

Summary
Acute infections are associated with a set of stereotypic behavioral responses, including anorexia, lethargy, and social withdrawal. Although these so called sickness behaviors are the most common and familiar symptoms of infections, their roles in host defense are largely unknown. Here we investigated the role of anorexia in models of bacterial and viral infections. We found that anorexia was protective while nutritional supplementation was detrimental in bacterial sepsis. Furthermore, glucose was necessary and sufficient for these effects. In contrast, nutritional supplementation protected against mortality from influenza infection and viral sepsis, while blocking glucose utilization was lethal. In both bacterial and viral models, these effects were largely independent of pathogen load and magnitude of inflammation. Instead, we identify opposing metabolic requirements tied to cellular stress adaptations critical for tolerance of differential inflammatory states.
Introduction
Sickness behaviors are a collection of prominent symptoms of acute illness that include anorexia, lethargy, fever, altered sleep patterns, depression, lack of grooming and social withdrawal. It has been long appreciated that sickness behaviors are motivational states, rather than a result of debilitation of physiological functions (Miller, 1964). Furthermore, sickness behaviors have been conceptualized as adaptive programs that promote survival of acute infections (Hart, 1988; Kluger et al., 1975). However, the mechanisms whereby different sickness behaviors contribute to survival remain largely unknown. This issue is particularly important because a popular treatment for acute infections is the use of non-steroidal anti-inflammatory drugs, which work primarily by suppressing the symptoms of sickness behaviors (Pecchi et al., 2009). Understanding the biology of sickness behaviors is also important for improved management of critically ill patients suffering from conditions with poorly defined pathogenesis, such as sepsis. Indeed, sepsis remains a largely intractable clinical problem with high mortality rates even in modern medical facilities (Angus and van der Poll, 2013). It is now increasingly appreciated that the mortality of sepsis can stem from at least three distinct phases of the disease: hemodynamic shock, multiple organ failure and prolonged immunosuppression (Hotchkiss et al., 2013). Furthermore, different forms of sepsis can result from bacterial, fungal and viral infections, each creating a distinct challenge and potentially requiring different management strategies.
Anorexia of infection is an aspect of sickness behavior that is conserved from humans to insects (Adamo, 2005), and much effort has been dedicated to understanding the role of nutrition in septic critical illness (Casaer and Van den Berghe, 2014). In the 1970s it was demonstrated that interfering with the anorexia induced by Listeria infection using caloric supplementation led to pronounced mortality in mice (Murray and Murray, 1979; Wing and Young, 1980). Importantly, Ayres et al found that in Drosophila, anorexia was beneficial in some but not all infections (Ayres and Schneider, 2009). Similarly, it was recently shown that the fasting metabolic state was critical to surviving bacterial sepsis (Feingold et al., 2012). The mechanism remains elusive, and clinically, the role of nutrition in managing patients with sepsis is unclear.
An important new paradigm that can help explain at least some benefits of sickness behavior is the notion of resistance and tolerance: survival of infections can be promoted by either reducing pathogen burden, or by increasing host tolerance to the damage of infection (Ayres and Schneider, 2012; Soares et al., 2014). During an infection, multiple physiological processes undergo dramatic alterations with often unknown rationale. Depending on the infection, these can include profound changes in metabolism and alterations in hepatic, renal and cardiovascular functions. In principle, these responses can be either beneficial, induced as part of a general defense program, or they can be unintended and detrimental but unavoidable consequences of infections. Furthermore, the physiological changes that are beneficial for host survival can contribute to elimination of pathogens (resistance) or to mitigation of tissue damage caused by infection (tolerance). These physiological responses are poorly understood aspects of infection biology, where most of the focus has been devoted to the immune response and microbial pathogenesis.
There are several examples where organismal metabolism has been shown to be regulated in order to deprive necessary substrates for pathogen viability as a strategy of enhancing host resistance (Liu et al., 2012; Nairz et al., 2015), but how organismal metabolic states, such as the fasted state of anorexia, contributes to host defense, is not understood. Indeed, the role of metabolic homeostasis during sepsis, which is directly impacted by nutritional status, is becoming increasingly recognized as critical in surviving sepsis in the clinical setting. In addition to the well-studied, but contentious role of glucose homeostasis in managing sepsis in the intensive care units (Investigators et al., 2009; van den Berghe et al., 2001), the largest proteomic and metabolomic screen of patients with sepsis to date identified fatty acid, glucose and beta-oxidation pathways as being discriminatory between survivors and non-survivors (Langley et al., 2013). Bacterial sepsis leads to a pro-lipolytic state, which affects the ability of target tissues to utilize glucose via glycolysis and alternative fuel sources such as ketone bodies (KB) and free fatty acids (FFA) via oxidative phosphorylation (Agwunobi et al., 2000). Growing evidence suggests that a shift from glucose to KB/FFA utilization is protective in bacterial sepsis and these studies largely rely on pharmacologic targeting of peroxisome proliferator-activated receptor alpha (PPARαα, the master regulator of the ketogenic program and fasting metabolism (Budd et al., 2007; Camara-Lemarroy et al., 2015). The role of substrate utilization in viral infections is even less understood (Greseth and Traktman, 2014). These metabolic changes are presumed to be protective but their mechanism of protection remains obscure.
Here we report that while nutritional supplementation increased mortality of Listeria monocytogenes infection, it protected against lethality of influenza virus infection. The causative component of food was determined to be glucose, and this effect was largely independent of inflammation or pathogen burden. To study the differential effects of glucose metabolism in bacterial and viral inflammation and sepsis generally, we utilized lipopolysaccharide (LPS) and Poly(I:C) models of sepsis, and found that while therapeutic blockade of glucose utilization with 2-deoxy-D-glucose (2DG) protected against LPS-mediated sepsis, it was uniformly lethal with Poly(I:C) sepsis. We found that whereas glucose was necessary for adaptation to and survival from the stress of anti-viral inflammation by preventing initiation of endoplasmic reticulum (ER) stress-mediated apoptotic pathways, glucose prevented adaptation to the stress of bacterial inflammation by inhibiting ketogenesis, which was necessary for limiting reactive oxygen species (ROS) induced by anti-bacterial inflammation. Our study elucidates how specific metabolic programs are coupled to different types of inflammation to regulate tolerance to inflammatory damage.
Results
Anorexia Protects Against L. monocytogenes Infection
To assess the role of anorexia in infection, we revisited the model of listeriosis used in prior studies (Murray and Murray, 1979; Wing and Young, 1980). We confirmed that upon infection with L. monocytogenes, mice exhibited a dose-dependent decrease in their food intake (Figure 1A), and that enteral supplementation by gavage of one kilocalorie twice daily (bis in die, BID) starting 8 hours post-infection uniformly killed L. monocytogenes infected mice (Figure 1B). The caloric content supplemented was one-fifth of healthy mouse daily food intake. Additionally, we found that gavage with an isocaloric isovolumetric amount of glucose alone was sufficient to cause 100% mortality in infected mice (Figure 1B). To exclude contributions from enteroendocrine incretin signaling, we injected glucose intraperitoneally (IP) at a dose iso-osmolar to PBS, which provided about 2% of normal daily caloric intake, and found that this was sufficient to recapitulate the lethal effects of enteral glucose (Figure 1C). To assess if glucose was necessary for lethality, we injected L. monocytogenes infected mice with 2DG and found that 2DG fully rescued mice from listeriosis-induced mortality (Figure 1C), consistent with previous work (Miller et al., 1998). Collectively, these data suggest that glucose is the component of food that is necessary and sufficient to mediate lethality in listeriosis when anorexia is blocked by force-feeding.
Figure 1. Glucose caloric supplementation during Listeria monocytogenes infection worsens survival, while 2DG promotes survival.

(A) Food consumption after infection with 5 × 104 and 5 × 105 CFU wild-type L. monocytogenes.
(B) Survival after infection with 5 × 104 L. monocytogenes. Mice were per os (PO) gavaged with Abbott Promote (Food), glucose, or PBS vehicle, and injected intraperitoneally (IP) with 2DG or PBS. PO PBS/IP PBS n=20, PO Food/IP PBS n= 15 (p=0.0011 vs PO PBS/IP PBS), PO Glucose/IP PBS n=19 (p=0.004 vs PO PBS/IP PBS), PO PBS/IP 2DG n=10 (p=0.0085 vs PO PBS/IP PBS).
(C-F) Mice were infected with 5 × 104 CFU L. monocytogenes, then treated with IP PBS, glucose, or 2DG. n=5/group.
(C) Survival after L. monocytogenes and indicated treatments. PBS vs Glucose p=0.0396, PBS vs 2DG p=0.0344, Glucose vs 2DG p=0.0017.
(D) Plasma IL-6 and IFNγ 24 and 48 hours after 5 × 104 L. monocytogenes infection. 24h plasma IL-6: PBS vs 2DG p=0.0005, Glucose vs 2DG p=0.0014; 24h plasma IFNγ: PBS vs Glucose, PBS vs 2DG, Glucose vs 2DG all p<0.0001. n=5/group.
(E) Listeria CFUs from spleen and liver 4 days post-infection.
(F) Flow cytometry analysis of CD45+ cells within the liver 4 days post-infection.
(G) CFU growth of L. monocytogenes after incubation in brain heart infusion broth with or without 15 mM 2DG for 18 hours.
(H) Bone marrow-derived macrophages (BMDM) were infected with 5 × 105 CFU of L. monocytogenes in the presence or absence of 15 mM 2DG for 24 hours. CFU of L. monocytogenes grown from the BMDM cell media supernatant and cell lysate.
Data are represented as mean ± SEM. *p<0.05, ***p<0.001, ****p<0.0001. See also Figure S1.
We found that glucose treatment did not significantly affect bacterial burden or immune infiltration, but did increase plasma interferon gamma (IFNɣ) 24 hours post-infection (Figure 1D-F and Figure S1). In contrast, 2DG-treated mice had significantly decreased bacterial load compared to controls (Figure 1E). The decreased bacterial burden was not due to heightened immune response, as plasma IFNɣ, plasma interleukin-6 (IL-6), and liver immune infiltrate were all decreased in 2DG-treated animals (Figure 1D-F and Figure S1). These data raised the possibility that 2DG was mediating clearance of L. monocytogenes through direct inhibition of bacterial growth. To address this, we tested the effect of 2DG on the growth of L. monocytogenes and on the antimicrobial activity of macrophages infected with L. monocytogenes. In both cases, 2DG administration did not affect bacterial growth (Figures 1G and 1H). Since the protective effect of anorexia during L. monocytogenes infection was neither mediated through conventional immune clearance mechanisms, nor was it due to inhibition of bacterial proliferation, we hypothesized that tissue protective mechanisms may be at play.
Inhibition of Glucose Utilization is Protective in Endotoxemia
To examine the role of tissue tolerance in mediating the protective effects of anorexia in bacterial sepsis, we used the LPS sepsis model, where mortality results entirely from a systemic inflammatory response. We found that gavaging mice with enteral nutrition starting one hour post-LPS injection led to significantly increased mortality, while fluid resuscitation improved survival (Figure 2A). To dissect the nutritional components that contribute to mortality, we gavaged mice with isocaloric isovolumetric amounts of glucose, olive oil, or casein and found that only glucose significantly increased mortality (Figure 2B). We then tested whether the effects of food intake on susceptibility to sepsis could be reversed with concurrent 2DG treatment. IP injection of 2DG concurrently with gavage of enteral nutrition in endotoxemic mice significantly improved survival (Figure 2C). As was the case in L. monocytogenes infection, IP injection of glucose was sufficient to uniformly kill endotoxemic mice while IP injection of 2DG was sufficient to fully rescue them (Figure 2D). To exclude contributions from carbohydrate-responsive element-binding protein (ChREBP) signaling, which would still be activated with 2DG, we utilized a glucose utilization inhibitor, D-mannoheptulose (DMH), which does not activate downstream ChREBP signaling (Li et al., 2010), and observed the same protective effects as 2DG (Figure S2A). Together, these data suggest that the component of nutritional intake that increased susceptibility to endotoxic shock was glucose and that inhibition of glucose utilization during sepsis was protective.
Figure 2. Glucose caloric supplementation during LPS sepsis worsens survival, while 2DG promotes survival.

(A and B) Survival after 15 mg/kg IP LPS and PO gavage with Abbott Promote (Food), glucose, olive oil, casein, or PBS vehicle. PO PBS n=10, PO Food n=10 (p=0.0002 vs PO PBS), PO Glucose n=8 (p<0.0001 vs PO PBS), no gavage n=10 (p=0.0679 vs PO PBS). Panel A is a subset of panel B, separated for clarity (the same PBS-treated, food-treated, and no gavage groups are shown in (A and B)).
(C) Survival after 15 mg/kg IP LPS and PO gavage with Food with IP injections of PBS or 2DG. p=0.0433
(D-F) Mice were given 15 mg/kg IP LPS, then treated with IP PBS, glucose, or 2DG.
(D) Survival after LPS and indicated treatments. IP PBS n=16, IP Glucose n=10 (p<0.0001 vs IP PBS), IP 2DG n=10 (p=0.01 vs IP PBS).
(E) Plasma TNFα and IL-6. (n=5-10/group)
(F) Liver mRNA expression 4 hours after LPS and treatment with PBS (LPS-PBS), glucose (LPS-glucose), and 2DG (LPS-2DG) compared to PBS alone. n=3-5/group.
Data are represented as mean ± SEM. See also Figure S2.
The advantage of the LPS sepsis model is that it isolates the inflammatory response, as opposed to direct pathogen toxicity, as the source of tissue damage. In this model, nutrients can affect survival by altering the magnitude of inflammatory response, or the tissue's ability to tolerate it. We found no difference in circulating levels of TNFα and IL-6 or hepatic expression of acute phase response genes in endotoxemic mice treated with PBS, glucose, or 2DG (Figure 2E,F). These findings suggest that glucose utilization does not affect the magnitude of the inflammatory response in endotoxic shock, but rather the ability of the tissues to tolerate inflammatory damage.
Glucose Utilization Promotes Tissue Damage in Endotoxemia
We observed that mice challenged with LPS and glucose displayed symptoms consistent with tonic-clonic seizure. Prior to death, animals would develop high-amplitude convulsions followed by decerebrate posturing. This is consistent with studies which have demonstrated neurologic deficits, including seizure, and neuronal apoptosis in animals suffering from endotoxic shock (Sayyah et al., 2003; Singer et al., 2016; Song et al., 2014). Hypoglycemia is a common cause of seizure, but we found that blood glucose did not differ significantly between groups over time (Figure S2B). We performed vital sign monitoring (body temperature, blood O2 saturation, respiratory rate, and heart rate) in endotoxemic mice treated with PBS, glucose, or 2DG 24 hours post-LPS injection, and found that 2DG-treated mice maintained their body temperature significantly better than glucose and PBS treated mice (Figure S2C). To assess other evidence of end-organ damage, we measured plasma markers of tissue injury (troponin-I, alanine aminotransferase, and creatinine), which were largely unchanged between treatments, with the exception that glucose treated mice had higher levels of plasma creatinine (Figure S2D). Finally, detailed histopathololgic analysis of hematoxylin & eosin (H&E) stained slides of brain, heart, lung, liver, kidney, pancreas, stomach, bone marrow, thymus, and spleen was performed to identify any pathologic changes including edema, hemorrhage, inflammation, necrosis, and apoptosis. The only difference in histopathologic changes was a decreased presence of dark, shrunken neurons in the brains of LPS mice given 2DG, compared to mice given LPS and PBS or glucose. Together, these data implicate neuronal dysfunction as a possible proximal cause of death in LPS endotoxemia.
Inhibition of Glucose Utilization is Lethal in Influenza Infection
We next asked whether viral infections, which induce a different type of immune response compared to bacterial infections, were also affected by caloric supplementation. We employed an influenza model in which mice are infected intranasally with influenza virus A/WSN/33. We observed that mice infected with influenza also exhibited anorexia, albeit less severely than in L. moncytogenes infection (Figure 3A). Surprisingly, we found that gavage of one kilocalorie BID of enteral nutrition starting eight hours post infection protected mice from influenza-associated mortality (Figure 3B). Gavage of isocaloric isovolumetric glucose partially recapitulated the effect of enteral nutrition, while IP injection of 2DG concurrently with feeding completely ablated the survival benefit (Figure 3C). Caloric supplementation with casein and olive oil provided little to no survival benefit (Figure S3A). With a lower dose of influenza infection, we found that 2DG alone was able to uniformly kill flu-infected mice compared to vehicle control (Figure 3D). These data together indicate that glucose availability and utilization are critical to surviving influenza infection.
Figure 3. Caloric supplementation and glucose utilization is required for surviving influenza infection.
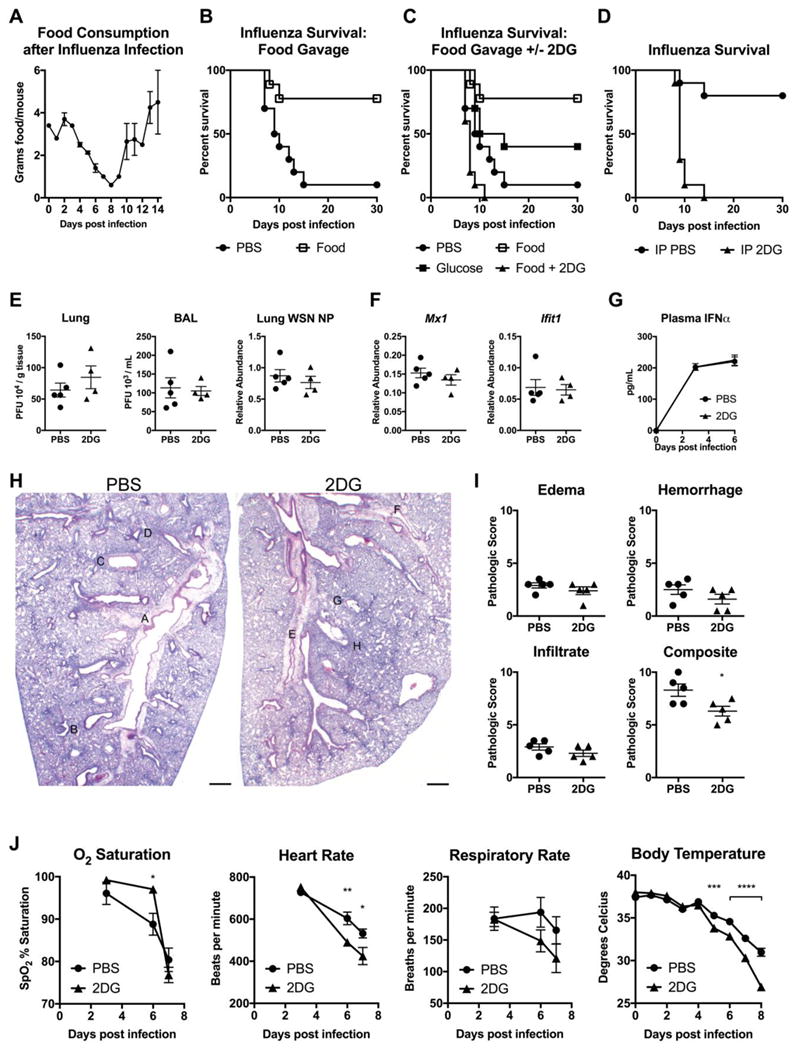
(A) Food consumption after infection with 450 plaque forming units (PFU) of Influenza strain A/WSN/33.
(B-C) Survival after infection with 800 PFU of influenza virus. Mice were gavaged with Abbott Promote (Food), glucose, or PBS vehicle. Mice gavaged with Food were also injected with IP PBS (Food) or 2DG (Food+2DG). PBS vs Food p=0.0047, PBS vs Glucose p=0.1058, Food vs Food+2DG p=0.0001, PBS vs Food+2DG p=0.0256. n=10/group. Panel B is a subset of panel C, separated for clarity (the same PBS-treated and food-treated groups are shown in (B and C)).
(D) Survival after infection with 375 PFU of influenza virus, and treatment with IP PBS or 2DG. p<0.0001, n=10/group.
(E-J) Mice were infected with 375 PFU influenza virus, and treated with IP PBS or 2DG. n=4-5/group
(E) Lung and broncho-alveolar lavage (BAL) viral load 6 days after influenza infection by PFU and quantitative PCR for WSN nucleoprotein (NP).
(F) mRNA expression of whole lung tissue at day 6.
(G) Plasma IFNα measured by ELISA.
(H and I) H&E staining of lung tissue 6 days post-infection and histologic scoring. Scale bar = 500 μm. For magnified views of insets please see Figure S3E.
(J) Vital signs after influenza infection.
Data are represented as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. See also Figure S3.
Next we assessed whether the effect of caloric supplementation on influenza infection was mediated through immune resistance of virus or tissue tolerance. Differences in viral burden and/or immune activation would implicate an effect of caloric supplementation on host resistance. Six days post-infection, we performed plaque assays using both lung homogenate and bronchoalveolar lavage fluid (BAL) from mice treated with PBS or 2DG. Additionally, we assayed gene expression of the influenza gene NP from lung tissue homogenate. In all three cases, we found no differences in viral load between groups (Figure 3E). We then assayed antiviral inflammatory mediators to determine if immunopathology could account for the lethality caused by 2DG treatment. We found that expression of interferon inducible genes as well as Cxcl1 and Il6 were similar between PBS and 2DG-treated groups (Figures 3F and S3B). Likewise, we found no difference in plasma IFNα levels or immune cell infiltration into the lung after infection (Figure 3G and Figure S3C).
Mortality from influenza infection is often linked to development of pneumonia (Taubenberger and Morens, 2008). To determine if 2DG impacted the extent of lung damage, we assessed the pathological outcomes of PBS versus 2DG treatment in influenza infection. Histopathologic examination of lung showed no difference in edema, hemorrhage, or inflammatory cell infiltrates (Figures 3H, 3I, and Figure S3D). To identify alternative causes of death, we assayed vital signs of PBS or 2DG-treated mice over the course of influenza infection. We found that 2DG-treated mice had no difference in blood O2 level, but had decreased heart rate, respiratory rate, and body temperature (Figure 3J). These findings are consistent with a derangement of central autonomic control. To verify that 2DG was not itself causing neuronal dysfunction and lethality, we administered the identical 2DG regimen utilized in influenza to mice infected with another pulmonary pathogen, Legionella pneumophila, and this did not cause mortality, indicating that the lethal effects of 2DG occurred only in the context of the viral inflammation induced by influenza infection (Figure S3E). Collectively, this suggests that the effect of caloric supplementation on influenza infection is mediated through availability of glucose utilization and its impact on tissue tolerance mechanisms, which are likely impaired in the brain.
Inhibition of Glucose Utilization is Lethal in Viral Inflammation
To generalize the findings from influenza infection, we next utilized intravenous Poly(I:C) injection as a model of systemic viral inflammation (Smorodintsev et al., 1978). Co-administration of Poly(I:C) and 2DG or DMH was uniformly lethal (Figure 4A and S4A). Since we were unable to generate a dose of Poly(I:C) that caused lethality by itself, we were unable to assess if glucose supplementation would be protective. To test if type I IFN was required for the effect of 2DG on Poly(I:C)-induced inflammation, we subjected IFNα-receptor (IFNaR) deficient (Ifnar1-/-) mice to Poly(I:C) and 2DG challenge. Ifnar1-/- mice were completely protected (Figure 4B), indicating that IFNaR signaling was required for mediating the lethal effects of 2DG.
Figure 4. Inhibition of glucose utilization is lethal in Poly(I:C) inflammation.

(A) Survival of mice after Poly(I:C) challenge and treatment with either IP PBS, glucose, or 2DG. IP PBS n=15, IP Glucose n=10 (p=0.2207 vs IP PBS), and IP 2DG n=15 (p<0.0001 vs IP PBS).
(B) Survival of B6 wild-type (WT) mice and Ifnar-/- mice after Poly(I:C) challenge and treatment with IP PBS, glucose, or 2DG. B6 vs Ifnar-/- p=0.0027, n=5/group.
(C-D) Mice were challenged with Poly(I:C), then treated with either IP PBS, glucose, or 2DG.
(C) Plasma IFNα. n=5/group.
(D) Vital signs measured 18 hours after Poly(I:C) administration. n=3-7/group.
(E) Averaged brain PET images after PBS vehicle (baseline), LPS, and Poly(I:C) administration. “A” is the brainstem and “B” is the hypothalamus. Anatomic atlas of regions-of-interest (ROI) and T2-weighted magnetic resonance images provided for reference. CT, computed tomography. n=3/group.
Data are represented as mean ± SEM. **p<0.01, ***p<0.001, ****p<0.0001. See also Figure S4.
To examine if the lethal effects of 2DG were mediated by differences in the magnitude of the inflammatory response, we assessed plasma cytokines and did not find significant differences in circulating IFNα (Figure 4C). Histopathologic analyses showed no differences between treatment groups. To identify the cause of mortality, we performed vital sign monitoring and found that mice challenged with Poly(I:C) and 2DG, like flu-infected mice, exhibited profound defects in the control of body temperature, respiratory and heart rate, but not oxygen saturation (Figure 4D). Thus, we reasoned that neuronal dysfunction and loss of autonomic control was responsible for mortality.
LPS and Poly(I:C) induced inflammation cause differential glucose uptake in the brain
To globally assess 2DG and glucose uptake and distribution following Poly(I:C) or LPS challenge, we subjected mice to 2-deoxy-2-[18F] fluorodeoxy-D-glucose-positron emission tomography-computed tomography (18F-FDG-PET-CT). We found that glucose was actively taken up by the brainstem after Poly(I:C), but not with LPS challenge. In contrast, LPS induced more glucose uptake in the hypothalamic area (Figure 4E). There were no other differences in glucose compartmentalization between Poly(I:C) and LPS (Figure S4B). Increased 2DG uptake in the hindbrain in Poly(I:C)-treated mice was associated with decreased levels of Il1b, Il6, and Tnfa and no difference in Mx1 in the hindbrain, indicating that neuronal dysfunction in brainstem was not due to increased inflammation (Figure S4C). Consistent with the PET data, we did not detect differences in blood glucose or measures of cardiac, liver, or renal dysfunction (Figure S4D).
These data suggest that the lethal effect of Poly(I:C) and 2DG co-administration was likely independent of the magnitude of inflammation, but rather was dependent on tissue tolerance to immunopathology in the hindbrain, downstream of IFNaR signaling.
Lethal effect of 2DG in viral inflammation is mediated by CHOP
Because we found that mice displayed signs of neuronal damage in both influenza infection and Poly(I:C)-induced viral inflammation, we further investigated the effect of inhibition of glucose utilization in viral inflammation. We reasoned that ER-stress mediated apoptotic pathways, which are integral to the cellular response to viral infection, might link viral inflammation to neuronal damage (Lin et al., 2008). In particular, we focused on the ER-stress induced transcription factor CHOP, which can induce apoptosis upon prolonged or excessive activation (Tabas and Ron, 2011). We found that expression of CHOP protein and its target gene Gadd34 was elevated in the hindbrains of mice treated with Poly(I:C) and 2DG, an effect that was IFNaR dependent (Figure 5A and 5B). Moreover, CHOP-deficient mice (Ddit3-/- mice) were completely protected from Poly(I:C) and 2DG challenge in a manner independent of inflammatory magnitude (Figures 5C and 5D). Autonomic dysfunction was also completely abrogated in CHOP-deficient mice challenged with Poly(I:C) and 2DG (Figure 5E). To test the contribution of CHOP to host tolerance and resistance, we utilized the influenza infection model. We found that CHOP-deficient mice were significantly protected from influenza and 2DG challenge in a manner independent of pathogen burden or degree of inflammation (Figure 5F-H and Figure S5A). This is in contrast to models of bacterial inflammation where CHOP deficiency promotes tissue injury and morbidity (Esposito et al., 2013). Interestingly, 2DG and IFNα together led to sustained and elevated CHOP expression and apoptosis in MEFs (Figure S5B and S5C). These data together suggest that glucose utilization is critical to tissue tolerance of virally-induced inflammation through maintenance of an appropriate ER stress response.
Figure 5. Inhibition of glucose utilization in Poly(I:C)-induced inflammation enhances ER stress.

(A) B6 WT and Ifnar-/- mice were challenged with Poly(I:C), then treated with either IP PBS or 2DG. Whole hindbrain lysates 24 hours after Poly(I:C) treatment immunoblotted for CHOP and β-Tubulin.
(B) Hindbrain mRNA expression of Gadd34 18 hours after Poly(I:C) challenge and treatment with IP PBS, glucose, or 2DG in WT mice. n=5/group (one death in Poly(I:C) group).
(C) Survival of B6 WT mice and Ddit3-/- mice after Poly(I:C) challenge and treatment with IP PBS or 2DG. WT vs Ddit3-/- p=0.0015 WT: Poly(I:C) + 2DG vs Ddit3-/-: Poly(I:C) + 2DG, n=5/group.
(D and E) B6 WT and Ddit3-/- mice were challenged with Poly(I:C) and treated with 2DG.
(D) Plasma IFNα after Poly(I:C) challenge and treatment with 2DG. n=5/group.
(E) Vital signs measured 18 hours after Poly(I:C) + 2DG. n=3-7/group (vital sign values of B6 WT mice same as those in Figure 4D)
(F-H) B6 WT mice and Ddit3-/- mice were infected with 700 PFU of influenza virus and treated with 2DG.
(F) Survival after influenza infection and treatment with 2DG. p=0.048 n=5/group (G) Plasma IFNα after influenza infection and treatment with 2DG. n=5/group.
(H) Lung and BAL viral load 5 days post-infection. n=5/group.
Data are represented as mean ± SEM. **p<0.01, ***p<0.001. See also Figure S5.
Glucose Promotes Neuronal ROS and Seizure-induced Death in Bacterial Inflammation
Prolonged fasting results in hypoglycemia accompanied by lipolysis and followed by ketogenesis. In anorexia following acute LPS challenge, there was a decrease in blood glucose and an increase in plasma FFA, plasma beta-hydroxybutyrate (BHOB), and the fasting hormone FGF21 (Figure 6A and S2B). The switch to this fasting metabolic profile was ablated by glucose administration (Figure 6A). There was no appreciable difference in the kinetics of blood glucose subsequent to treatment with glucose or 2DG (Figure S2B).
Figure 6. Role of ketogenic program in surviving bacterial, but not viral inflammation.

(A) Plasma nonesterified fatty acids (NEFA), beta-hydroxybutyrate (BHOB), and fibroblast growth factor 21 (FGF21) after LPS, with treatment with IP PBS or glucose. n=10/group.
(B) Survival after 8 mg/kg IP LPS and treatment with glucose. Mice were treated with vehicle, valproic acid (VA) or levetiracetam (Keppra) starting 6 hours after LPS. p<0.0001 for Glucose+VA vs Glucose and Glucose+VA vs Glucose+Keppra. n=15 Glucose, n=15 Glucose+VA, n=4 Glucose+Keppra.
(C) Survival after 12.5 mg/kg IP LPS in B6 WT, Fgf21-/- and Ppara-/- mice. n=10/group. p<0.0001 for WT vs Fgf21-/-; p=0.0003 for WT vs Ppara-/-.
(D) Plasma BHOB after 12.5 mg/kg IP LPS in WT, Fgf21-/- and Ppara-/- mice. n=4-6/group.
(E) Plasma TNFα and IL-6 in B6 WT, Fgf21-/-, and Ppara-/- mice after LPS. n=5/group *p<0.05.
(F) Survival after 12.5 mg/kg IP LPS in Fgf21-/- and Ppara-/- mice, treated with i.v. 5 ng recombinant mouse FGF21 (rmFGF21) twice daily starting 6 hours after LPS injection. n=5-6/group. p=0.0491 Fgf21-/- VEH vs Fgf21-/- rmFGF21. p=0.5767 Ppara-/- VEH vs Ppara-/- rmFGF21.
(G) Survival after 8 mg/kg IP LPS in B6 WT and Ppara-/- mice. 2DG treatment was initiated one hour after LPS. Valproic acid (VA) was initiated 6 hours after LPS. n=4-5/group. p=0.0177 Ppara-/- VEH vs Ppara-/- VA.
(H-I) WT and Ppara-/- mice were infected with 400 PFU of influenza virus.
(H) Survival after influenza infection. p=0.0074 WT n=6, Ppara-/- n=8, representative of three independent experiments.
(I) Lung and BAL viral load 5 days post-infection. n=6-7/group.
Data are represented as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. See also Figures S6 and S7.
We observed that the administration of glucose to LPS-challenged mice potentiated seizures, implicating neurotoxicity as a mechanism for death. We therefore asked if anti-epileptic drugs would be sufficient to rescue glucose-mediated death, and found that administration of valproic acid (VA), but not levetiracetam (two commonly used anti-epileptic agents) was able to completely rescue LPS-challenged mice treated with glucose (Figure 6B). The anti-epileptic effects of valproic acid are incompletely understood but appear to impact HDAC-inhibition (HDACi), GABA transduction, and PI3K and calcium handling (Terbach and Williams, 2009). Ketone bodies have also been implicated as HDACi of the same class as VA, and have recently been shown to coordinate gene expression programs that confer resistance to ROS-mediated damage (Shimazu et al., 2013). We thus hypothesized that the suppression of ketone bodies by glucose administration may be inhibiting these HDACi-mediated ROS adaptation pathways. To test this, we utilized dihydroethidium staining to measure ROS in situ in the brains of LPS-challenged mice treated with glucose, and observed increased ROS in the brains of these mice (Figure S6A). We also observed more TUNEL-positive nuclei in sections of mouse brain in glucose-treated mice compared to 2DG- or PBS-treated mice challenged with LPS (Figure S6B). All groups had TUNEL-positive nuclei in areas of lymphoid cell death (thymus, spleen), but there were no TUNEL-positive nuclei in any other tissues surveyed (heart, lung, liver, and kidney). The brain was the only tissue where differences in TUNEL-positive nuclei were seen between groups (Figure S6C and D); however, the amount of cell death, which was assessed in the hypothalamus – given PET localization – was not dramatically different, indicating that cellular dysfunction, and not necessarily cell death, was being potentiated by glucose administration. Together, these data suggest that the enhanced lethality caused by glucose supplementation in endotoxemia is likely mediated through increased ROS and neuronal dysfunction.
Inhibition of the Ketogenic Program in Bacterial Inflammation, but not Viral Inflammation, Results in Mortality
To test the role of ketogenesis in bacterial and viral inflammation, we subjected mice deficient in PPARα and FGF21 to LPS or influenza infection. Both PPARα and FGF21-deficient mice displayed enhanced mortality after LPS administration (Figure 6C). We verified that PPARα-deficient mice have severely impaired ketogenesis following LPS challenge, and did not observe significant changes in the level of BHOB in FGF21-deficient animals, consistent with findings observed in the fasting state (Potthoff et al., 2009) (Figure 6D). We also did not detect an increase in systemic cytokines in PPARα deficient mice (Figure 6E). We hypothesized that either the lack of FGF21 or the lack of alternative fuel sources, which were both suppressed after glucose supplementation, was the cause of mortality. Since FGF21 is a known downstream target of PPARα (Feingold et al., 2012; Inagaki et al.), we tested if defective FGF21 production was the causative lesion in PPARα-deficiency. We reconstituted PPARα-deficient and FGF21-deficient mice with recombinant FGF21 and found that while FGF21 was sufficient to rescue FGF21-deficient mice, it was not sufficient to rescue PPARα-deficient mice (Figure 6F), arguing that other aspects of the fasting program were necessary to mediate survival of LPS sepsis. Finally, VA, but not 2DG, was able to rescue PPARα mice challenged with LPS (Figure 6G), indicating that some aspect of VA action – likely its HDACi activity – was sufficient to rescue the lack of ketone bodies in PPARα mice, and also that the protective effects of 2DG required an intact ketogenic program.
Since ketotic pre-conditioning has been shown to improve other neurologic conditions such as epilepsy (Levy et al., 2012), we tested whether it would improve survival in sepsis. We found that mice pre-fasted for 24 hours or mice fed ketogenic diets for 3 days displayed enhanced sensitivity to LPS despite generating adequate levels of ketone bodies (Figure S7A and B). We excluded the possibility that ketoacidosis was driving death (Figure S7C). These data indicate that the activation of the ketogenic program must be temporally coupled to the course of the inflammatory challenge.
We hypothesized that, since our viral and bacterial models had opposite dependencies on glucose, they would also have opposite dependencies on ketogenesis. Therefore, we subjected PPARα-deficient mice to influenza challenge. Whereas PPARα deficiency was lethal following LPS challenge, it was protective in influenza infection in a manner independent of pathogen control (Figure 6H and 6I). This protective effect was not observed in FGF21-deficient animals (Figure S7D). Together, these data show that whereas impairment of ketogenesis, whether through genetic deletion of PPARα or glucose administration, was lethal in bacterial inflammation, it was protective in viral inflammation, in a manner independent of the magnitude of inflammation.
Discussion
Here we addressed the effect of anorexia during acute infection, and uncovered a surprising differential role for fasting metabolism in maintaining tissue tolerance in different infectious states. It is increasingly appreciated that inflammatory responses must be coupled to specific metabolic programs to support their energetic demands (Buck et al., 2015; Galvan-Pena and O'Neill, 2014). In this study, we observed that systemic metabolism appears to be coordinated to support tolerance to different inflammatory states. We found that whereas glucose utilization was required for survival in models of viral inflammation, it was lethal in models of bacterial inflammation. Concordantly, we found that whereas ketogenesis was required for survival in bacterial inflammation, it was dispensable in the case of viral inflammation. Unexpectedly, we found that these effects on mortality were largely independent of the degree of inflammation and pathogen clearance. In the case of viral inflammation, lethality subsequent to inhibition of glucose utilization appeared to be mediated by type I IFN signaling on target tissues – likely the brain – which require glucose to mitigate the ER stress response and CHOP-mediated cellular dysfunction. In the case of bacterial inflammation, lethality subsequent to glucose administration appeared to be mediated by suppression of ketogenesis, which lead to impaired resistance to ROS-mediated damage in the brain (Figure 7). Thus, our results suggest that distinct inflammatory responses may be coupled with specific metabolic programs in order to support unique tissue tolerance mechanisms, that, when uncoupled, lead to enhanced immunopathology leading to death.
Figure 7. Model of glucose utilization during viral and bacterial-mediated inflammation supporting unique tissue tolerance mechanisms.

(A) Glucose inhibits PPARα-dependent ketogenesis and cellular adaptation programs required for tissue tolerance during LPS and Listeria-mediated (bacterial) inflammation. Ketones act as fuel source and as HDACi allowing for cellular and tissue adaptation. Inhibition of glucose utilization during bacterial inflammation with 2DG protects against tissue dysfunction and organismal mortality.
(B) Glucose utilization is required for adaptation to Poly(I:C) and influenza mediated (viral) sepsis. Viral inflammation activates ER stress and the unfolded protein response (UPR) downstream of type I interferon signaling through IFNaR. Inhibition of glucose utilization in viral inflammation with 2DG enhances ER stress through a CHOP-dependent pathway leading to tissue dysfunction and death.
Host defense from infections involves both resistance and tolerance mechanisms. Whereas host resistance promotes pathogen clearance, host tolerance relies on adaptation to a given level of pathogen or a given level of inflammatory response (Raberg et al., 2007; Schneider and Ayres, 2008). Disease morbidity and mortality can be a result of either inadequate or impaired host resistance, characterized by high pathogen burden, or impaired host tolerance. Immunopathology falls into the latter category, and insufficient tissue protection is likely to be an important determinant in conditions characterized by excessive inflammation, such as sepsis. Tissue protection is likely a function of cellular stress adaptation pathways, which allow cells to survive noxious states such as increased free radicals and accumulation of unfolded proteins (Figueiredo et al., 2013; Larsen et al., 2010). When these adaptation pathways are overwhelmed, cells can undergo apoptotic cell death (Boison, 2013; Tabas and Ron, 2011). Thus, one important determinant of host tolerance may be related to the ability of cellular adaptation programs to tolerate noxious states found in infections (Medzhitov et al., 2012). Because different infections generate different inflammatory responses and noxious states, specific cellular adaption programs would need to be activated in distinct infectious contexts.
We found that interfering with the normal ketogenic state following LPS-mediated inflammation was lethal, likely by interfering with ROS adaption programs in the brain. Our findings are consistent with observations that PPARα agonism and inhibition of glucose utilization are generally protective in bacterial sepsis models (Budd et al., 2007; Camara-Lemarroy et al., 2015; Yoo et al., 2013). However, unlike many of these studies, we did not observe large differences in the magnitude of inflammation, likely because we administered 2DG and glucose after and not before infectious or inflammatory challenge. Thus, our observations are likely unrelated to the body of literature that supports a role for HIF1α, PKM2, and aerobic glycolysis in generating the LPS inflammatory response (Liu et al., 2016; Yang et al., 2014). Consistent with our inability to detect differences in inflammation in sterile inflammatory models, we did not detect differences in pathogen burden in live infection models where glucose administration led to lethality in L. monocytogenes infection in the absence of increased pathogen burden or bolstered immune response.
ROS-mediated cytotoxicity is a well-appreciated phenomenon in bacterial sepsis (Hoetzenecker et al., 2012; Kolls, 2006), and ROS-detoxification pathways have been implicated in mitigating tissue damage and mortality. Shimazu et al recently reported that BHOB functioned as an HDAC-1 inhibitor, and that this lead to transcription of ROS-detoxification pathways (Shimazu et al., 2013). Furthermore, we found that the timing of ketogenesis, an adequately nourished host, or both, are necessary for the protective effect of fasting that occurs as a coordinated response to bacterial inflammation. Instead of rescuing mice from LPS mortality, fasting and ketogenic pre-conditioning potentiated death. Taken together, we present evidence that the fasting response that occurs as part of the inflammatory response is required to maintain resistance to oxidative stress in LPS sepsis.
Although anorexia is a common response in both bacterial and viral infections, we find the opposite consequence of fasting metabolism in our models of bacterial and viral inflammation. Whereas in bacterial infection and LPS-induced inflammation we found a detrimental effect of glucose, a protective effect of 2DG, and a requirement for ketogenesis in order to maintain tolerance, in viral infection and Poly(I:C)-induced inflammation these effects were opposite. 2DG administration led to lethality in Poly(I:C)-induced inflammation in a manner that was independent of the magnitude of IFNα expression, but dependent on IFNaR signaling. Manipulation of fasting metabolism did not affect viral burden in influenza infection. Viral infections are known to stimulate the unfolded protein response mediated, in part, via the PERK-eIF2a-ATF4-CHOP pathway (Janssens et al., 2014). When this pathway is engaged, cells can either adapt to ER stress, or induce apoptosis through CHOP if ER stress cannot be managed (Tabas and Ron, 2011). Our data suggests that glucose utilization is required for the cytoprotective response in neurons in the setting of viral inflammation, as inhibition of glucose utilization lead to death, which was CHOP-dependent. Indeed, our PET studies indicate that glucose redistribution is largely the same between the early phases of bacterial and viral inflammatory challenge with the exception of sub-regions in the brain, where LPS and Poly(I:C) appear to regulate glucose uptake differentially. The purpose and mechanism underlying this observation remains to be elucidated. The precise mechanism whereby IFN signaling converges with glucose utilization programs also remains to be fully resolved, but recent studies demonstrated that interferon signaling leads to changes in glucose uptake, which is important for the antiviral response (Burke et al., 2014). Thus, whereas alternative fuel substrate availability is coupled to and necessary for adaptation to bacterial inflammation, glucose availability is coupled to and necessary for cellular adaptation to viral inflammation. The logic of their coupling is likely related to the substrate-dependence of the cellular adaptation programs that are engaged. These findings are consistent with our previous study where we found that synergistic lethality in mice co-infected with influenza and Legionella occurred in a manner independent of pathogen burden (Jamieson et al., 2013), and it is interesting to speculate here that perhaps one cause of lethality in this co-infection model is a result of metabolic incompatibility in the setting of both a viral and bacterial infection.
Given the conservation of cellular adaptation and metabolic programs in mouse and human, our findings likely have clinical implications. The role of nutrition in managing patients with sepsis is unclear at best, and multiple studies have failed to show differences in survival from feeding, including the most recent study which asked if lower caloric supplementation would improve outcomes (Arabi et al., 2015). There have been a series of studies exploring different feeding formulations with different caloric or micronutrient contents (Casaer and Van den Berghe, 2014). However, we could not find an example where different feeding formulations were targeted to different types of infections, as opposed to different types of organ failure (Seron-Arbeloa et al., 2013), or where post-hoc analyses was directed at pathogen class. Our study implicates a differential need for metabolic fuels as a function of infection (or inflammation) class, and sheds light on the biology behind the old adage “starve a fever, stuff a cold.” Much work will need to be done to identify how organismal metabolism is coordinated in other infectious and inflammatory states, and whether or not these findings can be extended to humans in the management of inflammatory diseases and critical illness.
Limitations, Caveats and Open Questions
One limitation of this study (common to most animal studies) is that it was performed on a single mouse strain (C57BL/6J) in one mouse facility. Thus, the roles of genetic background and facility-specific environment remain unknown. In addition, there are many caveats with extrapolating data on organismal biology from studies in unnatural settings of animal facilities. There are also many open questions raised by this study, including: why are different brain regions differentially affected by bacterial and viral inflammation? What are intrinsic properties of the affected neurons that make them susceptible to damage depending on inflammatory pathway and available metabolic fuels? Finally, it remains to be seen how these results apply to critical illness in humans.
Methods and Resources
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-β Tubulin | Santa Cruz | Cat# sc-9104; RRID: AB_2241191 |
| Mouse monoclonal anti-GADD153 (clone B-3) | Santa Cruz | Cat# sc-7351; RRID: AB_627411 |
| Hamster monoclonal anti-TNF alpha (clone TN3-19.12) | eBioscience | Cat# 14-7423; RRID: AB_468492 |
| Rat monoclonal anti-IL-6 (clone MP5-20F3) | eBioscience | Cat# 14-7061; RRID: AB_468423 |
| Biotin-conjugated rabbit polyclonal anti-TNF alpha | eBioscience | Cat# 13-7341; RRID: AB_466951 |
| Biotin-conjugated rat monoclonal anti-IL-6 (clone MP5-32C11) | BD | Cat# 554402; RRID: AB_395368 |
| Anti-mouse CD4 FITC (clone GK1.5) | eBioscience | Cat# 11-0041; RRID: AB_464893 |
| Anti-mouse CD4 BV421 (clone GK1.5) | BioLegend | Cat# 100438; RRID: AB_11203718 |
| Anti-mouse CD11c FITC (clone N418) | eBioscience | Cat# 11-0114; RRID: AB_464941 |
| Anti-mouse B220 PE (clone RA3-6B2) | eBioscience | Cat# 12-0452; RRID: AB_465673 |
| Anti-mouse Ly6G PE-Cy7 (clone RB6-8C5) | eBioscience | Cat# 25-5931; RRID: AB_469663 |
| Anti-mouse CD45 APC-eFluor 780 (clone 30-F11) | eBioscience | Cat# 47-0451; RRID: AB_1548781 |
| Anti-mouse NK1.1 PE (clone PK136) | eBioscience | Cat# 12-5941; RRID: AB_466051 |
| Anti-mouse CD11b BV421 (clone M1/70) | eBioscience | Cat# 12-0112; RRID: AB_465548 |
| Anti-mouse TCR beta FITC (clone H57-597) | eBioscience | Cat# 11-5961; RRID: AB_465324 |
| Anti-mouse Ly6C PE (clone AL-21) | BD | Cat# 560592; RRID: AB_1727556 |
| Anti-mouse CD8a APC (clone 53-6.7) | eBioscience | Cat# 17-0081; RRID: AB_469335 |
| Anti-mouse F4/80 PE-Cy5 (clone BM8) | BioLegend | Cat# 123112; RRID: AB_893482 |
| Anti-mouse CD16/32 (clone 93) | eBioscience | Cat# 16-0161; RRID: AB_468900 |
| Annexin V FITC | eBioscience | Cat# BMS500FI/300, RRID: AB_2575600 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-deoxy-D-glucose | Sigma-Aldrich | Cat# G8270; CAS: 154-17-6 |
| D-mannoheptulose | Cayman Chemical | Cat# 16548; CAS: 3615-44-9 |
| Valproic acid | Sigma-Aldrich | Cat# P4543; CAS: 1069-66-5 |
| Levetiracetam | Sigma-Aldrich | Cat# L8668; CAS: 102767-28-2 |
| Mouse recombinant FGF21 | R&D Systems | Cat# 8409-FG/CF; GenPept: Q9JJN1 |
| Lipopolysaccharide from E. coli strain 055:B5 | Sigma-Aldrich | Cat# L2880 |
| Poly(I:C) | Invivogen | Cat# tlrl-pic-5 |
| BHI broth | BD | Cat# 241830 |
| Thapsigargin | Cayman Chemical | Cat# 10522; CAS: 67526-95-8 |
| Mouse recombinant IFNα | R&D Systems | Cat# 12100-1; GenPept: NP_996753 |
| Dihydroethidium | Thermo Fisher Scientific | Cat# D1306; CAS: 28718-90-3 |
| DAPI | Thermo Fisher Scientific | Cat# D1168; CAS: 38483-26-0 |
| Propidium iodide | eBioscience | Cat# BMS500FI/300 |
| N-PER Neuronal Protein Extraction Reagent | Thermo Fisher Scientific | Cat# 87792 |
| RNA-Bee | Tel Test, Inc | Cat# CS-501B |
| SMART MMLV Reverse Transcriptase | Clontech | Cat# 639524 |
| Low ROX PerfeCTa SYBR Green SuperMix | Quanta | Cat# 95056 |
| Ethidium monoazide bromide | Biotium | Cat# 40015 |
| Critical Commercial Assays | ||
| Mouse IFNα ELISA kit | eBioscience | Cat# BMS6027 |
| Mouse IL-1β ELISA kit | eBioscience | Cat# 88-7013 |
| Mouse Cardiac Troponin-I ELISA kit | Life Diagnostics | Cat# CTNI-1-HSP |
| Alanine Transaminase Activity Assay Kit | Cayman Chemical | Cat# 700260 |
| Non-esterified Fatty Acid Assay Reagent | Wako Diagnostics | Cat# 999-34691, Cat# 995-34791, Cat# 991-34891, Cat# 993-35191 |
| β-hydroxybutyrate Assay Kit | Cayman Chemical | Cat# 700190 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| i-STAT CG4+ Cartridge | Abaxis Global Diagnostics | Cat# 600-9000 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No. 000664 |
| Mouse: CHOP KO: B6.129S(Cg)-Ddit3tm2.1Dron/J | The Jackson Laboratory | Stock No. 005530 |
| Mouse: PPARα B6;129S4-Pparatm1Gonz/J | The Jackson Laboratory | Stock No. 008154 |
| Mouse: IFNaR KO: B6.129S2-Ifnar1tm1Agt/Mmjax | The Jackson Laboratory | Stock No. 32045 |
| Mouse: FGF21 KO | David J. Mangelsdorf (Potthoff et al., 2009) | RRID: MGI_4354176 |
| Listeria monocytogenes: Strain 10403s | Daniel A. Portnoy (Auerbuch et al., 2004) | |
| Influenza: Strain A/WSN/33 | Akiko Iwasaki (Lund et al., 2004) | |
| Legionella pneumophila: serogroup 1 strain JR32 | Craig R. Roy (Archer and Roy, 2006) | |
| Sequence-based Reagents | ||
| Primers for qPCR, see Table S1 | This paper | |
| Software and Algorithms | ||
| Inveon Research Workplace | Siemens Healthineers | |
| IDL 8.0 | Exelis Visual Information Solutions | |
| Other | ||
| Medical food (Abbott Promote) | Abbott | Cat# 50774 |
| Ketogenic diet | Envigo | Cat# TD.96355.PWD |
| Standard mouse chow (Teklad Global 18% Protein Rodent Diets) | Envigo | Cat# 2018S |
| Pulse oximeter | Starr Life Sciences Corp. | MouseOx Plus |
| Rectal probe thermometer | Physitemp | TH-5 Thermalert |
| Handheld blood analyzer | Abaxis Global Diagnostics | VetScan i-STAT 1 |
| Lysing Matrix D, 2 mL Tube | MP Biomedicals | Cat# 116913500 |
| Plasma separator tubes | BD | Cat# 365985 |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Ruslan Medzhitov (ruslan.medzhitov@yale.edu).
Experimental Model and Subject Details
Mice
All animal experiments were performed in accordance with institutional regulations after protocol review and approval by Yale University's Institutional Animal Care and Use Committee. Male C57BL/6J mice (The Jackson Laboratory Stock No. 000664) between 7 and 10 weeks of age were used for all animal experiments in this study except where indicated. CHOP knockout mice are strain B6.129S(Cg)-Ddit3tm2.1Dron/J purchased from Jackson Laboratory (Stock No. 005530). PPARα knockout mice are strain B6;129S4-Pparatm1Gonz/J purchased from Jackson Laboratory (Stock No. 008154). FGF21 knockout mice were a generous gift from Dr. David J. Mangelsdorf. IFNaR knockout mice (strain B6.129S2-Ifnar1tm1Agt/Mmjax, Jackson Laboratory Stock No. 32045) were a generous gift from Dr. Akiko Iwasaki.
For infection of mice, Listeria monocytogenes strain 10403s was originally obtained from the laboratory of Dr. Daniel Portnoy. L. monocytogenes was grown to log phase in brain heart influsion (BHI) broth (BD), washed once with PBS, and resuspended in fresh BHI. Mice were injected retro-orbitally with 5×104 colony forming units (CFU) of L. monocytogenes diluted in PBS. Influenza virus strain A/WSN/33 was originally obtained from the laboratory of Dr. Akiko Iwasaki and was propagated using Madin-Darby Canine Kidney (MDCK) cells and titrated to determine concentration by plaque assay described below. For influenza infection, mice were anesthetized with a ketamine/xylazine mixture and indicated plaque forming units (PFU) of influenza in 30 μl PBS was administered intranasally dropwise. Legionella pneumophila (wild-type serogroup 1 strain JR32) was originally obtained from the laboratory of Dr. Craig Roy and was grown on charcoal-yeast extract agar (Sigma-Aldrich) plates for 2 days, then cultured overnight in N-(2-acetamido)-2-aminoethanesulfonic acid (ACES)-buffered yeast extract (AYE) broth (10 g/liter yeast extract, 10 g/liter ACES, 0.4 g/liter L-cysteine HCl-H2O, 0.135 g/liter ferric nitrate, all components from Sigma-Aldrich) and grown to an optical density at 600 nm of 1 in AYE broth. L. pneumophila culture was then diluted in PBS and 1×106 CFU in 40 μl PBS was administered intranasally dropwise to mice anesthetized with ketamine/xylazine.
For the LPS endotoxemia, mice were injected intraperitoneally with the indicated dose of LPS derived from Escherichia coli 055:B5 (Sigma-Aldrich) diluted in 100 μl PBS. For the Poly(I:C) viral inflammation model, mice were injected retro-orbitally with 30 mg/kg of high molecular weight Poly(I:C) (InvivoGen) diluted in 100 μl of normal saline provided by the manufacturer.
For feeding experiments, enteral nutrition/supplementation consisted of Abbott Promote (25%, 23%, and 52% calories from protein, fat, and carbohydrates, respectively) which is an enteral nutrition commonly used in supportive care of critically ill patients with a nearly identical nutritional profile to laboratory Global 2018 Teklad chow (24%, 18%, and 58% calories from protein, fat, and carbohydrates, respectively). Mice were gavaged PBS control or the equivalent of one kilocalorie of the indicated substance (glucose, casein, olive oil, or Abbott Promote) twice a day starting 8 hours post-infection in infection models and 1 hour post-injection in sterile inflammatory models.
For intraperitoneal administration of D-(+)-glucose (Sigma-Aldrich G8270), D-mannoheptulose (DMH, Cayman Chemical), and 2-deoxy-D-glucose (2DG, Sigma-Aldrich), mice were injected intraperitoneally with glucose (20 mg in 100 μl water), DMH (1 mg in 100 μl water) or 2DG (5 mg in 100 μl water) twice a day starting 8 hours post-infection in infection models and 1 hour post-injection in sterile inflammatory models. Valproic acid and levetiracetam were administered intraperitoneally starting 6 hours post-injection at 125 mg/kg and 18 mg/kg in 100 μL PBS, respectively. FGF21 supplementation was done by retro-orbital injection of recombinant mouse FGF21 (R&D Systems) twice daily at 5 ng in 100 μl PBS per injection.
Blood oxygen saturation, breath rate, and heart rate were measured by pulse oximetry using the MouseOx Plus (Starr Life Sciences Corp.). Core body temperature was measured by rectal probe thermometry (Physitemp TH-5 Thermalert). Fasting was performed by placing mice over wire-bottomed cages with food removed from the hopper. The ketogenic diet was purchased from Envigo. Venous pH was measured using an iSTAT 1 Handheld Analyzer (Abaxis) and the CG4+ cartridges as per manufacturers instruction.
Cell culture
To prepare bone marrow derived macrophages (BMDMs), C57BL/6J mice were euthanized by CO2 asphyxiation and femurs and tibias were isolated. The bones were cleansed with ethanol then washed with RPMI 1640 (Corning 10-040-CV), and pulverized in a mortar. Resulting contents were filtered through a 70 μm nylon cell strainer. This pulverizing and filtering process was repeated two more times, after which the resulting contents were treated with ACK lysing buffer (Lonza) for 5 minutes, then washed with RPMI, contents were centrifuged and the pellet was resuspended in macrophage growth medium (RPMI 1640 supplemented with 10% FBS (Gibco), 1% penicillin-streptomycin (Gibco), 2 mM L-glutamine (Gibco), 1 mM sodium pyruvate (Gibco), 0.01 M HEPES (AmericanBio), and 30% L929-conditioned media as a source of CSF-1), and plated on petri dishes. Macrophage growth medium was supplemented on day 3. Cells were plated for use on day 6. For assaying the effect of 2DG on L. monocytogenes replication in macrophages, BMDMs were plated at a density of 5×105 in 24-well tissue culture plates. BMDMs were infected with 5×105 of L. monocytogenes in the presence or absence of 2DG (2.5 mg/ml). After 24 hours, both supernatant and cell lysate were harvested for quantification of bacterial load.
To prepare mouse embryonic fibroblasts (MEFs), pregnant C57BL/6J female mice were euthanized by CO2 asphyxiation at 13.5 to 14.5 days post conception. Embryos were harvested and washed in PBS. After removal of the head and visceral organs, the remaining tissue was washed in PBS then minced in 0.05% Trypsin with EDTA (Gibco). After a 30-minute incubation at 37 °C, the minced tissue is washed with DMEM (Sigma-Aldrich), centrifuged and cell pellet re-suspended in complete MEF media (DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate, and 0.01 M HEPES). After overnight incubation, cells were trypisinized and filtered through 70 μm cell strainer and plated in complete MEF media. For assaying the effect of glucose and 2DG on various cell stress and cytokine treatments, MEFs were plated at a density of 1×105 cells per well in a 24 well plate. After overnight rest, cells were treated with the indicated chemicals or cytokines in the presence of additional glucose (final concentration 9 g/L), 15 mM 2DG, or vehicle control. Thapsigargin (Cayman Chemical) was administered at 1 μM. Poly(I:C) was given at 20 μg/mL. Recombinant mouse IFNα (R&D Systems) was used at 1000 U/ml.
Method Details
Quantification of bacterial and viral loads
L. monocytogenes CFU titers were determined by plating titrated amounts of liver and spleen homogenate on BHI plates. Briefly, liver and spleen were harvested at indicated times post-infection and weighed. Tissue homogenates were generated by pushing the tissue through a 70 μm cell strainer using the plunger of a 5 mL syringe. Titrated dilutions of tissue homogenate were generated in 1% Triton X-100 (Sigma-Aldrich), plated on BHI plates, and grown overnight at 37 °C. For in vitro assays testing the effect of 2DG on L. monocytogenes growth in macrophages, titrated amounts of culture supernatant and cell lysate were plated on BHI plates and grown overnight at 37 °C. To test the effect of 2DG on growth of L. monocytogenes in BHI broth, 5 ×103 L. monocytogenes was inoculated into 25 ml of BHI with or without 10 mg/ml of 2DG. Flasks were incubated overnight. Titrated volumes of L. monocytogenes containing BHI broth were plated the following day on BHI plates and incubated overnight for quantification.
To determine influenza virus strain A/WSN/33 titers in the lung, lung tissue was harvested at the indicated times post-infection, weighed, and disrupted by bead homogenization. For viral titers in the bronchoalveolar lavage (BAL) fluid, mice were euthanized and trachea exposed. BAL was performed via cannulation of the trachea and lungs were lavaged with 1 mL of PBS. Virus titer was determined by infection of MDCK cells with titrated amounts of lung homogenate and BAL followed by addition of an agar overlay for 48 hours. Cell monolayers were then stained with crystal violet and plaque numbers were determined.
Plasma cytokine, metabolite, and tissue injury marker analysis
Whole blood was harvested from mice by retro-orbital bleeding and plasma was isolated using lithium heparin coated plasma separator tubes (BD). Plasma TNFα and IL-6 concentration were assayed by sandwich ELISA using capture antibodies (eBioscience), biotin-conjugated detection antibodies (eBioscience and BD, respectively), HRP-conjugated streptavidin (BD), and TMB substrate reagent (BD). Plasma IFNα and IL-1β concentrations were assayed using kits according to the manufacturer's protocols (eBioscience). Plasma Tropinin-I concentration (Life Diagnostics) and Alanine Aminotransferase (ALT) activity (Cayman Chemical) were assayed using kits according to manufacturers' protocols. Plasma creatinine was assayed using HPLC by The George M. O'Brien Kidney Center at Yale. Plasma non-esterified fatty acid concentration was measured using a kit according the manufacturer's protocols (Wako Diagnostics). Plasma β-hydroxybutyrate concentrations were measured using a kit according the manufacturer's protocols (Cayman Chemical).
RNA extraction and quantification
For tissue RNA extraction, tissues were harvested into RNA Bee RNA isolation reagent (Tel Test, Inc) and disrupted by bead homogenization in Lysing Matrix D tubes using a FastPrep-24 5G homogenizer (MP Biomedicals). RNA was extracted using the RNeasy Kit according to manufacturer's protocol (Qiagen). For RNA extraction from cultured cells, RNA was harvested using phenol-chloroform extraction according to manufacturer's protocol (Tel Test, Inc). cDNA synthesis was performed using MMLV reverse transcriptase (Clontech) with oligo(dT) primers. qRT-PCR reactions were performed on either a CFX96 Real-Time System or CFX384 Real-Time System (Bio-Rad) using PerfeCTa SYBR Green SuperMix (Quanta Biosciences) and transcript levels were normalized to Rpl13a. Primers used for qRT-PCR are cataloged in Table S1.
Flow cytometry
Antibodies used for flow cytometry are cataloged in the antibodies section of the key resources table. Cell viability was determined using ethidium monoazide bromide (EMA) following manufacturer's suggested protocol (Biotium). Samples were Fc-blocked with functional grade mouse anti-CD16/32 antibody (93) (eBioscience). Annexin V-FITC and PI were used for apoptosis assays following the manufacturer's protocol (eBioscience). For tissue analyses, at least 1 × 105 cells were acquired on CD45+ cells within the singlet live gate, as defined by size, granularity and pulse-width. Samples were acquired on an LSRII flow cytometer (BD), and analyzed using FlowJo (Tree Star Technologies).
In vivo reactive oxygen species staining
24 hours after administration of indicated treatments, mice were injected with 0.2 mg of dihydroethidium in 100 μl PBS. After 30 minutes, mice were anesthetized with a ketamine/xylazine mixture and perfused with 4% paraformaldehyde. Brains were harvested and 50 micron sections were cut on a vibratome (Lancer Vibratome 1000 Plus). Sections were then stained with DAPI (ThermoFisher Scientific) for 48 hours and visualized immediately.
Positron-emission tomography and analyses
Mice were imaged on the Inveon small animal PET/CT scanner (Siemens Medical Solutions) using 5.3±3.9 MBq of 18F-FDG. Mice were injected with either LPS or Poly(I:C), and two hours after challenge, injected with 18F-FDG. Mice were scanned for 18F-FDG localization continuously over two hours under isoflurane anesthesia. To draw the lung, heart, liver and whole body ROIs, we used the Inveon Research Workplace (IRW) software (Siemens Healthineers). Regions-of-interest were manually drawn for the heart, liver and lung, and we used a template for the brain (Ma et al., 2005). To co-register the brain images with the template of brain regions, J.G. manually estimated translations and rotations, using a custom-built visualization tool written in IDL 8.0 (Exelis Visual Information Solutions). Standard uptake values (SUVs) at 40-60 min post-injection were used to assess glucose metabolism.
Histology
All mice were euthanized by carbon dioxide asphyxiation and perfused with PBS or fixative. Tissues were immersion-fixed in either 10% neutral buffered formalin or Bouin's fixative (Ricca Chemical Corporation). Tissues were trimmed, processed, embedded, and sectioned and stained for hematoxylin and eosin by routine methods. Tissues were evaluated by a veterinarian (C.J.B.) trained in veterinary pathology with extensive expertise in mouse pathology blinded to both experimental and genetic manipulations.
Digital light microscopic images were acquired using a Zeiss Axio Imager A1 microscope, an AxioCam MRc5 Camera, and AxioVision 4.8.3.0 imaging software (Carl Zeiss MicroImaging, Inc.). The resulting images were optimized using Adobe Photoshop 13.0.1× 64.
TUNEL staining was performed by the Yale Research Histology Service. TUNEL images were not captured by C.J.B. These images were captured on a Leica DMI6000B. All other images were captured by C.J.B. For TUNEL staining quantification, at least 7 non-overlapping 400× magnification fields per mouse brain were counted for total nuclei and TUNEL-stained nuclei to generate a final % TUNEL-positive count. Counts were performed by three independent, blinded investigators and averaged.
Western Blot
Hindbrain samples were harvested and snap frozen in liquid nitrogen. Frozen tissues were pulverized in liquid nitrogen, then protein extracted with N-PER Neuronal Protein Extraction Reagent (ThermoFisher Scientific), supplemented with HALT protease and phosphatase inhibitor cocktail (ThermoFisher Scientific), according to the manufacturer's protocols. Equal amounts of protein were loaded per well of a 10% Bis-Tris mini-gel (Invitrogen) and run in 1× MOPS buffer (Invitrogen). Protein was transferred onto activated PVDF membrane (Millipore), then blocked in 5% milk in TBST (20 mM Tris, 150 mM NaCl, 0.05% Tween 20) for 30 minutes. Membranes were incubated with primary antibodies mouse anti-GADD153 (B-3) and rabbit anti-β Tubulin (H-235), both from Santa Cruz, overnight at 4°C. Anti-GADD153 was incubated at 1:1000 dilution in SignalBoost reagent (Millipore). Anti-β Tubulin was incubated at 1:5000 dilution in 4% BSA TBST. Membranes were washed 3× with TBST, then incubated with secondary antibodies at 1:10,000 dilution for one hour at room temperature. After washing, protein was visualized using enhanced chemiluminescence reagent (ThermoFisher Scientific).
Quantification and Statistical Analysis
Results were statistically analyzed using Student's t test or an analysis of variance (ANOVA) test with multiple comparisons where appropriate using Prism 6.0 (GraphPad Software, Inc). Kaplan Meier survival curves were compared using log-rank Mantel-Cox test. A p value of < 0.05 was considered to be statistically significant.
Supplementary Material
Figure S1. The effect of glucose caloric supplementation or inhibition of glucose utilization on hepatic immune cell infiltration during L. monocytogenes infection, Related to Figure 1
Flow cytometry analysis of CD45+ cells within the liver 4 days after L. monocytogenes infection and treatment with PBS, glucose, or 2DG.
Data are represented as mean ± SEM. ***p<0.001, ****p<0.0001.
Figure S2. Systemic effects of glucose caloric supplementation or inhibition of glucose utilization during LPS sepsis, Related to Figure 2
(A) Mice were given 15 mg/kg IP LPS, then treated with PBS, 5 mg 2DG, 50 mg/kg D-mannoheptulose (DMH) given IP twice a day initiated one hour after LPS administration. n=5/group.
(B) Blood glucose at baseline, 2, 6 and 24 hours after 15 mg/kg IP LPS with PO gavage of PBS vehicle (LPS-PBS), glucose (LPS-Glucose), or Abbott Promote (LPS-Food) BID; or with IP 2DG (LPS-2DG). Blood glucose of mice treated with only 2DG IP are also shown.
(C-D) Mice were given 15 mg/kg IP LPS, then treated with IP PBS, glucose, or 2DG given IP.
(C) O2 saturation, respiratory rate, heart rate and body temperature 24 hours after LPS.
(D) Plasma troponin-I, ALT, and creatinine levels measured at 24 hours after LPS.
Data are represented as mean ± SEM. *p<0.05, **p<0.01, ****p<0.0001.
Figure S3. The effect of caloric supplementation or inhibition of glucose utilization on survival and lung inflammation after influenza virus infection, Related to Figure 3.
(A) Survival after infection with 800 PFU of influenza virus. Mice were gavaged with Abbott Promote (Food), casein, olive oil, or PBS vehicle. Figure 3B and 3C are a subset of Figure S3A, separated for clarity (the same PBS-treated and food-treated groups are shown).
(B) mRNA expression of whole lung tissue at day 6 after 375 PFU of influenza virus.
(C) Flow cytometric analysis of lung and BAL on day 6 after 700 PFU of influenza virus.
(D) H&E staining of lung tissue 6 days after influenza virus infection. Letters correspond to areas of the lung annotated in Figure 3. Scale bar = 50 μm.
(E) Survival after infection with 1×106 L. pneumophila, and treatment with IP PBS or 2DG. n=5/group.
Data are represented as mean ± SEM.
Figure S4. Systemic effects of glucose caloric supplementation or inhibition of glucose utilization during Poly(I:C) sepsis, Related to Figure 4.
(A) Survival of mice after Poly(I:C) challenge and treatment with either IP PBS, 2DG, or DMH.
(B) Averaged sagittal PET images after PBS vehicle (baseline), LPS, and Poly(I:C) administration. CT, computed tomography; FDG, 2-deoxy-2-[18F] fluorodeoxy-D-glucose. n=3/group.
(C-D) Mice were challenged with Poly(I:C), then treated with either IP PBS, glucose, or 2DG.
(C) Hindbrain mRNA expression 4 hours after Poly(I:C) administration and indicated treatments. n=3-5/group.
(D) Blood glucose measured at 2, 6, and 18 hours after Poly(I:C) and indicated treatments. Plasma troponin-I, ALT and creatinine measured at 24 hours after Poly(I:C).
Data are represented as mean ± SEM. *p<0.05,**p<0.01, ****p<0.0001. n.s., not significant.
Figure S5. The effect of inhibiting glucose utilization on inflammation and ER stress during viral inflammation, Related to Figure 5.
(A) Flow cytometric analysis of Lung and BAL on day 5 after infection with 700 PFU of influenza virus in B6 WT and Ddit3-/- mice.
(B) Mouse embryonic fibroblasts (MEFs) treated with vehicle, IFNα, 2DG, or IFNα and 2DG. mRNA expression at 0, 4, and 24 hours after treatment. n=3 replicates per group. Data representative of two independent experiments.
(C) MEFs treated with vehicle, IFNα, Poly(I:C), and Thapsigargin (Thaps), in the presence of vehicle, glucose or 2DG for 24 hours. Flow cytometric analysis for Annexin V. Two-three replicates per group. Data representative of three independent experiments.
Data are represented as mean ± SEM. *p<0.05,**p<0.01, ****p<0.0001
Figure S6. Histopathology of LPS sepsis, Related to Figure 6.
(A) Mice were given IP LPS then initiated with IP PBS, glucose, and 2DG treatment. Dihydroethidium staining of brain 24 hours after LPS.
(B) TUNEL-stained sections of brain 24 hours after LPS and treatment with PBS, glucose, or 2DG. 3,3′-Diaminobenzidine (DAB) hematoxylin, scale bars = 100 mm. Quantification of TUNEL positive cells per 400× power field. n=3/group.
(C) Representative images of TUNEL-stained sections of heart, lung, liver, and kidney 24 hours post-LPS. DAB Hematoxylin, scale bars = 100 mm.
(D) Representative sections of TUNEL-stained thalamus, cerebral cortex, and cerebellum 24 hours post-LPS in WT mice. DAB Hematoxylin, scale bars = 100 mm.
Data are represented as mean ± SEM.
Figure S7. Investigation of the ketogenic program in bacterial and viral inflammation, Related to Figure 6.
(A) Plasma BHOB levels after 3 days of ketogenic diet (KD) and 24 hours of fasting compared to control fed mice. Control vs KD diet p=0.0001; Control vs 24h Fast p=0.0011. n=5/group.
(B) Survival after 10 mg/kg IP LPS in mice on control diet, after 3 days of ketogenic diet (KD), and after 24 hours of fasting. Control vs KD p=0.0002; Control vs 24h Fast p=0.0007.
(C) Venous blood gas measured in mice on control diet and 3 days after ketogenic diet (KD).
(D) Survival after 375 PFU influenza in WT and Fgf21-/- mice.
Data are represented as mean ± SEM.
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Cxcl1 | 5′-ATCCAGAGCTTGAAGGTGTTGC-3′ | 5′-AAGGCAAGCCTCGCGACC-3′ |
| Ddit3 (Chop) | 5′-TCTCATCCCCAGGAAACGAAG-3′ | 5′- TGTGCGTGTGACCTCTGTTGG-3′ |
| Ifit1 | 5′-ACCCAGAGAACAGCTACCAC-3′ | 5′-GAACTGGACCTGCTCTGAG-3′ |
| Ifit2 (Isg54) | 5′-TCTGATTCTGAGGCCTTGCA-3′ | 5′-CTTGCTGACCTCCTCCATTCTC-3′ |
| Ifnb1 | 5′-GCACTGGGTGGAATGAGACTATTG-3′ | 5′-TTCTGAGGCATCAACTGACAGGTC-3′ |
| Il1b | 5′-CAGTTGTCTAATGGGAACGTCA-3′ | 5′- GCACCTTCTTTTCCTTCATCTTT-3′ |
| Il6 | 5′-GACTTCCATCCAGTTGCCTTCTTGG-3′ | 5′-CCAGTTTGGTAGCATCCATCATTTCT-3′ |
| Mx1 | 5′-TCCTCCCCAAATGTTTTCAG-3′ | 5′-ACTGAGATGACCCAGCACCT-3′ |
| Oasl1 | 5′-CAGAGGTCAGCTGGAAAGGA-3′ | 5′-CTTGAGTACCTTGAGCACGC-3′ |
| Ppp1r15a (Gadd34) | 5′-CAGCAAGGAAATGGACTG-3′ | 5′-GCGGCTCAGATTGTTCAAAG-3′ |
| Rpl13a | 5′-GAGGTCGGGTGGAAGTACCA-3′ | 5′-TGCATCTTGGCCTTTTCCTT-3′ |
| Rsad2 | 5′-CTTCAACGTGGACGAAGACA-3′ | 5′-ATTCAGGCACCAAACAGGAC-3′ |
| Saa3 | 5′-ATGCTCGGGGGAACTATGAT-3′ | 5′-TCCATGTCCCGTGAACTTCT-3′ |
| Tnf | 5′-TCTGTCTACTGAACTTCGGGGTG-3′ | 5′-ACTTGGTGGTTTGCTACGACG-3′ |
| Tnfsf10 | 5′-ACCTCAGCTTCAGTCAGCACTTC-3′ | 5′-TGTAAGTCACAGCCACAGACACAG-3′ |
| Trex1 | 5′-TTAGACCTGGAAGCCACTGG-3′ | 5′-GAGCAATGCACAGAGAGAGC-3′ |
Acknowledgments
We thank members of the Medzhitov lab for helpful discussions, Marya Shanabrough and Tamas Horvath for help with processing of brain specimens, and David Mangelsdorf and Steven Kliewer for making available the Fgf21-/- mice. This study was supported by the HHMI, Else Kröner Fresenius Foundation, The Blavatnik Family Foundation, and grants from the NIH (AI046688, AI089771, CA157461). A.W. was supported by NIH Grant T32 AR07107-39. S.C.H. was supported by American Heart Association Grant 13FTF17070000. H.H.L. was supported by the Gruber Science Fellowship. Plasma creatinine was processed through the Yale George M. O'Brien Kidney Center, NIH Grant P30-DK079310. Preparation of histology sections were performed by the Yale Research Pathology and Histology Core.
Footnotes
AUTHOR CONTRIBUTIONS: A.W., S.C.H., and H.H.L. contributed equally to this work. A.W., S.C.H., H.H.L., and R.M. designed the study, analyzed the data, and wrote the manuscript with input from the other authors. A.W., S.C.H., and H.H.L. performed the experiments with assistance from S.Y. and C.Z. J.G. performed all analyses for PET studies. C.J.B. evaluated tissue histology.
References
- Adamo SA. Parasitic suppression of feeding in the tobacco hornworm, Manduca sexta: Parallels with feeding depression after an immune challenge. Arch Insect Biochem Physiol. 2005;60:185–197. doi: 10.1002/arch.20068. [DOI] [PubMed] [Google Scholar]
- Agwunobi AO, Reid C, Maycock P, Little RA, Carlson GL. Insulin resistance and substrate utilization in human endotoxemia. J Clin Endocrinol Metab. 2000;85:3770–3778. doi: 10.1210/jcem.85.10.6914. [DOI] [PubMed] [Google Scholar]
- Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- Arabi YM, Aldawood AS, Haddad SH, Al-Dorzi HM, Tamim HM, Jones G, Mehta S, McIntyre L, Solaiman O, Sakkijha MH, et al. Permissive Underfeeding or Standard Enteral Feeding in Critically Ill Adults. N Engl J Med. 2015;372:2398–2408. doi: 10.1056/NEJMoa1502826. [DOI] [PubMed] [Google Scholar]
- Archer KA, Roy CR. MyD88-dependent responses involving toll-like receptor 2 are important for protection and clearance of Legionella pneumophila in a mouse model of Legionnaires' disease. Infect Immun. 2006;74:3325–3333. doi: 10.1128/IAI.02049-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–533. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS, Schneider DS. The Role of Anorexia in Resistance and Tolerance to Infections in Drosophila. PLoS Biol. 2009;7:e1000150. doi: 10.1371/journal.pbio.1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS, Schneider DS. Tolerance of infections. Annu Rev Immunol. 2012;30:271–294. doi: 10.1146/annurev-immunol-020711-075030. [DOI] [PubMed] [Google Scholar]
- Boison D. Chopping Out CHOP Chops the Fate of Neurons. Epilepsy Curr. 2013;13:219–220. doi: 10.5698/1535-7597-13.5.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212:1345–1360. doi: 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd A, Alleva L, Alsharifi M, Koskinen A, Smythe V, Mullbacher A, Wood J, Clark I. Increased survival after gemfibrozil treatment of severe mouse influenza. Antimicrob Agents Chemother. 2007;51:2965–2968. doi: 10.1128/AAC.00219-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JD, Platanias LC, Fish EN. Beta interferon regulation of glucose metabolism is PI3K/Akt dependent and important for antiviral activity against coxsackievirus B3. J Virol. 2014;88:3485–3495. doi: 10.1128/JVI.02649-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara-Lemarroy CR, Guzman DELAGFJ, Cordero-Perez P, Ibarra-Hernandez JM, Munoz-Espinosa LE, Fernandez-Garza NE. Gemfibrozil attenuates the inflammatory response and protects rats from abdominal sepsis. Exp Ther Med. 2015;9:1018–1022. doi: 10.3892/etm.2015.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaer MP, Van den Berghe G. Nutrition in the Acute Phase of Critical Illness. N Engl J Med. 2014;370:1227–1236. doi: 10.1056/NEJMra1304623. [DOI] [PubMed] [Google Scholar]
- Esposito V, Grosjean F, Tan J, Huang L, Zhu L, Chen J, Xiong H, Striker GE, Zheng F. CHOP deficiency results in elevated lipopolysaccharide-induced inflammation and kidney injury. Am J Physiol Renal Physiol. 2013;304:F440–450. doi: 10.1152/ajprenal.00487.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold KR, Grunfeld C, Heuer JG, Gupta A, Cramer M, Zhang T, Shigenaga JK, Patzek SM, Chan ZW, Moser A, et al. FGF21 Is Increased by Inflammatory Stimuli and Protects Leptin-Deficient ob/ob Mice from the Toxicity of Sepsis. Endocrinology. 2012;153:2689–2700. doi: 10.1210/en.2011-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo N, Chora A, Raquel H, Pejanovic N, Pereira P, Hartleben B, Neves-Costa A, Moita C, Pedroso D, Pinto A, et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity. 2013;39:874–884. doi: 10.1016/j.immuni.2013.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan-Pena S, O'Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greseth MD, Traktman P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog. 2014;10:e1004021. doi: 10.1371/journal.ppat.1004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–137. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- Hoetzenecker W, Echtenacher B, Guenova E, Hoetzenecker K, Woelbing F, Bruck J, Teske A, Valtcheva N, Fuchs K, Kneilling M, et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med. 2012;18:128–134. doi: 10.1038/nm.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, et al. Endocrine Regulation of the Fasting Response by PPARα-Mediated Induction of Fibroblast Growth Factor 21. Cell Metab. 5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Investigators NSS, Finfer S, Chittock DR, Su SY, Blair D, Foster D, Dhingra V, Bellomo R, Cook D, Dodek P, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360:1283–1297. doi: 10.1056/NEJMoa0810625. [DOI] [PubMed] [Google Scholar]
- Jamieson AM, Pasman L, Yu S, Gamradt P, Homer RJ, Decker T, Medzhitov R. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science. 2013;340:1230–1234. doi: 10.1126/science.1233632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Pulendran B, Lambrecht BN. Emerging functions of the unfolded protein response in immunity. Nat Immunol. 2014;15:910–919. doi: 10.1038/ni.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger MJ, Ringler DH, Anver MR. Fever and survival. Science. 1975;188:166–168. [PubMed] [Google Scholar]
- Kolls JK. Oxidative stress in sepsis: a redox redux. J Clin Invest. 2006;116:860–863. doi: 10.1172/JCI28111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley RJ, Tsalik EL, van Velkinburgh JC, Glickman SW, Rice BJ, Wang C, Chen B, Carin L, Suarez A, Mohney RP, et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci Transl Med. 2013;5:195ra195–195ra195. doi: 10.1126/scitranslmed.3005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza Fa, Japiassú AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A, et al. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med. 2010;2:51ra71. doi: 10.1126/scitranslmed.3001118. [DOI] [PubMed] [Google Scholar]
- Levy RG, Cooper PN, Giri P. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst Rev. 2012 doi: 10.1002/14651858.CD001903.pub2. CD001903. [DOI] [PubMed] [Google Scholar]
- Li MV, Chen W, Harmancey RN, Nuotio-Antar AM, Imamura M, Saha P, Taegtmeyer H, Chan L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP) Biochem Biophys Res Commun. 2010;395:395–400. doi: 10.1016/j.bbrc.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, Jellbauer S, Poe AJ, Ton V, Pesciaroli M, Kehl-Fie TE, Restrepo NA, Hosking MP, Edwards RA, Battistoni A, et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe. 2012;11:227–239. doi: 10.1016/j.chom.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Lu Y, Martinez J, Bi Y, Lian G, Wang T, Milasta S, Wang J, Yang M, Liu G, et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1alpha-dependent. Proc Natl Acad Sci U S A. 2016;113:1564–1569. doi: 10.1073/pnas.1518000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hof PR, Grant SC, Blackband SJ, Bennett R, Slatest L, McGuigan MD, Benveniste H. A three-dimensional digital atlas database of the adult C57BL/6J mouse brain by magnetic resonance microscopy. Neuroscience. 2005;135:1203–1215. doi: 10.1016/j.neuroscience.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ES, Bates RA, Koebel DA, Fuchs BB, Sonnenfeld G. 2-deoxy-D-glucose-induced metabolic stress enhances resistance to Listeria monocytogenes infection in mice. Physiol Behav. 1998;65:535–543. doi: 10.1016/s0031-9384(98)00199-1. [DOI] [PubMed] [Google Scholar]
- Miller NE. Some psychophysiological studies of motivation and of the behavioural effects of illness. Bull Br Psychol Soc. 1964;17:1–20. [Google Scholar]
- Murray MJ, Murray AB. Anorexia of infection as a mechanism of host defense. Am J Clin Nutr. 1979;32:593–596. doi: 10.1093/ajcn/32.3.593. [DOI] [PubMed] [Google Scholar]
- Nairz M, Ferring-Appel D, Casarrubea D, Sonnweber T, Viatte L, Schroll A, Haschka D, Fang FC, Hentze MW, Weiss G, et al. Iron Regulatory Proteins Mediate Host Resistance to Salmonella Infection. Cell Host Microbe. 2015;18:254–261. doi: 10.1016/j.chom.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecchi E, Dallaporta M, Jean A, Thirion S, Troadec JD. Prostaglandins and sickness behavior: old story, new insights. Physiol Behav. 2009;97:279–292. doi: 10.1016/j.physbeh.2009.02.040. [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA, et al. FGF21 induces PGC-1α and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci U S A. 2009;106:10853–10858. doi: 10.1073/pnas.0904187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raberg L, Sim D, Read AF. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science. 2007;318:812–814. doi: 10.1126/science.1148526. [DOI] [PubMed] [Google Scholar]
- Sayyah M, Javad-Pour M, Ghazi-Khansari M. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: involvement of proinflammatory factors: nitric oxide and prostaglandins. Neuroscience. 2003;122:1073–1080. doi: 10.1016/j.neuroscience.2003.08.043. [DOI] [PubMed] [Google Scholar]
- Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol. 2008;8:889–895. doi: 10.1038/nri2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seron-Arbeloa C, Zamora-Elson M, Labarta-Monzon L, Mallor-Bonet T. Enteral nutrition in critical care. J Clin Med Res. 2013;5:1–11. doi: 10.4021/jocmr1210w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, et al. Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer BH, Newstead MW, Zeng X, Cooke CL, Thompson RC, Singer K, Ghantasala R, Parent JM, Murphy GG, Iwashyna TJ, et al. Cecal Ligation and Puncture Results in Long-Term Central Nervous System Myeloid Inflammation. PLoS One. 2016;11:e0149136. doi: 10.1371/journal.pone.0149136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smorodintsev AA, Aksenov OA, Konstantinova IK, Vil'ner LM, Tinufanov AV. Comparative study of the toxicity of poly G-poly C and poly I-poly C in different objects. Vopr Virusol. 1978:201–206. [PubMed] [Google Scholar]
- Soares MP, Gozzelino R, Weis S. Tissue damage control in disease tolerance. Trends Immunol. 2014;35:483–494. doi: 10.1016/j.it.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Song Y, Shen J, Lin Y, Shen J, Wu X, Yan Y, Zhou L, Zhang H, Zhou Y, Cao M, et al. Up-regulation of podoplanin involves in neuronal apoptosis in LPS-induced neuroinflammation. Cell Mol Neurobiol. 2014;34:839–849. doi: 10.1007/s10571-014-0060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terbach N, Williams RS. Structure-function studies for the panacea, valproic acid. Biochem Soc Trans. 2009;37:1126–1132. doi: 10.1042/BST0371126. [DOI] [PubMed] [Google Scholar]
- van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345:1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- Wing EJ, Young JB. Acute Starvation Protects Mice Against Listeria monocytogenes. Infect Immun. 1980;28:771–776. doi: 10.1128/iai.28.3.771-776.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, Kang R, Lotze MT, Billiar TR, Wang H, et al. PKM2 regulates the Warburg effect and promotes HMGB1. release in sepsis Nat Commun. 2014;5 doi: 10.1038/ncomms5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Abdelmegeed MA, Song BJ. Activation of PPARalpha by Wy-14643 ameliorates systemic lipopolysaccharide-induced acute lung injury. Biochem Biophys Res Commun. 2013;436:366–371. doi: 10.1016/j.bbrc.2013.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The effect of glucose caloric supplementation or inhibition of glucose utilization on hepatic immune cell infiltration during L. monocytogenes infection, Related to Figure 1
Flow cytometry analysis of CD45+ cells within the liver 4 days after L. monocytogenes infection and treatment with PBS, glucose, or 2DG.
Data are represented as mean ± SEM. ***p<0.001, ****p<0.0001.
Figure S2. Systemic effects of glucose caloric supplementation or inhibition of glucose utilization during LPS sepsis, Related to Figure 2
(A) Mice were given 15 mg/kg IP LPS, then treated with PBS, 5 mg 2DG, 50 mg/kg D-mannoheptulose (DMH) given IP twice a day initiated one hour after LPS administration. n=5/group.
(B) Blood glucose at baseline, 2, 6 and 24 hours after 15 mg/kg IP LPS with PO gavage of PBS vehicle (LPS-PBS), glucose (LPS-Glucose), or Abbott Promote (LPS-Food) BID; or with IP 2DG (LPS-2DG). Blood glucose of mice treated with only 2DG IP are also shown.
(C-D) Mice were given 15 mg/kg IP LPS, then treated with IP PBS, glucose, or 2DG given IP.
(C) O2 saturation, respiratory rate, heart rate and body temperature 24 hours after LPS.
(D) Plasma troponin-I, ALT, and creatinine levels measured at 24 hours after LPS.
Data are represented as mean ± SEM. *p<0.05, **p<0.01, ****p<0.0001.
Figure S3. The effect of caloric supplementation or inhibition of glucose utilization on survival and lung inflammation after influenza virus infection, Related to Figure 3.
(A) Survival after infection with 800 PFU of influenza virus. Mice were gavaged with Abbott Promote (Food), casein, olive oil, or PBS vehicle. Figure 3B and 3C are a subset of Figure S3A, separated for clarity (the same PBS-treated and food-treated groups are shown).
(B) mRNA expression of whole lung tissue at day 6 after 375 PFU of influenza virus.
(C) Flow cytometric analysis of lung and BAL on day 6 after 700 PFU of influenza virus.
(D) H&E staining of lung tissue 6 days after influenza virus infection. Letters correspond to areas of the lung annotated in Figure 3. Scale bar = 50 μm.
(E) Survival after infection with 1×106 L. pneumophila, and treatment with IP PBS or 2DG. n=5/group.
Data are represented as mean ± SEM.
Figure S4. Systemic effects of glucose caloric supplementation or inhibition of glucose utilization during Poly(I:C) sepsis, Related to Figure 4.
(A) Survival of mice after Poly(I:C) challenge and treatment with either IP PBS, 2DG, or DMH.
(B) Averaged sagittal PET images after PBS vehicle (baseline), LPS, and Poly(I:C) administration. CT, computed tomography; FDG, 2-deoxy-2-[18F] fluorodeoxy-D-glucose. n=3/group.
(C-D) Mice were challenged with Poly(I:C), then treated with either IP PBS, glucose, or 2DG.
(C) Hindbrain mRNA expression 4 hours after Poly(I:C) administration and indicated treatments. n=3-5/group.
(D) Blood glucose measured at 2, 6, and 18 hours after Poly(I:C) and indicated treatments. Plasma troponin-I, ALT and creatinine measured at 24 hours after Poly(I:C).
Data are represented as mean ± SEM. *p<0.05,**p<0.01, ****p<0.0001. n.s., not significant.
Figure S5. The effect of inhibiting glucose utilization on inflammation and ER stress during viral inflammation, Related to Figure 5.
(A) Flow cytometric analysis of Lung and BAL on day 5 after infection with 700 PFU of influenza virus in B6 WT and Ddit3-/- mice.
(B) Mouse embryonic fibroblasts (MEFs) treated with vehicle, IFNα, 2DG, or IFNα and 2DG. mRNA expression at 0, 4, and 24 hours after treatment. n=3 replicates per group. Data representative of two independent experiments.
(C) MEFs treated with vehicle, IFNα, Poly(I:C), and Thapsigargin (Thaps), in the presence of vehicle, glucose or 2DG for 24 hours. Flow cytometric analysis for Annexin V. Two-three replicates per group. Data representative of three independent experiments.
Data are represented as mean ± SEM. *p<0.05,**p<0.01, ****p<0.0001
Figure S6. Histopathology of LPS sepsis, Related to Figure 6.
(A) Mice were given IP LPS then initiated with IP PBS, glucose, and 2DG treatment. Dihydroethidium staining of brain 24 hours after LPS.
(B) TUNEL-stained sections of brain 24 hours after LPS and treatment with PBS, glucose, or 2DG. 3,3′-Diaminobenzidine (DAB) hematoxylin, scale bars = 100 mm. Quantification of TUNEL positive cells per 400× power field. n=3/group.
(C) Representative images of TUNEL-stained sections of heart, lung, liver, and kidney 24 hours post-LPS. DAB Hematoxylin, scale bars = 100 mm.
(D) Representative sections of TUNEL-stained thalamus, cerebral cortex, and cerebellum 24 hours post-LPS in WT mice. DAB Hematoxylin, scale bars = 100 mm.
Data are represented as mean ± SEM.
Figure S7. Investigation of the ketogenic program in bacterial and viral inflammation, Related to Figure 6.
(A) Plasma BHOB levels after 3 days of ketogenic diet (KD) and 24 hours of fasting compared to control fed mice. Control vs KD diet p=0.0001; Control vs 24h Fast p=0.0011. n=5/group.
(B) Survival after 10 mg/kg IP LPS in mice on control diet, after 3 days of ketogenic diet (KD), and after 24 hours of fasting. Control vs KD p=0.0002; Control vs 24h Fast p=0.0007.
(C) Venous blood gas measured in mice on control diet and 3 days after ketogenic diet (KD).
(D) Survival after 375 PFU influenza in WT and Fgf21-/- mice.
Data are represented as mean ± SEM.