

Abstract
Background
Inflammation is believed to play an important role in the pathology of Alzheimer's disease (AD) and cytokine production is a key pathologic event in the progression of inflammatory cascades. The current study characterizes the cytokine expression profile in the brain of two transgenic mouse models of AD (TgAPPsw and PS1/APPsw) and explores the correlations between cytokine production and the level of soluble and insoluble forms of Aβ.
Methods
Organotypic brain slice cultures from 15-month-old mice (TgAPPsw, PS1/APPsw and control littermates) were established and multiple cytokine levels were analyzed using the Bio-plex multiple cytokine assay system. Soluble and insoluble forms of Aβ were quantified and Aβ-cytokine relationships were analyzed.
Results
Compared to control littermates, transgenic mice showed a significant increase in the following pro-inflammatory cytokines: TNF-α, IL-6, IL-12p40, IL-1β, IL-1α and GM-CSF. TNF-α, IL-6, IL-1α and GM-CSF showed a sequential increase from control to TgAPPsw to PS1/APPsw suggesting that the amplitude of this cytokine response is dependent on brain Aβ levels, since PS1/APPsw mouse brains accumulate more Aβ than TgAPPsw mouse brains. Quantification of Aβ levels in the same slices showed a wide range of Aβ soluble:insoluble ratio values across TgAPPsw and PS1/APPsw brain slices. Aβ-cytokine correlations revealed significant relationships between Aβ1–40, 1–42 (both soluble and insoluble) and all the above cytokines that changed in the brain slices.
Conclusion
Our data confirm that the brains of transgenic APPsw and PS1/APPsw mice are under an active inflammatory stress, and that the levels of particular cytokines may be directly related to the amount of soluble and insoluble Aβ present in the brain suggesting that pathological accumulation of Aβ is a key driver of the neuroinflammatory response.
Background
Alzheimer's disease is a progressive neurodegenerative disorder characterized by intra-cellular abnormally phosphorylated tau protein and extra-cellular beta amyloid plaques. It has been suggested that inflammation may be a key player in the pathophysiology of AD as evidenced by epidemiological studies which have revealed that the long term use of non-steroidal anti-inflammatory drugs reduces the risk of developing AD [1-3]. Transgenic mouse models of Alzheimer's disease that over-express β-amyloid (Aβ) exhibit significant cerebrovascular inflammation and microgliosis around areas of plaque deposition [4-7]. Chronic administration of ibuprofen can reduce plaque pathology and brain Aβ levels in these animal models of AD [8,9].
There are numerous reports of increased levels of cytokines in the brains of Alzheimer's disease patients, and in transgenic mouse models of Alzheimer's disease [10-12]. However, all these reports have focused on a small number of cytokines within the same sample. It is not clear which cytokines are key in promoting and maintaining the inflammatory environment in the AD brain. Furthermore, it is unclear which Aβ species (1–40, 1–42, soluble or insoluble) are most closely related to cytokine levels. Multiplex technology enables the simultaneous quantification of many cytokines within a single sample.
By examining different mouse models of AD using multiplex technology, it is possible to more clearly characterize the particular cytokines which maintain the inflammatory environment and to relate them to particular forms of Aβ (1–40, 1–42, soluble or insoluble).
There is considerable debate over which length of Aβ and which conformations are most potently toxic. Recently, specific oligomeric forms have been shown to be most toxic to neurons. These soluble species of Aβ differ from the higher-molecular-weight aggregated insoluble forms that are found precipitated in the AD patient and mouse brain. This study sought to determine whether soluble or insoluble Aβ fractions were most closely related to cytokine levels.
Materials and methods
Organotypic brain slice cultures
Mouse brain slice cultures were prepared as previously described [29]. Briefly, 15-month-old PS1 (M146L), TgAPPsw (K670M / N671L), PS1/APPsw and wildtype littermates were humanely euthanized and the brains extracted under sterile conditions. One-mm-thick brain slices were sectioned from co-ordinates 1 to -4 from bregma using a mouse brain slicer. Sections were cultured in neurobasal medium with 5% B27 supplement (Gibco-Invitrogen, CA) and Penicillin-Streptomycin-Fungizone mixture (Cambrex Corp., NJ). After 40 hours, media was collected for quantification of cytokine levels.
Multi-plex cytokine array analysis was performed using the Bio-plex protein multi-array system, which utilizes Luminex-based technology [13]. For the current experiments, a mouse 12-plex assay was used according to the recommendations of the manufacturer (BioRad, CA).
Measurement of Aβ levels in brain slices
Brain slices were washed with PBS (BioSource, CA), and 300 μl of lysis buffer was added. Lysis buffer consisted of mammalian protein extraction reagent (Pierce-Endogen, IL) with 1X protease inhibitor cocktail XI (Calbiochem, CA), 100 μM Sodium Orthovanadate, and 1 μM Phenylmethylsulfonyl Fluoride (PMSF) (Sigma-Aldrich, MO). The resulting mixture was sonicated using a sonic dismembrator (Fisher Scientific, PA)
Protein content in each slice was determined using the bicinchoninic acid (BCA) protein reagent kit (Pierce-Endogen, IL), as per the manufacturers protocol. Insoluble Aβ was extracted using 70% formic acid as previously published [14].
Aβ content in brain slices was determined using human Aβ 1–40 and Aβ 1–42 ELISA detection kits (Biosource, CA), as per the manufacturers protocol.
Statistical analyses
For statistical analyses, ANOVA and t-tests were performed where appropriate using SPSS for Windows release 10.1. Hierarchical cluster analysis of Aβ-cytokine data from brain slices were performed with the R program http://cran.r-project.org/. A correlation matrix was constructed using the raw data and subsequently converted to a distance matrix by subtracting each element in the correlation matrix from 1. The distance matrix was used as the dissimilarity matrix for building an hierarchical cluster using the averaging method. The resulting dendrogram consists of closely related members under the same node. The farther one needs to traverse across the tree to reach another member, the higher the dissimilarity represented. The distance from the base in the y-axis represents dissimilarity or 1-r, where r is the correlation co-efficient.
Results
Cytokine production by organotypic brain slice cultures
Cytokine production was evaluated by multi-plex cytokine array analysis using the cell culture supernatant of organotypic brain slice cultures from control, PS1 (Presenilin 1 mutant heterozygotes), TgAPPsw, and TgPS1/APPsw mice at 15 months of age. We chose non-transgenic littermates as controls for the TgAPPsw mice and the PS1 animals as controls for the PS1/APPsw mice as the PS1 animals were the littermates of the PS1/APPsw mice. There were no significant differences in cytokine production between control slices and PS1 slices showing that PS1 over-expression does not directly induce inflammatory events. Compared to control slices, production of IL-1α, TNF-α, GM-CSF and IL-6 was increased in TgAPPsw slices (figs. 1, 2). Compared to TgAPPsw slices, PS1/APPsw brain slices produced significantly more IL-12p40, IL-1β, IL-1α, TNF-α, GM-CSF and IL-6. Across control, TgAPPsw, and PS1/APP transgenic brain slices, there was a graduated increase in IL-1α, TNF-α, GM-CSF and IL-6.
Figure 1.

Cytokine production by brain slices from transgenic mouse models of AD at 15 months of age. Freshly harvested brain slices were incubated in neurobasal medium with B27 supplement. Media was collected after 24 hours, and cytokine levels measured. Mean concentrations (N = 15) +/- standard error are expressed in picograms per milligram of protein. P < 0.05 was considered statistically significant.
Figure 2.

Cytokine production by brain slices from transgenic mouse models of AD at 15 months of age. Freshly harvested brain slices were incubated in neurobasal medium with B27 supplement. Media was collected after 24 hours, and cytokine levels measured. Mean concentrations (N = 15) +/- standard error are expressed in picograms per milligram of protein. P < 0.05 was considered statistically significant.
Correlation between Aβ level and cytokine production by transgenic mouse brain slices
Quantification of amyloid levels in brain mouse slices revealed that PS1/APPsw mice produce significantly more total Aβ as compared to TgAPPsw mice at the same age, and levels of insoluble and soluble Aβ (both 1–40 and 1–42) correlated well with each other (Table 1). Analysis of the ratio of soluble:insoluble Aβ revealed a wide range of values across the TgAPPsw and PS1/APPsw mouse brain slices, with a 15.3-fold variance for Aβ 1–40 and a 5.4-fold variance for Aβ 1–42 (for Aβ 1–40, comparison of soluble:insoluble ratios revealed an average difference of 3.9 fold, and an average 1.7-fold difference for Aβ 1–42).
Table 1.
Quantification of Aβ levels in TgAPPsw and PS1/APPsw mouse brain slices. Data expressed as picograms/mg protein, mean ± S.E.M. for 13 determinations.
| TgAPPsw | PS1/APPsw | |
| Soluble Aβ1–40 | 331.15 ± 35.36 | 4957.79 ± 322.30 |
| Soluble Aβ1–42 | 68.11 ± 6.82 | 1644.29 ± 90.30 |
| Insoluble Aβ1–40 | 67619.38 ± 7089.61 | 4095442 ± 409212.3 |
| Insoluble Aβ1–42 | 6837.22 ± 2741.70 | 286463.3 ± 31395.63 |
Although all the cytokines that changed in the transgenic brain slices were correlated with increases in Aβ levels, some showed a closer relationship than others to Aβ levels (Figs. 3, 4, and 5). A table of r-correlation values is given in Additional file 1. It is important to note that the dendrograms depict the closeness of a correlation between a particular cytokine and Aβ levels, and that all the members in the dendrograms are in fact highly correlated with Aβ levels (1% significance was considered as r >= 0.496, and 5% significance was considered as r >= 0.388). IL-4 and IL-5 were not produced in detectable amounts, were therefore omitted from the dendrograms. Of all the cytokines, IL-12p40 showed the strongest correlation with levels of both Aβ1–40 and 42 (soluble or insoluble). IL-1α and IL-1β were also highly correlated with Aβ1–40 and 42 (soluble or insoluble).
Figure 3.
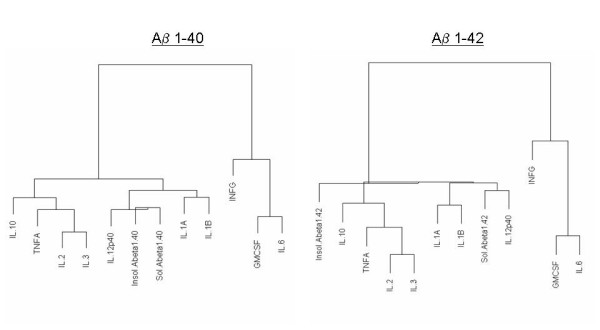
Dendrogram correlations of Aβ1–40 and Aβ1–42-cytokine relationships. Closely related members appear under the same node. The farther one needs to travel across the tree to reach another member, the greater the dissimilarity.
Figure 4.

Dendrogram correlations of Total Aβ (Aβ1–40+Aβ1–42)-cytokine relationships. Closely related members appear under the same node. Total Aβ levels were calculated by adding soluble and formic acid extracted Aβ. The farther one needs to travel across the tree to reach another member, the greater the dissimilarity.
Figure 5.

Dendrogram correlations of (Aβ1–42:40 ratio)-cytokine relationships. Total Aβ1–42:40 ratio's were calculated for both soluble and formic acid extracted Aβ. Closely related members appear under the same node. The farther one needs to travel across the tree to reach another member, the greater the dissimilarity.
Discussion
Levels of both peripheral and local CNS cytokines are elevated in AD patients, indicating that there is cellular activation occurring in response to inflammatory stimuli [15-20]. However, there is still considerable debate over exactly what is triggering this inflammation. Studies using mouse models of AD have shown that ibuprofen is effective in reducing plaque pathology and also in improving behavioral deficits characteristic of these transgenic models [8,21]. The transgenic mouse models used to study AD exhibit some of the pathological features seen in the AD patient brain and show an increased production of inflammatory markers such as COX-2, PGE2 and also increased levels of the pro-inflammatory cytokines IFN-γ and IL-12, TNF-α, IL-1α, IL-1β and IL-6 [12,22]. Pathological analysis of tissue from AD patients and from mouse models of AD shows that there is extensive astrocytic and microglial activation around areas of Aβ plaque deposition [6,7]. In addition, the chronic use of non-steroidal anti-inflammatory drugs (NSAIDs) has been associated with a reduced risk of developing AD [23,24], suggesting that inflammation is an important contributor to the pathophysiology of AD.
One aim of this study was to create a cytokine expression profile for organotypic brain slice cultures from transgenic mouse models of Alzheimer's disease, and to further relate this increase to the level of Aβ present in the brain. Another purpose of our study was to determine whether inflammatory events may be correlated with the accumulation of particular forms of Aβ; either soluble or insoluble.
In the current study, we used the organotypic brain slice culture model to assess multiple cytokine production in the culture medium surrounding brain slices from transgenic mice that are engineered to over-produce Aβ. Cytokine production from 15-month-old control, PS1, TgAPPsw and PS1/APPsw mouse brain slices was assessed using the Bioplex cytokine multi-array system. Cytokine levels were not significantly elevated in PS1 brain slices compared to control slices, indicating that the PS1 (M146L) mutation does not have a significant impact on cytokine production. No significant change in the production of IL-4 and IL-10 was observed in the brains of these transgenic mice compared to their respective controls, indicating the absence of an anti-inflammatory response. All of the cytokines that were increased in the TgAPPsw brain slices (IL-1α, TNF-α, GM-CSF and IL-6) were further increased in the PS1/APP brain slices. This suggests that the presence of these inflammatory molecules is related to the amount of β-amyloid protein present, in agreement with a pro-inflammatory effect of Aβ [25-29]. A recent report has also shown increases in IL-1β, IL-6 and TNFα in-vivo after intra-cerebral administration of fibrillar Aβ into rat brain [30].
In order to further understand the correlation between the amount of Aβ and cytokine levels in the brains of transgenic mice, levels of both soluble and insoluble (formic acid-extracted) Aβ1–40 and 1–42 were quantified in the same slices from which cytokine production was measured, allowing a direct correlation of Aβ-cytokine levels.
Levels of soluble and insoluble Aβ1–40 correlated well with each other, and the same was observed for Aβ1–42. As expected, quantification of Aβ levels generally revealed significantly higher amyloid levels in the PS1/APPsw mouse brain slices compared to TgAPPsw (for soluble Aβ, approximately 15 fold more Aβ1–40, and 20 fold more 1–42) but there was considerable slice-to-slice variation in soluble and insoluble Aβ levels within and between genotypes. The TgAPPsw and PS1/APPsw mice express equal levels of the APPsw molecule, but the PS1/APPsw model produces greater levels of Aβ and develops plaques at an earlier age (10 weeks) [31-33]. This increased deposition of Aβ in the PS1/APPsw mouse is due to a PS1 mutation, resulting in increased production of Aβ1–42 [34-36].
The Aβ data in the current report found a significant range of values for soluble:insoluble Aβ ratios between brain slices. This broad spread of values allowed correlation with equally wide ranges of cytokine production. This approach of examining Aβ-cytokine correlations within the same slices in the same aged animals eliminated the confounding factor of age related changes in cytokine production. Both Aβ1–40 and 1–42 correlated closely with all the cytokines that changed in the brain slices, but the correlation was particularly striking with IL-12p40. IL-12 is a hetero-dimeric cytokine which can comprise two subunits; IL-12p40 and IL-12p35. It is produced mainly by monocytes and macrophages and is a crucial factor in directing the T-cell response to infection, by inducing a Th1-type cytokine response. Our data agrees with that of previous reports showing that IL-12p40 is strongly up-regulated in-vitro (in response to an inflammatory stimulus) and in-vivo in the cerebral cortex of TgAPPsw mice [12,37,38].
IL-1, which was increased in the transgenic brain slices, is a major immune-response molecule functioning in the periphery and brain. The family comprises three related proteins (IL-1α, IL-1β and IL-1 receptor antagonist (IL-1ra)). IL-1α and IL-1β are two different isoforms of IL-1 that have similar affinities for their receptor IL-1R, and therefore have similar activities. Both are capable of inducing inflammatory cascades in-vivo and in-vitro, and it has been shown that they are capable of up-regulating expression of astrocyte-derived S100B and APP [39,40]. It has been shown that IL-1β can promote β-secretase cleavage of APP in human astrocytes and thereby increase production of Aβ1–40 and 1–42 [41,42]. It is also known that accumulation of plaques and the formation of neurofibrillary tangles are correlated with increased IL-1 levels in the AD brain [43-45]. Certain polymorphisms of IL-1A (the gene for IL-1α) are associated with late onset AD, although there is controversy as to whether all IL-1 gene polymorphisms represent risk factors for AD [46-50]. Microglia, in particular, have been shown to locally up regulate IL-1α at both the protein and mRNA level when inflamed, a situation that occurs in chronic disease states such as AD [51]. Both IL-1α and IL-1β can enhance the translation of APP mRNA in human astrocytes [52]; an up-regulation of IL-1α/β production in-vivo could therefore increase Aβ production, and an inflammatory cycle with increased Aβ levels may further increase IL-1α/β production.
The Aβ 1–42:40 ratio is also of considerable interest in relation to cytokine levels and although there are currently no studies correlating Aβ 1–42:40 ratio with cytokine levels in-vivo, certain reports have suggested that cytokines can modulate Aβ production [53-55]. PS1 mutations are known to cause a shift in the production of Aβ species, favoring the production of Aβ1–42 over 1–40 and causing an increase in the Aβ1–42:40 ratio [56]. Since TNF-α correlated better with the level of Aβ1–42 than with that of Aβ 1–40, and correlated particularly well with the Aβ1–42:40 ratio in our study, TNF-α levels may be partly determined by this ratio. Higher levels of Aβ1–42 can promote the formation of toxic oligomers [57-59], and it therefore seems possible that the increased level of Aβ oligomers in PS1/APP mice (compared to APPsw) and the level of oligomeric forms present in the brains of our transgenic mice may be related to the amount of TNF-α being produced.
It is important to consider the nature of the exact form of Aβ that may be most responsible for the inflammatory events seen in AD brains. Aβ can exist in various forms (monomeric, dimeric, oligomeric and fibrillar), but it is not yet clear which of these forms are most potent in inducing inflammatory cellular responses [57,60,61]. This is of interest because the oligomeric forms of Aβ which are thought to be the most toxic are produced more readily by Aβ1–42 (for review see [62]). Future studies will assess the relative proportions of monomers/dimers, oligomers or fibrils occurring in these mice brains and their relationship with the cytokine increases observed.
List of abbreviations
AD: Alzheimer's disease
APP: Amyloid precursor protein
APPsw: Amyloid precursor protein Swedish mutation
PS1: Presenilin 1
Aβ: Beta-amyloid
Tg: Transgenic
TNF: Tumor necrosis factor
IL-x: Interleukin-x
IL-1ra: Interleukin-1 receptor antagonist
GM-CSF: Granulocyte macrophage colony stimulating factor
PBS: Phosphate buffered saline
COX-2: Cyclo-oxygenase-2
PGE2: Prostaglandin E2
IFN: Interferon
NSAID: Non-steroidal anti-inflammatory drug
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
NP carried out the in-vitro brain slice assays, processed brain tissues, performed the Bio-plex assay, ELISAs and drafted the manuscript. DP conceived the design of the study, carried out Bio-plex assays, performed statistical analyses and aided in manuscript preparation. VM analyzed data and constructed dendrograms. AQ aided in ELISA and Bio-plex assays and collected mouse brain tissues. FC oversees management of the mouse colonies. MM aided in manuscript preparation and gave critical analysis of the manuscript.
Supplementary Material
Correlation table of levels of different β-amyloid species with cytokines in transgenic mouse models of Alzheimer's disease.
Acknowledgments
Acknowledgements
The authors would like to thank Bob and Diane Roskamp for their generous support.
Contributor Information
Nikunj S Patel, Email: npatel@rfdn.org.
Daniel Paris, Email: dparis@rfdn.org.
Venkatarajan Mathura, Email: vmathura@rfdn.org.
Amita N Quadros, Email: aquadros@rfdn.org.
Fiona C Crawford, Email: fcrawford@rfdn.org.
Michael J Mullan, Email: mmullan@rfdn.org.
References
- Anthony JC, Breitner JC, Zandi PP, Meyer MR, Jurasova I, Norton MC, Stone SV. Reduced prevalence of AD in users of NSAIDs and H2 receptor antagonists: the Cache County study. Neurology. 2000;54:2066–71. doi: 10.1212/wnl.54.11.2066. [DOI] [PubMed] [Google Scholar]
- Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer's disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327:128. doi: 10.1136/bmj.327.7407.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JC, Goodman SN. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. 2004;23:159–69. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–17. [PMC free article] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–84. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol Aging. 2001;22:49–61. doi: 10.1016/S0197-4580(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Vehmas AK, Kawas CH, Stewart WF, Troncoso JC. Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol Aging. 2003;24:321–31. doi: 10.1016/S0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–14. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23:7504–9. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlhorn G, Hollborn M, Schliebs R. Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int J Dev Neurosci. 2000;18:423–31. doi: 10.1016/S0736-5748(00)00012-5. [DOI] [PubMed] [Google Scholar]
- Apelt J, Schliebs R. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001;891:21–30. doi: 10.1016/S0006-8993(00)03176-0. [DOI] [PubMed] [Google Scholar]
- Abbas N, Bednar I, Mix E, Marie S, Paterson D, Ljungberg A, Morris C, Winblad B, Nordberg A, Zhu J. Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J Neuroimmunol. 2002;126:50–7. doi: 10.1016/S0165-5728(02)00050-4. [DOI] [PubMed] [Google Scholar]
- Prabhakar U, Eirikis E, Davis HM. Simultaneous quantification of proinflammatory cytokines in human plasma using the LabMAP assay. J Immunol Methods. 2002;260:207–18. doi: 10.1016/S0022-1759(01)00543-9. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–81. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo DM, Lapchak PA. Induction of immune system mediators in the hippocampal formation in Alzheimer's and Parkinson's diseases: selective effects on specific interleukins and interleukin receptors. Neuroscience. 1994;61:745–54. doi: 10.1016/0306-4522(94)90398-0. [DOI] [PubMed] [Google Scholar]
- Cacabelos R, Alvarez XA, Fernandez-Novoa L, Franco A, Mangues R, Pellicer A, Nishimura T. Brain interleukin-1 beta in Alzheimer's disease and vascular dementia. Methods Find Exp Clin Pharmacol. 1994;16:141–51. [PubMed] [Google Scholar]
- Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer's and de novo Parkinson's disease patients. Neurosci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Roberts GW, Mrak RE. Interleukin-1 expression in different plaque types in Alzheimer's disease: significance in plaque evolution. J Neuropathol Exp Neurol. 1995;54:276–81. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- Singh VK, Guthikonda P. Circulating cytokines in Alzheimer's disease. J Psychiatr Res. 1997;31:657–60. doi: 10.1016/S0022-3956(97)00023-X. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Wallin A, Regland B, Blennow K, Tarkowski A. Local and systemic GM-CSF increase in Alzheimer's disease and vascular dementia. Acta Neurol Scand. 2001;103:166–74. doi: 10.1034/j.1600-0404.2001.103003166.x. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao-Ashec K, Frautschy SA, Cole GM. Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging. 2001;22:983–91. doi: 10.1016/S0197-4580(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. 1999;20:581–9. doi: 10.1016/S0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- Andersen K, Launer LJ, Ott A, Hoes AW, Breteler MM, Hofman A. Do nonsteroidal anti-inflammatory drugs decrease the risk for Alzheimer's disease? The Rotterdam Study. Neurology. 1995;45:1441–5. doi: 10.1212/wnl.45.8.1441. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–32. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- Gitter BD, Boggs LN, May PC, Czilli DL, Carlson CD. Regulation of cytokine secretion and amyloid precursor protein processing by proinflammatory amyloid beta (A beta) Ann N Y Acad Sci. 2000;917:154–64. doi: 10.1111/j.1749-6632.2000.tb05379.x. [DOI] [PubMed] [Google Scholar]
- Rah JC, Kim HS, Kim SS, Bach JH, Kim YS, Park CH, Seo JH, Jeong SJ, Suh YH. Effects of carboxyl-terminal fragment of Alzheimer's amyloid precursor protein and amyloid beta-peptide on the production of cytokines and nitric oxide in glial cells. FASEB J. 2001;15:1463–5. doi: 10.1096/fj.00-0724fje. [DOI] [PubMed] [Google Scholar]
- Paris D, Townsend KP, Obregon DF, Humphrey J, Mullan M. Pro-inflammatory effect of freshly solubilized beta-amyloid peptides in the brain. Prostaglandins Other Lipid Mediat. 2002;70:1–12. doi: 10.1016/S0090-6980(02)00111-9. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, Pepeu G, Casamenti F. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis. 2002;11:257–74. doi: 10.1006/nbdi.2002.0538. [DOI] [PubMed] [Google Scholar]
- Quadros A, Patel N, Crescentini R, Crawford F, Paris D, Mullan M. Increased TNFalpha production and Cox-2 activity in organotypic brain slice cultures from APPsw transgenic mice. Neurosci Lett. 2003;353:66–8. doi: 10.1016/j.neulet.2003.08.076. [DOI] [PubMed] [Google Scholar]
- Rosales-Corral S, Tan DX, Reiter RJ, Valdivia-Velazquez M, Acosta-Martinez JP, Ortiz GG. Kinetics of the neuroinflammation-oxidative stress correlation in rat brain following the injection of fibrillar amyloid-beta onto the hippocampus in vivo. J Neuroimmunol. 2004;150:20–8. doi: 10.1016/j.jneuroim.2004.01.005. [DOI] [PubMed] [Google Scholar]
- McGowan E, Sanders S, Iwatsubo T, Takeuchi A, Saido T, Zehr C, Yu X, Uljon S, Wang R, Mann D, Dickson D, Duff K. Amyloid phenotype characterization of transgenic mice overexpressing both mutant amyloid precursor protein and mutant presenilin 1 transgenes. Neurobiol Dis. 1999;6:231–44. doi: 10.1006/nbdi.1999.0243. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T. Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol. 2000;157:331–9. doi: 10.1016/s0002-9440(10)64544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt MA, Davies DC, Kidd M, Duff K, Rolph SC, Jennings KH, Howlett DR. Neurodegenerative changes associated with beta-amyloid deposition in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Exp Neurol. 2001;171:59–71. doi: 10.1006/exnr.2001.7717. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–13. doi: 10.1016/S0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Yang Y, Han SH, Kim H, Kim C, Kim KY, Shin SM, Choi I, Pyun KH. Interleukin-12 p40 gene expression is induced in lipopolysaccharide-activated pituitary glands in vivo. Neuroendocrinology. 2002;75:347–57. doi: 10.1159/000059431. [DOI] [PubMed] [Google Scholar]
- Ichikawa D, Matsui A, Imai M, Sonoda Y, Kasahara T. Effect of various catechins on the IL-12p40 production by murine peritoneal macrophages and a macrophage cell line, J774.1. Biol Pharm Bull. 2004;9:1353–8. doi: 10.1248/bpb.27.1353. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Ito K, Skinner RD, Mrak RE, Rovnaghi CR, Van Eldik LJ, Griffin WS. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging. 1996;17:761–6. doi: 10.1016/0197-4580(96)00104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2001;22:915–22. doi: 10.1016/S0197-4580(01)00293-7. [DOI] [PubMed] [Google Scholar]
- Schmitt TL, Steiner E, Klinger P, Sztankay A, Grubeck-Loebenstein B. The production of an amyloidogenic metabolite of the Alzheimer amyloid beta precursor protein (APP) in thyroid cells is stimulated by interleukin 1 beta, but inhibited by interferon gamma. J Clin Endocrinol Metab. 1996;81:1666–9. doi: 10.1210/jc.81.4.1666. [DOI] [PubMed] [Google Scholar]
- Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1–40 and Abeta1–42 by human astrocytes. Neurobiol Dis. 2000;7:682–9. doi: 10.1006/nbdi.2000.0321. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, 3rd, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:7611–5. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WS. Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol. 1997;56:285–90. [PubMed] [Google Scholar]
- Griffin WS, Mrak RE. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer's disease. J Leukoc Biol. 2002;72:233–8. [PMC free article] [PubMed] [Google Scholar]
- Du Y, Dodel RC, Eastwood BJ, Bales KR, Gao F, Lohmuller F, Muller U, Kurz A, Zimmer R, Evans RM, Hake A, Gasser T, Oertel WH, Griffin WS, Paul SM, Farlow MR. Association of an interleukin 1 alpha polymorphism with Alzheimer's disease. Neurology. 2000;55:480–3. doi: 10.1212/wnl.55.4.480. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Mrak RE, Graham DI, Stewart J, Wilcock G, MacGowan S, Esiri MM, Murray LS, Dewar D, Love S, Moss T, Griffin WS. Association of interleukin-1 gene polymorphisms with Alzheimer's disease. Ann Neurol. 2000;47:365–8. [PMC free article] [PubMed] [Google Scholar]
- Grimaldi LM, Casadei VM, Ferri C, Veglia F, Licastro F, Annoni G, Biunno I, De Bellis G, Sorbi S, Mariani C, Canal N, Griffin WS, Franceschi M. Association of early-onset Alzheimer's disease with an interleukin-1alpha gene polymorphism. Ann Neurol. 2000;47:361–5. [PubMed] [Google Scholar]
- Fidani L, Goulas A, Mirtsou V, Petersen RC, Tangalos E, Crook R, Hardy J. Interleukin-1A polymorphism is not associated with late onset Alzheimer's disease. Neurosci Lett. 2002;323:81–3. doi: 10.1016/S0304-3940(02)00114-3. [DOI] [PubMed] [Google Scholar]
- Sciacca FL, Ferri C, Licastro F, Veglia F, Biunno I, Gavazzi A, Calabrese E, Martinelli Boneschi F, Sorbi S, Mariani C, Franceschi M, Grimaldi LM. Interleukin-1B polymorphism is associated with age at onset of Alzheimer's disease. Neurobiol Aging. 2003;24:927–31. doi: 10.1016/S0197-4580(03)00011-3. [DOI] [PubMed] [Google Scholar]
- Hetier E, Ayala J, Denefle P, Bousseau A, Rouget P, Mallat M, Prochiantz A. Brain macrophages synthesize interleukin-1 and interleukin-1 mRNAs in vitro. J Neurosci Res. 1988;21:391–7. doi: 10.1002/jnr.490210230. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN. Translation of the alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5'-untranslated region sequences. J Biol Chem. 1999;274:6421–31. doi: 10.1074/jbc.274.10.6421. [DOI] [PubMed] [Google Scholar]
- Del Bo R, Angeretti N, Lucca E, De Simoni MG, Forloni G. Reciprocal control of inflammatory cytokines, IL-1 and IL-6, and beta-amyloid production in cultures. Neurosci Lett. 1995;188:70–4. doi: 10.1016/0304-3940(95)11384-9. [DOI] [PubMed] [Google Scholar]
- Brugg B, Dubreuil YL, Huber G, Wollman EE, Delhaye-Bouchaud N, Mariani J. Inflammatory processes induce beta-amyloid precursor protein changes in mouse brain. Proc Natl Acad Sci U S A. 1995;92:3032–5. doi: 10.1073/pnas.92.7.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–32. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- Borchelt DR. Metabolism of presenilin 1: influence of presenilin 1 on amyloid precursor protein processing. Neurobiol Aging. 1998;19:S15–8. doi: 10.1016/S0197-4580(98)00026-8. [DOI] [PubMed] [Google Scholar]
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-assembly of Abeta(1–42) into globular neurotoxins. Biochemistry. 2003;42:12749–60. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid beta-protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc Natl Acad Sci U S A. 2003;100:330–5. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace EA, Rabiner CA, Busciglio J. Characterization of neuronal dystrophy induced by fibrillar amyloid beta: implications for Alzheimer's disease. Neuroscience. 2002;114:265–73. doi: 10.1016/S0306-4522(02)00241-5. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:S10–7. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Correlation table of levels of different β-amyloid species with cytokines in transgenic mouse models of Alzheimer's disease.