

Summary
Peptide immunoaffinity enrichment coupled with targeted mass spectrometry is a quantitative approach for the robust and reproducible quantification of peptide analytes. The approach is capable of multiplexed quantification of peptides, including post-translational modifications such as phosphorylation. Anti-peptide antibodies are used to enrich analytes and heavy stable isotope-labeled standards. The enriched peptides are directly measured by multiple reaction monitoring (MRM), a well characterized quantitative mass spectrometry-based method. Quantification is performed by measuring the analyte (light) peptide response relative to the heavy standard, which is spiked at a known concentration. Here, we describe the methodology for multiplexed measurement of phosphorylated peptides on the ATM kinase and their non-modified peptide analogs in cellular lysates. The method provides quantitative measurements of phospho-signaling and can be extended to a number of other phosphopeptides and sample types.
Keywords: Targeted proteomics, DNA damage response, multiplex, quantitation, LC/MS
1. Introduction
Targeted mass spectrometry techniques applied to proteomics have seen widespread growth in recent years [1, 2]. The advantages of targeted approaches, like multiple reaction monitoring (MRM; also known as selected reaction monitoring, SRM), include high specificity, precision, interlaboratory reproducibility, and the ability to multiplex measurements of peptides [3, 4]. Analyzing proteotypic peptides (i.e. peptides unique to a given protein of interest and analyzable by the mass spectrometer), released upon proteolysis of biospecimens, allows for quantification of proteins in a wide array of sample types. However, for many peptide analytes of interest, an enrichment step must be performed in order for MRM assays to have sufficient sensitivity to quantify endogenous levels of analyte in a complex biological matrix. This is particularly true for post-translational modifications, including phosphorylation. Coupling a peptide immunoaffinity enrichment step with MRM, resulting in an immuno-MRM assay, enables sufficient sensitivity for many low-level analytes [5–9]. The benefits of immuno-MRM include excellent specificity, a wide dynamic range, ease of sample handling, transferability of methods across laboratories, and the capability of multiplexing many analytes together.
We previously demonstrated the application of a multiplexed immuno-MRM assay targeting phospho-signaling in response to DNA damage [10]. The assay measured the response of 69 analytes (phosphorylated and non-modified peptides) to demonstrate: i) multiplexed quantitative analysis of signaling events, ii) simultaneous analysis of modified and non-modified isoforms of peptides, and iii) applicability to a variety of sample types and conditions. Here, we describe in detail the method for applying the multiplexed assay, with focus on measurement of two phosphopeptides derived from the ATM kinase. The two phosphopeptides targeted by the assay contain Ser 367 and Ser 2996 phosphosites. The method describes reagent preparation (e.g. cross-linking antibodies on magnetic beads) in addition to sample preparation (e.g. cell lysis, trypsin digestion, immunoaffinity enrichment, and analysis by targeted MRM mass spectrometry). While the method is focused on analysis of the ATM kinase, it is generally applicable to any multiplexed immuno-MRM assay applied to cell lysates.
2. Materials
2.1. Samples
Whole cell lysates prepared from cultured immortalized cell lines, primary human cells, and tissue specimens have been evaluated. Detection of endogenouos analyte depends on expression levels and treatment conditions in individual samples. The method has been tested using sufficient sample material to provide 500 μg of total protein as input material. The method describes application to cultured HeLa cell lines but can be adapted to alternative cell lines or inputs (e.g. tissue specimens) for cell lysis. The procedure assumes an adherent cell line but can also be used with cells grown in suspension, in which case the cells are harvested directly from growth medium without trypsinization. Cells were harvested 2 hours post-exposure to ionizing radiation to induce DNA damage and a phosphorylation response on ATM.
2.2. Cross-linking Solutions and Buffers
1×PBS (137 mM NaCl, 2.7 mM KCl, 11.9 mM Phosphate): Dilute from 10×PBS (ThermoFisher Scientific, Waltham, MA) using HPLC-grade water. Store at room temperature for 3 months.
Cross-linking solution: 20 mM dimethyl pimelimidate (DMP) in 200 mM triethanolamine (TEA), pH 8.5. To make 200 mM TEA, dissolve 10.6 mL of neat triethanolamine into 400 mL HPLC-grade water; adjust pH to 8.5 with 5 M HCl. Store in the dark in well-ventilated cabinet. Dissolve 1.04 g of DMP in 200 mL of 200 mM TEA. Make fresh and store at room temperature.
Cross-linking quench solution: 150 mM monoethanolamine, pH 9. Dissolve 3.62 mL of neat monoethanolamine in 400 mL HPLC-grade water; adjust pH to 9.0 with 5 M HCl. Store in the dark in well-ventilated cabinet.
Cross-linked antibodies wash solution: 1×PBS / 0.03% CHAPS. First, prepare 5% (w/v) CHAPS (3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate) by dissolving 1.0 mg CHAPS powder in 20 mL of LCMS-grade water. Then, prepare 1×PBS / 0.03% CHAPS by diluting 100 mL of 10×PBS and 6.0 mL of 5% CHAPS to 1 L using LCMS-grade water. Store at room temperature for 3 months.
Cross-linked antibodies storage solution: 1×PBS / 0.03% CHAPS / 0.1% sodium azide (NaN3). First, prepare 10% (w/v) NaN3 by dissolving 10 g of NaN3 in 100 mL of HPLC-grade water. Then, prepare 1×PBS / 0.03% CHAPS / 0.1% NaN3 by diluting 50 mL of 10×PBS, 3.0 mL of 5% CHAPS, and 5.0 mL of 10% NaN3 to 500 mL using HPLC-grade water. Store at room temperature.
2.3. Cell Lysis Solutions and Buffers
1×DPBS (Gibco, ThermoFisher Scientific) (138 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4-2H2O, 1.5 mM KH2PO4).
7.5 M Urea: Add 6 mL HPLC-grade water to a 15 mL Falcon conical tube. Add 4.50 g urea and mix until the urea is in solution. Bring final volume to 10 mL with HPLC-grade water. Must be made fresh daily. Store at room temperature.
5×Lysis buffer stock solution: To Make 100 mL: Add 12.5 mL 1 M Tris (pH 8.0), 1.0 mL 0.5 M EDTA, and 1.0 mL 0.5 M EGTA. Bring to 100 mL with HPLC-grade water. Filter with 0.22 μm filter. Store at room temperature for 6 months.
Lysis buffer: Add 4 Parts 7.5 M Urea, 1 Part 5×Lysis buffer stock solution, 1% Sigma phosphatase cocktail 2, 1% Sigma phosphatase cocktail 3, and 1% Sigma Protease Inhibitor to a final volume of 100 mL. Mix well. Must be made fresh daily, store on ice.
0.2 M Tris, pH 8.0. Add 4 parts HPLC-grade water and 1 part 1 M Tris, pH 8.0.
2.4. Digestion Solutions and Buffers
Reducing agent: 0.5 M TCEP (tris(2-carboxyethyl)phosphine) bond breaker solution (ThermoFisher Scientific).
Alkylating agent: 0.5 M Iodoacetamide (IAM). To one 56 mg vial of iodoacetamide (Sigma, St. Louis, MO), add 605 μL of 0.2 M Tris, pH 8.0 (made by diluting from 1.0 M Trizma hydrochloride buffer solution, pH 8.0 (Sigma, St. Louis, MO) using HPLC-grade water). Mix until dissolved. Prepare immediately before use and keep out of light.
Trypsin solution: Sequencing-grade trypsin (Promega, Madison, WI) at 0.5 μg/μL in 50 mM acetic acid solution.
Digestion quenching solution: 20% Formic acid. Add 4 parts HPLC-grade water and 1 part neat formic acid.
2.5. Immunoaffinity Enrichment Solutions and Buffers
1×PBS / 0.01% CHAPS: Prepare a 5% CHAPS stock solution by dissolving 5 mg CHAPS in 100 mL LCMS-grade water. Add 100 mL of 10×PBS and 2 mL of 5% CHAPS to LCMS-grade water, for a total of 1 L. Store at room temperature for 3 months.
0.1×PBS / 0.01%CHAPS. Add 10 mL of 10×PBS and 2 mL of 5% CHAPS to LCMS-grade water, for a total of 1L. Store at room temperature for 3 months.
Peptide elution solution: 3% v/v acetonitrile (ACN) / 5% v/v acetic acid (AcOH) / 50 mM citrate. First, prepare a 200 mM citric acid solution by dissolving 8.4 g of citric acid monohydrate in 200 mL of LCMS-grade water. Then, mix 3.0 mL LCMS-grade ACN, 5.0 mL AcOH (ACS reagent grade, ≥99.7%), and 25.0 mL of 200 mM citric acid in LC-MS-grade water, for a total of 100 mL.
Peptide storage solution: 30% acetonitrile (LCMS-grade) / 0.1% formic acid in LCMS-grade water.
2.6. Liquid Chromatography / Mass Spectrometry Solutions and Buffers
Mobile phase A: 0.1% formic acid. Mix 1.0 mL formic acid in 1 L LCMS-grade water. Store at room temperature for 3 months. Sonicate before use and degas regularly.
Mobile phase B: 0.1% formic acid in 90% acetonitrile in water. Mix 100 mL LCMS-grade water and 1.0 mL formic acid with LCMS-grade acetonitrile, for a total of 1 L. Store at room temperature for 3 months. Sonicate before use and degas regularly.
2.7. Equipment and Supplies
Magnet: A suitable magnet is required for separation. Formats vary depending on if tubes or plates are used. The protocol described requires tube-based (Antibody Cross-linking) and plate-based (Peptide Immunoaffinity Enrichment) magnets.
Refrigerated micro-centrifuge
Centrifuge for use with 96 well plates
Coulter Counter or hemocytometer
Sonic dismembrator, (model 550, ThermoFisher Scientific)
Lyophilizer
Positive Pressure-96 Processor Manifold (Waters, Milford, MA)
Autosample and nanoLC system
Column heater for capillary column (Phoenix S&T, Chester, PA)
Hybrid triple quadrupole-ion trap mass spectrometer (model 6500 QTRAP, Sciex, Framingham, MA)
(Optional) King-Fisher magnetic bead handling platform (ThermoFisher Scientific) equipped with a 96 well deep well magnet head and a 96 well PCR magnet head, with associated plastic tip combs
LabQuake tube rotator (Barnstead, ThermoFisher Scientific)
Oasis HLB 96-plate, 30 mg (30 μm), Waters
2 mL polypropylene 96 well deep well plates (ThermoFisher Scientific)
200 μL 96 well plates (ThermoFisher Scientific)
PCR plates (BioRad, Hercules, CA)
Square Matrix CapMats For 2 mL blocks (ThermoFisher Scientific)
Chemically resistant sealing foil (BioExpress, Kaysville, UT)
ThermalSeal® PCR Sealing Films (Genesee Scientific, San Diego, CA)
96-well Silicone Sealing Mat (Axygen Scientific, Union City, CA)
Protein-G magnetic beads, suspended as supplied by vendor (see Note 1).
2.8. Specialized Reagents, Standards, and Controls
-
Antibody master mixes (see Note 2).
Antibody stock solutions: Purified monoclonal antibody stocks from the vendor should be in 1×PBS / 0.1% sodium azide. Store at 4 °C (for long-term storage, use -20 °C or -80 °C).
Antibody working master mix: Combine equivalent amounts of antibodies cross-linked onto magnetic beads, with a final concentration of 1 μg of each antibody per 50 μL. Store in 1×PBS / 0.03% CHAPS / 0.1% sodium azide at 4 °C.
-
Heavy Stable Isotope-Labeled Synthetic Peptide Solutions and Mixtures
Heavy peptide stock solutions: Heavy peptides may be obtained from synthetic peptide vendors. Prepare stock solutions of individual heavy peptides at 100-500 μM (pmol/μL) in Peptide storage solution (30% acetonitrile / 0.1% formic acid) in sealed screw-top microcentrifuge tubes. Store aliquots at -80 °C for 6-12 months (see Note 3).
Heavy peptide working solution (SIS mix): Generate a master mix of the standard heavy peptides to yield an equal molar mix at 2 pmol/μL in 3% acetonitrile / 0.1% formic acid. Store aliquots at -80 °C for 3 months.
-
Quality control standards
MS QC System suitability standard: Prepare a mixture of standard peptides spanning the retention time range of the analytes (such as an iRT standard (Pierce or Biognosys) or mixture of digested proteins). Dilute to a final concentration of 2.5 fmol / μL in 0.1% formic acid immediately prior to analysis.
Process QC standard: A background matrix consisting of HeLa cells harvested 2 hours post-exposure to 10 Gy ionizing radiation (see Note 4). Prepare lysates as described below (Cell Lysis). Aliquots are stored in liquid nitrogen and are used as process controls and background matrix for characterization experiments.
Response curve standards: Prepare a series of samples with varying concentrations of heavy peptides by serially diluting the heavy peptide working solution in previously digested aliquots of Process QC standard. Prepare 7 or 8 concentration points ranging from 0.1 fmol to 1000 fmol in 500 μg of background digested lysate, suspended in 1×PBS / 0.01% CHAPS. Add equal amount (100 fmol) of light peptide to each sample. A blank (no heavy peptide added) and double blank (no light or heavy peptide added) are also prepared. Prepare sufficient samples to run each concentration point in triplicate. The same pipettor tip is used for the serial dilution.
3. Methods
3.1. Antibody Cross-linking
Antibodies are separately cross-linked to magnetic Protein G beads depending on the loading capacity of the magnetic beads used. For the beads described in this protocol, a ratio of 5 μg antibody to 1 μL beads is applied. This method describes cross-linking of 500 μg of an antibody to 100 μL beads, which provides enough material for approximately 500 enrichment experiments (1 μg of each antibody per capture).
Measure the concentration of antibody solution by bicinchoninic acid assay (BCA assay).
Wash 100 μL of the magnetic beads twice using 500 μL aliquots of 1×PBS (pH 7) in microcentrifuge tubes and a magnet. Remove 1×PBS and add 500 μg of the antibody to the washed beads. If the antibody concentration is very low, 5 or 15 mL tubes should be used.
Incubate the mixture overnight at 4 °C with tumbling.
Using a magnet, pull the beads to the side of the tube and remove the supernatant. Measure the protein concentration in the supernatant by BCA (see Note 5).
Add 900 μL of cross-linking solution (20 mM DMP in 200 mM triethanolamine, pH 8.5). Mix at room temperature for 30 min (see Notes 6 and 7).
Remove the supernatant and quench the coupling reaction by adding 900 μL of cross-linking quench solution (150 mM monoethanolamine, pH 9.0). Mix at room temperature for 30 min.
Remove the quench solution and wash the antibody-coupled beads three times for 5 min each time, using 900 μL aliquots of antibody wash solution 1 (1×PBS / 0.03% CHAPS)(see Note 8).
After the final wash, resuspend the antibody-coupled beads in 1000 μL antibody storage solution (1×PBS / 0.03% CHAPS / 0.1% sodium azide), at a final antibody concentration of approximately 0.5 μg/μL (disregarding the volume of the beads), and store at 4 °C in 1.5 mL screw-cap tubes (Sarstedt, Numbrecht, Germany) until use (see Note 9).
3.2. Cell Lysis
To prepare for cell lysis: turn on the refrigerated micro-centrifuge and cool to 4 °C, turn on benchtop centrifuge and cool to 4 °C, turn on Coulter Counter and prime aperture, thaw phosphatase and protease inhibitors, label and pre-cool 50 mL tubes, label and pre-cool micro-centrifuge tubes, label and pre-cool Cryo-vials, and make fresh Lysis buffer.
Remove media and rinse cells with half volume DPBS. Harvest cells by trypsinization and incubate cells at room temperature until the cells lift from the culture surface (as seen under the microscope).
Add Trypsin Neutralization Solution (TNS) and collect cells in a pre-cooled 50 mL centrifuge tube. Rinse plates with an additional aliquot of TNS to ensure all cells are collected. If needed, use a cell lifter to remove all cells from the culture surface.
Centrifuge cells at 400×g for 8 min at 4 °C. Remove the supernatant (i.e. TNS).
Resuspend and pool cells (if necessary) from the same cell line in ice-cold DPBS (note the volume), remove 150 μL for cell counting by Coulter Counter and check viability.
Add ice-cold DPBS to 50 mL and centrifuge cells at 400×g for 8 min at 4 °C while counting cells with Coulter Counter of hemocytometer. Remove supernatant.
Add ice cold Lysis buffer to a final concentration of 5×107 cells / mL.
Disperse cell pellet by dragging the tube along a microfuge tube rack. Do not pipette.
Sonicate cells 2 × 10 sec (Sonic Dismembrator knob set to 1). Wipe down probe with water and 70% ethanol between samples. Place lysate on ice for ∼30 seconds between sonications.
Transfer lysate by pipette to a micro-centrifuge tube, vortex 15 sec. Place the tube in a pre-cooled micro-centrifuge and spin 20k×g for 10 min at 4 °C.
Transfer supernatant to a 1.0 mL cryo-vial and store lysates in liquid nitrogen (see Note 10).
3.3. Trypsin Digestion
Adjust the cell lysate concentration to 5 mg/mL with Lysis buffer.
Transfer 100 μL (500 μg) of cell lysate to a 2 mL polypropylene 96 deep well plate. Digest duplicate aliquots of Process QC standard along with each batch of samples.
Add 6 μL of reducing agent (0.5 M TCEP).
Incubate with mixing at 700 rpm on a heater / shaker block for 30 min at 37 °C.
Add 14 μL of alkylating agent (0.5 M IAM). Incubate at room temperature for 30 min in the dark.
Add 880 μL of 0.2 M Tris, pH 8.0 to each sample to decrease the urea concentration to ∼0.6 M.
Add 20 μL trypsin solution to each digest to achieve a 1:50 enzyme-to-substrate ratio.
Incubate with mixing at 700 rpm on a heater /shaker for 2 hr at 37 °C.
Add 10 μL trypsin solution to each digest (a 1:100 enzyme-to-substrate ratio).
Incubate with mixing at 700 rpm on a heater / shaker for ∼16 hr (overnight) at 37 °C.
Add 50 μL of digestion quenching solution (20% formic acid) to each digest to quench the digestion (should have a final formic acid concentration of ∼1%).
Add 10 μL of SIS mix. Before spiking the heavy peptide master mix into the digested samples, dilute the master mix with 3% acetonitrile / 0.1% formic acid to a 10 fmol/μL concentration, to be able to spike 10 μL of the master mix into each sample (100 fmol of each heavy peptide per sample).
-
Desalt the digested peptides on an Oasis HLB 96 well plate, or equivalent solid phase extraction medium.
Condition the wells with 4 × 0.4 mL of 0.1% formic acid in 50% acetonitrile.
Equilibrate the wells using 4 × 0.4 mL of 0.1% formic acid in water.
Apply the digests to the wells.
Wash with 4 × 0.4 mL of 0.1% formic acid in water.
Elute peptides with 3 × 0.4 mL of 0.1% formic acid in 50% acetonitrile. Collect eluates in a 2 mL polypropylene 96 deep well plate.
Lyophilize the digests and store frozen (-70 °C) as dried powder until the next step.
3.4. Peptide Immunoaffinity Enrichment
Resuspend the digested lysate in 200 μL of 1×PBS / 0.01% CHAPS.
Adjust the pH to between 7.5-8.0 with 1 M Tris, pH 8.0. Check pH by pipetting 1 μL onto a pH 5-10 test strip. Verify that pH is between 7.5-8.0 before continuing to addition of antibody-beads.
Transfer the resuspended digests to 200 μL 96 well plates (the plates actually hold up to 400 μL of liquid).
Add the Antibody working master mix to each sample well to deliver an equivalent of 1 μg of each antibody (see Note 11).
Cover the plate with a Square Matrix CapMat and seal it by pressure to ensure that no liquid can leak from any of the wells (see Note 12).
Incubate samples overnight at 4 °C with tumbling (see Note 13).
Remove samples from the tumbler and centrifuge at 800 × g for 30 seconds to remove liquid that may be on the mat surface. Then carefully remove the mat.
-
Wash the beads and elute the peptides (see Note 14).
Wash the beads twice with 200 μL of 1×PBS / 0.01% CHAPS and once with 200 μL of 0.1×PBS / 0.01% CHAPS by mixing beads for 1.5 min in each wash solution and using a magnet to separate the beads from the supernatant in between washes.
The peptides are eluted in PCR plates by mixing the beads for 5 min in 26 μL of Peptide Elution Solution (3% acetonitrile / 5% acetic acid / 50 mM citrate). Separate the beads from the eluate using a magnet (see Note 15).
Cover the plate containing the eluates with ThermalSeal® PCR Sealing Film and spin the plate at 800 × g for 30 seconds.
Place the elution plate on a plate magnet for 5 minutes.
Transfer and split the eluates to two clean PCR plates by carefully drawing up 2 × 12 μL of the supernatant using a multi-channel pipet, without touching the bottom or sides of the wells, and transferring into corresponding wells of two clean PCR plates. Cover one plate with chemically resistant sealing foil, spin down the plate briefly and store it at -80 °C.
If analyzing samples immediately, proceed to next step using the second plate. Otherwise, cover the plate with chemically resistant sealing foil, spin it down briefly, and store at -80 °C.
3.5. Analysis of Samples by LC-MRM
When ready for analysis, thaw the samples (if necessary) and spin the plate at 800 × g for 30 seconds.
Add MS QC system suitability standards and blank injections (3% ACN / 5% acetic acid / 50 mM citrate) to empty wells, and response curve standards to regions of the plate not containing samples.
Cover the plate with a 96-well Silicone Sealing Mat, spin it down briefly, and place it on the autosampler (see Note 16).
-
Set up the HPLC instrument configuration according to Table 1.
Degas solvents for mobile phases A and B. Purge pumps. Calibrate flow rates.
Autosampler method: Pick up 2 μL of mobile phase A (0.1 % formic acid in water). Pick up 11 μL of sample. Briefly rinse the outside of the needle by inserting in a separate vial of 0.1% formic acid in water. Pick up 2 μL of mobile phase A. Switch autosampler valve to inject.
Perform the HPLC Gradient method according to Table 1.
-
Set up the MS instrument configuration and method according to Table 2 (see Note 17).
Set up an analysis batch file and submit samples for analysis (see Note 18).
Table 1. HPLC and autosampler parameters.
| HPLC parameters | |||
|---|---|---|---|
| Autosampler loop volume | 10 μL | ||
| Autosampler needle volume | 2.6 μL | ||
| Autosampler tray temperature | 4-8 °C | ||
| Trap column | 5 × 0.3 mm, PepMap 100 C18, 5 μm particle, 100 Å (Dionex, ThermoFisher Scientific) | ||
| Analytical column | 100 × 0.075 mm, ReproSil-Pur C18-AQ, 3 μm particle (Dr. Maisch GmbH) | ||
| Column temperature | 45 °C | ||
|
| |||
| Gradient | Time | Mobile phase B (%) | Flow rate |
|
| |||
| (Loading) | 0 | 2 | 10 |
| 1.5 | 2 | 10 | |
| (Elution) | 1.5 | 2 | 0.3 |
| 3 | 2 | 0.3 | |
| 18 | 40 | 0.3 | |
| 19 | 90 | 0.3 | |
| 20 | 90 | 0.3 | |
| 21 | 2 | 0.3 | |
| 30 | 2 | 0.3 | |
Table 2. Mass spectrometer settings and parameters.
| Mass spec parameters | |||||||
|---|---|---|---|---|---|---|---|
| Ion mode | Positive ion | ||||||
| IonSpray voltage | 1900-2200V | ||||||
| CAD gas | Medium | ||||||
| Interface heater temperature | 150 °C | ||||||
| Q1/Q3 resolution | Unit resolution | ||||||
| Settling time | 0 msec | ||||||
| Pause between masses | 3 msec | ||||||
| Declustering potential | 100 V | ||||||
| MRM detection window | 120 sec | ||||||
| Target scan time | 0.5 sec | ||||||
|
| |||||||
| Compound dependent parameters | |||||||
|
| |||||||
| Peptide | Light Q1 | Light Q3 | Heavy Q1 | Heavy Q3 | CE (V) | Fragment ion | |
|
| |||||||
| ATM.SLEISQSYTTTQR++ (1) | 757.38 | 1071.51 | 762.38 | 1081.51 | 36.8 | y9+ | |
| ATM.SLEISQSYTTTQR++ (2) | 757.38 | 856.42 | 762.38 | 866.42 | 33.8 | y7+ | |
| ATM.SLEISQSYTTTQR++ (3) | 757.38 | 984.47 | 762.38 | 994.48 | 34.8 | y8+ | |
| ATM.SLEISQSYTTTQR++ (4) | 757.38 | 1184.59 | 762.38 | 1194.60 | 32.8 | y10+ | |
| ATM.SLEISQSYTTTQR++ (5) | 757.38 | 769.38 | 762.38 | 779.39 | 34.8 | y6+ | |
| ATM.SLEIpSQSYTTTQR++ (1) | 797.36 | 856.42 | 802.37 | 866.42 | 41.9 | y7+ | |
| ATM.SLEIpSQSYTTTQR++ (2) | 797.36 | 583.79 | 802.37 | 588.80 | 36.9 | y10-98++ | |
| ATM.SLEIpSQSYTTTQR++ (3) | 797.36 | 606.32 | 802.37 | 616.33 | 39.9 | y5+ | |
| ATM.SLEIpSQSYTTTQR++ (4) | 797.36 | 769.38 | 802.37 | 779.39 | 42.9 | y6+ | |
| ATM.SLEIpSQSYTTTQR++ (5) | 797.36 | 1151.47 | 802.37 | 1161.48 | 34.9 | y9+ | |
| ATM.NLSDIDQSFNK++ (1) | 640.81 | 1053.49 | 644.82 | 1061.50 | 27.7 | y9+ | |
| ATM.NLSDIDQSFNK++ (2) | 640.81 | 966.45 | 644.82 | 974.47 | 28.7 | y8+ | |
| ATM.NLSDIDQSFNK++ (3) | 640.81 | 738.34 | 644.82 | 746.36 | 26.7 | y6+ | |
| ATM.NLSDIDQSFNK++ (4) | 640.81 | 851.43 | 644.82 | 859.44 | 28.7 | y7+ | |
| ATM.NLSDIDQSFNK++ (5) | 640.81 | 495.26 | 644.82 | 503.27 | 33.7 | y4+ | |
| ATM.NLpSDIDQSFNK++ (1) | 680.79 | 1133.45 | 684.80 | 1141.47 | 26.8 | y9+ | |
| ATM.NLpSDIDQSFNK++ (2) | 680.79 | 738.34 | 684.80 | 746.36 | 32.8 | y6+ | |
| ATM.NLpSDIDQSFNK++ (3) | 680.79 | 1035.47 | 684.80 | 1043.49 | 33.8 | y9-98+ | |
| ATM.NLpSDIDQSFNK++ (4) | 680.79 | 966.45 | 684.80 | 974.47 | 32.8 | y8+ | |
| ATM.NLpSDIDQSFNK++ (5) | 680.79 | 851.43 | 684.80 | 859.44 | 33.8 | y7+ | |
3.6. Data Analysis
Typical results for peptides captured from the Process QC sample are shown in Figure 1. Peak profiles are similar and retention times are identical for the light and heavy peptides. The relative areas of transitions for each set of peptides should be within 20% when comparing the light and heavy peptides.
Import the raw data into the Skyline document containing the targeted peptides.
Check the integration and retention time alignment of the endogenous peptides with the heavy peptide standards and relative peak areas of transitions. Annotate samples according to sample type and treatment (if applicable).
Export the results from Skyline to a csv file. Analysis can be performed in any quantitative analysis package (e.g. R platform) or with flat files (e.g. Excel files). Endogenous light peptide concentrations in fmol/mg are determined by multiplying the light-to-heavy peak area ratios by the known, spiked-in heavy peptide concentration (see Note 19).
Figure 1. Chromatograms for targeted ATM peptides.
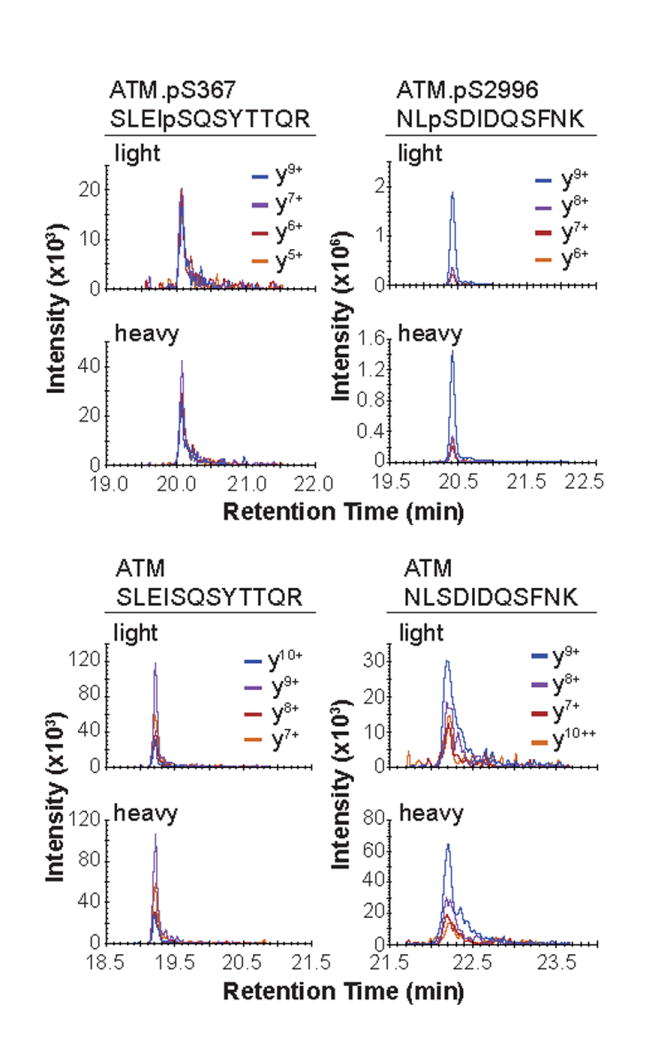
Chromatgorams for the phosphorylated (top panel) and non-modified analogs (bottom panel) for the targeted peptides as measured by MRM and analyzed from 500 μg total protein of the Process QC standard sample described in the method.
4. Notes
Protein-G magnetic beads are recommended to provide solid support for antibodies in optimum orientation for binding. Several sizes and capacities are commercially available. The protocol described uses Protein G Mag Sepharose beads (GE Healthcare, Little Chalfont, UK) which have been tested in the protocol and feature a large binding capacity.
It is necessary for the user to obtain suitable antibodies for peptide immunoaffinity enrichment. One source for monoclonal antibodies is the CPTAC Antibody Portal (antibodies.cancer.gov), which contains a repository of publicly-available and characterized antibodies. Custom made polyclonals can also be generated by most antibody vendors. Typically, the peptide antigen is coupled to KLH via a C-terminal cysteine linker and injected into animals. Polyclonal antibodies are affinity-purified from the antisera. Other sources may include commercially available antibodies capable of peptide immunoprecipitation.
Heavy stable isotope-labeled standards and matched light versions are synthesized and purified to >95% purity by HPLC. Heavy peptides incorporate a fully atom labeled 13C and 15N isotope at the C-terminal lysine (K) or arginine (R) position of each (tryptic) peptide, resulting in a mass shift of +8 or +10 Da, respectively. Peptides are quantified by amino acid analysis and aliquots are stored as described until use. Synthetic peptide characterization and quality control is an important component of quantitative studies. The degree to which care is taken in quantitative studies may depend on the goal of the research [12], however, it is recommended that laboratories characterize the purity of peptide standards and adhere to best practices when storing and handling peptides. Storage and handling of synthetic peptides may vary depending on the peptide sequence. We have found that general recommendations for peptide standards as outlined in Hoofnagle et. al. [13] are suitable for the ATM peptides measured in this method.
A suitable background matrix that closely replicates the nature of the intended sample with confirmed expression of the target can be substituted for the HeLa cell lysate.
The approximate coupling efficiency can be determined by comparing the amount of antibody in the supernatant pre- and post-coupling to magnetic beads. Coupling efficiencies are typically in the range of 60-80% using the procedure outlined.
Bead handling steps can be performed manually or using an automated bead handling system.
Due to the toxicity of DMP, triethanolamine, and monoethanolamine, it is advisable to perform steps using these reagents in a fume hood or well-ventilated area.
The method describes coupling of purified monoclonal antibodies. When coupling purified polyclonal antibodies, a slight modification is made to the procedure to remove any peptide that may be bound to the antibody. Wash the beads twice with 3% acetonitrile / 5% acetic acid and then wash with 1×PBS / 0.03% CHAPS.
Check the crosslinking step by subjecting an aliquot of antibody cross-linked beads to elution with 3% acetonitrile / 5% acetic acid. Run the eluate using a 4-20% gradient SDS-PAGE gel system and silver stain. The lanes should be void of bands corresponding to antibody.
If storing aliquots of a lysate, first transfer the lysate to a fresh micro-centrifuge tube to ensure homogeneous mixing of the lysate before aliquoting. In addition, an aliquot is used to determine protein concentration by BCA to inform the digestion step and the amount of trypsin to add. Lysates can be stored in liquid nitrogen for up to one month or more. For extended storage, it is recommended to lyophilize the lysate and store the dried powder frozen at -80 °C or in a liquid nitrogen dewar.
Ensuring the antibody-coupled beads are evenly distributed and well-mixed is important to ensure consistent application of equivalent amount of antibody and ultimately the reproducibility of the method. Vortexing, tumbling, agitating, or vigorously pipetting up-and-down are preferred methods for mixing beads.
Performance can vary depending on the type of beads used in the assay. Some beads do not interact with adhesive film and other types of beads can adhere to adhesive sealing film.
This is a critical step to the reproducibility of the method. We have found that tumbling the beads end-over-end is the most effective way to achieve good mixing. This is accomplished by sealing the plate and fixing it to a Labquake tube rotator using lab tape, rubber bands, or Velcro strips. Mixing is conducted for 16 hours at 4 °C.
Like previous bead handling steps, the wash and elution steps can be performed by an automated magnetic particle processor (e.g. KingFisher). If using a KingFisher (ThermoFisher Scientific), we recommend using the PCR magnetic head and a PCR deep well tip comb.
Following elution, the initial samples (i.e. analyte-depleted protein digests) can be saved for potential sequential capture experiments [14] and the antibody-coupled magnetic beads can be saved for potential recycling [15]. Transfer or resuspend used beads in 100 μL PBS / 0.03% CHAPS / 0.1% sodium azide. Seal the sample incubation plate and the collected antibody-bead plate with adhesive foil and store at -80 °C and 4 °C, respectively. The utility of sequential capture or recycling experiments should be validated prior to use.
It is advisable to use a plate magnet on the autosampler tray to keep any residual beads out of the HPLC system.
The following settings and configuration are reported for a Sciex 6500 QTRAP. Adjust settings to corresponding values for alternative triple-quadrupole systems. Table 2 provides the top 5 most intense transitions for the peptides determined on the 6500 QTRAP. Transitions may vary slightly depending on the instrument used and interferences arising from different sample matrices. Collision energy values were determined using Skyline optimization of the heavy peptides.
Inject the MS QC system suitability standard with an unscheduled MRM method a minimum of once per day. Injection of samples is dependent on satisfactory response of the MS QC system suitability standard compared to historical values. Inject the Process QC standard a minimum of twice per sample batch (i.e. twice per day), once before and once after samples, and monitor the results. Carryover can be determined by running a blank injection following the Process QC standard and monitoring the signal in the light and heavy peptide channels. Carryover for the ATM peptides is typically low (i.e. less than 1 percent). If carryover is a concern, inject wash runs using a mixture of methanol:acetonitrile:water (50:40:10) between samples.
If desired, annotate peaks and replicates for downstream analysis. SampleGroup, Treatment, Concentration, IS Spike, and Replicate annotations are useful for response curve and sample analysis. Peak annotations can be incorporated for split peaks, peaks not found, or unstable spray. Response curves can be analyzed directly in Skyline or by using add-ons such as QuaSAR [16] for analysis of response curves and identifying potential interferences. Additional tools for analysis directly in Skyline are continually being developed. Check with https://skyline.gs.washington.edu/ for updates.
Acknowledgments
This work was funded by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) of the US National Cancer Institute (U24CA160034).
References
- 1.Picotti P, Bodenmiller B, Aebersold R. Proteomics meets the scientific method. Nat Methods. 2013;10:24–27. doi: 10.1038/nmeth.2291. [DOI] [PubMed] [Google Scholar]
- 2.Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods. 2013;10:28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Addona TA, Abbatiello SE, Schilling B, et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27:633–641. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennedy JJ, Abbatiello SE, Kim K, et al. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat Methods. 2014;11:149–155. doi: 10.1038/nmeth.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whiteaker JR, Paulovich AG. Peptide immunoaffinity enrichment coupled with mass spectrometry for peptide and protein quantification. Clin Lab Med. 2011;31:385–396. doi: 10.1016/j.cll.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madian AG, Rochelle NS, Regnier FE. Mass-linked immuno-selective assays in targeted proteomics. Anal Chem. 2013;85:737–748. doi: 10.1021/ac302071k. [DOI] [PubMed] [Google Scholar]
- 7.Ackermann BL, Berna MJ. Coupling immunoaffinity techniques with MS for quantitative analysis of low-abundance protein biomarkers. Expert Rev Proteomics. 2007;4:175–186. doi: 10.1586/14789450.4.2.175. [DOI] [PubMed] [Google Scholar]
- 8.Boström T, Takanen JO, Hober S. Antibodies as means for selective mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2015 doi: 10.1016/j.jchromb.2015.10.042. [DOI] [PubMed] [Google Scholar]
- 9.Anderson NL, Anderson NG, Haines LR, et al. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3:235–44. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 10.Whiteaker JR, Zhao L, Yan P, et al. Peptide Immunoaffinity Enrichment and Targeted Mass Spectrometry Enables Multiplex, Quantitative Pharmacodynamic Studies of Phospho-Signaling. Mol Cell Proteomics. 2015;14:2261–2273. doi: 10.1074/mcp.O115.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinforma Oxf Engl. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carr SA, Abbatiello SE, Ackermann BL, et al. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics. 2014;13:907–917. doi: 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoofnagle AN, Whiteaker JR, Carr SA, et al. Recommendations for the Generation, Quantification, Storage, and Handling of Peptides Used for Mass Spectrometry-Based Assays. Clin Chem. 2016;62:48–69. doi: 10.1373/clinchem.2015.250563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whiteaker JR, Zhao L, Lin C, et al. Sequential multiplexed analyte quantification using peptide immunoaffinity enrichment coupled to mass spectrometry. Mol Cell Proteomics. 2012;11:M111.015347. doi: 10.1074/mcp.M111.015347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao L, Whiteaker JR, Voytovich UJ, et al. Antibody-Coupled Magnetic Beads Can Be Reused in Immuno-MRM Assays To Reduce Cost and Extend Antibody Supply. J Proteome Res. 2015;14:4425–4431. doi: 10.1021/acs.jproteome.5b00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mani DR, Abbatiello SE, Carr SA. Statistical characterization of multiple-reaction monitoring mass spectrometry (MRM-MS) assays for quantitative proteomics. BMC Bioinformatics. 2012;13(Suppl 16):S9. doi: 10.1186/1471-2105-13-S16-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]