

Abstract
Na+/H+ exchanger isoform 9, NHE9, finely tunes the pH within the endosomal lumen to regulate cargo trafficking and turnover. In patients with autism, genetic approaches have revealed deletions, truncations and missense mutations in the gene encoding NHE9 (SLC9A9). To help establish causality, functional evaluation is needed to distinguish pathogenic mutations from harmless polymorphisms. Here, we evaluated three previously uncharacterized NHE9 variants, P117T, D496N, and Q609K reported in patients with autism and epilepsy. We show that NHE9-DsRed localizes to recycling endosomes in HEK293 cells where it significantly alkalinizes luminal pH, and elevates accumulation of transferrin. All three NHE9 variants were expressed and localized to endosomal compartments, similar to wild-type NHE9. In contrast to previously characterized NHE9 variants, we observed no loss-of-function with respect to endosomal pH homeostasis and transferrin endocytosis. These findings suggest that the three NHE9 substitutions analyzed in our study are either benign polymorphisms or may have a cell-type specific or regulatory function not detected in our cell culture model. Our findings highlight the importance of combining the use of cellular studies of function with sequencing technologies that capture genomic variation in patients.
Keywords: Endosomal PH, Autism
Objective
Given the importance of functional evaluation of genetic variants to establish causal disease associations, we sought to evaluate three previously uncharacterized NHE9 missense substitutions that were identified in patients with autism and epilepsy.
Introduction
Autism spectrum disorder (ASD) has emerged as a major public health concern worldwide, with a prevalence of 1 in 68 children and a growing adult population with an urgent, unmet therapeutic need [3] . Although autism is highly heritable, the identification of candidate genes has been complicated by extreme genetic heterogeneity, polygenic inheritance, occurrence of de novo mutations and rare variants that individually do not contribute to more than 1% of cases [4] . Furthermore, most genes remain candidates because of a lack of functional validation. Among these candidates is the endosomal Na+/H+ exchanger NHE9 (SLC9A9), also named as autism susceptibility candidate 16 (AUTS16) (OMIM: 613410) [5] . Homozygosity mapping in autism pedigrees with shared ancestry identified a large deletion upstream from the 5′ end of NHE9. Further analysis of NHE9 sequence in non-consanguineous autism patients revealed a nonsense mutation and 6 rare, missense variants not found in controls [1] . The nonsense change in NHE9 was phenotypically similar to an adjacent nonsense mutation in a related isoform, NHE1 that is causal to slow wave epilepsy in mouse [6] . However, in the absence of functional evaluation, it was not possible to distinguish whether the remaining variants were benign polymorphisms or pathological mutations causal to autism phenotypes. Previously, loss of function was demonstrated in three of these variants (V176I, L236S and S438P) [2] ; here, we evaluated the remaining three variants (P117T, D496N, and Q609K) reported in patients with autism and epilepsy [1] . When expressed in HEK293 cells, all three were similar to wild type in endosomal expression and function. We discuss the implications of our findings on future diagnosis and treatment of autism patients.
Results & Discussion
NHE9 missense substitutions that were identified in patients with autism and epilepsy localize throughout the coding frame, including the membrane embedded NHE-homology domain and the C-terminal cytoplasmic tail [1] (Figure 1A). Of these, P117T localizes to an extracellular loop in NHE9 that corresponds to a region between transmembrane helices I and II in E. coli NhaA crystal structure [2] . The other two variants, D496N and Q609K, are localized to the C-terminal tail. As a starting point, we calculated evolutionary conservation (Consurf) score for these three residues, using a scale ranging from 1 (highly variable) to 9 (invariant) [7] . Whereas Gln609 is poorly conserved with a Consurf score of 1, Pro117 and Asp496 are moderately conserved with score of 4 and 6, respectively (Supplemental table 1). We then evaluated the physiochemical effects of these substitutions and predicted the functional consequences using several standard bioinformatic approaches (Supplemental table 1). The P117T substitution exhibits a shift in polarity from non-polar to polar and displays an increase in Kyte-Doolitle hydrophobicity from -1.6 to -0.7. The D496N substitution exhibits a shift in polarity from negatively charged to polar and displays no change in hydrophobicity (-3.5). The Q609K variation exhibits a shift in polarity from polar to positively charged and displays a decrease in hydrophobicity from -3.5 to -3.9. The outcome predictions from bioinformatic efforts varied from being neutral to disease causing (Supplemental table 1). To experimentally test these predictions on endosomal function, we expressed NHE9 variants with C-terminal DsRed tag in HEK293 cells and evaluated protein expression by western analysis, using an anti-DsRed antibody. NHE9-DsRed migrated as prominent high molecular weight smear (>200 kDa) that was resolved into discrete band(s) with dithiothreitol (DTT) treatment, suggesting oligomerization by disulfide bonds (Figure 1B). Densitometric scan of immunoblot normalized to tubulin showed that the DsRed-tagged variants, P117T, D496N, and Q609K, were expressed at levels ∼60-90% of wild-type (WT) NHE9, suggesting that autism variants might result in modest changes in expression (Figure 1C). To compare subcellular localization of NHE9 variants, we determined DsRed fluorescence overlap with Alexa Flour 488 labelled transferrin (TFN), following 60-min incubation with live cells (Figure 1D). NHE9 (WT and variants) showed characteristic distribution to vesicular compartments overlapping with TFN as previously described [2] . Fractional colocalization determined using Manders' coefficient revealed no significant differences (WT: 0.83±0.03, P117T: 0.80±0.04, D496N: 0.88±0.03, Q609K: 0.87±0.03), suggesting no deleterious effects of these autism-associated NHE9 variants on endosomal trafficking. We took advantage of the excellent overlap of NHE9 (WT and variants) with TFN to determine endosomal pH as a measure of Na+/H+ exchange activity [2] [8] . Endosomal pH was quantitatively measured by flow cytometry using pH-sensitive fluorescein (FITC) tagged TFN. Uptake of TFN into endosomes was normalized using pH-insensitive Alexa Fluor 633 tag and calibrated in four buffers with fixed-pH for a ratiometric determination of lumen pH [8] (R2, square of the Pearson correlation coefficient=0.969; Figure 1E). Consistent with proton-leak function of NHE9, we documented alkalization of endosomal pH from 6.10 ± 0.09 in vector-transformed control, to 7.05 ± 0.04 in cells expressing WT NHE9 (mean ± S.D.; n=three biological replicates; ***,P<0.001; two-tailed t test; Figure 1F). To rule out nonspecific overexpression effects, we examined an autism with epilepsy associated S438P mutation within the membrane-embedded transporter domain of NHE9 (Figure 1A), which was previously shown to result in loss of function [2] . NHE9 carrying the S438P mutation failed to elevate endosomal pH in HEK293 cells, confirming loss of function as predicted using a structure-function approach (mean ± S.D.; n=three biological replicates; NS, not significant; P=0.155; two-tailed t test; Figure 1F). We used this gain-of-function phenotype to score NHE9 variants. In contrast to previously characterized NHE9 autism variants [2], we observed significant alkalization of endosomes with expression of P117T, D496N, and Q609K substitutions, relative to empty vector transfection (mean ± S.D.; n=three biological replicates; **,P<0.01; ***,P<0.001; two-tailed t test; Figure 1G). No significant difference in endosomal pH was noted in cells expressing either WT or the three NHE9 variants (Figure 1G). Steady-state accumulation of TFN is dependent on endosomal NHE function: cells expressing NHE9 were shown to accumulate more TFN relative to vector-transformed control [2] . We used flow cytometry of ∼10,000 cells per sample gated for NHE9 positivity in biological triplicates to quantify endocytosed TFN in NHE9 variants. All three variants significantly increased TFN accumulation relative to levels comparable to WT NHE9 (mean ± S.D.; n=three biological replicates; ***,P<0.001; two-tailed t test; Figure 1H). Taken together, our findings indicate that all three autism-associated variants were not deleterious to ion transport function of NHE9.
Figure 1.
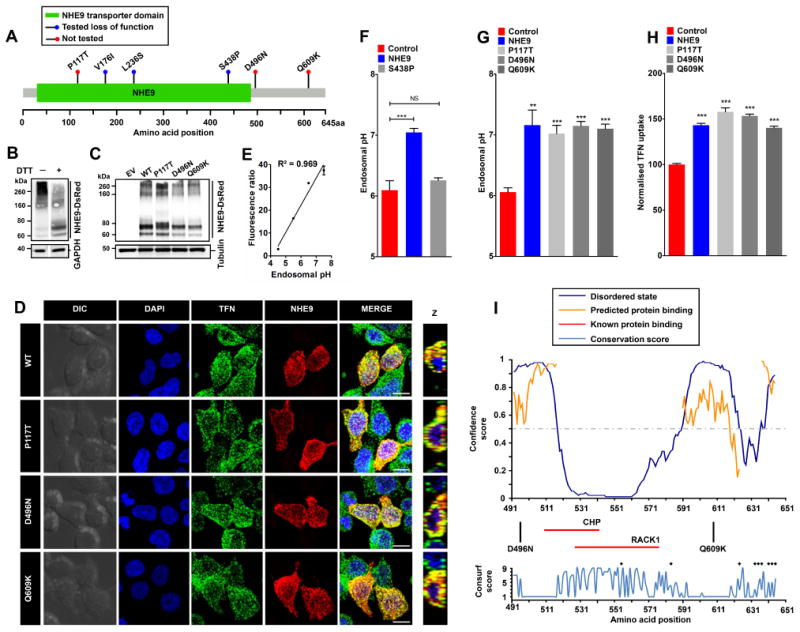
A. Gene distribution of autism associated rare missense variants in NHE9 in membrane embedded transporter domain and the C-terminal regulatory cytoplasmic domain [1] . The x-axis indicates amino acid locations of the NHE9 protein. Three of these variants (V176I, L236S and S438P) were reported as loss of function previously [2] and the remaining three (P117T, D496N, and Q609K) variants are evaluated in this study. B. Immunoblot of cell lysate from HEK293 cells expressing NHE9-DsRed using an Anti-DsRed antibody without and with dithiothreitol (DTT) treatment run on the same Western blot. Note prominent high molecular weight smear (>200 kDa) of NHE9 that was resolved into discrete band(s) with DTT treatment. C. Expression levels of NHE9 and autism-associated missense variants in HEK293 cells. Immunoblot of total lysate from cells with empty vector (EV) transfection and from cells expressing NHE9-DsRed (WT), P117T-DsRed, D496N-DsRed, and Q609K-DsRed using Anti-DsRed antibody. D. Localization of NHE9 and autism-associated variants to transferrin (TFN)-positive endosomes in HEK293 cells following 60 minutes of uptake. Confocal fluorescence images of DsRed-tagged NHE9 (WT and variants) (red) localize with Alexa Fluor 488-tagged TFN (green). Prominent colocalization can be seen in merged images and their orthogonal views (Z) by the presence of yellow puncta. Scale bar, 10 μm. E. Linear calibration curve of endosomal pH from fluorescence ratio of internalized FITC tagged TFN and Alexa Fluor 633 tagged TFN. Cells were loaded with TFN for 1 hour and then exposed to buffers with fixed pH values in the presence of 10 μM K+/H+ ionophore nigericin and 10 μM K+ ionophore valinomycin and quantified using flow cytometry (R2, square of the Pearson correlation coefficient=0.969). F. Expression of NHE9, but not the previously characterized autism-associated S438P mutation, resulted in significant alkalization of endosomal pH (mean ± S.D.; n=three biological replicates; NS, not significant; ***,P<0.001; two-tailed t test). G. Expression of NHE9 (WT and variants) resulted in significant alkalization of endosomal pH, relative to control (mean ± S.D.; n=three biological replicates; **,P<0.01; ***,P<0.001; two-tailed t test). No significant difference in endosomal pH was noted in cells expressing either WT or the three NHE9 variants. H. Expression of all three variants significantly increased steady state accumulation Alexa Fluor 633-tagged TFN to levels comparable to WT NHE9 (mean ± S.D.; n=three biological replicates; ***,P<0.001; two-tailed t test), indicating that all three autism-associated variants were not deleterious to ion transport function of NHE9. I. Upper panel shows in silico disorder (solid blue line) prediction for the NHE9 C-terminal tail. The orange line shows the confidence of disordered residues being involved in protein binding activity. The threshold above which amino acids are regarded as disordered is shown as a grey dashed horizontal line. The x-axis indicates amino acid locations of the NHE9 protein and the y-axis indicates confidence scores. The middle panel shows location of the two autism variants in the tail and binding residues for two known interacting proteins, RACK1 and CHP (horizontal red lines). The lower panel shows Consurf conservation scores for amino acids in the C-terminal tail. Intriguingly, most disordered amino acids showed poor evolutionary conservation. Black asterisks indicate amino acid residues with Consurf scores below the confidence cut-off.
Our findings may be interpreted in two ways. First, the variants could be benign polymorphisms and not causal to autism phenotype. This is especially likely for residues with low conservation scores, such as Q609. Second, the variants could be involved in regulatory functions not detectable in this cell culture model. This may be relevant for variants that map to the C-terminal tail as only the membrane-spanning helices are thought to be required for ion transport. All NHE isoforms have an extra-membranous C-terminal domain known to bind regulatory proteins [5] . In silico prediction revealed the presence of disordered elements in NHE9 C-terminal tail, consistent with experimental observations in NHE1 [9] (Figure 1I). The D496N and Q609K variants lying partly within disordered regions might alter binding to the CHP or RACK1 proteins [10] [11] , previously shown to interact with NHE9 tail. These possibilities remain be investigated further.
As sequencing technology improves and costs decline, more rare variants will be identified that may be scored as pathogenic because they occur in autism patients. For instance, in another autism related gene, NHE6, a potentially harmless Ala9Ser variant was reported in a male with Angelman-like phenotype [12] . However, his sister also presented with intellectual disability, but did not carry the Ala9Ser variant, strongly suggesting that the underlying neuropathology might be due to an unrelated brain defect or a mutation in a different autism gene. This is recently well appreciated in the field of cancer biology, wherein about a third of variants identified in clinically actionable cancer genes in patient tumors were false positives or passenger mutations [13] . Functional evaluation of disease-associated NHE9 variants as we described here will therefore be essential to predict clinical outcome, as a prerequisite to personalized therapy in patients with autism and other neurological diseases.
Three autism-associated variants in NHE9, P117T, D496N, and Q609K, show no loss of function defects in a cell culture model and may well be benign polymorphisms, although we cannot rule out cell type specific, or subtle, regulatory function. Our findings highlight the need for functional analysis of all variants in autism-associated genes to distinguish between harmless polymorphisms and disease-associated mutations. We argue that performing genetic tests on autism patients without careful cellular studies of function could lead to inaccurate false positive diagnoses and poor treatment decisions resulting in unnecessary tests and unneeded therapies.
The functional analysis of NHE9 variants was conducted using a gain-of-function scoring system performed in cell culture. Although no functional deficits were observed for these variants in HEK293 cells, we cannot rule out cell type specific effects that may alter CNS function e.g., in neurons or astrocytes.
Although our data suggest that the variants P117T, D496N, Q609K tested here do not significantly affect transporter function of NHE9, it remains to be determined if these variants are involved in transporter regulation. It will be interesting to determine if NHE9 tail has a disordered structure as we predict here and whether autism variants in the C-terminal tail alter protein binding and regulation. For example, C-terminal NHE9 variants in a rodent model of ADHD appear to show increased binding to CHP [10] , although functional consequences were not examined. Further characterization of these variants in a neurobiological model might also be warranted.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health R01 DK108304 to R.R. H.P. was a Fulbright Fellow supported by the International Fulbright Science and Technology Award.
Full Open Access: Supported by the Velux Foundation, the University of Zurich, and the EPFL School of Life Sciences.
Footnotes
Triple Blind Peer Review: The handling editor, the reviewers, and the authors are all blinded during the review process.
Ethics Statement: All work was performed according to the Johns Hopkins University regulations regarding use of recombinant DNA and cell lines.
Citations
- 1.Morrow EM, et al. Identifying Autism Loci and Genes by Tracing Recent Shared Ancestry. Science. 2008 Jul;321(5886):218–223. doi: 10.1126/science.1157657. url: https://doi.org/10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kondapalli Kalyan C, et al. Functional evaluation of autism-associated mutations in NHE9. Nature Communications. 2013 Sep;4:2510. doi: 10.1038/ncomms3510. url: https://doi.org/10.1038/ncomms3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christensen Deborah L, et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveillance Summaries. 2016 Apr;65(3):1–23. doi: 10.15585/mmwr.ss6503a1. url: https://doi.org/10.15585/mmwr.ss6503a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Devlin Bernie, Scherer Stephen W. Genetic architecture in autism spectrum disorder. Current Opinion in Genetics and Development. 2012 Jun;22(3):229–237. doi: 10.1016/j.gde.2012.03.002. url: https://doi.org/10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Kondapalli Kalyan C, Prasad Hari, Rao Rajini. An inside job: how endosomal Na+/H+ exchangers link to autism and neurological disease. Frontiers in Cellular Neuroscience. 2014 Jun;8 doi: 10.3389/fncel.2014.00172. url: https://doi.org/10.3389/fncel.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox Gregory A, et al. Sodium/Hydrogen Exchanger Gene Defect in Slow-Wave Epilepsy Mutant Mice. Cell. 1997 Oct;91(1):139–148. doi: 10.1016/s0092-8674(01)80016-7. url: https://doi.org/10.1016/s0092-8674(01)80016-7. [DOI] [PubMed] [Google Scholar]
- 7.Haim Ashkenazy, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Research. 2016 May;44(W1):W344–W350. doi: 10.1093/nar/gkw408. url: https://doi.org/10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prasad Hari, Rao Rajini. The Na+/H+Exchanger NHE6 Modulates Endosomal pH to Control Processing of Amyloid Precursor Protein in a Cell Culture Model of Alzheimer Disease. Journal of Biological Chemistry. Jan 2015;290(9):5311–5327. doi: 10.1074/jbc.m114.602219. url: https://doi.org/10.1074/jbc.m114.602219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hendus-Altenburger Ruth, et al. The human Na+/H+ exchanger 1 is a membrane scaffold protein for extracellular signal-regulated kinase 2. BMC Biology. 2016 Apr;14(1) doi: 10.1186/s12915-016-0252-7. url: https://doi.org/10.1186/s12915-016-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang-James Yanli, et al. SLC9A9 mutations, gene expression, and protein-protein interactions in rat models of attention-deficit/hyperactivity disorder. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2011 Aug;156(7):835–843. doi: 10.1002/ajmg.b.31229. url: https://doi.org/10.1002/ajmg.b.31229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohgaki R, et al. Cell Surface Levels of Organellar Na+/H+ Exchanger Isoform 6 Are Regulated by Interaction with RACK1. Journal of Biological Chemistry. 2007 Dec;283(7):4417–4429. doi: 10.1074/jbc.m705146200. url: https://doi.org/10.1074/jbc.m705146200. [DOI] [PubMed] [Google Scholar]
- 12.Fichou Yann, et al. Mutation in the SLC9A6 gene is not a frequent cause of sporadic Angelman-like syndrome. European Journal of Human Genetics. 2009 May;17(11):1378–1380. doi: 10.1038/ejhg.2009.82. url: https://doi.org/10.1038/ejhg.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones S, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Science Translational Medicine. 2015 Apr;7(283):283ra53–283ra53. doi: 10.1126/scitranslmed.aaa7161. url: https://doi.org/10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stenson Peter D, et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human Genetics. 2013 Sep;133(1):1–9. doi: 10.1007/s00439-013-1358-4. url: https://doi.org/10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones DT, Cozzetto D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics. 2014 Nov;31(6):857–863. doi: 10.1093/bioinformatics/btu744. url: https://doi.org/10.1093/bioinformatics/btu744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.