise of better risk management. Ann Surg Oncol. 2009;4:801–3. doi: 10.1245/s10434-008-0298-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarilin AA, Belyakov IM, Kusmenok OI, et al. Late T cell deficiency in victims of the Chernobyl radiation accident:possible mechanisms of induction. Int J Radiat Biol. 1993;63:519, 28. doi: 10.1080/09553009314550681. [DOI] [PubMed] [Google Scholar]