

Abstract
Here we report the first demonstration of near-complete sequence coverage of intact proteins using activated ion-electron transfer dissociation (AI-ETD), a method that leverages concurrent infrared photo-activation to enhance electron-driven dissociation. AI-ETD produces mainly c/z-type product ions and provides comprehensive (77–97%) protein sequence coverage, outperforming HCD, ETD, and EThcD for all proteins investigated. AI-ETD also maintains this performance across precursor ion charge states, mitigating charge state dependence that limits traditional approaches.
Graphical Abstract
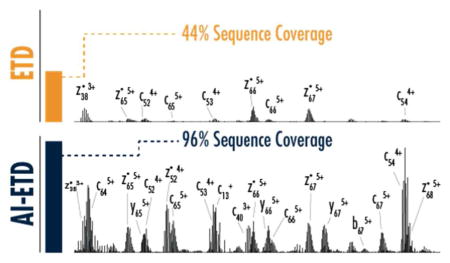
Top-down proteomics, a technique that interrogates intact proteins, can provide several potential benefits, including the ability to characterize sequence truncations, splice variants, single nucleotide polymorphisms, and combinatorial patterns of post-translational modifications.1 Realization of these benefits, however, is predicated on the ability to generate extensive fragmentation for unambiguous sequence elucidation of various proteoforms. Due to limitations in tandem mass spectrometry dissociation methods near-complete sequence coverage (>75%) is still difficult to achieve for proteins larger than 10 kDa.2 Slow-heating methods such as collision-activated dissociation (CAD) and infrared multi-photon dissociation (IRMPD) often fail to produce extensive fragmentation due to their proclivity to break only the most labile bonds in protein ions.3–6
Electron-driven dissociation methods have been a valuable alternative to collision-based fragmentation, especially for top-down proteomics. Electron capture dissociation (ECD) was first described as a method for generating more random and extensive backbone bond cleavage from intact proteins, and electron transfer dissociation (ETD) was described shortly after, making electron-driven dissociation accessible on a diverse set of instrument platforms.7–10 Despite their value for top-down proteomics, ECD and ETD exhibit a strong dependency on precursor ion charge state, limiting their ability to provide extensive fragmentation and sequence information on all analytes.11–13 Several strategies to combat this charge state dependence and improve the utility of ECD and ETD have been described and include collisional and photo-activation before, during, and after reactions, raised ambient temperatures, and higher energy electrons.14–21
Two of the most recent developments for improved ETD fragmentation of intact proteins include higher-energy collisional activation of all ions after an ETD reaction (EThcD) and infrared photo-activation concurrent with ETD reactions (activated ion ETD, AI-ETD).22–24 Both were shown to improve characterization over standard ETD, but neither has been shown to generate near-complete sequence coverage in their previously described implementations despite the theoretical capability of both to do so. AI-ETD produces more sequence information from ETD reactions by mitigating non-dissociative electron transfer (ETnoD), a process by which backbone cleavage occurs but product ions are held together in a complex by non-covalent interactions. These non-covalent interactions are more prevalent in low-charge density precursors where secondary gas-phase structure is more compact.25–29 The energy from irradiation with IR photons in AI-ETD disrupts this structure, partially unfolding precursors as they undergo ETD, which promotes formation of sequence-informative product ions.23,30
We recently implemented AI-ETD on a quadrupole-Orbitrap-linear ion trap hybrid MS system (Orbitrap Fusion Lumos),31 and here we report the first demonstration of near-complete sequence coverage of intact proteins using AI-ETD. Focusing on proteins in molecular weight range seen in standard top-down proteomic experiments (<20 kDa),32,33 we show that AI-ETD provides comprehensive sequence coverage for all precursor ion charge states investigated, and with the first head-to-head comparison of AI-ETD and EThcD, we show that AI-ETD outperforms EThcD as a supplemental activation method for ETD. Notably, this work also demonstrates that the impressive sequence coverage recently described using ultraviolet photo-dissociation (UVPD)34 is not exclusive to photo-dissociation approaches and that near-complete sequence coverage can be achieved for a variety of intact proteins using electron-driven dissociation.
MATERIALS AND METHODS
Sample Preparation
Ubiquitin from bovine erythrocytes (UniProt Accession P62992), lysozyme from chicken egg whites (UniProt Accession P00698), and myoglobin from horse heart (UniProt Accession P68082) were purchased as standards from Protea Biosciences (Morgantown, WV), product numbers PS-143, PS-123, and PS-124, respectively). Trypsin inhibitor from glycine max (UniProt Accession P01070) was purchased from Sigma Aldrich (St. Louis, MO), product number T9003. Due to the presence of multiple disulfide bonds, lysozyme and trypsin inhibitor were resuspended in buffer (8 M urea, 50 mM Tris, pH 8), reduced with 5 mM dithiothreitol for 45 minutes at 58 °C, alkylated with 15 mM iodoacetamide for 45 minutes at room temperature in the dark, and desalted using a C2 SepPak (Waters, Milford, MA). All samples were resuspended at ~10 pmol/μL in 49.9:49.9:0.2 acetonitrile/water/formic acid prior to infusions.
Mass Spectrometry
All experiments were performed on a quadrupole-Orbitrap-linear ion trap hybrid MS system (Orbitrap Fusion Lumos) that outfitted with Firestar ti60 Synrad 60-W CO2 continuous wave laser (10.6 μm) as previously described.31 Proteins were infused at 5–10 μL/min using the instrument’s syringe pump, precursors were ionized using electrospray ionization at 4–5 kV with respect to ground, and the inlet capillary was held at 275 °C. Survey scans of precursor ion charge states were collected at 240,000 resolution with 10–100 microscans. Data were acquired using intact protein mode, which lowers the ion routing multipole nitrogen bath gas setting to 3 mTorr (originally 8 mTorr). All MS/MS scans were conducted at 240,000 resolution and precursor ion targets of 800,000. For ETD, EThcD, and AI-ETD experiments, the reagent anion population was set to 300,000 ions and reaction times varied for each precursor, ranging from 10–35 ms. For EThcD, normalized collision energies of 8, 10, 12, and 15 were tested for each precursor to determine the optimum setting. For AI-ETD, laser power settings of 18, 24, 30, and 36 Watts were tested for each precursor to determine the optimum setting. For HCD, normalized collision energies of 15, 20, and 25 were tested for each precursor. For each precursor the optimum dissociation conditions were collected for each dissociation method (HCD, ETD, EThcD, and AI-ETD) using 50, 150, 200, and 300 microscans for ubiquitin, lysozyme, myoglobin, and trypsin inhibitor, respectively.
Data Analysis
MS/MS spectra were deconvoluted with XTRACT (Thermo Fisher Scientific) using default parameters and a S/N threshold of three. Peak lists from XTRACT and protein sequences from UniProt were loaded into ProSight Lite35 to determine the number of matched fragments generated for each precursor and dissociation method. “EThcD”, which matched b/y- and c/z-type product ions, was used for ETD, EThcD, and AI-ETD spectra, while “HCD”, which matched b/y-type product ions, was used for HCD spectra. All matches were made within a 10 ppm tolerance. No sequence modifications were used for ubiquitin or myoglobin spectra, while carbamidomethylation of cysteines (+57.021 Da) was used for lysozyme and trypsin inhibitor.
RESULTS AND DISCUSSION
AI-ETD increases both the number and intensity of sequence-informative product ions generated from a given precursor compared to ETD. Figure 1 compares ETD and AI-ETD spectra from the z =+7 precursor ion of ubiquitin. Charge-reduced precursors, denoted with asterisks (*) in Figure 1a, contain ETnoD products, and their intensities are substantially reduced when using AI-ETD (i.e., precursor ion channels account for ~59% total ion current in the ETD spectrum compared to ~18% for AI-ETD). Additionally, dissociation efficiency, i.e., product ion generation, is markedly increased with AI-ETD, where nearly 52% of total ion current is in sequence-informative product ion channels versus only ~6% in the ETD spectrum. Figure 1b and 1c show zoomed-in portions of the spectra in panel 1 respectively. Besides producing higher totals of product ions that ETD, AI-ETD also increases the signal of product ions shared between the two for both lower and higher charged product ions, making sequence elucidation straight-forward even for this low charge-density precursor. AI-ETD does increase the presence of y-type products ions, too, although this is from a combination of both enhanced ETD dissociation and small degree of IRMPD-like fragmentation (as also indicated by a few b-type ions).31 Supplemental Figure 1 shows a similar comparison for the z = +14 precursor ion of myoglobin. Again, ETnoD/charge reduced precursor ions are diminished in intensity in AI-ETD compared to ETD, and the generation of more numerous and more intense product ions is easy to observe.
Figure 1. Comparison ETD and AI-ETD spectra of ubiquitin (1 spectral acquisition, 50 uScans).

a) A comparison of the MS/MS spectra illustrates the substantial increase in fragmentation and decrease in charge-reduced precursor signal, which translates to more than triple the number of fragments and more than double the sequence coverage for the z = +7 precursor. Peaks with the asterisk (*) show remaining precursor and charge-reduced precursor ions, which are five-fold more intense than they are shown. Panels (b) and (c) compare the indicated regions of the spectra, demonstrating that AI-ETD increases the number and signal of both higher and lower charge state product ions. Note, for each comparison ETD and AI-ETD spectra are on the same intensity scale. Base peak intensities are 4e6, 2e5, and 1e5 for panels (a), (b), and (c), respectively.
Figure 2 compares AI-ETD performance with ETD, EThcD, and HCD for multiple precursor ion charge states for four proteins: ubiquitin (B. taurus, 8.6 kDa), lysozyme (G. gallus, 14.7 kDa with carbamidomethylation of cysteines), myoglobin (E. caballus, 16.9 kDa), and trypsin inhibitor (G. max, 20.2 kDa with carbamidomethylation of cysteines). The precursor ion charge state distributions and those charge states selected are highlighted in Supplemental Figure 2. Charge states were chosen to cover a diverse set of precursor ions centered around the middle of the a given charge state distribution that would be standard to investigate in top-down experiments. Precursor ions were mass selected using a mass filtering quadrupole, and all ETD reactions occurred in the high pressure trap of the dual cell linear ion trap using a helium bath gas. Importantly, all AI-ETD activation occurred at standard pressures and were conducted just as any other ETD reaction. HCD and the re-activation of products in EThcD both occurred in the ion routing multipole, which uses nitrogen as a bath gas. All four dissociation methods generate good sequence coverage for ubiquitin, although AI-ETD is the only method to maintain 95% sequence coverage or greater for all seven precursor ion charge states investigated. Note, protein sequence coverage, reported as a percentage, is defined here as the number of inter-residue positions explained by product ions divided by the total number of inter-residue positions, (100% maximum). Coverage drops sharply with ETD for the z = +8 and z = +7 charge states due to the low-charge density of those precursor ions. AI-ETD and EThcD both effectively increase sequence coverage over ETD for these charge states, although AI-ETD outperforms EThcD by ~10% coverage for each.
Figure 2. AI-ETD provides near-complete protein sequence coverage.

Percent sequence coverage achieved for (a) ubiquitin, (b) lysozyme, (c) myoglobin, and (d) trypsin inhibitor is shown for HCD, ETD, EThcD, and AI-ETD fragmentation methods. AI-ETD provides the greatest sequence coverage for all precursors investigated.
Interestingly, HCD generates the least sequence coverage for most precursors of lysozyme, myoglobin, and trypsin inhibitor with its best performance usually occurring for the lowest charge state precursor ions of each protein. Thus, these data highlight the general utility that ETD maintains for top-down proteomics, although it fails to approach near-complete sequence coverage for any of these three proteins. EThcD never performs worse than ETD, although it fails to offer much benefit over ETD for the higher charge state precursor ions for each protein. This is contrasted to AI-ETD, which improves sequence coverage over ETD most markedly for low charge state precursor ions, as expected, but also offers considerable boosts in coverage even for high charge state precursors. Al-ETD consistently outperforms EThcD in sequence coverage achieved, as well, indicating it is the superior approach to improving ETD fragmentation. In all AI-ETD maintains 95–97% sequence coverage for seven charge states of ubiquitin, 82–93% sequence coverage for three charge states of lysozyme, 89–92% sequence coverage for five charge states of myoglobin, and 71–77% sequence coverage of three charge states of trypsin inhibitor. This is the most comprehensive sequence coverage achieved across multiple precursor ion charge states for multiple proteins for any electron-driven dissociation method and represents the state-of-the-art in AI-ETD protein characterization. Indeed, comparing sequence coverage of ubiquitin and myoglobin from our original AI-ETD study shows achievement of approximately 30% higher sequence coverage or greater for a given precursor (i.e., 92% vs. 59% for the z = +18 precursor ion of myoglobin) for both proteins (Supplemental Figure 3).
The outstanding performance of AI-ETD for protein sequence coverage is explained by the extensive generation of sequence-informative product ions detected across all precursor ion charge states. Figure 3 displays heat maps of the number of matching fragment ion counts (blue) that were returned by ProSight Lite35 using a 10 ppm tolerance. Note, ±1 errors common in deconvolution of MS/MS spectra of intact proteins were not accounted for, meaning hand annotation could improve further upon the results discussed here. AI-ETD generates approximately 45–60% more fragments for a given precursor ion charge state for all proteins except ubiquitin, where it ranges from ~10–70% (and nearly three-fold for the z = +7 charge state). The majority of product ions generated by AI-ETD are c/z•-type, as well (Figure 3, gold heat map). AI-ETD only slightly, if at all, increases the percentage of total product ions seen that are b/y-type over ETD. In ETD of the ~10–20% of product ions that are b/y-type, the vast majority of them are y-type ions, while in AI-ETD there is a small increase in the number of b-type ions. EThcD increases the number of product ions that are b/y-type more than AI-ETD (~25–35%), but this is expected as EThcD seeks to generate collisional dissociation product ions in addition to ETD product ions while the IR photo-activation of AI-ETD is mainly to augment electron-drive dissociation rather than to generate IRMPD-type fragments. This is interesting to note, as well, seeing as using the laser only (i.e., IRMPD) at the tested AI-ETD laser powers generated virtually no precursor ion dissociation when performed in the high pressure cell of the linear ion trap. This is expected because the collisional cooling of the background gas in the high pressure cell would mitigate increases in internal energy sufficient to cause backbone dissociation.36,37 Thus, the additional energy imparted by the concurrent IR photo-activation in AI-ETD serves to disrupt weaker, non-covalent interactions to improve electron-driven fragmentation but rarely drives vibrational dissociation of backbone bonds.
Figure 3. Number and type of fragment ions generated by AI-ETD and other dissociation methods.

The heat map to the left (blue) show the number of matched fragments generated for each dissociation method for each precursor charge state from the four proteins investigated in this study. The percentage of these fragments that are b/y-type (rather than c/z•-type) for each condition is shown in the right (orange) heat maps, with the darker regions indicating a higher proportion of b/y-type fragments. By default, only b/y-type ions were considered for HCD, so it was omitted from the right heat map.
This is an especially interesting comparison when considering UVPD, which generates significant populations of a/x-, b/y, and c/z•-type product ions in each MS/MS event. The benefit of having rich spectra with all six product ion types is that each inter-residue position can often be explained by multiple fragment ion types. This can lend further confidence to sequence assignment, but the signal is then split between several fragment ion channels. This has the potential to be detrimental by diluting available signal and lowering product ion signal-to-noise (S/N), which is already a challenge in top-down proteomics.38 AI-ETD avoids this problem by concentrating the majority of its product ion signal into c/z•-type fragment ions, but still provides comparable near-complete sequence coverage to UVPD.34,39
Another challenge with product ion S/N in top down experiments, especially with ETD and UVPD, is low precursor-to-product ion conversion, i.e., a significant proportion of original precursor signal is not converted to sequence information. Supplemental Figure 4 shows AI-ETD increases reaction efficiency in generating sequence-informative product ions (i.e., c/z•- and b/y-type) over ETD, especially as charge density decreases. AI-ETD also outperforms EThcD for all data here, with the only exception of lysozyme. Supplemental Figure 5 shows, however, that AI-ETD provides more measured signal in sequence-informative product ions than EThcD. These data confirm that AI-ETD improves generation of product ions over ETD by increasing the fragment ion yield, channeling more available signal into ions that inform protein sequence determination. For EThcD at higher charge states, the percent total ion current (TIC) accounted for by sequencing ions is often similar to ETD, indicating that post-reaction activation often does not aid in product ion generation for ETD when more charge dense precursors are dissociated. By comparison, AI-ETD improves signal in product ion channels by 10–15% even for the most highly charge precursors, suggesting that (1) there is still some degree of significant gas-phase structure in more highly charge precursors and (2) that activation of the ion population during ETD reactions can improve reaction efficiency even for precursors with high charge density. Interestingly, HCD, which is considered to be a higher efficiency dissociation process, is often the lowest in percent TIC in matched fragments. We hypothesize this is largely due to internal fragments that are common during HCD fragmentation that contribute to the total ion current in an HCD spectrum but do not contribute to protein sequence information. Perhaps this percentage could be increased if internal fragments were considered, although that calculation is not straight-forward. Regardless, this reiterates that HCD can be valuable for top-down proteomics, but it often fails to generate the degree of fragmentation that can be achieved by electron-driven methods.
The performance of AI-ETD in this work also highlights the benefits afforded both by improvements to the underlying instrumentation (i.e., Orbitrap Fusion Lumos) and a more robust control of photon flux in this iteration of AI-ETD. Several key differences in the previous proof-of-principle study from 201524 and the data shown here include where the ion-ion reactions are conducted and how the laser is focused into the reaction cell. Most notably, the previous AI-ETD work required modification to the HCD cell in an Orbitrap Elite to enable ion-ion reactions to be conducted in that region of the instrument.24,40,41 The so-called multi-purpose dissociation cell (MDC) took the place of the HCD cell and provided an avenue to introduce the IR photon beam concentric to an ion-ion reaction cell where ETD could be performed. This device required additional electronic boards and instrument control that made operation very specialized. Converse to the extensive modifications required for AI-ETD on the Elite, the newest implementation on the Lumos capitalizes on the accessible location of the linear ion trap (i.e., the traditional ETD reaction cell) at the back on the instrument. This permits more straightforward AI-ETD modifications that necessitate only hardware additions to affix the laser rather than modifications for both laser and reaction cell.31 Additionally, gains in ion manipulation and transmission, more robust ETD reagent anion generation, and high capacity ETD that are tenants of the Lumos platform serve to increase performance over the breadboard system that originally housed AI-ETD reactions. Furthermore, our initial AI-ETD work on the Elite did not include laser focusing optics, but the need became apparent because gas pressures in the MDC had to be lowered to get effective photo-activation to improve ETD fragmentation. AI-ETD on the Lumos is implemented with focusing lenses to focus the beam to a ~1mm waist and then columnate the beam before it enters the linear ion trap. This increases the energy that is introduced to the trapping volume where ETD occurs, eliminating the need for lower gas pressures, lowering the photon flux requirements (i.e., laser power) needed for effective AI-ETD, and increasing production generation. Thus the improvements for intact protein ion dissociation described in this work reflect a combination of a better control of infrared photo-activation for AI-ETD and the boosts of moving AI-ETD to a more highly developed instrument platform.
CONCLUSION
Near-complete protein sequence coverage is readily achievable with AI-ETD. AI-ETD significantly improves sequence information that can be gleaned from a given precursor ion charge state over ETD, EThcD, and HCD, and the method offers these benefits without any additional time cost or ion manipulation over standard ETD. Moreover, with the new implementation on the quadrupole-Orbitrap-linear ion trap hybrid MS system, AI-ETD can now be performed for top down experiments with minimal instrument modification and while keeping all instrument setting identical to standard ETD functions. Unlike EThcD, AI-ETD improves sequence information obtained from all precursor ions, including high charge states where ETD also performs well. AI-ETD has been shown to benefit phosphosite localization in intact proteins,42 making it a powerful method to map post-translational modifications in a variety of proteoforms with the comprehensive sequence coverage demonstrated here. This work also shows that electron-driven fragmentation methods in general remain very valuable in top-down proteomics, as ETD, EThcD, and AI-ETD all outperformed HCD for the majority of precursor ion charge states investigated. Interestingly, other alternative fragmentation methods like UVPD and electron induced dissociation have had success in analyzing native proteins and protein complexes,43–47 and AI-ETD offers a combination of photon- and electron-driven fragmentation that may prove valuable for that arena as well. As such, future work will investigate how AI-ETD can improve dissociation of native proteins and protein complexes and also how AI-ETD can benefit analysis of larger (>20 kDa) proteins cations.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge support from Thermo Fisher Scientific and R35 GM118110. N.M.R. was funded through an NIH Predoctoral to Postdoctoral Transition Award (F99 CA212454).
Footnotes
Supplemental Material that contains five figures further describing this data is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Toby TK, Fornelli L, Kelleher NL. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu Rev Anal Chem. 2016;9(1):499–519. doi: 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catherman AD, Skinner OS, Kelleher NL. Top Down proteomics: Facts and perspectives. Biochem Biophys Res Commun. 2014;445(4):683–693. doi: 10.1016/j.bbrc.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Little DP, Speir JP, Senko MW, O’Connor PB, McLafferty FW. Infrared multiphoton dissociation of large multiply charged ions for biomolecule sequencing. Anal Chem. 1994;66(18):2809–2815. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 4.Wysocki VH, Tsaprailis G, Smith LL, Breci LA. Journal of Mass Spectrometry. John Wiley & Sons, Ltd; Dec, 2000. Mobile and localized protons: A framework for understanding peptide dissociation; pp. 1399–1406. [DOI] [PubMed] [Google Scholar]
- 5.Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Müller M, Viner R, Schwartz J, Remes P, Belford M, et al. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics. 2012;11(3):O111.013698. doi: 10.1074/mcp.O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahlf DR, Compton PD, Tran JC, Early BP, Thomas PM, Kelleher NL. Evaluation of the compact high-field orbitrap for top-down proteomics of human cells. J Proteome Res. 2012;11(8):4308–4314. doi: 10.1021/pr3004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zubarev R, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 8.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations. Anal Chem. 2000;72(3):563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 9.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc Natl Acad Sci U S A. 2005;102(27):9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zubarev RA. Electron-capture dissociation tandem mass spectrometry. Curr Opin Biotechnol. 2004;15(1):12–16. doi: 10.1016/j.copbio.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance characteristics of electron transfer dissociation mass spectrometry. Mol Cell Proteomics. 2007;6(11):1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Coon JJ. Collisions or electrons? Protein sequence analysis in the 21st century. Anal Chem. 2009;81(9):3208–3215. doi: 10.1021/ac802330b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breuker K, Oh H, Horn DM, Cerda BA, McLafferty FW. Detailed Unfolding and Folding of Gaseous Ubiquitin Ions Characterized by Electron Capture Dissociation. J Am Chem Soc. 2002;124(22):6407–6420. doi: 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]
- 15.Horn DM, Ge Y, McLafferty FW. Activated ion electron capture dissociation for mass spectral sequencing of larger (42 kDa) proteins. Anal Chem. 2000;72(20):4778–4784. doi: 10.1021/ac000494i. [DOI] [PubMed] [Google Scholar]
- 16.Sze SK, Ge Y, McLafferty FW. Plasma Electron Capture Dissociation for the Characterization of Large Proteins by Top Down Mass Spectrometry. Anal Chem. 2003;75(7):1599–1603. doi: 10.1021/ac020446t. [DOI] [PubMed] [Google Scholar]
- 17.Horn DM, Breuker K, Frank AJ, McLafferty FW. Kinetic Intermediates in the Folding of Gaseous Protein Ions Characterized by Electron Capture Dissociation Mass Spectrometry. J Am Chem Soc. 2001;123(40):9792–9799. doi: 10.1021/ja003143u. [DOI] [PubMed] [Google Scholar]
- 18.Tsybin YO, Witt M, Baykut G, Kjeldsen F, Håkansson P. Combined infrared multiphoton dissociation and electron capture dissociation with a hollow electron beam in Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun Mass Spectrom. 2003;17(15):1759–1768. doi: 10.1002/rcm.1118. [DOI] [PubMed] [Google Scholar]
- 19.Bourgoin-Voillard S, Leymarie N, Costello CE. Top-down tandem mass spectrometry on RNase A and B using a Qh/FT-ICR hybrid mass spectrometer. Proteomics. 2014;14(10):1174–1184. doi: 10.1002/pmic.201300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mikhailov VA, Cooper HJ. Activated Ion Electron Capture Dissociation (AI ECD) of proteins: synchronization of infrared and electron irradiation with ion magnetron motion. J Am Soc Mass Spectrom. 2009;20(5):763–771. doi: 10.1016/j.jasms.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sze SK, Ge Y, Oh H, McLafferty FW. Top-down mass spectrometry of a 29-kDa protein for characterization of any posttranslational modification to within one residue. Proc Natl Acad Sci U S A. 2002;99(4):1774–1779. doi: 10.1073/pnas.251691898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunner AM, Lossl P, Liu F, Huguet R, Mullen C, Yamashita M, Zabrouskov V, Makarov A, Altelaar AFM, Heck AJR. Benchmarking multiple fragmentation methods on an Orbitrap Fusion for top-down phosphoproteoform characterization. Anal Chem. 2015;87(8):4152–4158. doi: 10.1021/acs.analchem.5b00162. [DOI] [PubMed] [Google Scholar]
- 23.Ledvina AR, McAlister GC, Gardner MW, Smith SI, Madsen Ja, Schwartz JC, Stafford GC, Syka JEP, Brodbelt JS, Coon JJ. Infrared photoactivation reduces peptide folding and hydrogen-atom migration following ETD tandem mass spectrometry. Angew Chem Int Ed Engl. 2009;48(45):8526–8528. doi: 10.1002/anie.200903557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riley NM, Westphall MS, Coon JJ. Activated Ion Electron Transfer Dissociation for Improved Fragmentation of Intact Proteins. Anal Chem. 2015;87(14):7109–7116. doi: 10.1021/acs.analchem.5b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lermyte F, Williams JP, Brown JM, Martin EM, Sobott F. Extensive Charge Reduction and Dissociation of Intact Protein Complexes Following Electron Transfer on a Quadrupole-Ion Mobility-Time-of-Flight MS. J Am Soc Mass Spectrom. 2015;26(7):1068–1076. doi: 10.1007/s13361-015-1124-z. [DOI] [PubMed] [Google Scholar]
- 26.Laszlo KJ, Munger EB, Bush MF. Folding of Protein Ions in the Gas Phase after Cation-to-Anion Proton-Transfer Reactions. J Am Chem Soc. 2016;138(30):9581–9588. doi: 10.1021/jacs.6b04282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clemmer DE, Hudgins RR, Jarrold MF. Naked Protein Conformations: Cytochrome c in the Gas Phase. J Am Chem Soc. 1995;117(40):10141–10142. [Google Scholar]
- 28.Zhang Z, Browne SJ, Vachet RW. Exploring Salt Bridge Structures of Gas-Phase Protein Ions using Multiple Stages of Electron Transfer and Collision Induced Dissociation. J Am Soc Mass Spectrom. 2014;25(4):604–613. doi: 10.1007/s13361-013-0821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loo RRO, Loo JA. Salt Bridge Rearrangement (SaBRe) Explains the Dissociation Behavior of Noncovalent Complexes. J Am Soc Mass Spectrom. 2016;27(6):975–990. doi: 10.1007/s13361-016-1375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood TD, Chorush RA, Wampler FM, Little DP, O’Connor PB, McLafferty FW. Gas-phase folding and unfolding of cytochrome c cations. Proc Natl Acad Sci U S A. 1995;92(7):2451–2454. doi: 10.1073/pnas.92.7.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riley NM, Westphall MS, Hebert AS, Coon JJ. Implementation of Activated Ion Electron Transfer Dissociation on a quadrupole-Orbitrap-linear ion trap hybrid mass spectrometer. Anal Chem. 2017 doi: 10.1021/acs.analchem.7b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480(7376):254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Catherman AD, Durbin KR, Ahlf DR, Early BP, Fellers RT, Tran JC, Thomas PM, Kelleher NL. Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Mol Cell Proteomics. 2013;12(12):3465–3473. doi: 10.1074/mcp.M113.030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS. Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J Am Chem Soc. 2013;135(34):12646–12651. doi: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fellers RT, Greer JB, Early BP, Yu X, LeDuc RD, Kelleher NL, Thomas PM. ProSight Lite: Graphical software to analyze top-down mass spectrometry data. Proteomics. 2015;15(7):1235–1238. doi: 10.1002/pmic.201570050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gardner MW, Smith SI, Ledvina AR, Madsen JA, Coon JJ, Schwartz JC, Stafford GC, Brodbelt JS, Brodbelt JS. Infrared multiphoton dissociation of peptide cations in a dual pressure linear ion trap mass spectrometer. Anal Chem. 2009;81(19):8109–8118. doi: 10.1021/ac901313m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madsen JA, Gardner MW, Smith SI, Ledvina AR, Coon JJ, Schwartz JC, Stafford GC, Brodbelt JS. Top-down protein fragmentation by infrared multiphoton dissociation in a dual pressure linear ion trap. Anal Chem. 2009;81(21):8677–8686. doi: 10.1021/ac901554z. [DOI] [PubMed] [Google Scholar]
- 38.Riley NM, Mullen C, Weisbrod CR, Sharma S, Senko MW, Zabrouskov V, Westphall MS, Syka JEP, Coon JJ. Enhanced Dissociation of Intact Proteins with High Capacity Electron Transfer Dissociation. J Am Soc Mass Spectrom. 2016;27:520–531. doi: 10.1007/s13361-015-1306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holden DD, McGee WM, Brodbelt JS. Integration of Ultraviolet Photodissociation with Proton Transfer Reactions and Ion Parking for Analysis of Intact Proteins. Anal Chem. 2016;88(1):1008–1016. doi: 10.1021/acs.analchem.5b03911. [DOI] [PubMed] [Google Scholar]
- 40.Ledvina AR, Rose CM, McAlister GC, Syka JEP, Westphall MS, Griep-Raming J, Schwartz JC, Coon JJ. Activated ion ETD performed in a modified collision cell on a hybrid QLT-Oribtrap mass spectrometer. J Am Soc Mass Spectrom. 2013;24(11):1623–1633. doi: 10.1007/s13361-013-0621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rose CM, Russell JD, Ledvina AR, McAlister GC, Westphall MS, Griep-Raming J, Schwartz JC, Coon JJ, Syka JEP. Multipurpose dissociation cell for enhanced ETD of intact protein species. J Am Soc Mass Spectrom. 2013;24(6):816–827. doi: 10.1007/s13361-013-0622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riley NM, Hebert AS, Dürnberger G, Stanek F, Mechtler K, Westphall MS, Coon JJ. Phosphoproteomics with Activated Ion Electron Transfer Dissociation. Anal Chem. 2017 doi: 10.1021/acs.analchem.7b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Brien JP, Li W, Zhang Y, Brodbelt JS. Characterization of Native Protein Complexes Using Ultraviolet Photodissociation Mass Spectrometry. J Am Chem Soc. 2014;136(37):12920–12928. doi: 10.1021/ja505217w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cammarata MB, Brodbelt JS, Suzuki H, Shen S, Ruan J, Kurgan L, Nagai K, Olson JS, Kelleher NL, Brodbelt JS. Structural characterization of holo- and apo-myoglobin in the gas phase by ultraviolet photodissociation mass spectrometry. Chem Sci. 2015;6(2):1324–1333. doi: 10.1039/c4sc03200d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cammarata MB, Thyer R, Rosenberg J, Ellington A, Brodbelt JS. Structural Characterization of Dihydrofolate Reductase Complexes by Top-Down Ultraviolet Photodissociation Mass Spectrometry. J Am Chem Soc. 2015;137(28):9128–9135. doi: 10.1021/jacs.5b04628. [DOI] [PubMed] [Google Scholar]
- 46.Morrison LJ, Brodbelt JS, Botelho MM, Sawyer L, Ferreira ST, Polikarpov I, Chipot C, Skeel RD, Kale L, Schulten K. Charge site assignment in native proteins by ultraviolet photodissociation (UVPD) mass spectrometry. Analyst. 2016;141(1):166–176. doi: 10.1039/c5an01819f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Sheng Y, McGee W, Cammarata M, Holden D, Loo JA. Structural Characterization of Native Proteins and Protein Complexes by Electron Ionization Dissociation-Mass Spectrometry. Anal Chem. 2017;89(5):2731–2738. doi: 10.1021/acs.analchem.6b02377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.