

Abstract
RATIONALE
Phenethylamides are a large group of naturally occurring molecules found both in the plant and animal kingdoms. In addition, they are used as intermediates for the synthesis of pharmaceutically important dihydro- and tetrahydroisoquinolines. To enable efficient characterization of this class of molecules, a detailed mass spectrometric fragmentation study of a broad series of analogs was carried out.
METHODS
The test compounds were synthesized using standard methods for amide bond formation. Low energy high resolution tandem mass spectra were acquired on a hybrid quadrupole/time-of-flight mass spectrometer using positive ion electrospray ionization.
RESULTS
A total of 26 analogs were investigated in the study. Fragmentation of phenethylamides was found to proceed via intermediate ion-neutral complexes. The complexes can break down via multiple pathways including dissociation, proton transfer, Friedel-Crafts acylation, and single electron transfer. The relative contribution of each of these pathways strongly depends on the structure of the coupling amine and acid.
CONCLUSIONS
A general scheme for the fragmentation of phenethylamides was developed. This study further extends the knowledge base of the ion-neutral complex by discovering Friedel-Crafts acylation as a novel reaction. The strong influence of minor structural modifications on the fragmentation patterns highlights the importance of testing many analogs in order to fully predict a fragmentation pattern of a particular class of molecules.
Keywords: Phenethylamides, collision-induced dissociation, ion-neutral complex, cinnamides, gas-phase chemistry
INTRODUCTION
Phenethylamides are a large group of naturally occurring molecules found both in the plant and animal kingdoms. Chemically, they can be characterized as amides where the amine moiety is phenethylamine or, more commonly, its oxygenated derivatives such as tyramine, dopamine or their O-methylated analogs (Figure 1), and where the carboxylic acid is any of a myriad of known aliphatic and aromatic acids. In humans, long chain fatty acid phenethylamides such as N-arachidonoyl, oleoyl, palmitoyl and stearoyl dopamine have been identified in the central nervous systems where their biological role(s) is actively being investigated[1]. Since these amides chemically resemble endocannabinoids, much of the research is focused on their role in this system. In plants, phenethylamides represent a subgroup of a broad class of compounds called alkamides[2, 3]. The most widely occurring phenethylamides are those that couple tyramine or dopamine to any of the common cinnamic acids such as coumaric, caffeic or ferulic acid. In addition to their occurrence in nature, phenethylamides are often used as synthetic intermediates for the preparation of biologically important dihydro- and tetrahydroisoquinolines via Bischler-Napieralski reaction.
Figure 1.

Chemical structures of natural phenethylamines
Tandem mass spectrometry is an indispensable tool for the characterization of small molecules. Understanding fragmentation pathways of small molecules is not only a subject of basic research but also has important practical applications as many active fields of research such as dereplication of natural products or drug metabolism studies rely on understanding the correlation between the fragmentation pattern and the structure of a molecule. As a part our interest in studying nitrogenous plant metabolites, we recently reported tandem mass spectra of several naturally occurring cinnamides[4] but did not carry out a thorough investigation of their fragmentation pathways. To better understand the fragmentation pattern of this class of molecules, we investigated the fragmentation behavior of 26 synthetic and naturally occurring phenethylamides under low energy CID. The results reveal that fragmentation proceeds via a transient ion-neutral complex whose subsequent decomposition is strongly influenced by the structure of the molecule.
EXPERIMENTAL
Chemicals and reagents
All reagents for synthesis were from Sigma-Aldrich (St. Louis, MO). HPLC-grade or better solvents were from Fisher Scientific (Fair Lawn, NY). Deuterated solvents were from Cambridge Isotope Laboratories (Andover, MA).
Test compounds were synthesized using established procedures for amide bond formation. Compounds 3, 7 and 8 were prepared by treating tyramine[5], 3-O-methyldopamine[6] and dopamine[6] and with acetic anhydride, respectively; compounds 5, 12, and 26 were prepared by treating amine with 2-methyl-6-nitrobenzoic acid anhydride. Nicotinyl dopamine (10), feruloyl tyramine (6) and feruloyl dopamine (11) were synthesized by coupling the amine and the corresponding acid using 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide as the coupling agent, as described previously[4]. All other amides were synthesized by treating the amine with an appropriate acyl chloride under Schotten-Baumann conditions[7]. Chemical structures of tested compounds are shown in Table 1.
Table 1.
Chemical structures of the investigated compounds.
| Compound | Structure | Protonated molecule (m/z) |
|---|---|---|

|
||
| 1 | R= Me | 164 |
| 2 | Phe | 226 |

|
||
| 3 | R= Me | 180 |
| 4 | Phe | 242 |
| 5 | 2-Methyl-6-nitrophenyl | 301 |
| 6 | Feruloyl | 314 |
| 7 |

|
210 |
| 8 | R= Me | 196 |
| 9 | n-Pentyl | 252 |
| 10 | 3-Pyridyl | 259 |
| 11 | Feruloyl | 330 |
| 12 | 2-Methyl-6-nitrophenyl | 317 |

|
||
| 13 | R= Me | 224 |
| 14 | n-Pentyl | 280 |
| 16 | Phe | 286 |
| 17 | p-(NO2)Phe | 331 |
| 18 | p-(Cl)Phe | 320 |
| 19 | p-(F)Phe | 304 |
| 20 | p-(CN)Phe | 311 |
| 21 | p-(Me)Phe | 300 |
| 22 | p-(OH)Phe | 302 |
| 23 | Benzyl | 300 |
| 24 | (3,4-diOMe)Phe | 346 |
| 25 | Cinnamoyl | 312 |
| 26 | 2-Methyl-6-nitrophenyl | 345 |
Mass spectrometry
High resolution mass spectra (resolving power 10,000 FWHM) and product ion tandem mass spectra were acquired using a Waters (Milford, MA) SYNAPT hybrid quadrupole/time-of-flight mass spectrometer operated in positive ion electrospray mode. Compounds were dissolved in 50% MeCN/1% acetic acid at approximately 5 μM and infused into the ion source at 10 μL/min. Typical source parameters were: capillary 3.5 kV; cone: 25V; source temperature 100°C; desolvation gas temperature: 250 ºC; gas flow: 500 L/hr. For acquisition of deuterium exchanged spectra compounds were dissolved in 50%MeCN/D2O containing 1% of CD3COOD. Product ion tandem mass spectra were recorded at collision energies from 6–25 eV using argon as collision gas.
RESULTS AND DISCUSSION
Low energy CID tandem mass spectra of three N-acetyl phenethylamines are shown in Figure 2, the tandem mass spectra of selected aromatic amides of homoveratrylamine are shown in Figure 3, while the tandem mass spectra of other analogs are summarized in Table 2. Of particular interest for this study is the observation of a fragment ion corresponding to the loss of ammonia from the protonated molecule (ion c in Figures 2 and 3). During interpretation of tandem mass spectra, loss of ammonia from the protonated molecule is often interpreted as an indication of the presence of a primary or secondary amino group. However, there is no amino group in these compounds, thus formation of this ion must include some sort of skeletal rearrangement. It was interesting to note that this fragment was observed only when the phenethylamine ring is strongly activated by the presence of at least two hydroxy or methoxy groups as was the case for dopamine and homoveratrylamine (Figure 2c and Figure 3). The origin of this and other major ions in these tandem mass spectra is discussed below.
Figure 2.

Product ion tandem mass spectra of selected N-acetyl phenethylamines. For ion labels see Scheme 1. Note that loss of ammonia (ion c) was observed only for the highly activated phenethylamine ring. Spectra were obtained at 10 eV collision energy.
Figure 3.

Product ion tandem mass spectra of selected aromatic phenethylamides of homoveratrylamine. Note how the relative ratios of the acylium ion (ion a) and ion b vary depending on the stability of the a ion. Spectra were obtained at 10 eV collision energy.
Table 2.
Summary of the low energy (10 eV) high resolution product ion tandem mass spectra of phenethylamides investigated in this study.
| Product ions: m/z (relative intensity)
| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd. | [M+H]+ | c | b1 | b2 | a | MH-H2O | Other |
| 2 | 226.1223 (100) | - | - | 105.0688(4) | 105.0321(45) | - | 77.0391 (5) |
| 4 | 242.1197(100) | - | - | 121.0647(28) | 105.0334(40) | - | 103.054(4);93.070(4); 77.0378(5) |
| 6 | 314.1392(100) | - | - | - | 177.0540(94) | - | 145.027(18);117.032(10) |
| 7 | 210.1130(21) | 193.0848(6) | 168.1012(11) | 151.0742(100) | - | - | 119.048(15);91.053(16) |
| 8 | 196.0981(20) | 179.0810(6) | 154.0887(20) | 137.0618(100) | - | - | 119.051(15);91.056(21) |
| 9 | 252.1600(60) | 235.1345(2) | 154.0860(69) | 137.0592(100) | 99.0804 (1) | - | 119.049(9);91.054(12) |
| 10 | 259.1082(100) | - | - | 137.0614(3) | - | - | |
| 11 | 330.1341(96) | - | - | - | 177.0541(100) | - | 145.027(28);117.032(8) |
| 14 | 280.1935(55) | 263.1667(8) | 182.1182(40) | 165.0924(100) | - | 150.0692(9) | |
| 18 | 320.1070(100) | 303.0805(12) | - | 165.0927(32) | 138.9957(16) | 302.0956(2) | 111.0047(2) |
| 19 | 304.1349(100) | 287.1097(15) | - | 165.0907(52) | 123.0237(23) | 286.1243(2) | 150.0673(5) |
| 20 | 311.1396(89) | 294.1136(14) | - | 165.0901(100) | 130.0284(2) | 293.1491(1) | 150.0663(10) |
| 21 | 300.1600(100) | 283.1348(6) | - | 165.0907(14) | 119.0484(61) | 282.1507(1) | 91.0533(8) |
| 22 | 302.1392(100) | 285.1114(2) | 182.1160(19) | 165.0898(35) | 121.0269(51) | 284.1385(3) | 93.0326(3) |
| 23 | 300.1602(100) | 283.1337(8) | 182.1169 (19) | 165.0920(63) | - | 282.1130(1) | 150.0676(5) |
| 25 | 312.1604 (100) | 295.1346 (7) | 182.1182 (3) | 165.0925 (7) | 131.04999 (67) | - | 103.0550(15) |
The formation of major fragment ions in the tandem mass spectra of phenethylamides can be rationalized according to Scheme 1. It is well established that the most favorable protonation site in amides is the carbonyl oxygen[8], but the dissociating species of amides has been shown to be N-protonated[9]. That protonation of the amide nitrogen is important for fragmentation is supported by an observation that when the ionizing proton was sequestered by an alternative protonation site, such as strongly basic pyridine nitrogen in 10, very little fragmentation was observed (Table 2). The formation of the N-protonated amide is in principle possible by a direct 1,3-H+ transfer from oxygen to nitrogen. However, this transfer has been shown to have a high energy barrier and is not mechanistically relevant[10]. We propose an alternative route for the proton transfer which involves the aromatic ring of the phenethylamines as shown in Scheme 2A. This mechanism is similar to the one proposed for the H/D scrambling observed during fragmentation of protonated tryptophan[11, 12] and protonated alkyl dihydrocinnamates[13] and is consistent with the H/D scrambling observed in our study (Figure 4). Note that the scrambling observed in the spectra rules out a direct O to N proton transfer, which would result in no observable scrambling.
Scheme 1.

Summary of proposed fragmentation pathways of phenethylamides. A key intermediate in the Path A is ion-neutral complex (INC-1) between the acylium ion and the base. The complex can break down via simple dissociation (Path A1, ion a), proton transfer (Path A2, ion b1) or Friedel-Crafts acylation (Path A3, ion c). For the path A2, the transferred proton originates from a labile position (shown for p-hydroxybenzoyl analog on the right). In the proposed structure of the ion c the position of the acyl group is drawn arbitrarily for illustration; Path B represents inductive cleavage of the Cα-N bond. It could proceed either via 1,2-hydride assisted cleavage or by neighboring group participation in which aromatic ring serves as the nucleophile to produce a phenonium-type structure.
Scheme 2.

Proposed mechanisms for H/D scrambling observed during fragmentation of A) aliphatic and aromatic phenethylamides, and B) amides of with cinnamic acid derivatives.
Figure 4.

Product ion mass spectra of selected phenethylamides analogs in deuterated solvent.
While for acetylated derivatives (Figure 4a), the migrating proton does not have an alternative route from the carbonyl side, this route is in principle possible for aromatic and conjugated aromatic analogs. From deuterium exchange experiments with unconjugated aromatic amides, there was no evidence for the proton transfer to the aromatic ring as the corresponding acylium ions (see below) did not increase in mass even for the highly activated 3,4-dimethoxyphenyl analog (Figure 4b). However, evidence for an alternative proton migration pathway that involves the carbonyl side was found in the case of conjugated amides.
Figure 4c and 4d shows tandem mass spectra of deuterated feruloyl tyramine and feruloyl dopamine, two common natural products. The ion of m/z 179 represents an acylium ion that had retained one extra deuterium in addition to the deuterium from the exchangeable position. Two possible mechanisms for this transfer can be envisioned as shown in Scheme 2B. One mechanism involves two 1,3-proton transfers involving the α-carbon. The first proton transfer creates a resonantly stabilized benzylic cation, which is likely the driving force for this pathway. In the second step, the double bond is restored by proton transfer to the amide nitrogen. Scrambling occurs because either deuteron or proton can be transferred in this step. The second potential mechanism involves proton transfer to the aromatic ring following prior cis-trans isomerization that is necessary to bring the reaction partners into proper orientation. It is interesting to note that the ion of m/z 179 was less abundant in the spectrum of feruloyl dopamine compared to that of feruloyl tyramine (Figure 4c and 4d). This suggests that proton transfer to the carbonyl side competes with the proton transfer to the phenethylamine ring. Two strong electron donating hydroxyl groups on dopamine apparently provide a lower energy pathway for the proton transfer due to increased stabilization of the resulting cation compared to the tyramine case which has only one hydroxyl group.
Once the N-protonated species has been formed the consequence of such protonation have been well studied in the literature. Protonation of the amido nitrogen induces elongation and weakening of both of the amide and the Cα-N bond making them amenable to cleavage[14, 15]. Hence, phenethylamides can produce fragments that originate from bond cleavages arising from either side of the amido nitrogen.
We will first discuss the Path A in Scheme 1 which represents cleavage of the amide bond. As shown in the seminal work by Tu and Harrison[9], cleavage of the amide bond results in the formation of a transient ion-neutral complex (INC-1) consisting of the amine and the acylium ion held together by electrostatic forces (Scheme 1). This complex can then break down via multiple pathways. A simple dissociation of the two partners (Path A1) produces an acylium ion (ion a) and an uncharged amine. The relative contribution of this pathway to the overall fragmentation depends strongly on the stability of the resulting acylium ion. For example, for alkyl derivatives such as acetyl and hexanoyl, the acylium ion was a minor fragment. In case of aromatic acylium ions, when an electron withdrawing group was present at the para position, the ion a was barely detectable (Figure 3b), but when an electron donating group(s) was present (Figure 3c) or the ion could be stabilized by extended resonance as was the case with cinnamic acid derivatives 6, 11 and 25, the formation of the resulting acylium ion was the dominant fragmentation pathway.
The second breakdown pathway for the ion-neutral complex occurs via proton transfer (Path A2) to produce a protonated base (ion b1). It has been shown that the intermediate species in this process is a proton-bound complex [9]. This process can obviously occur only if the acylium ion has available hydrogen that can be transferred to the base as was the case with aliphatic analogs and aromatic analogs containing a hydroxyl group. For example, the tandem mass spectrum of deuterium exchanged 22 (Figure 4e) shows that it is the labile hydrogen that was transferred to the base to produce the fragment ion of m/z 185. In comparison, this ion appears at m/z 184 in the spectrum of 13 (Figure 4a). The occurrence of ion of m/z 185 in the spectrum of 22 shows that the partners in the ion-neutral complex have considerable mobility as the acylium ion needs to rotate to align its exchangeable hydrogen with the base (Scheme 1). This so-called reorientation criterion is often used as a proof of existence of ion-neutral complex[16, 17]. It is interesting to note that the b1 ion was not observed in the spectra of cinnamides 6 and 11 which also have exchangeable hydrogen. Apparently, enhanced stabilization afforded by the two activating groups in 6 and 11 makes the acylium ion so stable that proton transfer cannot compete with simple dissociation. In addition, the feruloyl ion is bulky, thus the ion-neutral complex might not provide enough mobility for the hydroxyl group to align properly with the base. It was also interesting to note that proton transfer was not observed from the para-methyl benzoyl cation formed from compound 21. This is in contrast to the para-methyl benzyl cation from which proton transfer to amines has been observed[18].
Another interesting and peculiar observation in the deuterium exchanged spectra of 6, 11 and 22 in Figure 4c–e is the presence of an acylium ion in which labile deuterium had been replaced with hydrogen (ions at m/z 177 and m/z 121). One possible explanation for this observation is that H/D exchange occurred in the proton bound complex. As noted above, deuterium scrambling of the ionizing proton with hydrogens from the phenethylamine ring results in a distribution of N-protonated amide species, namely ND2+, NDH+ and NH2+. In the proton-bound complex, the exchange between one of the two hydrogen-containing species and the labile hydrogen in the acylium ion could occur resulting in the replacement of deuterium from the exchangeable position.
An alternative explanation might be the participation of a water molecule in the exchange process. Presence of water in the collision cell gases and its influence on the appearance of tandem mass spectra has been well established[19–22], and we have previously observed formation of water adducts of fragment ions of isoferulic acid in this particular instrument[4]. Acylium ions are particularly prone to reversible reaction with residual water as demonstrated in several reports[23–25]. Adventitious water molecules may simply exchange hydrogen with the labile proton on the acylium ion in much the same manner as to what occurs in solution. Alternatively, a water molecule might function as a proton shuttle between the base and the acylium ion in the ion-neutral complex. In any case, this is an interesting observation that deserves additional studies to be fully elucidated as none of the proposed mechanisms have been demonstrated before.
Finally, path A3 corresponds to the electrophilic attack of the acylium ion onto the aromatic ring which can be considered as gas-phase Friedel-Crafts acylation. Electrophilic aromatic substitution within an ion-neutral complex is a well-documented phenomenon[18, 26–32]. However, most of the reports deal with the benzyl cation transfer. More recently, reports have appeared demonstrating sulfonyl cation transfer during fragmentation of protonated sulfonamides[33–35], but we are unaware of similar reports describing Friedel-Crafts acylation. Reported ion-molecule reactions of acylium ions within ion-neutral complex mostly include intramolecular transacylation reaction in which the acylium ion migrates to attack another nucleophilic center in the molecule[36–39]. Migration of an acetyl ion to a remote nucleophilic site via multiple benzene rings has been observed in the MIKES mass spectrometry studies of protonated aromatic carbonyl compounds[40].
As noted above, the Friedel-Crafts pathway requires a highly activated aromatic ring with a minimum of two hydroxyl groups, probably due to low reactivity of the attacking acylium electrophile (Figure 2). The single hydroxyl group in tyramine apparently does not provide sufficient activation for this reaction to occur. Following proton transfer from the resulting sigma complex to the amino group, a ring acylated phenethylamine species is formed that can easily lose ammonia to produce the ion c in Scheme 1. The loss of ammonia could proceed via two possible pathways, either by the C1 attack to produce a phenonium-type ion, or by inductive cleavage assisted by 1,2-hydride migration to produce a stable benzylic type ion (see more discussion below).
As implied from the preceding discussion, the Friedel-Crafts acylation and simple dissociation are competing processes for the breakdown of the ion-neutral complex. The relative ratios of these two pathways depend on the stability/reactivity of the acylium ion. When the acylium ion is stabilized by electron-donating substituents, it is not sufficiently reactive to attack the phenethylamine ring. In contrast, acylium ions with electron-withdrawing substituents become sufficiently reactive to undergo electrophilic substitution. This observation can be represented by a Hammet plot as shown in Figure 5 in which a good correlation was observed between the logarithmic values of abundance ratios of the a and c ions versus the Brown-Okatomoto substituent constant.
Figure 5.
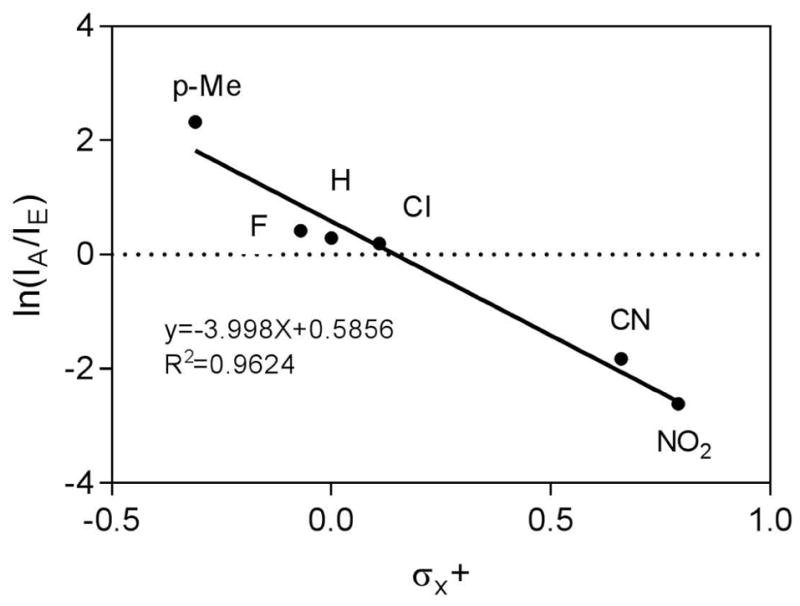
The Hammet plot of abundance ratios of ion a (IA) and ion c (IE) versus Brown-Okamoto substituent constant σx+ for a series of aromatic homoveratrylamides. The abundances of the ion a are adjusted to include secondary loss of CO which was observed in some cases. The values for the constants were taken from the comprehensive review by Hansch et al. [54]
Particularly interesting fragmentation pathways were observed for phenethylamides containing 2-methyl-6-nitrobenzoic acid (Figure 6). An acylium ion (ion a) at m/z 164.03 was the dominant peak in the product ion mass spectra of tyramine and dopamine analogs and the only peak in the spectrum of an unsubstituted phenethylamine analog (not shown). At first glance, this is surprising considering that for the p-NO2 analog 17, the acylium ion was barely detectable (Figure 3b) and for the p-Me analog 21, the b2 ion was also present. However, the most striking observation was that the tandem mass spectrum of 26 (Figure 6c) does not resemble the spectrum of any other compound evaluated in this study. In particular, ion b2 (at m/z 165), which was the dominant fragment in most other homoveratrylamine analogs, is only a minor fragment ion present at <2% relative intensity. Instead, the most intense peaks at m/z 181 and 152 represent the even electron species C10H15NO2 and C9H12O2, respectively, as determined by accurate mass measurements. The ion of m/z 181 can be considered an odd electron-ion equivalent of the b1 ion, that is, it represents a cation radical of homoveratrylamine.
Figure 6.

Product ion mass spectra of phenethylamides containing 2-methyl-6-nitro benzoic acid. Note a dramatic change in the appearance of the spectra depending on the number of electron-donating substituents on the phenethylamine ring. Mass spectra were obtained at 10 eV collision energy.
The main features of these spectra can be rationalized according to the mechanism proposed in Scheme 3. After initial protonation on the carbonyl oxygen, we propose that for these compounds the neighboring nitro group serves as an acceptor site to shuttle the proton to the amide nitrogen. Such a transfer mechanism has been proposed for o-nitro anilides[9]. Initial protonation on the nitro group itself is also plausible. After rupture of the amide bond, an ion-neutral complex (INC-2) is formed between the base and the o-methyl, o-nitro benzoyl ion. Fission of the amide bond is likely assisted by the neighboring nitro group to create a cyclic structure of the resulting acylium as shown in Scheme 3. This cyclized form is likely the main reason behind the great stability of this ion. A small population of ions is likely in an open form considering that a small, but detectable peak for ion c is present in the spectrum of 26 (Figure 6c). In contrast to the tyramine and dopamine analogs, the INC-2 complex for the homoveratrylamine analog undergoes a single electron transfer to produce homoveratrylamine cation radical at m/z 181, which subsequently loses methyleneimine (CH2=NH; 29 Da) to produce a McLafferty rearrangement product ion at m/z 152.0825 (Figure 6c).
Scheme 3.

Proposed fragmentation pathways for compounds 26 (3A) and 12 (3B). For these analogs, the nitro group may serve as a proton acceptor to transfer the ionizing proton to the amido nitrogen. In contrast to other tested analogs, the ion-neutral complex intermediate (INC-2) can undergo a single electron transfer due to favorable combination of electron donating ability of the activated aromatic ring and electron accepting ability of the o-nitro benzoyl ion. In case of 12 an additional hydrogen abstraction step from the hydroxyl group is proposed to occur resulting in the formation of the ion of m/z 152.068.
There are several examples in the literature describing electron transfer within ion-neutral complexes. Most of these examples involve either a benzyl cation[41, 42] or a sulfonyl cation/aniline complex formed during fragmentation of sulfonamides[34, 43], but we were unable to find any reports describing this phenomenon for phenethylamines. As noted by Chai et al., single electron transfer processes within ion-neutral complexes are much more energetically favorable compared to simple homolytic cleavage[41]. Electron transfer processes are affected by the electron affinity of the acceptor and the ionization energy of the electron donor. In our case, the resulting o-methyl, o-nitro benzoyl radical is an extremely stable species[44]. This stabilization has been shown to originate from intramolecular trapping of the carbon centered radical by the neighboring nitro group to produce the cyclic acyloxy aminoxyl structure shown in Scheme 3. In solution phase, the lifetime of this radical is measured in days[44]. On the other hand, the ability of phenethylamines to serve as electron donors depends on the energy of their highest occupied molecular orbital which is related to the ionization energy. Hu et al. found good correlation between ionization energies of electron donor and single electron transfer process in the ion-neutral complex[43]. Ionization energies of phenethylamines have been measured using photoelectron spectroscopy and were determined to be 8.99, 8.41, 8.18 and 8.03 eV for phenethylamine, tyramine, dopamine, and homoveratrylamine, respectively[45, 46]. These data at least qualitatively agree with the notion that ionization energy of the donor plays an important role in single electron transfer. It has also been shown that the single electron transfer process occurs if the products are more stable than the separation products. In our case, it is clear that two methoxy groups provide greater stabilization to the homoveratrylamine cation than can one hydroxyl group in tyramine.
Decomposition of the dopamine INC-2 complex was slightly different from that of homoveratrylamine (Figure 6b vs 6c). The dopamine radical cation which should appear at m/z 153 was not detected; instead the even electron species of m/z 152.068 was present with the elemental composition of C8H10NO2. We propose that this ion is formed by a reaction sequence shown in Scheme 3B. In this mechanism, single electron transfer still occurs in the ion-neutral complex, but the resulting dopamine radical cation (which has labile hydrogens) undergoes hydrogen abstraction by the o-methyl, o-nitro benzoyl radical to produce a protonated dopaminechrome-like structure, which can subsequently lose methyleneimmine by a McLafferty-type rearrangement similarly to what is described for homoveratrylamine. The resulting ion can then rearrange to a stable 3,4-dihydroxybenzyl cation. It is interesting to note that an ion of m/z 123 was detected in photoionization spectra of gas-phase dopamine, but neither the structure nor the mechanism of formation of this ion were proposed[46]. The examples shown in Figure 6 highlight not only the strong influence of the ortho effect on the appearance of the spectra, but also the fact that seemingly small changes in the structure of the molecule can have profound influence on the appearance of the spectrum.
The other main fragmentation pathway of phenethylamides (Scheme 1, Path B) is a loss of neutral amide by direct cleavage of the aliphatic C-N bond resulting in the formation of ion b2. Except for analogs that produced stable acylium ions, ion b2 was the base peak for all other cases. The mechanistic considerations for the formation of this ion are the same as those for the ion c (Scheme 1), that is, it can proceed via 1,2-hydride assisted inductive cleavage or via intramolecular nucleophilic attack to produce a phenonium-type structure.
There has been considerable work done to address this issue in similar model systems using molecular orbital calculations. It has been originally proposed, for example, that loss of ammonia from protonated phenylalanine proceeds via phenonium pathway[47]. Subsequently, Lioe and O’Hair examined in detail the elimination of ammonia from protonated phenylalanine derivatives and concluded that although 1,2-hydride migration pathway is thermodynamically favored (produces a more stable benzylic cation) the transition state barrier for this pathway is higher than the corresponding barrier for the phenonium pathway[48]. This energy gap was more pronounced when electron donating substituents were present on the aromatic ring. A similar conclusion was reached when loss of ammonia from Ag+-phenylalanine complex was examined[49]. Based on these studies, it would appear that ions b2 and c have a phenonium structure. It should also be noted that ion b2 could also be formed by further fragmentation of ion b1 via loss of ammonia. Loss of ammonia from dopamine and its derivatives is a facile process. A recent study examined the structures of protonated dopamine fragments using IR multiple photon dissociation along with molecular orbital calculations and concluded that loss of ammonia most likely proceeds through the 1,2-hydride pathway, which in contrast with the above mentioned studies on phenylalanine[50].
Another interesting fragmentation pathway observed was loss of water, particularly from analogs with electron withdrawing group on the aromatic ring (see Figure 3b). Although this pathway was minor (~ 2% of the base peak) it is mechanistically interesting considering that loss of water in tandem mass spectra is typically associated with the presence of a hydroxyl group. Loss of water from N-(3-aminophenyl) benzamide was reported recently by Zu et al. and has been shown to involve a water-nitrilium ion complex[51]. While this mechanism might be operational for our compounds, we propose another mechanism that involves electrophilic attack of the protonated amideonto the aromatic ring as shown in Scheme 4. After proton transfer and rearomatization, water is eliminated to produce a protonated dihydroisoquinoline structure. This mechanism is analogous to that of the well-known Bishner-Naprielski reaction that is a standard method for preparation of dihydroisoquinolies from acylated phenethylamines. It is also consistent with the observation that this pathway was promoted by electron withdrawing groups which increase electrophilicity of the attacking electrophile. The factors that promote formation of this ion are the same as those observed for the Friedel-Crafts pathway, namely it requires an activated phenethylamine ring and is promoted by electron-withdrawing substituents on the acyl moiety.
Scheme 4.

Proposed mechanism for the loss of water from protonated phenethylamides. In this mechanism, protonated amide itself acts as an electrophile that attacks activated aromatic ring of the phenethylamine. After proton transfer and rearomatization, water is eliminated to produce a protonated dihydroisoquinoline structure. This pathway is promoted when R is an electron-withdrawing group.
Recently, there has been a considerable interest in studying the gas phase chemistry of ion-neutral complexes[52, 53]. This intermediate species has been invoked to explain origins of fragment ions that at first glance appeared strange. In addition to known pathways such as simple dissociation, proton transfer and hydrogen transfer, we identified Friedel-Crafts acylation as a new pathway. As the ion-neutral complex is a common intermediate, seemingly small changes in the structures of reactants can make profound differences as to which decomposition pathway will be primarily followed. For example, Friedel-Crafts acylation was not observed for tyramine amides. A “minor” change of addition of an extra hydroxyl group allows this reaction to proceed resulting in a formation of a new fragment ion that could not be predicted beforehand. Similarly, going from two hydroxyl groups as in 12 to two methoxy groups in 26 opens single electron transfer as an energetically favorable pathway resulting in spectra that could hardly be recognized as belonging to the same class of molecules.
This study also highlights that these subtleties in fragmentation behavior can only be discovered when many structural analogs are investigated and underscores the need that such studies be conducted before one can start to predict how other analogs from the same class of compounds might fragment. Currently, the main drawback of in silico prediction programs is in their inability to take into account these unusual fragmentation channels. The mechanisms proposed in this study also imply that other arylalkylamides, where the aryl group is activated for electrophilic substitution, should fragment similarly. Examples from the natural products world would include indolamines such as serotonin and melatonin. Indeed, in preliminary experiments, we observed that amides of these indoleamines also display the same fragmentation channels as observed in this study.
Conclusions
Low energy collision-induced dissociation tandem mass spectra of a broad range of phenethylamides were studied using high resolution mass spectrometry. The results demonstrate that fragmentation proceeds via an ion-neutral complex, which can decompose through multiple pathways depending on the nature of the coupling partners. In addition to the well-established decomposition pathways of ion-neutral complexes, Friedel-Crafts acylation was identified as a novel reaction fragmentation pathway. The study further demonstrated that seemingly small changes in the structure of the compounds may produce dramatic changes in the appearance of tandem mass spectra. The pathways described in this study provide a solid foundation for the prediction of fragmentation of other analogs belonging to this class of compounds.
Acknowledgments
This work was supported by NIH grant P50 AT000155 from the NIH Office of Dietary Supplements and the National Center for Complementary and Integrative Health. We thank Dr. Victor Ryzhov and Michael Lesslie, Northern Illinois University for helpful discussion.
References
- 1.Connor M, Vaughan CW, Vandenberg RJ. N-acyl amino acids and N-acyl neurotransmitter conjugates: neuromodulators and probes for new drug targets. Br J Pharmacol. 2010;160:1857. doi: 10.1111/j.1476-5381.2010.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boonen J, Bronselaer A, Nielandt J, Veryser L, De Tre G, De Spiegeleer B. Alkamid database: Chemistry, occurrence and functionality of plant N-alkylamides. J Ethnopharmacol. 2012;142:563. doi: 10.1016/j.jep.2012.05.038. [DOI] [PubMed] [Google Scholar]
- 3.Rios M. Drug Discovery Research in Pharmacognosy. InTech; Rijeka: 2012. p. 107. www.intechopen.com. [Google Scholar]
- 4.Nikolic D, Godecke T, Chen SN, White J, Lankin DC, Pauli GF, van Breemen RB. Mass spectrometric dereplication of nitrogen-containing constituents of black cohosh (Cimicifuga racemosa L) Fitoterapia. 2012;83:441. doi: 10.1016/j.fitote.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Antonio M, Doria F, Richter SN, Bertipaglia C, Mella M, Sissi C, Palumbo M, Freccero M. Quinone Methides Tethered to Naphthalene Diimides as Selective G-Quadruplex Alkylating Agents. J Am Chem Soc. 2009;131:13132. doi: 10.1021/ja904876q. [DOI] [PubMed] [Google Scholar]
- 6.Seo JW, Srisook E, Son HJ, Hwang O, Cha YN, Chi DY. Syntheses of NAMDA derivatives inhibiting NO production in BV-2 cells stimulated with lipopolysaccharide. Bioorg Med Chem Lett. 2005;15:3369. doi: 10.1016/j.bmcl.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 7.Moreno L, Cabedo N, Boulange A, Parraga J, Galan A, Leleu S, Sanz MJ, Cortes D, Franck X. Synthesis of pyrido[2,1-a]isoquinolin-4-ones and oxazino[2,3-a]isoquinolin-4-ones: new inhibitors of mitochondrial respiratory chain. Eur J Med Chem. 2013;69:69. doi: 10.1016/j.ejmech.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 8.Fraenkel G, Franconi C. Protonation of Amides. J Am Chem Soc. 1960;85:4478. [Google Scholar]
- 9.Tu YP, Harrison AG. Fragmentation of protonated amides through intermediate ion-neutral complexes: Neighboring group participation. J Am Soc Mass Spectrom. 1998;9:454. [Google Scholar]
- 10.Lin HY, Ridge DP, Uggerud E, Vulpius T. Unimolecular Chemistry of Protonated Formamide - Mass-Spectrometry and Ab-Initio Quantum-Chemical Calculations. J Am Chem Soc. 1994;116:2996. [Google Scholar]
- 11.Lioe H, O’Hair RAJ, Reid GE. Gas-phase reactions of protonated tryptophan. J Am Soc Mass Spectrom. 2004;15:65. doi: 10.1016/j.jasms.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Cai T, Wang D, Xu XY, Fang DM, Qi HY, Jiang Y, Wu ZJ. New evidence for H/D scrambling of tryptophan and its analogues in the gas phase. Int J Mass Spectrom. 2015;385:26. [Google Scholar]
- 13.Xu SH, Zhang Y, Errabelli R, Attygalle AB. Ambulation of Incipient Proton during Gas-Phase Dissociation of Protonated Alkyl Dihydrocinnamates. J Org Chem. 2015;80:9468. doi: 10.1021/acs.joc.5b01390. [DOI] [PubMed] [Google Scholar]
- 14.Bouchoux G. Gas-phase basicities of polyfunctional molecules. part 1: Theory and methods. Mass Spectrom Rev. 2007;26:775. doi: 10.1002/mas.20151. [DOI] [PubMed] [Google Scholar]
- 15.Bouchoux G. From the mobile proton to wandering hydride ion: mechanistic aspects of gas-phase ion chemistry. J Mass Spectrom. 2013;48:505. doi: 10.1002/jms.3204. [DOI] [PubMed] [Google Scholar]
- 16.Morton TH. The Reorientation Criterion and Positive-Ion Neutral Complexes. Org Mass Spectrom. 1992;27:353. [Google Scholar]
- 17.Longevialle P. Ion-neutral complexes in the unimolecular reactivity of organic cations in the gas phase. Mass Spectrom Rev. 1992;11:157. [Google Scholar]
- 18.Chai Y, Wang L, Sun H, Guo C, Pan Y. Gas-phase chemistry of benzyl cations in dissociation of N-benzylammonium and N-benzyliminium ions studied by mass spectrometry. J Am Soc Mass Spectrom. 2012;23:823. doi: 10.1007/s13361-012-0344-8. [DOI] [PubMed] [Google Scholar]
- 19.Beuck S, Schwabe T, Grimme S, Schlorer N, Kamber M, Schanzer W, Thevis M. Unusual Mass Spectrometric Dissociation Pathway of Protonated Isoquinoline-3-Carboxamides Due to Multiple Reversible Water Adduct Formation in the Gas Phase. J Am Soc Mass Spectrom. 2009;20:2034. doi: 10.1016/j.jasms.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 20.Neta P, Farahani M, Simon-Manso Y, Liang YX, Yang XY, Stein SE. Unexpected peaks in tandem mass spectra due to reaction of product ions with residual water in mass spectrometer collision cells. Rapid Commun Mass Spectrom. 2014;28:2645. doi: 10.1002/rcm.7055. [DOI] [PubMed] [Google Scholar]
- 21.Neta P, Simon-Manso Y, Liang YX, Stein SE. Loss of H-2 and CO from protonated aldehydes in electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom. 2014;28:1871. doi: 10.1002/rcm.6968. [DOI] [PubMed] [Google Scholar]
- 22.Tuytten R, Lemiere F, Van Dongen W, Esmans EL, Witters E, Herrebout W, Van der Veken B, Dudley E, Newton RP. Intriguing mass spectrometric behavior of guanosine under low energy collision-induced dissociation: H2O adduct formation and gas-phase reactions in the collision cell (vol 16, pg 1904, 2005) J Am Soc Mass Spectrom. 2005;16:1904. doi: 10.1016/j.jasms.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 23.Guan ZQ, Liesch JM. Salvation of acylium fragment ions in electrospray ionization quadrupole ion trap and Fourier transform ion cyclotron resonance mass spectrometry. J Mass Spectrom. 2001;36:264. doi: 10.1002/jms.124. [DOI] [PubMed] [Google Scholar]
- 24.Van Stipdonk M, Kullman M, Berden G, Oomens J. IRMPD and DFT study of the loss of water from protonated 2-hydroxynicotinic acid. Int J Mass Spectrom. 2012;330:134. [Google Scholar]
- 25.Neta P, Godugu B, Liang YX, Simon-Manso Y, Yang XY, Stein SE. Electrospray tandem quadrupole fragmentation of quinolone drugs and related ions. On the reversibility of water loss from protonated molecules. Rapid Commun Mass Spectrom. 2010;24:3271. doi: 10.1002/rcm.4769. [DOI] [PubMed] [Google Scholar]
- 26.Bialecki J, Ruzicka J, Attygalle AB. An unprecedented rearrangement in collision-induced mass spectrometric fragmentation of protonated benzylamines. J Mass Spectrom. 2006;41:1195. doi: 10.1002/jms.1089. [DOI] [PubMed] [Google Scholar]
- 27.Sun H, Chai Y, Xu X, Pan Y. Intramolecular benzyl cation transfer in the fragmentation of Cinchona alkaloid-based quaternary ammonium cations. Int J Mass Spectrom. 2013;335:16. [Google Scholar]
- 28.Sun H, Chai Y, Pan Y. Dissociative Benzyl Cation Transfer versus Proton Transfer: Loss of Benzene from Protonated N-Benzylaniline. J Org Chem. 2012;77:7098. doi: 10.1021/jo301011e. [DOI] [PubMed] [Google Scholar]
- 29.Shen S, Chai Y, Weng G, Pan Y. Intramolecular Electrophilic Aromatic Substitution in Gas-phase Fragmentation of Protonated N-Benzylbenzaldimines. J Am Soc Mass Spectrom. 2014;25:1662. doi: 10.1007/s13361-014-0935-7. [DOI] [PubMed] [Google Scholar]
- 30.Li F, Wu Y, Zhang N, Jiang J, Jiang K. Competing benzyl cation transfers in the gas-phase fragmentation of the protonated benzyl phenylalaninates. Int J Mass Spectrom. 2014;369:23. [Google Scholar]
- 31.Guo C, Yue L, Guo M, Jiang K, Pan Y. Elimination of Benzene from Protonated N-Benzylindoline: Benzyl Cation/Proton Transfer or Direct Proton Transfer? J Am Soc Mass Spectrom. 2013;24:381. doi: 10.1007/s13361-012-0561-1. [DOI] [PubMed] [Google Scholar]
- 32.Guo C, Jiang K, Zheng S. Fragmentation reactions of N-benzyltetrahydroquinolines in electrospray ionization mass spectrometry: the roles of ion/neutral complex intermediates. Rapid Commun Mass Spectrom. 2014;28:1381. doi: 10.1002/rcm.6918. [DOI] [PubMed] [Google Scholar]
- 33.Wang SS, Yu L, Wu YQ, Guo C, Zhang NW, Jiang KZ. Gas-Phase Fragmentation of Protonated N,2-Diphenyl-N′-(p-Toluenesulfonyl)Ethanimidamides: Tosyl Cation Transfer Versus Proton Transfer. J Am Soc Mass Spectrom. 2015;26:1428. doi: 10.1007/s13361-015-1156-4. [DOI] [PubMed] [Google Scholar]
- 34.Wang SS, Guo C, Zhang NW, Wu YQ, Zhang HR, Jiang KZ. Tosyl oxygen transfer and ion-neutral complex mediated electron transfer in the gas-phase fragmentation of the protonated N-phenyl p-toluenesulfonamides. Int J Mass Spectrom. 2015;376:6. [Google Scholar]
- 35.Wang SS, Dong C, Yu L, Guo C, Jiang KZ. Dissociation of protonated N-(3-phenyl-2H-chromen-2-ylidene)-benzenesulfonamide in the gas phase: cyclization via sulfonyl cation transfer. Rapid Commun Mass Spectrom. 2016;30:95. doi: 10.1002/rcm.7420. [DOI] [PubMed] [Google Scholar]
- 36.Yan Z, Tounge B, Caldwell GW. An unusual intramolecular transfer of the fluorobenzyl cation between two remote amidic nitrogen atoms induced by collision in the gas phase. Rapid Commun Mass Spectrom. 2012;26:49. doi: 10.1002/rcm.5297. [DOI] [PubMed] [Google Scholar]
- 37.Tu YP, Huang YY, Atsriku C, You Y, Cunniff J. Intramolecular transacylation: fragmentation of protonated molecules via ion-neutral complexes in mass spectrometry. Rapid Commun Mass Sp. 2009;23:1970. doi: 10.1002/rcm.4108. [DOI] [PubMed] [Google Scholar]
- 38.Tu YP. Dissociative protonation and fragmentation: Retro-Friedel-Crafts reactions of heterocyclic drug and metabolite molecules in mass spectrometry. Int J Mass Spectrom. 2012;316:40. [Google Scholar]
- 39.Guo ZQ, Qi HY, Jiang Y, Fang DM, Zhang GL, Wu ZJ. Analysis of a caffeic acid derivative by ESI-MS/MS: unexpected product ions formed by ‘internal residue loss’. J Mass Spectrom. 2014;49:428. doi: 10.1002/jms.3352. [DOI] [PubMed] [Google Scholar]
- 40.Thielking G, Filges U, Grutzmacher HF. Remote Fragmentations of Protonated Aromatic Carbonyl-Compounds Via Internal Reactions in Intermediary Ion-Neutral Complexes. J Am Soc Mass Spectrom. 1992;3:417. doi: 10.1016/1044-0305(92)87069-B. [DOI] [PubMed] [Google Scholar]
- 41.Chai YF, Sun HZ, Pan YJ, Sun CR. N-Centered Odd-Electron Ions Formation from Collision-Induced Dissociation of Electrospray Ionization Generated Even-Electron Ions: Single Electron Transfer via Ion/Neutral Complex in the Fragmentation of Protonated N, N′-Dibenzylpiperazines and Protonated N-Benzylpiperazines. J Am Soc Mass Spectrom. 2011;22:1526. doi: 10.1007/s13361-011-0176-y. [DOI] [PubMed] [Google Scholar]
- 42.Guo C, Jiang KZ, Zheng S. Fragmentation reactions of N-benzyltetrahydroquinolines in electrospray ionization mass spectrometry: the roles of ion/neutral complex intermediates. Rapid Commun Mass Spectrom. 2014;28:1381. doi: 10.1002/rcm.6918. [DOI] [PubMed] [Google Scholar]
- 43.Hu N, Tu YP, Jiang KZ, Pan YJ. Intramolecular Charge Transfer in the Gas Phase: Fragmentation of Protonated Sulfonamides in Mass Spectrometry. J Org Chem. 2010;75:4244. doi: 10.1021/jo100761k. [DOI] [PubMed] [Google Scholar]
- 44.Janzen EG, Oehler UM. An Electron-Spin-Resonance and Endor Study of Intramolecular and Intermolecular Addition-Reactions of Benzoyl and Substituted Benzoyl Radicals to Nitroaromatic Compounds - a 2-Step Oxygen Atom Abstraction Reaction. Tetrahedron Lett. 1983;24:669. [Google Scholar]
- 45.Domelsmith LN, Munchausen LL, Houk KN. Photoelectron-Spectra of Psychotropic-Drugs 1. Phenethylamines, Tryptamines, and Lsd. J Am Chem Soc. 1977;99:4311. doi: 10.1021/ja00455a018. [DOI] [PubMed] [Google Scholar]
- 46.Vorsa V, Willey KF, Winograd N. Photoionization of gas-phase versus ion-beam-desorbed dopamine with femtosecond laser pulses. Anal Chem. 1999;71:574. doi: 10.1021/ac980774m. [DOI] [PubMed] [Google Scholar]
- 47.Dookeran NN, Yalcin T, Harrison AG. Fragmentation reactions of protonated alpha-amino acids. J Mass Spectrom. 1996;31:500. [Google Scholar]
- 48.Lioe H, O’Hair RAJ. Neighbouring group processes in the deamination of protonated phenylalanine derivatives. Org Biomol Chem. 2005;3:3618. doi: 10.1039/b503355a. [DOI] [PubMed] [Google Scholar]
- 49.Shoeib T, Cunje A, Hopkinson AC, Siu KWM. Gas-phase fragmentation of the Ag+-phenylalanine complex: Cation-pi interactions and radical cation formation. J Am Soc Mass Spectrom. 2002;13:408. doi: 10.1016/s1044-0305(02)00353-7. [DOI] [PubMed] [Google Scholar]
- 50.Lagutschenkov A, Langer J, Berden G, Oomens J, Dopfer O. Infrared spectra of protonated neurotransmitters: dopamine. Phys Chem Chem Phys. 2011;13:2815. doi: 10.1039/c0cp02133d. [DOI] [PubMed] [Google Scholar]
- 51.Zu CL, Mukhopadhyay S, Hanley PS, Xia SJ, Bell BM, Grigg D, Gilbert JR, O’Brien JP. Fragmentation of Protonated N-(3-Aminophenyl)Benzamide and Its Derivatives in Gas Phase. J Am Soc Mass Spectrom. 2016;27:917. doi: 10.1007/s13361-016-1342-z. [DOI] [PubMed] [Google Scholar]
- 52.Bowen RD. Ion-Neutral Complexes. Acc Chem Res. 1991;24:364. [Google Scholar]
- 53.Longevialle P. Ion-Neutral Complexes in the Unimolecular Reactivity of Organic Cations in the Gas-Phase. Mass Spectrom Rev. 1992;11:157. [Google Scholar]
- 54.Hansch C, Leo A, Taft RW. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem Rev. 1991;91:165. [Google Scholar]