

A new strategy is used to reveal nanometer-scale single-molecule dynamics within protein assemblies to study the eisosome: a stable, linear cluster of proteins on the yeast plasma membrane. The BAR-domain protein Pil1p binds and unbinds at eisosome ends, supporting a new model of eisosomes as dynamic oligomeric filaments.
Abstract
Molecular assemblies can have highly heterogeneous dynamics within the cell, but the limitations of conventional fluorescence microscopy can mask nanometer-scale features. Here we adapt a single-molecule strategy to perform single-molecule recovery after photobleaching (SRAP) within dense macromolecular assemblies to reveal and characterize binding and unbinding dynamics within such assemblies. We applied this method to study the eisosome, a stable assembly of BAR-domain proteins on the cytoplasmic face of the plasma membrane in fungi. By fluorescently labeling only a small fraction of cellular Pil1p, the main eisosome BAR-domain protein in fission yeast, we visualized whole eisosomes and, after photobleaching, localized recruitment of new Pil1p molecules with ∼30-nm precision. Comparing our data to computer simulations, we show that Pil1p exchange occurs specifically at eisosome ends and not along their core, supporting a new model of the eisosome as a dynamic filament. This result is the first direct observation of any BAR-domain protein dynamics in vivo under physiological conditions consistent with the oligomeric filaments reported from in vitro experiments.
INTRODUCTION
The eisosome is a multimolecular assembly on the cytoplasmic face of the plasma membranes of fungi, a structure similar to caveolae in mammals. It consists of a stable assembly of proteins clustered on a small invagination of membrane (Malínská et al., 2003; Walther et al., 2006; Strádalová et al., 2009; Douglas and Konopka, 2014), whose various functions in cell membrane organization and lipid regulation remain questions of study (Aguilar et al., 2010; Fröhlich et al., 2014; Kabeche et al., 2015a,b). Fission yeast eisosomes are highly stable, linear domains (50 nm wide and 1–2 µm long), whereas budding yeast eisosomes appear as diffraction-limited puncta. The main protein component of the eisosome, Pil1p in fission yeast, contains a Bin/amphiphysin/Rvs (BAR) domain, which facilitates its organization in vivo (Olivera-Couto et al., 2011; Ziółkowska et al., 2011) and its oligomerization into filaments in vitro (Kabeche et al., 2011; Karotki et al., 2011), features conserved in budding yeast Pil1. Other BAR-domain proteins, common throughout eukaryotes, play critical roles in membrane-remodeling events and similarly form filaments in vitro, but the extent of oligomerization in cells remains unclear (Suetsugu, 2016). Because Pil1p is closely related in structure to classical N-BAR proteins such as endophilin (Ziółkowska et al., 2011) and the fission yeast eisosome is highly stable and observable at nanometer to micrometer length scales, it provides an interesting model to study BAR domain oligomerization dynamics in live cells.
Methods such as fluorescence recovery after photobleaching (FRAP) have been invaluable for characterizing cellular organization and dynamics at the micrometer scale. However, detecting spatial heterogeneities at the nanometer scale and dynamics within multimolecular assemblies in cells is still challenging. In physiological conditions, eisosomes are essentially immobile and exhibit no dynamics in FRAP experiments on time scales up to 20 min (Walther et al., 2006; Kabeche et al., 2011), and are therefore considered to be static microdomains.
Here we present a strategy to monitor nanometer-scale single-molecule dynamics within dense macromolecular assemblies in live cells called single-molecule recovery after photobleaching (SRAP). By labeling only a small fraction of Pil1p molecules, we visualized whole eisosomes, and after photobleaching, we observed isolated Pil1p molecules binding to existing eisosomes. This strategy allows us to measure with high precision the positions and the on- and off-rates of dynamic Pil1p molecules in eisosomes in live cells. We show that binding and exchange of Pil1p occurs specifically at the ends of eisosomes and not along the filament body. By comparing our data with computer simulations, we reject simple models of the eisosome as a static or uniformly dynamic microdomain and show that our data support a model of the eisosome as a dynamic filament. This result is, to our knowledge, the first report of a BAR-domain protein as a membrane-bound oligomeric filament in normal cellular conditions. We expect that our studies of the eisosome will enable further insights into BAR protein oligomerization and function in other organisms.
RESULTS AND DISCUSSION
Quantitative analysis of number of Pil1p molecules and density at eisosomes
We used quantitative microscopy (Wu and Pollard, 2005; Wu et al., 2008) of live fission yeast to directly determine the cellular concentration of Pil1p as well as the local density of Pil1p at eisosomes in cells for the first time. By comparing the fluorescence intensity of cells expressing Pil1p fused to monomeric enhanced green fluorescent protein (mEGFP) to a calibrated standard strain expressing Fim1p-mEGFP, we determined the total expression of Pil1p-mEGFP to be 619,000 ± 60,300 molecules/cell, or 38.2 ± 3.7 µM global concentration (mean ± SD across six images, 150 cells). This result is comparable to that reported by mass spectrometry experiments (Carpy et al., 2014), confirming that Pil1p is one of the most highly expressed proteins in fission yeast. In addition, we determined the cytoplasmic concentration of Pil1p-mEGFP to be 22.8 ± 4.7 µM; ∼40% of the total protein is bound to the membranes in eisosomes. The local density of Pil1p-mEGFP at eisosomes is 2890 ± 680 molecules/µm along the linear eisosome axis, or approximately seven dimers per 5-nm length, remarkably consistent with the lattice dimensions of in vitro–reconstituted filaments (Karotki et al., 2011). Assuming a hemicylindrical geometry as seen in electron micrographs, this corresponds to a surface density of 28,700 ± 6700 Pil1p dimers/µm, or a membrane surface area of ∼35 nm2 per Pil1p dimer.
This density is similar to the theoretical close-packed limit and much higher than BAR protein scaffolds necessary to generate membrane tubules in experiments in vitro (Bhatia et al., 2009; Sorre et al., 2012; Shi and Baumgart, 2015). We interpret this remarkably high local density as evidence that the lattice organization of Pil1p in filaments observed in vitro indeed also exists in eisosomes in live cells, facilitating their high stability through extensive protein–protein interactions.
Single-molecule recovery after photobleaching of Pil1p
To determine whether any subpopulations within eisosomes are dynamic, we devised a general strategy called SRAP that extends concepts from other microscopy methods, such as TOCCSL (Brameshuber and Schütz, 2012), sptPALM (Manley et al., 2008), and speckle microscopy (Danuser and Waterman-Storer, 2006). We fluorescently label only a small fraction of a protein of interest in the cell with an organic fluorophore at sufficient density that the overall shape of the multimolecular assembly is visible. In a continuous microscopy movie under normal imaging illumination, we allow the structure to photobleach completely and then visualize reappearance of spots corresponding to single fluorescently tagged proteins recruited to dynamic regions of the multimolecular assembly.
To perform SRAP experiments in live fission yeast, we sparsely labeled Pil1p by fusing a SNAP-tag to the protein C-terminus and incubating cells with low concentration (0.5 µM) of benzylguanine-conjugated silicon-rhodamine 647 dye (SiR647; Keppler et al., 2002; Lukinavičius et al., 2013) and then imaged cells in total internal reflection fluorescence (TIRF) microscopy. This protocol yielded sufficient density of Pil1p-SNAP labeled with SiR647 (referred to as Pil1p-SiR; typically 3–5% of cellular Pil1p-SNAP) to visualize long, linear eisosomes on the cell membrane (Figure 1A and Supplemental Figure S1A). Pil1p-SiR structures were similar in shape, size, and number to structures in cells expressing Pil1p-mEGFP (Figure 1, F–H), and we observed very low nonspecific fluorescence (Supplemental Figure S1B).
FIGURE 1:

Single-molecule recovery after photobleaching of SNAP-tag labeled Pil1p. Cells expressing Pil1p-SNAP were labeled with SNAP-SiR647 at 0.5 µM for 15 h, washed, and imaged in TIRF. (A) Average intensity projection of the first 5 frames (0.5 s) of a movie reveals linear eisosomes. (B) Maximum intensity projection of frames 50–200 (5–20 s) of the same movie shows single-molecule recovery events. Orange lines i–iv are the line traces used for the kymographs in E. (C) Example intensity traces of SRAP spots show stepwise photobleaching and single-molecule recovery of Pil1p-SiR. (D) The positions of single-molecule recovery events (SRAP spots, magenta) are mapped on the visible eisosomes. (E) Kymographs of line traces along eisosomes (i–iv as labeled in A and B), with bars indicating the time spans for the projection images. (F) Cells expressing Pil1p-mEGFP imaged in TIRF are similar in appearance to SNAP-tag–labeled cells in A. (G) Comparison of eisosome lengths measured in cells with Pil1p-mEGFP (black, 1250 ± 650 nm for 304 eisosomes) or labeled Pil1p-SiR (gray, 1240 ± 580 nm for 275 eisosomes) show no significant difference by two-sample Kolmogorov–Smirnov test (p = 0.33). (H) Comparison of number of eisosome objects visible in cells expressing Pil1p-mEGFP (black, 15.7 ± 2.2) or labeled Pil1p-SiR (gray, 14.8 ± 2.7) shows no significant difference by two-sample Kolmogorov–Smirnov test (p = 0.89). Scale bar, 5 µm (A, B, D, F). Cell outlines are drawn in orange dashed lines. Mean ± SD reported for at least 16 cells in each measurement.
After ∼5 s of imaging under low-power TIRF illumination (∼20 W/cm2), the fluorescently labeled eisosomes visible in the first few frames photobleached. Because TIRF imaging illuminates only molecules within ∼500 nm above the coverslip, unbleached Pil1p-SiR molecules in the cytoplasm or on the membrane beyond the TIRF field may diffuse into the illumination field in later frames of the movie (Figure 1, A and B, and Supplemental Movie S1). Because only a small fraction of Pil1p molecules were fluorescently labeled, fluorescence reappeared as isolated spots. Intensity traces of recovery spots over the length of the movie revealed stepwise increases and decreases (Figure 1C) characteristic of single fluorescent molecules binding and unbinding or photobleaching. Although it is conceivable that fluorophore blinking could also give rise to recovery events, such events would appear uniformly along the eisosome. Our observation that recovery is localized at eisosome ends implies that the fraction of recovery events due to blinking is negligible, a conclusion supported by the fact that SiR647 has been shown to be very stable and usually requires high laser intensity or additives to enhance blinking (Uno et al., 2014).
In addition, SRAP spots at the sites of eisosomes were immobile, suggesting that they were not freely diffusing on the membrane surface and indeed corresponded to fluorescent Pil1p-SiR incorporated into eisosomes. Our SRAP method revealed that new Pil1p molecules bind at eisosomes within a few seconds after initial photobleaching of the labeled structure.
Pil1p recruitment is not uniformly distributed
Further inspection of the recovery events suggested that eisosome ends are hot spots of Pil1p exchange (Figure 1D). Kymographs of lines drawn along eisosomes showed that fluorescence signal at eisosome ends persisted longer and recovered after photobleaching more frequently than along the interior (Figure 1E). To calculate precisely the distance of SRAP spots to the eisosome end, we determined the position of each spot with superresolution localization and determined the position of each eisosome end by fitting a sigmoidal curve to the intensity profile of the eisosome end extracted from initial frames (see Materials and Methods and Figure 2A). We found that 92% of SRAP spots were within 250 nm from their corresponding eisosome end, with an average position of 97 ± 119 nm (mean ± SD, 191 spots in 20 cells; Figure 2B).
FIGURE 2:

Image analysis for localization of SRAP spots at eisosomes. (A) Schematic of the measurement of distance to eisosome end. (i) The end of an eisosome is traced in the average projection of the first 5 frames (0–0.5 s); (ii) the line intensity profile at the eisosome end is fitted to determine the position of the diffraction-limited end (red line); (iii) a SRAP spot position is determined with the PeakFit plug-in for ImageJ in the movie frame when it appeared, and superresolution localizations from multiple frames are averaged to calculate the position of the SRAP event; (iv) the distance, d, is calculated from the SRAP spot along the eisosome line trace to the end; in i, iii, and iv, one image pixel is 70 nm. (B) Measured SRAP spot positions relative to the eisosome end, average 97 ± 119 nm SD (191 spot/filament pairs across 20 cells). (C) Spot localization precision is determined as SD calculated for each SRAP spot that included multiple localizations in time, average 27.9 ± 15.9 nm SD (73 sets). (D) Errors in fitting of simulated sparsely labeled, dynamic eisosome ends. Mock eisosome end intensity profiles (as in Aii) were generated according to a 3% labeling efficiency, with three extra emitters added to the end position and fitted as described in Materials and Methods. Average difference between fitted end position and simulated true end is 67.8 ± 56 nm SD (1000 simulations).
To interpret more clearly this distribution of positions, we simulated data sets based on hypothetical models for Pil1p dynamics, including any possible sources of experimental noise or errors. In a first model (referred to as the uniform model), we assumed that binding events occur uniformly along the eisosome (Figure 3, blue). The simulations included noise terms to mimic the uncertainty in the localizations for spots and eisosome ends (Figure 2, C and D, and Supplemental Figure S2) and took into account the observed distribution of eisosome lengths (Figure 1G). The simulated positions were broadly distributed, with an average position of 346 ± 254 nm, clearly disagreeing with our SRAP data (Figure 3).
FIGURE 3:

Single-molecule recovery of Pil1p-SiR occurs at eisosome filament ends. (A) Probability distributions of measured distances (black squares, 191 spot/filament pairs across 20 cells) and simulation results (uniform dynamics model, blue; end dynamics model, magenta; 275,000 runs for each tested model). The simulation models are illustrated in schematic form. For the end model simulations, the dashed line represents simulations using an unbiased Gaussian noise distribution for eisosome end localizations, and the solid line represents simulations using the true noise predicted from fitting a dynamic end (as in Figure 2D). (B) Table of mean and SD of distributions for simulated data sets and measured SRAP spots.
In a second model (referred to as the end model), we assumed that binding of new Pil1p occurs only at eisosome ends, as in a dynamic oligomeric filament. The simulated distribution followed a shape more similar to our experimental data but with a mean position of 0 ± 67 nm (Figure 3, dashed magenta). The slight offset of our SRAP spot localizations toward the interior of the eisosome (97 ± 119 nm) seems to contradict a model of dynamics strictly confined to the end. This shift cannot be explained by our spot localization precision, as the SD of recurrent localizations at a given SRAP site was 27.9 ± 15.9 nm (Figure 2C). We wondered whether the offset could be explained by the accuracy of our localization of eisosome ends and whether a dynamic end could introduce a systematic bias.
Localization accuracy of sparsely labeled, dynamic eisosome ends
We evaluated the accuracy of our eisosome-end localizations by fitting simulated data mimicking linear filaments labeled with low density, similar to our experimental data (Supplemental Figure S2A). First, we found that fitting the intensity profile with an error function—a model that assumes a continuous distribution of emitters—overestimates the end position beyond the true position by a significant distance, depending on the number of fluorophores present. In simulations corresponding to 3% labeling efficiency, the average error of the fitted eisosome-end position is 38.6 ± 61.8 nm (Supplemental Figure S2C).
However, because we extracted intensity profiles from an image averaged over a short time, if Pil1p recruitment is indeed localized to the eisosome end, then any new labeled molecules that bind during the recording time would skew the intensity profile toward the end (Supplemental Figure S2B). Indeed, in kymographs of sparsely labeled eisosomes (Figure 1E), the signal at eisosome ends persisted longer than the signal along the eisosome body, likely due to additional Pil1p-SiR molecules binding before the initial labeled molecules have photobleached. We simulated this effect by adding extra emitters at the true end position before fitting the intensity profile. Simulations with three or six extra emitters (numbers as expected based on our estimates of Pil1p binding rate; see later discussion) resulted in net fitting errors of 67.8 ± 56.1 or 84.6 ± 52.1 nm, respectively (Figure 2D and Supplemental Figure S2C), mirroring the offset in our measured SRAP spot positions.
Eisosome ends are specific sites of single-molecule recovery events
We repeated simulations of the end model for Pil1p-SiR recovery incorporating this biased localization error for the eisosome end. Based on the more conservative bias estimated from simulations of three extra fluorophores in the initial fluorescence trace, the result resembled our SRAP data (70 ± 63 nm; Figure 3, solid magenta). Of importance, the only assumption of this model is that the eisosome end is the specific site of Pil1p binding; the bias in the eisosome-end localization arises from the sparse labeling of the sample.
As a putative alternative hypothesis, we considered a model in which Pil1p binding occurs on a “ragged end” or a dynamic region at the eisosome end rather than a flat end. Simulations of a ragged-end model using various sizes for the dynamic region showed that a 125-nm region at the eisosome end was necessary to produce a result similar to our SRAP data (97 ± 76 nm; Supplemental Figure S3). However, a ragged or tapered filament end is difficult to quantify from electron micrographs (Karotki et al., 2011) but might span only a few nanometers—not sufficient to cause the distribution of localizations we observed experimentally. In addition, the SD of recurrent localizations at the same SRAP site (27.9 ± 15.9 nm; Figure 2C) indicated that binding events occur at a fixed position on each eisosome, in contradiction with a large dynamic region. We conclude that the measured distribution of SRAP data is consistent with Pil1p binding only at the ends of eisosome filaments, but the sparse labeling introduces a slight error in conventional fluorescence image-fitting models.
Characterization of Pil1p kinetics using SRAP data
In addition to localization, a variety of other single-molecule analyses can be applied to SRAP spots, such as lifetimes analysis to determine rates of binding and unbinding. We first measured the lifetimes of SRAP spots and fitted the distribution with an exponential curve to determine an off-rate. The apparent off-rate, 2.4 ± 0.2 s–1, was much faster than the overall rate of photobleaching in the images, 0.48 ± 0.03 s–1 (fitted value ± 95% confidence intervals; Figure 4A), suggesting that Pil1p-SiR molecules are not only photobleaching but also unbinding from the eisosomes. By subtracting the photobleaching rate from the spots’ disappearance rate, we estimate the unbinding rate of Pil1p to be 2.0 ± 0.2 s–1, consistent with the findings of Olivera-Couto et al. (2015).
FIGURE 4:

Analysis of Pil1p unbinding and binding kinetics. (A) The distribution of SRAP spot lifetimes (dark gray, 433 events) was fitted with a single-exponential decay (red line) to determine the apparent off-rate of SRAP spots, 2.4 ± 0.2 s–1. Inset, photobleaching of whole cells. Ten curves of normalized whole-cell intensity were averaged (black, gray lines, ±1 SD), and the average curve (starting at frame 5) was fitted with a single exponential (red line) to determine the photobleaching rate, 0.48 ± 0.03 s–1. To determine Pil1p-SiR unbinding rate at eisosomes, the photobleaching rate was subtracted from the total off-rate, resulting in an unbinding rate of 2.0 ± 0.2 s–1. (B) The distribution of dark times between SRAP spot appearances (dark gray, N = 189) was fitted with a single-exponential decay (red line) to determine the apparent binding rate of Pil1p-SiR, 1.2 ± 0.2 s–1. The very short dark times (t < 0.4 s, light gray) were excluded as mostly artifacts of missed localizations causing artificial blinks. Fitted rate parameters are given with 95% confidence intervals.
We then measured the distribution of wait times between SRAP events to determine the apparent binding rate of Pil1p-SiR to an eisosome end (Figure 4B). Assuming mass-action kinetics, the distribution can be fitted to a single exponential. We noticed that our spot localization algorithm caused the bins for very short wait times (0.1–0.3 s) to be artificially overpopulated because it occasionally missed a localization for a spot that actually persisted over many frames. We excluded these bins and fitted the distribution of wait times between binding events with an exponential curve; we found an apparent on-rate of 1.2 ± 0.2 s–1 (Figure 4B). This apparent on-rate is the product of a binding rate constant and the concentration of Pil1p-SiR (0.9 µM, i.e., the product of the labeling efficiency [4%] and the cytoplasmic Pil1p concentration [22.8 µM]). Therefore the binding rate constant for Pil1p binding to the end of an eisosome is 1.3 ± 0.9 µM–1 s–1. We used single-exponential fits for binding and unbinding because we did not expect multiple populations of different rates, such as in a polar filament with unique kinetics at each end. Pil1p exists primarily as a symmetric dimer, which would result in a filament with no polarity (Karotki et al., 2011; Olivera-Couto et al., 2011), and indeed we observed a number of filaments with Pil1p recruitment at both ends (Figure 1D). Taken together, our data indicate that Pil1p is undergoing fast single-molecule exchange at eisosome ends, even in the absence of large-scale eisosome remodeling.
SRAP reveals heterogeneities at the nanometer scale in vivo
Pil1p exchange at the eisosome has previously been unobservable using conventional imaging approaches. FRAP experiments (Walther et al., 2006; Kabeche et al., 2011) were unable to observe this dynamic subpopulation because detecting single fluorescent proteins is challenging when fully labeled structures are imaged in the same frame. One study using fluorescence fluctuation techniques detected a subpopulation of Pil1 oligomers exchanging between the cytoplasm and plasma membrane (Olivera-Couto et al., 2015), but this method lacked the spatial resolution to determine the precise location and role of dynamic Pil1p molecules relative to the nanoscale structure of the eisosome.
Our SRAP method was critical for revealing the behavior of individual protein molecules in the context of the larger eisosome structure. We expect that our SRAP method will be easily and broadly applicable to reveal localized single-molecule dynamics and heterogeneities within other multimolecular assemblies because it requires only sparse labeling and a TIRF microscope with single-molecule detection capabilities. Although similar sparse fluorescence conditions might be achieved by partial photobleaching of the sample (Brameshuber and Schütz, 2012) or photoswitching of fluorescent proteins (Manley et al., 2008), our SRAP protocol has several advantages over existing methods. We avoid high-intensity laser illumination required for FRAP methods, which can be damaging to cells. We use organic fluorophores that are brighter and more photostable than fluorescent proteins, enabling better localization precision. By using a single fluorophore to characterize both the initial structure and the recovery dynamics, we avoid challenges of multichannel imaging and alignment. Of importance, we demonstrated that sparse labeling is sufficient to determine the overall shape of a macromolecular assembly but may require a minor adjustment from conventional fluorescence image-fitting models. Future applications of SRAP imaging for large cellular assemblies should consider this factor when modeling a structure of interest.
Filament model for the eisosome
Our results demonstrate that the eisosome is highly dynamic, with continuous and fast exchange of Pil1p at its ends, even in the absence of perturbation. Models of the eisosome as a membrane microdomain (Walther et al., 2006; Kabeche et al., 2011; Karotki et al., 2011) would predict Pil1p exchange to occur uniformly around its edges. Instead, our data support a new model for the eisosome as a membrane-bound filament with a stable body and dynamic ends (Figure 5).
FIGURE 5:
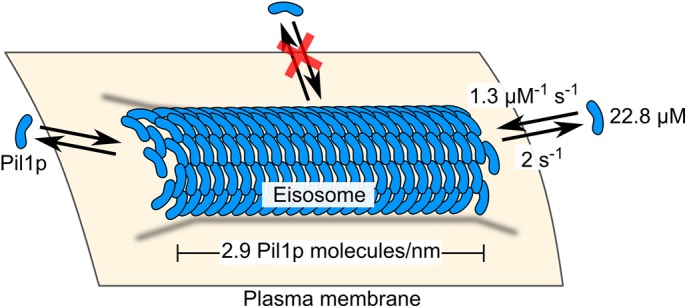
Model of the eisosome as a dynamic filament. Pil1p dimers assembled into a filament on the cytoplasmic surface of the plasma membrane are free to associate and dissociate at the ends but not at the interior. Binding and unbinding rate constants, cytoplasmic concentration of Pil1p, and density of Pil1p in the eisosome are given as reported in the text.
Pil1p and other BAR-domain proteins have been observed to oligomerize and form filaments and membrane tubules in vitro, but it has been unclear to what extent this oligomerization exists in vivo or whether instead BAR proteins are loosely clustered on patches of curved membranes (Adam et al., 2015; Daum et al., 2016; McDonald and Gould, 2016; Suetsugu, 2016). Recent in vitro studies of BAR proteins propose that binding at low or moderate surface density is sufficient to generate membrane tubes (Simunovic et al., 2016). Our quantitative analysis of Pil1p-mEGFP eisosomes indicates that Pil1p exists at extremely high density, consistent with the lattice structure of filaments reconstituted in vitro (Karotki et al., 2011). Although a polymer filament model has been previously hypothesized for eisosomes (Moseley, 2013), our results are the first experimental evidence of dynamic behavior that supports a filament model.
Reconsidering the eisosome as a membrane-bound oligomeric filament enables several predictions and poses new questions for future investigation. Of interest, electron micrographs of eisosomes in cells show that the membrane adopts a hemicylindrical furrow instead of a full tube as observed in vitro (Karotki et al., 2011). The physical or biochemical means by which a hemicylindrical scaffold of proteins is stabilized remain open questions, but our results clearly indicate that the filament body and long edge do not provide suitable binding sites for new Pil1p molecules.
Our measured rate constants predict a net growth of eisosome filaments of ∼28 Pil1p molecules/s, or ∼0.6 µm/min. This high rate of polymerization is surprising, considering that eisosomes grow very slowly throughout the cell cycle, ∼1 µm/h, and we do not observe large distances between successive Pil1p-SiR spots (Figure 2C). A likely reason for this discrepancy is that some eisosome ends might be capped, limiting the number of actively polymerizing filaments in the cell at any time. Indeed, we observed that ∼70% of filament ends did not have any SRAP events over the 15-s movies, whereas if, instead, all ends were active, this proportion would be equal to
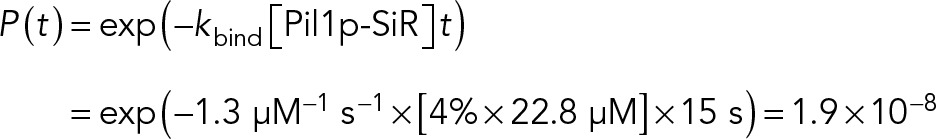 |
where P(t) is the probability of observing a wait time of length t, kbind is the binding rate constant, and [Pil1p-SiR] is the concentration of free Pil1p-SiR available.
Of importance, a filament model predicts that rapid eisosome remodeling could occur in response to physical or biochemical cues by simply modulating the rates of Pil1p binding and/or unbinding to achieve polymerization or depolymerization, just like other cytoskeletal filaments such as actin and microtubules (Gardner et al., 2011; Pollard, 2016). One recent study found that in yeast cells lacking a cell wall, eisosomes rapidly disassemble in response to high osmotic stress, within minutes (Kabeche et al., 2015a), which could easily occur for a dynamically regulated filament. A precise physical understanding of the eisosome not only will improve our knowledge of the cell biology and stress responses of fungi and pathogens, but it will also add to our understanding of BAR-domain protein assemblies in other organisms. We expect that filament-like oligomerization may be a feature of other BAR-domain proteins, even in diffraction-limited clusters or transient complexes, which have been difficult to study using conventional microscopy approaches in cells.
MATERIALS AND METHODS
Yeast strains and SNAP labeling
We tagged the pil1 gene at its C-terminus with SNAP-tag (Addgene plasmid 87024) or mEGFP (Addgene plasmid 87023) in its native locus in a wild-type Schizosaccharomyces pombe strain by homologous recombination (Bähler et al., 1998). Strains used in this study are listed in Supplemental Table S1. Cells were grown at 32°C in liquid YE5S medium to exponential phase (OD595nm between 0.4 and 0.8) and then diluted into liquid EMM5S and grown for 12–24 h at 25°C before labeling with SNAP fluorophore.
Although the SNAP-tag has been used successfully in a variety of applications (Stagge et al., 2013; Bosch et al., 2014; Klein et al., 2014), labeling cellular SNAP fusion proteins in live yeast is difficult because the cell wall impedes entry of the fluorophore substrate and multidrug exporters prevent its accumulation in the cytoplasm (McMurray and Thorner, 2008; Stagge et al., 2013). These issues can be avoided by enzymatically digesting the cell wall, deleting the multidrug exporter genes (McMurray and Thorner, 2008), or using electroporation to allow a large amount of dye to enter the cells (Stagge et al., 2013). However, such approaches may be problematic if the structure of interest is sensitive to cell integrity, as is the case with the eisosome. To avoid these difficulties, we used a minimally disruptive approach, adding a low concentration of SNAP substrate fluorophore in the medium for a long incubation.
To label SNAP-tag protein in live cells, 0.5 ml of cells at OD595nm of 0.5 were incubated at 25°C on a rotator in liquid EMM5S containing 0.1, 0.5, or 2.5 µM silicon-rhodamine benzylguanine derivative SNAP-SiR647 or SNAP-Alexa 647 (SNAP-Cell 647-SiR and SNAP-Surface Alexa Fluor 647; New England Biolabs) for 0.5, 5, or 15 h. For samples incubated for 15 h, the cells were initially diluted to OD595nm of 0.1 to avoid overgrowing during the incubation time. Cells were washed three times by centrifuging at 900 × g for 3 min and resuspending in 0.5 ml of EMM5S, and then additionally incubated at 25°C for 1 h in 0.5 ml of EMM5S, washed three times again by centrifuging at 900 × g for 3 min, and resuspending in 0.5 ml of EMM5S. Cells were finally resuspended in 50–100 µl of 0.22-µm filtered EMM5S to achieve suitable cell density for imaging.
We estimated the extent of labeling by dividing the total intensity of cells in the first frame by the mean pixel intensity of the late-appearing single-molecule spots to determine the number of fluorophores per cell. We then determined the fraction of labeled Pil1p-SNAP molecules by dividing the number of fluorophores per cell by the expected visible membrane-bound fraction of total number of Pil1p molecules determined by our quantitative microscopy analysis. The samples we used for SRAP analysis (labeled 15 h at 0.5 µM SNAP-Sir647) consistently had labeling efficiencies between 3 and 5%. Future applications of SRAP imaging should aim for a similarly low labeling efficiency, but the precise value is not critical as long as the overall shape of the structure is visible and a single-molecule regime can be reached after a short time of illumination and photobleaching. This protocol is the first reported use of SNAP-tag in live fission yeast, but similar protocols should be easy to adapt in other organisms, especially those lacking a cell wall.
Our protocol still requires use of a cell-permeable fluorophore conjugate, as incubation with SNAP-Alexa 647 yielded poor labeling (Supplemental Figure S1C). Incubation with 2.5 µM SiR647 for 15 h achieved a higher density of labeled Pil1p-SiR (≥15–20%; Supplemental Figure S1A), but short incubations yielded only sparse labeling with greater cell-to-cell variability (Supplemental Figure S1A).
Microscopy
Live cells were imaged on 25% gelatin pads in 0.22-µm filtered EMM5S with coverslips that had been washed in ethanol for 20 min and plasma treated for 2 min to avoid nonspecific attachment of dyes and other autofluorescent particles on the surface. Cells were imaged with an inverted fluorescence microscope (Ti Eclipse; Nikon) equipped with a 60×/1.49 numerical aperture (NA) objective (Nikon), illuminated with a 642-nm laser (for imaging SiR647 samples) or 488-nm laser (for imaging mEGFP samples) directed through the objective to achieve TIRF, and recorded with an electron-multiplying charge-coupled device (EMCCD) camera (iXon DU897; Andor). Samples labeled with SiR647 were imaged under low illumination intensity, ∼20 W/cm2. Movies were recorded at a single focal plane near the cell base at 10 frames/s.
For quantitative microscopy of mEGFP-tagged proteins, cells were imaged on an inverted fluorescence microscope (Ti Eclipse; Nikon) equipped with a 60×/1.4 NA Plan Apochromat Lambda objective (Nikon), coupled with a CSU-W1 spinning-disk confocal system (Yokogawa), illuminated with 488-nm laser, and recorded with an EMCCD camera (iXon Ultra888; Andor). Cells expressing Pil1p-mEGFP or Fim1p-mEGFP were imaged in z-stacks spanning the entire cell height, with 21 z-slices in 500-nm steps.
Image analysis and quantification
Image analysis was carried out in the Fiji distribution of ImageJ (Schindelin et al., 2012; Schneider et al., 2012), and further quantification was performed in Matlab (MathWorks), using built-in tools as well as self-written macros and scripts (see the Supplemental Material).
We first measured the lengths of filaments in the average intensity projection of frames 1–5 (AVG1–5) of Pil1p-SiR and Pil1p-mEGFP movies by drawing a line along the full length of visible fluorescence for each filament. We then identified SRAP spots in the maximum intensity projection of frames 50–200 (MAX50–200), after labeled eisosomes had photobleached. We first generated a preliminary list of SRAP spot positions from the MAX50–200 image by using the Find Maxima command and determining the brightness-weighted centroid of a 3-pixel-diameter circle at each point.
To determine the end position of the underlying eisosome for each point in this list, we manually traced the eisosome filament in the AVG1–5 image with a 3-pixel-wide line spanning past the spot position to extend beyond the end of the filament (Figure 2A) and analyzed the intensity profile along this line in Matlab. Spots >4 pixels away (280 nm) from the nearest eisosome were discarded (<10% of detected spots). To find the position of the end of the eisosome underlying the diffraction-limited image, we fitted the intensity profile with the following step-like function:
 |
This equation is equivalent to the cumulative intensity of a continuous distribution of Gaussian emitters, where I(x) is the intensity along the line coordinate x, A is the amplitude, er f (⋅) is the error function, x0 is the position of the underlying step corresponding to the end of the labeled structure, and σ is the SD of the diffraction-limited Gaussian spot. Measured intensity profiles were fitted in Matlab using a nonlinear fitting algorithm, with A and x0 as independent variables and σ fixed to 1.85 pixels (130 nm), representing the diffraction-limited spot width.
We used the PeakFit plug-in for Fiji (www.sussex.ac.uk/gdsc/intranet/microscopy/imagej/smlm_plugins) to determine superresolution localizations of the spots that appeared in frames 50–200, calibrated with pixel size 70 nm, wavelength 642 nm, objective NA 1.49, objective proportionality factor 1.4, and electron-multiplying gain 37.7, resulting in an estimated point-spread function width of 1.837 pixels. This generated a list of localizations with precision <40 nm. From the list of SRAP events’ spot centroids determined in the MAX50–200 projection, we matched each SRAP event with all localizations within a 1-pixel radius from the SRAP spot centroid. We calculated the distance from each localization to the fitted eisosome-end position projected along the filament line trace (Figure 2Aiv) and calculated the average distance to the end of all of the associated localizations for each SRAP event. For spot centroids that did not have any associated localization spots of sufficiently high precision, we used the brightness-weighted centroid of the SRAP spot in the projection image to determine its distance from the eisosome end.
To determine the lifetimes of SRAP events, we measured the intensity of a 3-pixel-diameter circle centered on the SRAP spot position through the length of the movie after subtracting the median-filter background. We processed these intensity time traces in Matlab with a Chung–Kennedy filter (Reuel et al., 2012) to highlight discrete intensity steps (Figure 1C). We computed the lengths of time between steps above and below a threshold intensity and then fitted the distribution of lifetimes with a single-exponential curve.
To determine the photobleaching rate, we measured the mean intensity of a region of interest (ROI) containing an entire cell through the length of the movie. For each ROI’s intensity decay profile, we subtracted the minimum baseline, normalized the intensities to the maximum value, and then computed the average across all movies. We fitted the average photobleaching profile with a single-exponential curve, starting at frame 5 to avoid biasing the fit with the fast-bleaching autofluorescence component. We estimated the protein-unbinding rate by subtracting the bulk photobleaching rate from the SRAP spot lifetime decay rate.
To determine the Pil1-binding rate, we computed the length of time between recurrent localizations in the single-molecule localization data set (from PeakFit results). We analyzed all of the interevent dark times, as well as the initial dark time before the first appearance. This approach is more error-prone because missing localizations of suboptimal spots could cause an artificially high number of very short dark times. We therefore fitted a subset of the data with a single-exponential curve, excluding events <0.4 s. We considered fitting with alternative models, accounting for photobleaching of the limited pool of free Pil1p-SiR or multiple rates or other processes. However, more complex analysis yielded little improvement and would require much larger data sets to be justified.
To quantify the number of Pil1p-mEGFP, we used quantitative microscopy approaches (Wu and Pollard, 2005; Wu et al., 2008; Berro and Pollard, 2014). We first corrected the raw z-stacks for camera offset noise and uneven illumination. We measured the integrated intensity of sum projections of z-stacks spanning whole cells expressing Fim1p-mEGFP or Pil1p-mEGFP or wild-type cells. We subtracted the autofluorescence intensity of wild-type cells and calibrated the brightness per mEGFP molecule in cells expressing Fim1p-mEGFP (using 86,500 ± 9100 molecules of Fim1p-mEGFP per cell as reported in Wu and Pollard [2005]) to determine the total number of Pil1-mEGFP molecules per cell. To determine the local density of Pil1p-mEGFP at eisosomes, we used sum projections of z-stacks spanning only the lower half of the cell. We measured the integrated intensity of rectangular ROIs drawn across eisosomes and subtracted the local cytoplasmic background intensity as measured in an adjacent ROI. Using the intensity per molecule calibrated from Fim1p-mEGFP stacks, we converted these intensities to number of Pil1p-mEGFP molecules per ROI. We calculated a linear density along the length of the eisosome axis (without assuming any geometry for the structure), as well as the membrane surface area density (assuming the geometry of a half-cylinder with radius 16 nm, as determined from electron micrographs; Karotki et al., 2011).
Characterization of eisosome-end localization
We performed simulations to estimate the precision of our method of fitting an error function to the intensity traces of sparsely labeled eisosomes to localize their ends. Indeed, this continuum model might not find the eisosome ends accurately when the structures are only sparsely labeled. From our quantitative microscopy of Pil1p-mEGFP filaments, we estimated that there are ∼2.8 Pil1p proteins per nanometer of eisosome lattice. Therefore, for a 350-nm region (equivalent to the typical 5-pixel region of eisosome body covered by the line profile extracted earlier), we expect 980 possible Pil1p sites. With our estimated 3% labeling efficiency, there are most likely between 20 and 50 fluorescently tagged Pil1p-SiR in this region. For each simulation, we first calculated a set of expected numbers of emitters according to a binomial distribution and then simulated each number of “emitter positions” on a uniform distribution along a 350-nm line. We added a Gaussian profile of intensity at each emitter position (mean xi, SD 135 nm, peak height of 1 AU) to mimic the point spread function of the microscope, added signal from emitters outside the simulated region to account for other fluorophores on the rest of the eisosome body, and also added noise to the sum traces (random value of mean 0, SD 1 AU at each x value, ∼10–20% of the simulated fluorescence signal). We fitted the resulting intensity profile (10 pixels long, including the 5-pixel region of simulated fluorophores plus 5-pixel tail region) with the error function model described earlier. We determined the distance from the fitted end position (position x0) to the true end of the eisosome (position 350 nm) in each simulation.
To determine a full population average of these errors, we simulated 1000 filaments. We repeated a similar set of simulations with a number of added fluorophores at the end position to account for the possibility of additional Pil1p-SiR binding during the imaging time, which leads to a characteristic bias in fitting (Figure 2D and Supplemental Figure S2C).
Eisosome dynamics model simulations
We compared the distribution of our experimentally measured distances to data sets simulated under different hypotheses. In one model (referred to as the uniform model), Pil1p SRAP events occur uniformly along the eisosome; in a second model (referred to as the end model), events occur exclusively at the end of the filament (Figure 3). For all models, each simulation was initialized by picking one of the eisosome lengths experimentally measured in Pil1p-SiR cells (10,000 runs with each of 275 filaments; Figure 1G). For the uniform model, the true SRAP spot positions were simulated by picking a number following a uniform distribution between zero and half the filament length, and for the end model, the true SRAP spot position was taken as the true end position of the eisosome end (position 0); a number following a Gaussian distribution (mean 0, SD 30 nm) was added to represent the spot localization uncertainty as measured experimentally (Figure 2C). For each simulation, we added a number following a Gaussian distribution with mean 0 and SD 60 nm to the true position of the eisosome end (position 0) to simulate the unbiased localization precision of the experimental fit of the eisosome end in our image analysis. Each simulated SRAP spot position was subtracted from the simulated end position to determine the relative distance from the end. In a second set of simulations to account for the fitting bias arising from a dynamic filament end, we used for the eisosome end position distribution a Gaussian distribution with mean position -70 nm and SD 55 nm (as in Figure 2D).
We also simulated a third class of models (referred to as ragged-end models) in which events occur uniformly within a zone of defined length at the eisosome end. For the ragged-end models, the true SRAP spot position was simulated by picking a number following a uniform distribution between 0 and the length of the end zone (e.g., 200 nm), and the end position and noise terms were generated with unbiased Gaussian distributions as described earlier.
Statistics and reproducibility
Unless otherwise noted, all reported measurements are given as mean ± SD. Values derived from curve fitting are given as value ± 95% confidence interval, and the type of curve fit is noted in the text. Numbers of cells analyzed, data points, and simulation runs are given in the text and figure legends with each reported measurement. Imaging experiments used single biological replicates, and the number of technical replicates (images, cells) is given in the text and figure legends. Any statistical tests used are noted in Results and figure legends. Statistical tests were not used to compare data and simulations because the outcome of the simulations may vary significantly, depending on user-defined parameters.
Code availability
Matlab and ImageJ scripts we used to analyze data and generate simulated data sets are available as supplemental files, with brief descriptions in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank Ronan Fernandez for assistance in creating yeast strains, and members of the Berro lab for helpful discussions. We thank E. de la Cruz, J. Bewersdorf, and S. Ferguson for reading earlier versions of the manuscript and providing helpful feedback. This research was supported in part by National Institutes of Health/National Institute of General Medical Sciences Grant R01GM115636. M.M.L. was supported by National Institutes of Health Training Grant T32GM008283. We also acknowledge support from the Raymond and Beverly Sackler Institute for Biological, Physical and Engineering Sciences at Yale University.
Abbreviations used:
- BAR
Bin/amphiphysin/Rvs domain
- FRAP
fluorescence recovery after photobleaching
- mEGFP
monomeric enhanced green fluorescent protein
- SiR647
silicon-rhodamine 647
- TIRF
total internal reflection fluorescence.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-04-0238) on June 28, 2017.
REFERENCES
- Adam J, Basnet N, Mizuno N. Structural insights into the cooperative remodeling of membranes by amphiphysin/BIN1. Sci Rep. 2015;5:15452. doi: 10.1038/srep15452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar PS, Fröhlich F, Rehman M, Shales M, Ulitsky I, Olivera-Couto A, Braberg H, Shamir R, Walter P, Mann M, et al. A plasma-membrane E-MAP reveals links of the eisosome with sphingolipid metabolism and endosomal trafficking. Nat Struct Mol Biol. 2010;17:901–908. doi: 10.1038/nsmb.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Berro J, Pollard TD. Synergies between Aip1p and capping protein subunits (Acp1p and Acp2p) in clathrin-mediated endocytosis and cell polarization in fission yeast. Mol Biol Cell. 2014;25:3515–3527. doi: 10.1091/mbc.E13-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia VK, Madsen KL, Bolinger P-YY, Kunding A, Hedegård P, Gether U, Stamou D. Amphipathic motifs in BAR domains are essential for membrane curvature sensing. EMBO J. 2009;28:3303–3314. doi: 10.1038/emboj.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch P, Corrêa I, Sonntag M, Ibach J, Brunsveld L, Kanger J, Subramaniam V. Evaluation of fluorophores to label SNAP-tag fused proteins for multicolor single-molecule tracking microscopy in live cells. Biophys J. 2014;107:803–814. doi: 10.1016/j.bpj.2014.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brameshuber M, Schütz GJ. Detection and quantification of biomolecular association in living cells using single-molecule microscopy. Methods Enzymol. 2012;505:159–186. doi: 10.1016/B978-0-12-388448-0.00017-6. [DOI] [PubMed] [Google Scholar]
- Carpy A, Krug K, Graf S, Koch A, Popic S, Hauf S, Macek B. Absolute proteome and phosphoproteome dynamics during the cell cycle of Schizosaccharomyces pombe (fission yeast) Mol Cell Proteomics. 2014;13:1925–1936. doi: 10.1074/mcp.M113.035824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danuser G, Waterman-Storer CM. Quantitative fluorescent speckle microscopy of cytoskeleton dynamics. Annu Rev Biophys Biomol Struct. 2006;35:361–387. doi: 10.1146/annurev.biophys.35.040405.102114. [DOI] [PubMed] [Google Scholar]
- Daum B, Auerswald A, Gruber T, Hause G, Balbach J, Kühlbrandt W, Meister A. Supramolecular organization of the human N-BAR domain in shaping the sarcolemma membrane. J Struct Biol. 2016;194:375–382. doi: 10.1016/j.jsb.2016.03.017. [DOI] [PubMed] [Google Scholar]
- Douglas LM, Konopka JB. Fungal membrane organization: the eisosome concept. Annu Rev Microbiol. 2014;68:377–393. doi: 10.1146/annurev-micro-091313-103507. [DOI] [PubMed] [Google Scholar]
- Fröhlich F, Christiano R, Olson D, Alcazar-Roman A, DeCamilli P, Walther T. A role for eisosomes in maintenance of plasma membrane phosphoinositide levels. Mol Biol Cell. 2014;25:2797–2806. doi: 10.1091/mbc.E13-11-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner MK, Charlebois BD, Jánosi IM, Howard J, Hunt AJ, Odde D J. Rapid microtubule self-assembly kinetics. Cell. 2011;146:582–592. doi: 10.1016/j.cell.2011.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche R, Baldissard S, Hammond J, Howard L, Moseley JB. The filament-forming protein Pil1 assembles linear eisosomes in fission yeast. Mol Biol Cell. 2011;22:4059–4067. doi: 10.1091/mbc.E11-07-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche R, Howard L, Moseley JB. Eisosomes provide membrane reservoirs for rapid expansion of the yeast plasma membrane. J Cell Sci. 2015a;128:4057–4062. doi: 10.1242/jcs.176867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche R, Madrid M, Cansado J, Moseley JB. Eisosomes regulate phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) cortical clusters and mitogen-activated protein (MAP) kinase signaling upon osmotic stress. J Biol Chem. 2015b;290:25960–25973. doi: 10.1074/jbc.M115.674192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karotki L, Huiskonen JT, Stefan CJ, Ziółkowska NE, Roth R, Surma MA, Krogan NJ, Emr SD, Heuser J, Grünewald K, et al. Eisosome proteins assemble into a membrane scaffold. J Cell Biol. 2011;195:889–902. doi: 10.1083/jcb.201104040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2002;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- Klein T, Proppert S, Sauer M. Eight years of single-molecule localization microscopy. Histochem. Cell Biol. 2014;141:561–575. doi: 10.1007/s00418-014-1184-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicˇius G, Umezawa K, Olivier N, Honigmann A, Yang G, Plass T, Mueller V, Reymond L, Corrêa IR, Luo Z-G, et al. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat Chem. 2013;5:132–139. doi: 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- Malínská K, Malínský J, Opekarová M, Tanner W. Visualization of protein compartmentation within the plasma membrane of living yeast cells. Mol Biol Cell. 2003;14:4427–4436. doi: 10.1091/mbc.E03-04-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-Schwartz J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- McDonald NA, Gould KL. Linking up at the BAR: oligomerization and F-BAR protein function. Cell Cycle. 2016;15:1977–1985. doi: 10.1080/15384101.2016.1190893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Thorner J. Septin stability and recycling during dynamic structural transitions in cell division and development. Curr Biol. 2008;18:1203–1208. doi: 10.1016/j.cub.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley JB. An expanded view of the eukaryotic cytoskeleton. Mol Biol Cell. 2013;24:1615–1618. doi: 10.1091/mbc.E12-10-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera-Couto A, Graña M, Harispe L, Aguilar PS. The eisosome core is composed of BAR domain proteins. Mol Biol Cell. 2011;22:2360–2372. doi: 10.1091/mbc.E10-12-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera-Couto A, Salzman V, Mailhos M, Digman MA, Gratton E, Aguilar PS. Eisosomes are dynamic plasma membrane domains showing Pil1-Lsp1 heteroligomer binding equilibrium. Biophys J. 2015;108:1633–1644. doi: 10.1016/j.bpj.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Actin and actin-binding proteins. Cold Spring Harb Perspect Biol. 2016;8:a018226. doi: 10.1101/cshperspect.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuel NF, Bojo P, Zhang J, Boghossian AA, Ahn J-HH, Kim J-HH, Strano MS. NoRSE: noise reduction and state evaluator for high-frequency single event traces. Bioinformatics. 2012;28:296–297. doi: 10.1093/bioinformatics/btr632. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Baumgart T. Membrane tension and peripheral protein density mediate membrane shape transitions. Nat Commun. 2015;6:5974. doi: 10.1038/ncomms6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic M, Evergren E, Golushko I, Prévost C, Renard H-FF, Johannes L, McMahon HT, Lorman V, Voth GA, Bassereau P. How curvature-generating proteins build scaffolds on membrane nanotubes. Proc Natl Acad Sci USA. 2016;113:11226–11231. doi: 10.1073/pnas.1606943113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorre B, Callan-Jones A, Manzi J, Goud B, Prost J, Bassereau P, Roux A. Nature of curvature coupling of amphiphysin with membranes depends on its bound density. Proc Natl Acad Sci USA. 2012;109:173–178. doi: 10.1073/pnas.1103594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagge F, Mitronova G, Belov V, Wurm C, Jakobs S. Snap-, CLIP- and Halo-Tag labelling of budding yeast cells. PLoS One. 2013;8:e78745. doi: 10.1371/journal.pone.0078745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strádalová V, Stahlschmidt W, Grossmann G, Blazíková M, Rachel R, Tanner W, Malinsky J. Furrow-like invaginations of the yeast plasma membrane correspond to membrane compartment of Can1. J Cell Sci. 2009;122:2887–2894. doi: 10.1242/jcs.051227. [DOI] [PubMed] [Google Scholar]
- Suetsugu S. Higher-order assemblies of BAR domain proteins for shaping membranes. Microscopy (Oxf) 2016;65:201–210. doi: 10.1093/jmicro/dfw002. [DOI] [PubMed] [Google Scholar]
- Uno S-NN, Kamiya M, Yoshihara T, Sugawara K, Okabe K, Tarhan MC, Fujita H, Funatsu T, Okada Y, Tobita S, et al. A spontaneously blinking fluorophore based on intramolecular spirocyclization for live-cell super-resolution imaging. Nat Chem. 2014;6:681–689. doi: 10.1038/nchem.2002. [DOI] [PubMed] [Google Scholar]
- Walther TC, Brickner JH, Aguilar PS, Bernales S, Pantoja C, Walter P. Eisosomes mark static sites of endocytosis. Nature. 2006;439:998–1003. doi: 10.1038/nature04472. [DOI] [PubMed] [Google Scholar]
- Wu JQ, McCormick CD, Pollard TD. Chapter 9: Counting proteins in living cells by quantitative fluorescence microscopy with internal standards. Methods Cell Biol. 2008;89:253–273. doi: 10.1016/S0091-679X(08)00609-2. [DOI] [PubMed] [Google Scholar]
- Wu JQ, Pollard TD. Counting cytokinesis proteins globally and locally in fission yeast. Science. 2005;310:310–314. doi: 10.1126/science.1113230. [DOI] [PubMed] [Google Scholar]
- Ziółkowska NE, Karotki L, Rehman M, Huiskonen JT, Walther TC. Eisosome-driven plasma membrane organization is mediated by BAR domains. Nat Struct Mol Biol. 2011;18:854–856. doi: 10.1038/nsmb.2080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.