

Abstract
The ability to localize phosphosites to specific amino acid residues is crucial to translating phosphoproteomic data into biological meaningful contexts. In a companion manuscript we described a new implementation of activated ion electron transfer dissociation (AI-ETD) on a quadrupole-Orbitrap-linear ion trap hybrid MS system (Orbitrap Fusion Lumos), which greatly improved peptide fragmentation and identification over ETD and other supplemental activation methods. Here we present the performance of AI-ETD for identifying and localizing sites of phosphorylation in both phosphopeptides and intact phosphoproteins. Using 90-minute analyses we show that AI-ETD can identify 24,503 localized phosphopeptide spectral matches enriched from mouse brain lysates, which more than triples identifications from standard ETD experiments and outperforms ETcaD and EThcD as well. AI-ETD achieves these gains through improved quality of fragmentation and MS/MS success rates for all precursor charge states, especially for doubly protonated species. We also evaluate the degree to which phosphate neutral loss occurs from phosphopeptide product ions due to the infrared photo-activation of AI-ETD and show that modifying phosphoRS (a phosphosite localization algorithm) to include phosphate neutral losses can significantly improve localization in AI-ETD spectra. Finally, we demonstrate the utility of AI-ETD in localizing phosphosites in α-casein, a ~23.5 kilodalton phosphoprotein that showed eight of nine known phosphorylation sites occupied upon intact mass analysis. AI-ETD provided the greatest sequence coverage for all five charge states investigated and was the only fragmentation method to localize all eight phosphosites for each precursor. Overall, this work highlights the analytical value AI-ETD can bring to both bottom-up and top-down phosphoproteomics.
Graphical Abstract
Protein phosphorylation is a dynamic post-translational modification (PTM) that is involved in a diverse array of biological regulation.1,2 Modern phosphoproteomic technology relies on mass spectrometry (MS) to characterize the diverse roles of phosphorylation in a wide array of biological systems.3–6 A principal advantage of MS-based phosphoproteomics is the ability to localize phosphorylation events with single residue resolution and to do so in a high-throughput and unbiased manner.7–9 To achieve this goal, peptides and proteins that contain phosphoryl group(s) must undergo extensive backbone fragmentation while retaining the modification to reveal both sequence and phosphosite information. Canonical slow-heating methods like collisionally activated dissociation (CAD) have traditionally struggled with phosphorylated sequences, mainly because the phosphate loss is a low-energy dissociation pathway.10–12 Higher-energy collisional activation (HCD) has somewhat addressed this challenge,13 but alternative fragmentation methods, including electron transfer dissociation (ETD) and several photo-dissociation approaches, have also proven valuable for phosphoproteomic experiments.14–24
ETD has become especially ubiquitous as an alternative dissociation method, largely because it cleaves N-Cα bond along the peptide backbone to form c- and z•-type ions while preserving labile bonds like phosphorylation.25–29 ETD is well-suited for fragmenting highly charged precursors, but fragmentation efficiency suffers for precursor ions with low charge density,30 which can be problematic when requiring extensive fragmentation to confidently localize phosphosites. Poor fragmentation efficiency occurs because non-covalent intramolecular interactions are more prevalent in the more compact gas-phase structures of low charge density precursors,31–34 and these interactions hold product ions together even in the event of successful backbone cleavage, a process called non-dissociative electron transfer (ETnoD).35–38
There are several methods to mitigate ETnoD and increase product ion yield from ETD reactions, including gentle collisional activation of ETnoD products (ETcaD),39 beam-type collisional activation of all products of the ETD reaction (EThcD),40 and infrared photo-activation concurrent with ETD reaction (activated ion-ETD, AI-ETD).41 AI-ETD is a particularly favorable approach because it significantly boosts sequence-informative fragment ion generation without increasing scan duration due to secondary activation events, and it also minimizes hydrogen rearrangements that can occur when activating the ETnoD products post-reaction.39,41–46 We recently implemented AI-ETD on a quadrupole-Orbitrap-linear ion trap (q-OT-QLT, Orbitrap Fusion Lumos) and showed that it significantly improves peptide identifications and quality of fragmentation in ETD experiments (described in a companion manuscript).47 With other ETD-specific improvements also incorporated on the Lumos platform, including a front-end ETD reagent source,48 high capacity ETD for improved product ion signal-to-noise (S/N),49 and calibrated ETD reaction times for optimal data acquisition in shotgun proteomic analyses,50 this system represents the state-of-the art for ETD technology.
With this work we demonstrate how AI-ETD on the Lumos system can improve both shotgun analyses of phosphopeptides and top-down fragmentation of intact phosphoproteins. Both approaches are valuable in modern phosphoproteomics; shotgun phosphoproteomics enables the identification and quantification of thousands to tens of thousands of phosphosites in just hours of analysis time,51–66 and top-down phosphoproteomics allows comprehensive proteoform characterization to localize phosphosites on a protein and show which sites are co-modified in a given proteoform.67–75 Here we show that AI-ETD can triple the number of localized phosphopeptides sequenced with ETD in shotgun phosphoproteomic analyses and that it also outmatches other supplemental activation techniques, i.e., ETcaD and EThcD. Furthermore, we demonstrate the first application of AI-ETD to intact phosphoproteins, showing it to be superior to other fragmentation methods for localizing multiple sites of phosphorylation on α-casein (~23.5 kilodaltons). In all, this study presents the significant potential AI-ETD has for advancing phosphoproteomics from both the shotgun and top-down perspectives.
MATERIALS AND METHODS
Shotgun LC-MS/MS
Phosphopeptides enriched from tryptic digests of mouse brain lysates were analyzed using 90-minuted LC-MS/MS analyses. The same mouse brain lysate used in the companion manuscript was used for phosphopeptide enrichments described herein, making comparisons between the two possible, as discussed in the text.47 Phosphopeptides were enriched using MagResyn Ti-IMAC Ti4+-functionalized magnetic microspheres (ReSyn Biosciences, Edenvale, South Africa). Liquid chromatography conditions were identical to those described in the companion manuscript, and one microliter resuspended phosphopeptides was injected onto the column. Calibrated charge dependent ETD parameters were enabled to determine ETD reagent ion AGC and ETD reaction times,50 and all MS/MS were mass analyzed in the Orbitrap with a resolution of 15,000 K at 200 m/z. Tandem mass spectra were searched using Proteome Discoverer 1.4 software and the SEQUEST HT algorithm.76 Precursor peaks within a ±1 Da window, charge reduced precursors with a ±0.5 Da window, and neutral losses from the charge reduced precursor with a ±0.5 Da window and a maximum neutral loss of 60 Da were cleaned from spectra.77,78 A UniProt mouse (mus musculus) database (canonical and isoforms) was used for searching with b-, y-, c-, and z-type fragment ion types included for all but ETD where b-type were not included and HCD experiments where only b- and y-type fragments were used. The Percolator node was used to filter results to a 1% false discovery rate.79,80 phosphoRS version 3.1 was used to localize phosphosites with a 0.05 Da fragment mass tolerance,81 only phosphosites with localization probabilities of 75% and higher were considered as localized and used for further analysis, and the phosphoRS algorithm was modified to include phosphate neutral losses in ETD spectra as discussed in the text. Three technical replicate analyses were batched together for each method.
Top-Down Analysis
A-casein was purchased as a mass spectrometry grade standards from Protea Biosciences (Morgantown, WV) and was resuspended at 10 pmol/μL in 50% ACN/49.8% H2O, and 0.2% FA. Intact protein mode was enabled to reduce nitrogen pressure in the ion-routing multipole to 3 mTorr, and five precursors were interrogated using HCD, ETD, EThcD, and AI-ETD. MS/MS spectra were deconvoluted with XTRACT (Thermo Fisher Scientific) using default parameters and a S/N threshold of three. ProSight Lite was used to generate matched fragments using a 10 ppm tolerance.82 Phosphoserines modified in α-casein were identified using known sites at the UniProt resource (accession P02663). See Supplemental Materials for more details.
RESULTS AND DISCUSSION
AI-ETD for bottom-up phosphoproteomics
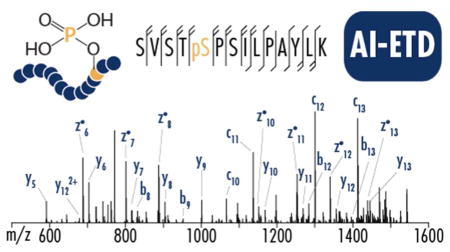
AI-ETD improves ETD dissociation efficiency by using IR photo-activation concurrent with the ion-ion reaction to disrupt non-covalent interactions and drive the formation of sequence-informative product ions. To evaluate AI-ETD for shotgun phosphoproteomics, 90-minute LC-MS/MS analyses were conducted on complex mixtures of tryptic phosphopeptides enriched from mouse brain lysate. Figure 1 shows an example comparison of ETD and AI-ETD spectra for the doubly protonated precursor of a phosphopeptide having the sequence SVSTpSPSILPAYLK, which was identified by both fragmentation methods in the LC-MS/MS analyses.
Figure 1. ETD and AI-ETD spectra of a phosphopeptide with several potential phosphosites.

ETD produces moderate fragmentation of doubly protonated precursor of peptide SVSTpSPSILPAYLK but fails to confidently localize the modified phosphoserine (Xcorr 1.09, ~46% backbone sequence coverage). Conversely, AI-ETD generates extensive fragmentation with c/z and b/y ion series to localize the phosphosite with high confidence (Xcorr 5.97, 100% backbone sequence coverage). Spectra are on the same intensity scale, and the grey regions are scaled to be ten percent of original intensity.
This 14 residue phosphopeptide has one phosphorylated residue but six potential sites of modification (S/T/Y). ETD generated seven total sequence-informative fragment ions that correspond to cleavage of six of the 13 total inter-residue positions (~46% sequence coverage), eliminating three potential phosphosites but ultimately failing to localize the correct site of modification. AIETD, on the other hand, provided comprehensive fragmentation, producing 25 b-, y-, c- and z•-type product ions. AI-ETD is known to produce c, z•-, and y-type fragments (also seen in standard ETD) with minimal addition of b-type products for unmodified peptides,47 but b- and y-type ions are more prevalent in AI-ETD spectra of phosphopeptides, as seen here. Below we discuss this phenomenon and how to make use of it in data analysis phosphopeptide AI-ETD spectra. Overall, AI-ETD generates product ions for every inter-residue position for 100% sequence coverage and confident localization of the phosphosite to the serine at position five in the peptide sequence.
Figure 2 summarizes the data from the shotgun analyses for ETD, ETcaD, EThcD, AI-ETD at two different laser powers, and with AI-ETD+. Described in the companion manuscript, AI-ETD+ is a novel hybrid fragmentation approach that combines AI-ETD with a second short (several ms) photo-activation in the low pressure cell of the QLT. This secondary activation can induce infrared multi photon dissociation (IRMPD) to produce b- and y-type fragments in addition to the c, z•-, and y-type fragments generated by AI-ETD. Overall AI-ETD (15 W) provides the greatest number of localized PSMs and unique phosphopeptides (Figure 2a and b), more than tripling the number of phospho peptide spectral matches (phospho PSMs) generated by ETD. EThcD, known to perform well for phosphopeptide fragmentation and phosphosite localization,83,84 and ETcaD, also both significantly increase phospho PSM identifications over ETD with increases of 2.6-fold and 1.9-fold, respectively. EThcD and AI-ETD generate approximately equivalent median SEQUEST Xcorr values, a measure of spectral match quality. Both improve upon the median score seen with ETD even with the large increase in number of phospho PSMs, illustrating that the phospho PSMs gained are high quality spectra. Surprisingly, AI-ETD+, which proved to be the optimal supplemental activation approach for non-modified peptides, underperformed compared to AI-ETD, indicating that the additional IRMPD activation may have contributed to over-fragmentation. We note that HCD outperformed all ETD-based analyses, but AI-ETD provided the greatest number of unique phosphopeptides when comparing the overlap of HCD and ETD-based methods (Figure S1). AI-ETD also provided 1,405 unique phosphopeptide identifications when evaluating the overlap in identifications between ETD, EThcD, and AI-ETD, compared to 153 and 471 unique phosphopeptides with ETD and EThcD, respectively (Figure S2).
Figure 2. Performance of AI-ETD and other ETD activation methods for phosphoproteome analysis.

a) The number of confidently localized phospho peptide spectral matches (phospho PSMs) for each activation method is shown, with singly- versus doubly-phosphorylated peptides being delineated (light blue versus dark blue, respectively). b) The median XCorr of PSMs and the number of unique phosphopeptides sequenced by each ETD activation methods is given. In (c) and (d), the distribution of peptide length (top) and peptide sequence coverage (bottom) is given for doubly (c) and triply (d) protonated peptides using ETD (gold) and AI-ETD at 15 W (blue). Data represents confidently localized phospho PSMs (part a). Here peptide sequence coverage is defined as the number of inter-residue bonds accounted for by b-, c-, y-, and z-type fragment ions compared to the total number of inter-residue bonds in the peptide sequence, calculated as a percentage.
To further evaluate the improvements of AI-ETD over ETD, we compared the length of phosphopeptides sequenced by both methods and the sequence coverage achieved for both doubly and triply protonated precursors (Figure 2c and d, respectively). Here and throughout this manuscript sequence coverage is defined as the ratio of [inter-residue positions that can be explained by observed product ions (b-, y-, c- or z•-type)] to [the total number of inter-residue positions (i.e., the number of residues minus 1)]. This number is reported as a percentage and the maximum sequence coverage attainable is by default 100%. AI-ETD generated ~10,000 more phospho PSMs for doubly protonated precursors than ETD and significantly increased the length of doubly charged phosphopeptides that were successfully sequenced. The majority of z = +2 phosphopeptides identified with AI-ETD also had sequence coverages of 80–100%. ETD performed more favorably for z = +3 precursors, but it still produced less than half of the identifications of AI-ETD, which successfully sequenced longer peptides than ETD while maintaining >80% sequence coverage for the majority of precursors.
To understand why AI-ETD soundly outperformed ETD, we compared LC-MS/MS data from non-enriched “whole” proteome samples to the phosphoproteome samples described here (Figure 3). Note, the whole proteome data comes from the companion manuscript on AI-ETD on the Lumos,47 and the peptides in that data set come from the same mouse brain lysate that was used for phosphopeptide enrichment for this study, making comparisons straight-forward. The charge state distribution of precursors selected for MS/MS fragmentation shifts to a wider range of charge states in the phosphoproteomic experiments (Figure 3a), which should benefit ETD; however, precursor m/z also shifts substantially to higher m/z values for doubly protonated precursors, with a less significant but still noticeable shift to higher m/z values for z = +3 precursors (Figure 3b). Thus, even with a wider range of charge states observed, precursors are shifted to lower charge densities overall, ultimately hindering ETD efficiency. These changes in precursor populations have been seen in other studies and are likely due not only to the addition of the modification mass but also to longer peptides that occur because of phosphorylation-based hindrance of trypsin digestion.85 Furthermore, if a phosphate group is deprotonated, the net charge of a peptide may be reduced, generating longer peptides at lower charge states (i.e., more peptides with lower charge density).
Figure 3. AI-ETD successfully fragments phosphopeptides that challenge standard ETD.

a) Precursor ion charge state distributions differ between whole proteome analysis and phosphopeptide-enriched samples, with the phosphoproteome shifting to higher charge states that should be favorable to ETD. b) The shift is charge states between whole proteome and phosphopeptide-enriched samples, however, is concomitant with a shift in precursor ion m/z distribution. Doubly protonated precursor ions (z= 2) are largely 350–800 Th in whole proteome samples (dark blue), where ETD is more effective at dissociating peptides. The precursor ion m/z distribution in phosphoproteome samples is distinctly shifted to higher m/z values (light blue), which limits the success of ETD. A shift to higher m/z values in the phosphoproteome also occurs for triply protonated precursor ions (z= 3), but the change is less dramatic. (c) The higher m/z values of phosphopeptides in this data set (light blue, part b) account for the low success rate of ETD on z= 2 precursors. (d) In contrast, AI-ETD performs remarkably well for z= 2 precursors compared to ETD and provides higher MS/MS success rates for all precursor charge states, which explains how the number of phospho PSMs sequenced can be more than tripled by AI-ETD over standard ETD (Figure 2a). Here MS/MS success rate represents the percentage of MS/MS scans that were successfully sequenced at PSMs for precursors at a given charge state.
Having the majority of precursors > 600 m/z is a consequential disadvantage for ETD but does not affect AI-ETD, which is reflected in the MS/MS success rates for precursors with different charge states between the two methods (Figure 3c and d). MS/MS success rate, reported as a percent, is defined as the number of PSMs successfully identified divided by the total number of MS/MS scans. AI-ETD maintains success rates above 60% for all precursor charge states, with a greater than 6-fold increase in success rate for z = +2 precursors. The clear improvement in success rates for all precursor charge states explains the more than 3-fold increase in phospho PSMs with AI-ETD.
Evaluating phosphopeptide fragmentation with AI-ETD
We systematically investigated the ETD, ETcaD, EThcD, AI-ETD, and AI-ETD+ fragmentation of phosphopeptides identified from tens of thousands of MS/MS spectra from our shotgun experiments, focusing on z = +2 precursors (Figure 4). Phosphoryl groups are chromophores for the 10.6 μm IR photons used in AI-ETD, making phosphopeptide fragmentation with AIETD particularly interesting.86 IRMPD studies have shown that phosphopeptides can be selectively fragmented over non-modified peptides because the phosphoryl moieties readily absorb IR photons, and the b- and y-type product ions formed by this fragmentation can both retain phosphoryl groups or undergo neutral loss.86–92 Previous work has also shown that AI-ETD on phosphopeptides with lowered bath gas pressures can generate band y-type ions from IRMPD-like activation.43 Here we show that b- and y-type products from IRMPD are present in AI-ETD spectra of phosphopeptides under normal operating conditions on the Lumos system, and we demonstrate that phosphate neutral losses caused by IRMPD can be used by a localization algorithm to improve phosphosite localization.
Figure 4. AI-ETD produces more sequence-informative product ions but also more phosphate losses from fragment ions.

a) The average number of product ions generated per peptide spectral match (PSM) is shown for each of the ETD fragmentation conditions. The total number of product ions is delineated by ion type, and phosphate neutral losses are included in this count. b) The average percent of total ion current (TIC) that is accounted for by b-, c-, y-, and z-type product ions shows that AI-ETD and AI-ETD+ generate the most total signal for product ions, but more of this signal resides in phosphate neutral losses ions than in other supplemental activation methods. These values represent the average percent TIC accounted for by product ions per PSM. c) Percent phosphate retention indicates the proportion of all product ions (b-, c-, y-, and z-type) that retained their phosphate group out of all product ions detected, i.e., those with and without phosphate neutral losses. The calculation for this metric is provided, and the percent phosphate retention from HCD analyses of phosphopeptides is show with a dotted line. d) Percent phosphate loss is the complement to percent phosphate retention (% phosphate loss = 100 - % phosphate retention). Here percent phosphate loss is calculated for each product ion type, showing that phosphate neutral losses from c- and z-type ions especially are a unique component of AI-ETD and AI-ETD+ spectra. All data in this figure are from doubly protonated precursors (z = 2).
Figure 4a presents the average number of product ions identified in MS/MS spectra of the different fragmentation methods, with the count being delineated by fragment ion type. EThcD significantly increases the number of b- and y-type ions generated while marginally increasing the number of c- and z•-type products, matching previous studies.40,83 AI-ETD (15 W) further improves upon c- and z•-type ion generation and produces even more b- and y-type fragments than EThcD. This differs from AI-ETD of non-modified peptides47 and is likely due to the increased internal energy of the precursors as phosphoryl groups absorb IR photons. AI-ETD+, which includes a short IRMPD activation step in the low pressure cell of the QLT, produces similar numbers of c- and z•-type ions to AI-ETD but generates the highest number of b- and y-type ions. Note, the fragment ion count includes intact b-, y-, c- and z•-type products and phosphate neutral losses from those ions. Figure 4b shows the average percent of total signal (total ion current) accounted for in b-, y-, c- and z•-type products and indicates how much of that signal comes from product ions that show phosphate neutral loss.
Clearly AI-ETD and AI-ETD+ generate considerable phosphate neutral loss from fragment ions, but a significant population of product ions retain their phosphoryl groups as well, as is expected with ETD-like fragmentation. We calculated a percent of total phosphoryl group retention, analogous to the calculations performed for UVPD spectra of phosphopeptides,23 to understand what fraction of product ions retain their phosphate moiety under different conditions (Figure 4c). Percent phosphoryl group retention represents the ratio of product ions (i.e., b-, y-, c- and z•-type) that retain the phosphoryl group to the total number of product ions that have the phosphorylation modification based on position in the phosphopeptide sequence. Nearly all product ions retain phosphoyl groups in ETD, while ~80% of product ions retain the modification in ETciD, EThcD, and lower laser power AI-ETD. AI-ETD (15 W), however, shows a more significant drop in product ion phosphoryl group retention, and the phosphoryl group retention in AI-ETD+ is lowest, nearly matching the amount of loss seen in HCD spectra. Figure 4d shows the percent phosphate loss (i.e., the complement to phosphate retention, or 100 – percent phosphoryl group retention) delineated by product ion type. For all fragmentation methods, if b-type ions are present, they are more likely to show phosphate loss than any other ion. Surprisingly, c- and z•-type product ions show the most significant phosphate loss in AI-ETD and AI-ETD+, a phenomenon that has not been reported.
Although phosphate neutral loss from product ions is not ideal, it is not debilitating. HCD spectra have considerable phosphate neutral losses from b- and y-type product ions, but phosphosite localization is still very much achievable in these cases.81,93,94 In fact, many phosphosite localization algorithms use phosphate neutral losses to aid in the localization process (Figure S1). The phosphoRS algorithm was modified for EThcD fragmentation to include phosphate neutral losses from b- and y-type ions in the localization calculation,81,83 but no algorithm to date has had reason to include phosphate losses from c- and z•-type fragments.
We reasoned we could improve our localization in AI-ETD spectra by modifying the phosphoRS algorithm to utilize neutral losses from all four fragment ion types in its calculation, analogous to how it currently uses neutral losses for HCD-type spectra. Figure 5a presents the percent gain in phospho PSMs for AI-ETD and AI-ETD+ when considering phosphate neutral loss from three different sets for product ions compared to localizing phosphosites without any consideration of phosphate neutral losses. Moderate gains were seen for AI-ETD (12 W) for each of the three fragment ion sets (c and z; b and y; and all four), but more considerable increases were achieved for AI-ETD (15 W) and AI-ETD+, matching the trends in Figure 4. Figure 5b shows the difference in localized phospho PSMs for ETD and the different supplemental activation methods when considering phosphate neutral losses from all four fragment ion types (dark blue) or when not considering any neutral losses (light blue). Note, the results from considering neutral losses is what is presented in Figure 2a. The gains in number of localized phospho PSMs are significant for AI-ETD and AI-ETD+, demonstrating that modifications to localization software to consider these features of AI-ETD spectra of phosphopeptides is of value.
Figure 5. Using phosphate losses from fragment ions improves phosphosite localization with AI-ETD.

a) phosphoRS software was modified to use phosphate losses from product ions in ETD spectra to aid phosphosite localization. The percent gain in the number of localized phospho PSMs after modifying phosphoRS is shown for AI-ETD conditions. Using phosphate losses from different combinations of production types (c- and z-type ions only, b- and y-type ions only, and all four ion types) provides varying gains, with the biggest increase achieved when considering losses from all four ion types. b) The number of localized phospho PSMs for ETD, ETciD, EThcD, and AI-ETD conditions are compared when not considering any neutral losses in the phosphosite localization calculation or when enabling use of phosphate losses from product ions in ETD spectra.
AI-ETD for top-down phosphoproteomics
Extensive backbone fragmentation is equally, if not more, important when trying to localize phosphosites on intact proteins as the number of potential modification sites greatly increases with longer amino acid chains. AI-ETD improves fragmentation and sequence coverage for top-down proteomics,95–97 but it has yet to be applied to intact phosphoproteins. To investigate the utility of AI-ETD for top-down phosphoproteomics, we fragmented five charge states of α-casein spanning the charge state envelope of the protein (z = +22, +20, +18, +16, and +14) and compared performance to HCD, ETD, and EThcD. The most dominant precursor species contained eight sites of phosphorylation, although nine known sites of phosphorylation and 31 potential sites (S/T/Y) exist.
Figure 6a reports the sequence coverage achieved with each fragmentation method for each of the five precursors selected. AI-ETD provides the highest sequence for each charge state, even outperforming EThcD, which is known to aid in sequence coverage and phosphosite localization.74 AI-ETD is the only fragmentation method to provide unambiguous localization of all eight sites of phosphorylation for each of the five precursors, although ETD also performed well (Figure 6b). AI-ETD generates more fragment ions than ETD and EThcD for all precursors, too (Figure 6c–e, blue line plots, left axes). Knowing how AI-ETD affected product ion generation in phosphopeptides, we investigated the percentage of matching fragments (i.e., the number of fragments matched by ProSight Lite) that were c/z•-type versus b/y-type (Figure 6c–e, gold bars, right axes). Nearly all fragment ions formed by ETD were c/z•-type, i.e., less than ~5% of the total sequence-informative product ions were b/y-type ions. EThcD substantially increase the ratio of b/y-type ions formed, with ~30–40% of all matched fragments being b/y-type and 52% of fragments of being b/y-type for the z = +14 precursor, which agrees with previous investigations.74 AI-ETD noticeably increases the ratio of b/y-type ions (~10–20%) over ETD, but is only roughly half of the ratio seen with EThcD. This differs from the phosphopeptide fragmentation data above, likely because the increase in internal energy that occurs from IR photon absorption is distributed along a much longer amino acid sequence for intact proteins, meaning the overall energy of the precursor ions is not increased enough to induce as much collisional activation into b/y-type products.
Figure 6. AI-ETD enables confident characterization of intact phosphoprotein α-casein.

a) AI-ETD provides the greatest sequence coverage of α-casein for all charge states investigated. b) The phosphor isoform of α-casein investigated had 8 phosphosites (out of nine previously reported sites). Only AI-ETD confidently localized all eight phosphosites for each precursor fragmented. The number of fragments (blue line) and the percentage of these fragments that are b- and y-type product ions (gold bars) are shown from ETD (c), EThcD (d), and AI-ETD (e).
The benefits of AI-ETD are clear when comparing ETD and AI-ETD spectra (Figure 7). AI-ETD decreases the amount of charge-reduced precursor (dominant peaks from 1400–2000 m/z) compared to ETD while boosting the number and abundance of product ions (Figure 7a). A zoom in of the spectra (Figure 7b, representative of the blue region in part a) shows that AI-ETD increases signal of product ions detected in ETD while also generating new product ions. Formation of new c-, z-, and y-type ions are easily distinguished in the AI-ETD spectrum, too. The product ions highlighted in yellow (c454+, c464+, and c474+) were critical in localizing the phosphosite on serine 46. All three c-type fragments are present in the AI-ETD spectrum, but c454+ and c464+ are either not present or have too low S/N for effective deconvolution in the ETD spectrum. Note, ETD and AI-ETD spectra are on the same intensity scale for both parts a and b. Figure 7c depicts a sequence coverage map for AI-ETD of z = +16 precursor of α-casein, showing the confident localization of all eight phosphosites of the modified precursor, and Figure S3 and S4 compare sequence coverage maps from ETD to AI-ETD data. The ability of AI-ETD to produce high quality data across a wide range of precursor charge states and m/z is extremely beneficial because it mitigates the reliance on exclusively high charge density (i.e., highly charged, low m/z) precursors while still providing extensive sequence coverage that is a hallmark of ETD for intact proteins.
Figure 7. Comparing ETD and AI-ETD for the z = 16 precursor of α-casein with 8 phosphosites.

a) Full spectra of ETD and AI-ETD fragmentation show more signal in fragment ions and less signal in charge reduced precursor ions with AI-ETD, with AI-ETD providing significantly more matching fragments and protein sequence coverage. Both spectra are on the same intensity scale. b) A zoom in of the blue highlighted region of the spectra in part a show that AI-ETD generates more product ions with higher abundance than ETD. Increased product ion signal in AI-ETD also provided easy deconvolution of c45–c47 product ions that were crucial in localizing phosphosite S46. Low signal-to-noise in the ETD spectrum prevented identification of two of these three product ions. These spectra are also on the same intensity scale. c) A sequence coverage map of α-casein using AI-ETD shows confident localization of all 8 phosphoserines. This product ion map is from the AI-ETD spectrum in part a. Dark blue marks denote c- and z-type ions while light blue show b- and y-type products.
CONCLUSION
In a companion manuscript we showed that AI-ETD can be implemented in a straight-forward and robust manner on a quadrupole-Orbitrap-linear ion trap hybrid MS system (Orbitrap Fusion Lumos), and here we have shown that AI-ETD can significantly improve phosphoproteomic analyses for both shotgun and top-down approaches. AI-ETD more than triples the number of localized phospho PSMs sequenced by ETD, which corresponds to a nearly 2.5-fold increase in unique localized phosphopeptides identified. These gains are realized because AI-ETD improves phosphopeptide fragmentation at all precursor charge states compared to ETD, producing substantially more product ions and providing much greater peptide sequence coverage. AI-ETD of phosphopeptides was also shown to produce more b- and y-type ions than expected, in addition to increased generation of c- and z•-type fragments. Phosphate neutral losses from all four fragment ion types were observed with AI-ETD, including losses from c- and z•-type fragments, which was surprising. These neutral loss products can be used by modified version of phosphoRS to improve phosphosite localization in AI-ETD spectra, which provides a blueprint for modifying other localization software to best utilize the rich fragmentation produced by AI-ETD. Finally, we have demonstrated that AI-ETD is the superior fragmentation method for sequencing the multiply-phosphorylated protein α-casein, where AI-ETD provided the highest sequence coverage and best ability to localize phosphosites over HCD, ETD, and EThcD. We conclude, AI-ETD is poised to be a premier method for top-down phosphoproteomics.
Looking forward, several studies have shown the negative mode to be an interesting avenue to pursue for characterizing the phosphoproteome.23,88,98–100 Negative electron transfer dissociation (NETD) is a valuable method for sequencing peptide anions in shotgun proteomic experiments, but it suffers from analogous charge-density limitations as positive ETD.101–103 This is especially noteworthy because doubly deprotonated precursors (i.e., low charge density precursors) are the main ion population in negative electrospray, even more so than in positive mode. Activated ion NETD (AI-NETD) has been shown to effectively mitigate this problem to improve peptide anion sequence characterization and enable whole proteome analysis in the negative mode.104,105 As such, AI-NETD represents an exciting new dimension to phosphoproteome analysis. The front-end ETD source on the q-OT-QLT system is capable of producing reagent cation for NETD reactions,48 and this work moves the field one step closer to implementing activated ion negative electron transfer dissociation for in-depth phosphoproteomic studies. In all, the work presented here both shows that AI-ETD is becoming a more accessible tool for the proteomics community and also opens the door to new frontiers of research with this technique. It can bring tangible benefits to challenging problems in phosphoproteomics and other PTM analyses and serve as another avenue for translating MS-based phosphoproteomic analyses into valuable biological contexts.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge support from Thermo Fisher Scientific and NIH grant R35 GM118110 awarded to J.J.C. N.M.R. was funded through an NIH Predoctoral to Postdoctoral Transition Award (F99 CA212454). The authors also thank Jae Schwartz, John E. P. Syka, and Chris Mullen for helpful discussions.
Footnotes
Additional information available in supporting documents, including expanding Materials and Methods descriptions. Raw data and information about identified spectra are available on Chorus (Project ID 1288).
References
- 1.Johnson LN. Biochem Soc Trans. 2009;37:627–641. doi: 10.1042/BST0370627. [DOI] [PubMed] [Google Scholar]
- 2.Thorner J, Hunter T, Cantley LC, Sever R. Cold Spring Harb Perspect Biol. 2014;6:a022913. doi: 10.1101/cshperspect.a022913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solari FA, Dell’Aica M, Sickmann A, Zahedi RP. Mol Biosyst. 2015;11:1487–1493. doi: 10.1039/c5mb00024f. [DOI] [PubMed] [Google Scholar]
- 4.Engholm-Keller K, Larsen MR. Proteomics. 2013;13:910– 931. doi: 10.1002/pmic.201200484. [DOI] [PubMed] [Google Scholar]
- 5.Olsen JV, Mann M. Mol Cell Proteomics. 2013;12:3444– 3452. doi: 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doll S, Burlingame AL. ACS Chem Biol. 2015;10:63–71. doi: 10.1021/cb500904b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riley NM, Coon JJ. Anal Chem. 2016;88:74–94. doi: 10.1021/acs.analchem.5b04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macek B, Mann M, Olsen JV. Annu Rev Pharmacol Toxicol. 2009;49:199–221. doi: 10.1146/annurev.pharmtox.011008.145606. [DOI] [PubMed] [Google Scholar]
- 9.von Stechow L, Francavilla C, Olsen JV. Expert Rev Proteomics. 2015;12:469–487. doi: 10.1586/14789450.2015.1078730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boersema PJ, Mohammed S, Heck AJR. J Mass Spectrom. 2009;44:861–878. doi: 10.1002/jms.1599. [DOI] [PubMed] [Google Scholar]
- 11.Brodbelt JS. Anal Chem. 2016;88:30–51. doi: 10.1021/acs.analchem.5b04563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JJ. Anal Chem. 2004;76:3590–3598. doi: 10.1021/ac0497104. [DOI] [PubMed] [Google Scholar]
- 13.Nagaraj N, D’Souza RCJ, Cox J, Olsen JV, Mann M. J Proteome Res. 2010;9:6786–6794. doi: 10.1021/pr100637q. [DOI] [PubMed] [Google Scholar]
- 14.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JEP, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Proc Natl Acad Sci U S A. 2007;104:2193– 2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Proc Natl Acad Sci. 2007;104:2199–2204. doi: 10.1073/pnas.0611217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith SA, Kalcic CL, Safran KA, Stemmer PM, Dantus M, Reid GE. J Am Soc Mass Spectrom. 2010;21:2031–2040. doi: 10.1016/j.jasms.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Shaffer CJ, Slováková K, Tureček F. Int J Mass Spectrom. 2015 doi: 10.1002/jms.3551. [DOI] [PubMed] [Google Scholar]
- 18.Lemoine J, Tabarin T, Antoine R, Broyer M, Dugourd P. Rapid Commun Mass Spectrom. 2006;20:507–511. doi: 10.1002/rcm.2333. [DOI] [PubMed] [Google Scholar]
- 19.Park S, Ahn W-K, Lee S, Han SY, Rhee BK, Oh H Bin. Rapid Commun Mass Spectrom. 2009;23:3609–3620. doi: 10.1002/rcm.4184. [DOI] [PubMed] [Google Scholar]
- 20.Madsen JA, Cheng RR, Kaoud TS, Dalby KN, Makarov DE, Brodbelt JS. Chemistry. 2012;18:5374–5383. doi: 10.1002/chem.201103534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim TY, Reilly JP. J Am Soc Mass Spectrom. 2009;20:2334–2341. doi: 10.1016/j.jasms.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 22.Fort KL, Dyachenko A, Potel CM, Corradini E, Marino F, Barendregt A, Makarov AA, Scheltema RA, Heck AJR. Anal Chem. 2016;88:2303–2310. doi: 10.1021/acs.analchem.5b04162. [DOI] [PubMed] [Google Scholar]
- 23.Robinson MR, Taliaferro JM, Dalby KN, Brodbelt JS. J Proteome Res. 2016;15:2739–2748. doi: 10.1021/acs.jproteome.6b00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayfield JE, Robinson MR, Cotham VC, Irani S, Matthews WL, Ram A, Gilmour DS, Cannon JR, Zhang YJ, Brodbelt JS. ACS Chem Biol. 2016 doi: 10.1021/acschembio.6b00729. acschembio.6b00729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc Natl Acad Sci U S A. 2004;101:9528– 9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coon JJ. Anal Chem. 2009;81:3208–3215. doi: 10.1021/ac802330b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim MS, Pandey A. Proteomics. 2012;12:530–542. doi: 10.1002/pmic.201100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhurov KO, Fornelli L, Wodrich MD, Laskay Üa, Tsybin YO. Chem Soc Rev. 2013;42:5014–5030. doi: 10.1039/c3cs35477f. [DOI] [PubMed] [Google Scholar]
- 29.Sarbu M, Ghiulai RM, Zamfir AD. Amino Acids. 2014;46:1625–1634. doi: 10.1007/s00726-014-1726-y. [DOI] [PubMed] [Google Scholar]
- 30.Good DM, Wirtala M, McAlister GC, Coon JJ. Mol Cell Proteomics. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Laszlo KJ, Munger EB, Bush MF. J Am Chem Soc. 2016;138:9581–9588. doi: 10.1021/jacs.6b04282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Little DP, Speir JP, Senko MW, O’Connor PB, McLafferty FW. Anal Chem. 1994;66:2809–2815. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 33.Clemmer DE, Hudgins RR, Jarrold MF. J Am Chem Soc. 1995;117:10141–10142. [Google Scholar]
- 34.Breuker K, Oh H, Horn DM, Cerda BA, McLafferty FW. J Am Chem Soc. 2002;124:6407–6420. doi: 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]
- 35.Pitteri SJ, Chrisman PA, McLuckey SA. Anal Chem. 2005;77:5662–5669. doi: 10.1021/ac050666h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lermyte F, Williams JP, Brown JM, Martin EM, Sobott F. J Am Soc Mass Spectrom. 2015;26:1068–1076. doi: 10.1007/s13361-015-1124-z. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, McLuckey SA. Int J Mass Spectrom. 2012;330:174–181. doi: 10.1016/j.ijms.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gunawardena HP, He M, Chrisman PA, Pitteri SJ, Hogan JM, Hodges BDM, McLuckey SA. J Am Chem Soc. 2005;127:12627–12639. doi: 10.1021/ja0526057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swaney DL, McAlister GC, Wirtala M, Schwartz JC, Syka JE, Coon JJ. Anal Chem. 2007;79:477–485. doi: 10.1021/ac061457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frese CK, Altelaar AFM, van den Toorn H, Nolting D, Griep-Raming J, Heck AJR, Mohammed S. Anal Chem. 2012;84:9668–9673. doi: 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- 41.Ledvina AR, McAlister GC, Gardner MW, Smith SI, Madsen JA, Schwartz JC, Stafford GC, Jr, Syka JE, Brodbelt JS, Coon JJ. Angew Chem Int Ed Engl. 2009;48:8526–8528. doi: 10.1002/anie.200903557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ledvina AR, Beauchene Na, McAlister GC, Syka JEP, Schwartz JC, Griep-Raming J, Westphall MS, Coon JJ. Anal Chem. 2010;82:10068–10074. doi: 10.1021/ac1020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ledvina AR, Rose CM, McAlister GC, Syka JEP, Westphall MS, Griep-Raming J, Schwartz JC, Coon JJ. J Am Soc Mass Spectrom. 2013;24:1623–1633. doi: 10.1007/s13361-013-0621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Connor PB, Lin C, Cournoyer JJ, Pittman JL, Belyayev M, Budnik BA. J Am Soc Mass Spectrom. 2006;17:576–585. doi: 10.1016/j.jasms.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 45.Sun R-X, Dong M-Q, Song C-Q, Chi H, Yang B, Xiu L-Y, Tao L, Jing Z-Y, Liu C, Wang L-H, Fu Y, He S-M. J Proteome Res. 2010;9:6354–6367. doi: 10.1021/pr100648r. [DOI] [PubMed] [Google Scholar]
- 46.Xia Y, Han H, McLuckey SA. Anal Chem. 2008;80:1111– 1117. doi: 10.1021/ac702188q. [DOI] [PubMed] [Google Scholar]
- 47.Riley NM, Westphall MS, Hebert AS, Coon JJ. Anal Chem. 2017 doi: 10.1021/acs.analchem.7b00213. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Earley L, Anderson LC, Bai DL, Mullen C, Syka JEP, English AM, Dunyach JJ, Stafford GC, Shabanowitz J, Hunt DF, Compton PD. Anal Chem. 2013;85:8385– 8390. doi: 10.1021/ac401783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riley NM, Mullen C, Weisbrod CR, Sharma S, Senko MW, Zabrouskov V, Westphall MS, Syka JEP, Coon JJ. J Am Soc Mass Spectrom. 2016;27:520–531. doi: 10.1007/s13361-015-1306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rose CM, Rush MJP, Riley NM, Merrill AE, Kwiecien NW, Holden DD, Mullen C, Westphall MS, Coon JJ. J Am Soc Mass Spectrom. 2015 doi: 10.1007/s13361-015-1183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV. Nat Commun. 2012;3:876. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Humphrey SJ, Yang G, Yang P, Fazakerley DJ, Stöckli J, Yang JY, James DE. Cell Metab. 2013;17:1009–1020. doi: 10.1016/j.cmet.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJR, Mohammed S. J Proteome Res. 2013;12:260– 271. doi: 10.1021/pr300630k. [DOI] [PubMed] [Google Scholar]
- 55.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA. Nat Methods. 2013;10:634–637. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monetti M, Nagaraj N, Sharma K, Mann M. Nat Methods. 2011;8:655–658. doi: 10.1038/nmeth.1647. [DOI] [PubMed] [Google Scholar]
- 57.Rigbolt KTG, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, Kassem M, Mann M, Olsen JV, Blagoev B. Sci Signal. 2011;4:rs3. doi: 10.1126/scisignal.2001570. [DOI] [PubMed] [Google Scholar]
- 58.Sharma K, D’Souza RCJ, Tyanova S, Schaab C, Wiśniewski JR, Cox J, Mann M. Cell Rep. 2014;8:1583– 1594. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 59.Kelstrup CD, Jersie-Christensen RR, Batth TS, Arrey TN, Kuehn A, Kellmann M, Olsen JV. J Proteome Res. 2014;13:6187–6195. doi: 10.1021/pr500985w. [DOI] [PubMed] [Google Scholar]
- 60.Batth TS, Francavilla C, Olsen JV. J Proteome Res. 2014;13:6176–6186. doi: 10.1021/pr500893m. [DOI] [PubMed] [Google Scholar]
- 61.de Graaf EL, Giansanti P, Altelaar AFM, Heck AJR. Mol Cell Proteomics. 2014;13:2426–2434. doi: 10.1074/mcp.O113.036608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bian Y, Song C, Cheng K, Dong M, Wang F, Huang J, Sun D, Wang L, Ye M, Zou H. J Proteomics. 2014;96:253–262. doi: 10.1016/j.jprot.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 63.Ruprecht B, Koch H, Medard G, Mundt M, Kuster B, Lemeer S. Mol Cell Proteomics. 2015;14:205–215. doi: 10.1074/mcp.M114.043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giansanti P, Aye TT, van den Toorn H, Peng M, van Breukelen B, Heck AJR. Cell Rep. 2015;11:1834–1843. doi: 10.1016/j.celrep.2015.05.029. [DOI] [PubMed] [Google Scholar]
- 65.Humphrey SJ, Azimifar SB, Mann M. Nat Biotechnol. 2015 doi: 10.1038/nbt.3327. advance on. [DOI] [PubMed] [Google Scholar]
- 66.Marx H, Lemeer S, Schliep JE, Matheron L, Mohammed S, Cox J, Mann M, Heck AJR, Kuster B. Nat Biotechnol. 2013;31:557–564. doi: 10.1038/nbt.2585. [DOI] [PubMed] [Google Scholar]
- 67.Garcia BA. J Am Soc Mass Spectrom. 2010;21:193–202. doi: 10.1016/j.jasms.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 68.Smith LM, Kelleher NL. Nat Methods. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sze SK, Ge Y, Oh H, McLafferty FW. Proc Natl Acad Sci U S A. 2002;99:1774–1779. doi: 10.1073/pnas.251691898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ge Y, Rybakova IN, Xu Q, Moss RL. Proc Natl Acad Sci. 2009;106:12658–12663. doi: 10.1073/pnas.0813369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang J, Guy MJ, Norman HS, Chen YC, Xu Q, Dong X, Guner H, Wang S, Kohmoto T, Young KH, Moss RL, Ge Y. J Proteome Res. 2011;10:4054–4065. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zabrouskov V, Ge Y, Schwartz J, Walker JW. Mol Cell Proteomics. 2008;7:1838–1849. doi: 10.1074/mcp.M700524-MCP200. [DOI] [PubMed] [Google Scholar]
- 73.Gregorich ZR, Peng Y, Cai W, Jin Y, Wei L, Chen AJ, McKiernan SH, Aiken JM, Moss RL, Diffee GM, Ge Y. J Proteome Res. 2016;15:2706–2716. doi: 10.1021/acs.jproteome.6b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brunner AM, Lossl P, Liu F, Huguet R, Mullen C, Yamashita M, Zabrouskov V, Makarov A, Altelaar AFM, Heck AJR. Anal Chem. 2015;87:4152–4158. doi: 10.1021/acs.analchem.5b00162. [DOI] [PubMed] [Google Scholar]
- 75.Toby TK, Fornelli L, Kelleher NL. Annu Rev Anal Chem. 2016;9:499–519. doi: 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eng JK, McCormack AL, Yates JR. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 77.Good DM, Wenger CD, McAlister GC, Bai DL, Hunt DF, Coon JJ. J Am Soc Mass Spectrom. 2009;20:1435– 1440. doi: 10.1016/j.jasms.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Good DM, Wenger CD, Coon JJ. Proteomics. 2010;10:164–167. doi: 10.1002/pmic.200900570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Nat Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 80.Spivak M, Weston J, Bottou L, Käll L, Noble WS. J Proteome Res. 2009;8:3737–3745. doi: 10.1021/pr801109k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taus T, Köcher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K. J Proteome Res. 2011;10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- 82.Fellers RT, Greer JB, Early BP, Yu X, LeDuc RD, Kelleher NL, Thomas PM. Proteomics. 2014 doi: 10.1002/pmic.201570050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frese CK, Zhou H, Taus T, Altelaar AFM, Mechtler K, Heck AJR, Mohammed S. J Proteome Res. 2013;12:1520–1525. doi: 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mommen GPM, Frese CK, Meiring HD, van Gaansvan den Brink J, de Jong APJM, van Els CACM, Heck AJR. Proc Natl Acad Sci U S A. 2014;111:4507–4512. doi: 10.1073/pnas.1321458111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dickhut C, Feldmann I, Lambert J, Zahedi RP. J Proteome Res. 2014;13:2761–2770. doi: 10.1021/pr401181y. [DOI] [PubMed] [Google Scholar]
- 86.Crowe MC, Brodbelt JS. J Am Soc Mass Spectrom. 2004;15:1581–1592. doi: 10.1016/j.jasms.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 87.Crowe MC, Brodbelt JS. Anal Chem. 2005;77:5726–5734. doi: 10.1021/ac0509410. [DOI] [PubMed] [Google Scholar]
- 88.Flora JW, Muddiman DC. Anal Chem. 2001;73:3305–3311. doi: 10.1021/ac010333u. [DOI] [PubMed] [Google Scholar]
- 89.Flora JW, Muddiman DC. J Am Chem Soc. 2002;124:6546–6547. doi: 10.1021/ja0261170. [DOI] [PubMed] [Google Scholar]
- 90.Vasicek La, Ledvina AR, Shaw J, Griep-Raming J, Westphall MS, Coon JJ, Brodbelt JS. J Am Soc Mass Spectrom. 2011;22:1105–1108. doi: 10.1007/s13361-011-0119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stephenson JL, Booth MM, Boué SM, Eyler JR, Yost RA. 1996:512–564. [Google Scholar]
- 92.Flora JW, Muddiman DC. J Am Soc Mass Spectrom. 2004;15:121–127. doi: 10.1016/j.jasms.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 93.Savitski MM, Lemeer S, Boesche M, Lang M, Mathieson T, Bantscheff M, Kuster B. Mol Cell Proteomics. 2011;10:M110.003830. doi: 10.1074/mcp.M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beausoleil SA, Villén J, Gerber SA, Rush J, Gygi SP. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 95.Riley NM, Westphall MS, Coon JJ. Anal Chem. 2015;87:7109–7116. doi: 10.1021/acs.analchem.5b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhao Y, Riley NM, Sun L, Hebert AS, Yan X, Westphall MS, Rush MJP, Zhu G, Champion MM, Medie FM, Champion PAD, Coon JJ, Dovichi NJ. Anal Chem. 2015;87:5422–5429. doi: 10.1021/acs.analchem.5b00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bourgoin-Voillard S, Leymarie N, Costello CE. Proteomics. 2014;14:1174–1184. doi: 10.1002/pmic.201300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Robinson MR, Moore KL, Brodbelt JS. J Am Soc Mass Spectrom. 2014;25:1461–1471. doi: 10.1007/s13361-014-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huzarska M, Ugalde I, Kaplan DA, Hartmer R, Easterling ML, Polfer NC. Anal Chem. 2010;82:2873– 2878. doi: 10.1021/ac9028592. [DOI] [PubMed] [Google Scholar]
- 100.Madsen JA, Kaoud TS, Dalby KN, Brodbelt JS. Proteomics. 2011;11:1329–1334. doi: 10.1002/pmic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McAlister GC, Russell JD, Rumachik NG, Hebert AS, Syka JEP, Geer LY, Westphall MS, Pagliarini DJ, Coon JJ. Anal Chem. 2012;84:2875–2882. doi: 10.1021/ac203430u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shaw JB, Madsen JA, Xu H, Brodbelt JS. J Am Soc Mass Spectrom. 2012;23:1707–1715. doi: 10.1007/s13361-012-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rush MJP, Riley NM. J Am Soc Mass Spectrom. 2017 doi: 10.1007/s13361-017-1600-8. [DOI] [Google Scholar]
- 104.Riley NM, Rush MJP, Rose CM, Richards AL, Kwiecien NW, Bailey DJ, Hebert AS, Westphall MS, Coon JJ. Mol Cell Proteomics. 2015;14:2644–2660. doi: 10.1074/mcp.M115.049726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Riley NM, Bern M, Westphall MS, Coon JJ. J Proteome Res. 2016;15:2768–2776. doi: 10.1021/acs.jproteome.6b00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.