

Abstract
Host microbiota plays important roles in providing colonization resistance to pathogens and instructing development and function of the immune system. Antibiotic treatments intended to target pathogens further weaken the host defenses and may paradoxically increase the risk of systemic infections. This consequence is especially problematic in patients undergoing hematopoietic stem cell transplantation, where the mucosal defenses are already weakened by the conditioning regimens. This review discusses the roles that indigenous microbiota plays in protecting the host and maintaining immune homeostasis. In addition, we highlight possible strategies that are being developed to allow targeted antimicrobial therapy against pathogens, while minimizing the harm to indigenous microbiota.
INTRODUCTION
Conditioning regimens involving various chemotherapeutic agents with or without radiation constitute critical elements for successful engraftment after hematopoietic stem cell transplantation (HCT). However, the resulting immunosuppression, coupled with widespread damage to the intestinal mucosa, creates severe vulnerability to infections leading to morbidity and mortality. The risk of bloodstream infections in patients undergoing HCT is greatest during periods of neutropenia before engraftment, and routine antibacterial prophylaxis (most commonly fluoroquinolones) is the current standard of care to reduce that risk.1 Additional broad-spectrum antibiotics are given if neutropenic patients develop a fever or other signs of suspected infection. Diarrhea is a common occurrence in HCT patients because of toxicities and infections. Nearly one-third of HCT patients are colonized with Clostridium difficile at admission for HCT, and the incidence of symptomatic C. difficile infection (CDI) may be as high as 10%–20%.2–4
The current paradigm of approaching infectious disease challenges in these highly immunocompromised patients rests entirely within the pathogen-centric framework that disregards the symbiotic relationship between the host and its microbiota. Commensal microbiota is an integral part of the human body and plays critical roles in providing colonization resistance to pathogens and instructing the immune system. Both of these functions are especially critical during the period of immune reconstitution when the risk of graft-vs-host disease (GVHD) is greatest. In addition, the current approach has had only limited success in curbing even the short-term infectious complications because of increasing emergence of multiple drug- resistant pathogens. Therefore, there is an urgent need to re-evaluate the current antimicrobial strategies and consider therapeutic alternatives that can be rapidly developed.
OVERVIEW OF HOST–MICROBIOTA INTERACTIONS AND CONSEQUENCES OF THEIR DISRUPTION IN HCT
The human body is a superorganism that incorporates highly specialized complex microbial communities, also called “microbiota,” present on all its surfaces.5 Microbiota and its host have a symbiotic relationship, which is generally (although not always) mutualistic. The microbes benefit from this relationship because they gain access to nutrients and in turn contribute to the health of the host. Not surprisingly, the vast majority of the microbiota is resident within the digestive tract, and specifically the colon. Humans are hindgut fermenters and colon microbiota participates in completing digestion that cannot be accomplished by the host alone, given its limited catabolic capacity.6,7 One of the benefits for the human host is harvest of the small fraction of energy liberated by microbial digestion, as well as some essential nutrients, for example, vitamin K and group B vitamins. However, the host–microbiota relationship is integral to many other aspects of human physiology, reflective of their long coevolutionary history that extends to the origin of eukaryotic organisms.8,9
One of the major functions of human microbiota is colonization resistance. In the setting of homeostasis, potential pathogens have to face intense competition for nutrients with the indigenous microbiota and penetrate formidable barriers imposed by the mucosal immune system, which are constantly reinforced by signaling from the indigenous microbiota. However, when the host microbiota is suppressed by antibiotics, more nutrients become available to pathogens, and intestinal barrier defenses are lowered because of reduction of the commensal-derived tonic signaling (Fig 1).
Fig 1.
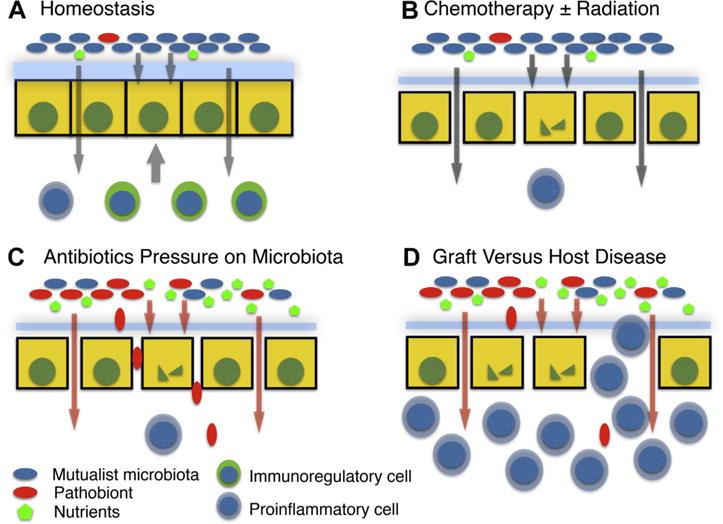
Overview of the roles of microbiota after HCT. (A) During steady state, the gut microbiota is kept compartmentalized from the host by a number of mechanisms, including the thick inner mucus in the colon fortified with antimicrobial peptides and secreted immunoglobulin. Microbiota-derived products provide trophic signals to the epithelium and promote development of noninflammatory elements of the immune system. Potentially pathogenic microbes (pathobionts) are constrained by the multiple mechanisms, including intense competition for nutrients. (B) Myeloablative regimens used to condition patients for HCT have toxic effects on the intestinal epithelium and the mucosal immune system. However, repair is facilitated by healthy microbiota-derived trophic signals. (C) Broad-spectrum antibiotics disrupt the normal microbial community structure of gut microbiota and lead to expansion of pathobionts, which are more likely to be antibiotic-resistant and more able to translocate across the compromised gut barrier leading to blood-borne infections. Loss of trophic microbiota-derived signals also limits repair of the epithelial barrier and the mucosal immune system. (D) Loss of mutualist microbiota and greater abundance of pathobionts results in altered signaling to the developing immune system and rise of an inflammatory host response leading to GVHD. HCT, hematopoietic stem cell transplantation; GVHD, graft-vs-host disease.
Another major function of human microbiota is instruction of immune system development. Increasingly intense attention to this function is being focused in the context of the first months and years after birth, when the composition of microbiota has the potential to determine responsiveness of the immune system in subsequent years. There is growing concern that various environmental factors such as caesarian birth, formula feeding, increased sanitation, and common antibiotic usage alter the composition of early microbiota that leads to unfavorable programming of the immune system that contributes to the development of autoimmunity, allergies and atopic diseases, and various inflammatory bowel diseases.10 However, the immune system undergoes another round of development after HCT, and under current standard of care this occurs in the context of the drastically distorted microbiota composition. It is possible that antibiotic-induced dysbiosis contributes to the development of GVHD, which is the major cause of morbidity and mortality after engraftment is achieved (Fig 1). The problem is likely further exacerbated by the delayed repair of the mucosal barrier breakdown caused by the absence of trophic signals from microbiota and exacerbation of adverse instruction of the immune system.
INDIGENOUS MICROBIOTA PROMOTES COLONIZATION RESISTANCE AND ENHANCES GUT BARRIER FUNCTION
The phenomenon of antibiotic-associated increased susceptibility to enteric infections has been known for many decades. For example, in 1950s Bohnhoff et al11 noted that a single pretreatment of mice with streptomycin lowered the infectious dose of Salmonella enteritidis from approximately 1 million organisms to fewer than 10. The same investigators determined that it was the anaerobic fraction of gut microbiota that provided the protection.12 These early researchers understood that host microbiota–mediated colonization resistance could be achieved to 2 main nonmutually exclusive pathways: (1) direct microbe–microbiota interactions, for example, competition for nutrients, microbicidal effects, and (2) indirect interactions involving the host, for example, immune system mediated. Multiple examples of both of these mechanisms have been defined over the past decade.
The indigenous microbes inhabit various ecological niches within the host that are in part defined by access to nutrients. Moreover, individual microbial community members form interdependent relationships defined by sequential division of labor resulting in complex metabolic networks.5,13 For example, a prominent commensal organism Bacteroides thetaiotaomicron encodes the sialidase required to liberate free sialic acid from mucin glycoproteins in the intestine. However, B. thetaiotaomicron does not consume sialic acid, which in turn becomes an energy source for other community members. Some pathogens, which include Salmonella typhimurium and C. difficile, possess a sialic acid catabolic operon, although they still have to compete with other members of microbiota for the pool of free sialic acid. Antibiotic regimens can suppress the competition and increase availability of sialic acid to the antibiotic-resistant enteric pathogens.13 Gut inflammation can further escalate competition for critical nutrients. The host deploys an array of responses to limit availability of transition metals such as iron, zinc, and manganese, in a process called nutritional immunity.14 Both pathogens and commensals compete intensely for these restricted resources by secreting high-affinity chelating compounds, for example, siderophores to capture iron, and expressing various cation transporter systems, for example, heme transporters for iron and ABC-type transporters for zinc and manganese. However, this strategy backfires when antibiotic treatments suppress indigenous competition and tilt the balance in favor of the pathogens.
Even when pathogens succeed in securing a foothold within the host microbial ecosystem, they still face formidable barrier immunity that consists of mucus fortified with antimicrobial peptides and secreted immunoglobulin covering the epithelial layer. The host main homeostatic strategy that allows a mutualistic relationship of the host with its microbiota is compartmentalization. In fact, the barriers are reinforced and optimized by signaling generated by microbiota. Thus, although some antimicrobial peptides, for example, α-defensins, are constitutively expressed, other antimicrobial peptides like RegIIIγ require signaling from the commensals for optimal expression.15 Antibiotic treatments lower expression of RegIIIγ and collapse the spatial separation maintained by the mucus layer between the epithelial cells and microbiota.16 Normally, the host can compensate by increasing other defense mechanisms, for example, augment secretion of immunoglobulin A (IgA). However, these options may not be fully functional in immunocompromised patients following myeloablative therapy.
The host microbiota further enhances the gut barrier function through direct trophic and tonic signals to the epithelial compartment. Short-chain fatty acids (SCFAs), which are produced by bacterial fermentation of complex carbohydrates, provide the major source of fuel for the colonocytes and upregulate the expression of tight junction proteins, which strengthen the physical epithelial barrier.6,17,18 In addition, all intestinal epithelial cells express different pattern recognition receptors capable of sensing the microbial status within the intestine. These include Nod-like receptors and Toll-like receptors (TLRs). The intracellular microbial sensor Nod2 is highly expressed in intestinal crypt stem cells, where its stimulation promotes epithelial survival and restitution after chemotherapy-induced damage.19 Seminal work over a decade ago by Medzhitov’s group showed that reduction in tonic TLR signaling either by genetic deletion of specific TLRs or MyD88 (a downstream adapter for most TLRs), or simply treating animals with antibiotics impaired the ability of epithelia to proliferate and repair after radiation or chemical damage.20 Isolation of MyD88 deficiency to the nonhematopoietic compartment in studies using bone marrow chimeras suggested that microbial signals enhance production of several epidermal growth factor ligands important for gut epithelial restitution, including such as amphiregulin and epiregulin.21
Finally, microbiota interacts with the hematopoietic cells within the mucosa and gut-associated lymphoid tissues, which produce molecules that play critical roles in epithelial homeostasis. One such master mediator is cytokine interleukin (IL)-22, which upregulates the expression of various genes involved in tight and gap junction formation, stimulates production of membrane-bound mucins, increases production of antimicrobial peptides, and promotes epithelial proliferation and wound repair.22 It is produced by multiple cell types, including elements of both adaptive and innate immune systems such as Th17 and Th22 cells and group 3 innate lymphoid cells (ILC3). These cells in turn may be induced by select members of microbiota, for example, segmented filamentous bacteria in mice,23 or respond to products of microbiota, for example, aryl hydrocarbon receptor ligands.24 It is notable that IL-22 has been found to contribute to the host defense against a number of bacterial and fungal infections, including Klebsiella pneumoniae, C. difficile, Aspergillus fumigatus, and Candida albicans, all of which are highly relevant in the care of HCT patients.1,22,25
HCT IS ASSOCIATED WITH SEVERE INTESTINAL DYSBIOSIS
Conditioning chemotherapy, radiation, mucosal damage, and use of antibiotics all contribute to disruption of microbial community structure in patients undergoing HCT. The reduction in microbial diversity is often dramatic, and single species can dominate the fecal microbiome in some patients.26 Not surprisingly, the organisms that gain in relative abundance in the intestines are typically the ones equipped with the most antibiotic resistance genes and include various representatives of the family enterobacteriaceae, viridans-group streptococci, and vancomycin-resistant enterococcus (VRE). Furthermore, the bloom of specific bacteria in the intestine is highly correlative with the specific organisms causing blood-borne infections.26
The types of bacteria that come to dominate are mostly reflective of specific antibiotic usage, rather than resource competition among the pathogens themselves. Thus, VRE and carbapenem-resistant K. pneumoniae occupy the same physical regions in the colon but exist within distinct metabolic niches.27 Routine prophylactic use of fluoroquinolones in neutropenic patients likely mitigates against the expansion of enterobacteriaceae, although growing antibiotic resistance remains a concern.28 Prophylactic use of vancomycin may contribute to the expansion of VRE. Most antibiotics suppress the generally protective intestinal anaerobes and often trigger CDI, which necessitates use of additional oral antibiotics with broad anaerobic activity, such as metronidazole and vancomycin. Thus, it is easy to envision how the current antibiotic usage commonly turns into a desperate whack-a-mole strategy until the surviving microbiota no longer resembles any normal configuration or structure and becomes dominated by opportunistic pathogenic commensals or “patho-bionts.”29
DYSBIOSIS AND IMMUNE DYSREGULATION AFTER HCT
Microbiota–immune system interactions may be particularly critical during developmental stages of the immune system. This consideration is fundamental to the concerns articulated by the hygiene hypothesis, which links early life antibiotic exposures and increased sanitation practices to increased incidence of autoimmunity.30 Indeed, multiple association studies correlate early life antibiotic exposure to the development of autoimmunity, asthma, allergies, and inflammatory bowel disease in later years. More recently, this hypothesis has incorporated the notion of disappearing commensal microbiota caused by altered microbial inoculation at birth (caesarian section delivery), common use of antibiotics, and dietary changes that may be less nutritive to the intestinal microbes.31,32
Animal models offer potential mechanisms that could explain the sustained effects of early host–microbiota interactions on the immune system. For example, germ-free animals have greater numbers of invariant natural killer T (iNKT) cells, which can drive increased severity of hapten-induced colitis and antigen-triggered asthma.33 Delayed microbial colonization of germ-free mice does not rescue their predisposition to these immunopathologies, but microbial colonization of germ-free mice at birth does.33 Proliferation of iNKT cells early in development is driven by bacterial sphingolipids produced primarily by members of the bacteroidetes phylum, 1 of the 2 dominant bacterial phyla in the colon.34 In fact, monocolonization of germ-free mice with just 1 species of this group, Bacteroides fragilis, is sufficient to inhibit iNKT cell proliferation during early development. Interestingly, B. fragilis is also a source of polysaccharide A, which is able to induce IL-10-producing regulatory T cells (Tregs) via engagement of TLR2.35
Germ-free mice and mice treated with antibiotics during the neonatal period also have heightened susceptibility to food antigen sensitization relative to animals maintained in typical specific pathogen-free housing conditions.36 Different members of microbiota have varying abilities in preventing these abnormalities. In this case, clostridia consortia prepared by chloroform extraction, which yields spores consisting predominantly of members of Clostridium clusters XIVa, XIVb, and IV, protect against sensitization. These clostridia lead to significant increases in fecal IgA levels, numbers of intestinal Foxp3+ Tregs, and production of IL-22 by Th17 and ILC3 cells, all contributing to improved gut barrier function. In contrast, Bacteroides uniformis, also an anaerobe, was only able to increase IgA levels but had no effects on Tregs or IL-22. The spore-forming clostridia in these preparations, both mouse and human, promote Treg induction at least in part via SCFAs signaling via G protein-coupled free fatty acid receptors and eliciting transforming growth factor beta 1 (TGFβ1) production.37 SCFAs and especially butyrate can inhibit histone deacetylases, which exert epigenetic control over gene expression that is involved in stimulating induction and boosting fitness of Tregs.38,39 The same clostridia consortia and immunoregulatory mechanisms have been described to inhibit multiple forms of experimental inflammatory bowel disease.40 Notably, the capacity of intestinal epithelial cells to uptake butyrate is reduced in the setting of inflammation, and treatment of mice after allogeneic bone marrow transplantation with exogenous butyrate or butyrate-producing consortia of clostridia mitigates GVHD.41
The immune system undergoes another round of development after HCT. However, this time it occurs in the context of a damaged gut barrier and severe dysbiosis, as described previously. In addition, the transient lymphopenic state in the recipient creates a homeostatic pressure for mature T cells, both passenger cells from the donor and residual recipient cells, to undergo proliferation. This situation favors oligoclonal expansion of T cells receiving cognate antigen stimulation, ultimately leading to a restricted T cell repertoire enriched for autoreactive, proinflammatory T cells that may drive GVHD.42 The contribution of gut dysbiosis in this setting has not yet been fully evaluated. However, it is likely that an increased proportion of cognate antigens may be derived from constituents of gut microbiota that breach the gut barrier during this time. In addition, loss of microbiota that normally drives and potentiates immunoregulatory circuits may tilt that balance toward more inflammatory T cell phenotypes.
The potential of differential interactions with various members of microbiota has not been incorporated into the current clinical management of GVHD. Historically, many practitioners and investigators believed that the risk of GVHD was essentially proportional to the overall microbial load present in the gut. In fact, a significant reduction in GVHD has been shown in germ-free mice and several animal models that make use of aggressive “gut-decontamination” protocols using combinations of antibiotics.43–45 Results of human clinical trials, however, have been equivocal. Several factors complicate interpretation of these trials. First, HCT is performed for a variety of diseases that may be broadly divided into malignant and nonmalignant conditions, which differ greatly in antibiotic exposure before administration of the stem cell–containing graft. Second, at least some prophylactic antibiotics, for example, quinolones, are now used routinely after allogeneic HCT, which make comparisons possible only between less-aggressive and more-aggressive antibiotic regimens. Some studies suggested that more- aggressive regimens reduced severity of GVHD46–48 but others did not.49 Importantly, the effectiveness of antibiotic regimens (when measured at all in these studies) has generally relied on conventional, culture- dependent microbiological techniques, which fail to capture many gut microbes that are otherwise detectable with molecular methods. Recently, Shono et al50 retrospectively analyzed specific antibiotic usage for neutropenic fevers in 857 allo-HCT recipients and correlated it with GVHD-related mortality at 5 years. They found increased GVHD-related mortality associated with the usage of imipenem–cilastatin and piperacillin–tazobactam; in contrast, increased GVHD-related mortality was not seen in association with aztreonam or cefepime used as alternatives primarily in penicillin-allergic patients. Notably, piperacillin–tazobactam was found to cause more profound suppression on the anaerobic fraction of gut microbiota compared with aztreonam or cefepime.
Recent animal studies also support more nuanced roles of gut microbiota in modulating GVHD. Thus, treatment of recipient mice with ampicillin caused more-aggressive acute GVHD in an allogeneic HCT model.51 Interestingly, GVHD alone in this experimental system resulted in significant shifts in the ileum and cecum within the phylum firmicutes, specifically expansion of lactobacillales and decreases in clostridiales and other firmicutes. Ampicillin inhibited the expansion of lactobacillales and allowed clostridiales and enterobacteriales to expand instead. Reintroduction of lactobacillales in the form of a probiotic Lactobacillus johnsonii into ampicillin-exposed mice reversed the microbial composition shifts and worsening of GVHD induced by ampicillin. Murine studies also corroborated the human experience of worsened GVHD after allo-HCT in association with imipenem–cilastatin compared with aztreonam.50 Imipenem–cilastatin treatment resulted in greater suppression of representative taxa of the dominant obligate anaerobe phyla, bacteroidetes and firmicutes, but also was correlated with a relatively increased abundance of Akkermansia muciniphila, a member of verrucomicrobia phylum that uses mucin as its carbon and nitrogen source. It was further noted that animals treated with imipenem–cilastatin had decreased thickness of the inner mucus in the colon and increased gut permeability.50
We performed a number of exploratory experiments in a preclinical xenogeneic model of acute GVHD (human T cells → mouse) to explore the role of gut microbiota in this disease. Briefly, NOD/SCID/γc−/− mice, which are defective in development of T-, B-, and NK cells, are injected with 20 × 106 human peripheral blood mononuclear cells. In this model, the human T cells cause classic GVHD with intestinal inflammation, weight loss, and ultimate mortality.52 To model the effects of antibiotic-induced dysbiosis experienced by human patients, we treated some of the animals with broad-spectrum antibiotics before and after injection of human T cells. The mice consistently developed more-aggressive GVHD when treated with antibiotics (Fig 2). These results are consistent with the recent studies demonstrating ampicillin- and imipenem-cilastatin–induced worsening of GVHD after allogeneic HCT,50,51 although contradicting early studies that showed beneficial effects of antibiotics in allogeneic HCT.43 There are, of course, multiple variables that differ between these models. However, it is notable that major changes took place in veterinary care and housing conditions of laboratory mice over the period of 40 years separating the early and recent experiments, including elimination of many potential pathogens in mouse colonies. In addition, we have far greater ability to characterize the gut microbiota using molecular techniques that were simply not available decades ago. Therefore, it may be productive to re-evaluate older conclusions and perform more detailed assessment of the effects of different antibiotics on GVHD development in model systems.
Fig 2.
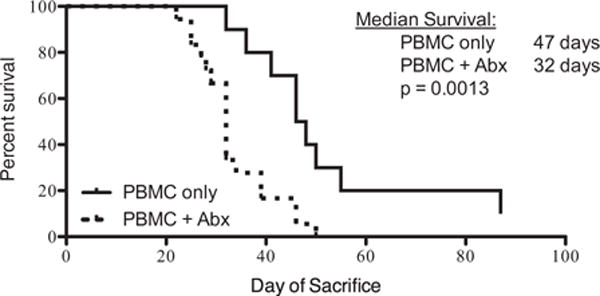
Deleterious effect of antibiotics in a model of xenogeneic GVHD. Human mononuclear cells from peripheral blood were injected intravenously into NOD/SCID/γc−/− mice, 20 × 106 cells per animal. Some of the mice (n = 20) were given ampicillin (1 mg/mL), clindamycin (1 mg/mL), vancomycin 1 mg/mL, and cefoperazone (0.5 mg/mL) in their drinking water. Control mice did not receive antibiotics (n = 10). The animals were monitored for diarrhea and weight loss and sacrificed when weight decreased 30% from the starting weight. The antibiotics in drinking water alone without the cell injection did not cause weight loss. The shown experiment is representative of 3 independent experiments. GVHD, graft-vs-host disease; PBMC, peripheral blood mononuclear cells.
RECONSIDERING THE STANDARD PARADIGMS AND FUTURE DIRECTIONS
The antibiotic-induced dysbiosis after HCT contributes to the perfect storm of events that also include disruption of the physical gut barrier and severe compromise of the immune barrier created by the conditioning therapy. Various considerations described previously suggest that the simplistic notion of trying to achieve sterilization of the gut in the setting of HCT is both unachievable and probably unwise as it ultimately leads to emergence of multiple drug-resistant pathogens. In fact, progressive loss of microbial diversity in patients undergoing HCT correlates with increasing overall mortality.53 The gut microbiota is an integral part of the digestive system that contributes to colonization resistance, reconstitution of the gut barrier, and instruction of the mucosal immune system. We think it is likely that the most beneficial constituents of the gut microbiota are contained within the obligate anaerobe fraction, as suspected by early investigators decades ago.54 Different strategies can be considered and tested in the future, including consideration of more selective antibiotic regimens, microbiota reconstitution treatments, and direct administration of protective microbial products.
In clinical practice, the choice of antibiotics is generally guided by consideration of potential pathogens rather than protection of the beneficial elements of the indigenous microbiota. Admittedly, the available choices that could preferentially target pathogens and spare the more beneficial microbes are few. The pharmaceutical industry has generally focused its efforts on developing antibiotics that have broader spectrum of activity and high bioavailability after oral administration. There are few antibiotics that can be administered intravenously or intramuscularly that are not distributed inside the gut lumen either directly or via biliary secretion. Aminoglycosides are one of the notable exceptions. We use intramuscular or intravenous aminoglycosides, such as gentamicin and amikacin, in treatment of urinary tract infections in non-HCT patients suffering from recurrent CDI syndrome—a situation where progressive gut dysbiosis is a known trigger of infection recurrence (manuscript in preparation). Current guidelines in treatment of CDI itself generally call for initial use of oral metronidazole, followed by oral vancomycin in cases of multiple recurrences. Both of these antibiotics are highly suppressive to gut anaerobes and likely contribute to perpetuation CDI recurrence. Fidaxomicin is a more selective antibiotic that targets the phylum firmicutes, whereas sparing most of the phylum bacteroidetes.55 This antibiotic has been shown to have less CDI recurrence after initial infection.56 However, in current practice, it is generally used only as a third-line antibiotic after demonstrated failure of metronidazole and vancomycin, at which point the benefits of its more selective activity are likely no longer relevant. Although this sequence of usage may appear to be more cost effective, this may not be the case in many patients in the long term and especially individuals who are candidates for HCT or may be HCT recipients already.
A microbiota restorative approach, for example, fecal microbiota transplantation (FMT), may be developed as an option in the setting of HCT. FMT is a highly effective rescue therapy for the recurrent CDI syndrome, which is already being used widely in treatment of this condition.57 It results in prompt and sustained engraftment of donor microbiota and donor-like normalization of the fecal microbial community structure.57–61 The notion of microbiota reconstitution in HCT setting dates was explored anecdotally in 1970s, primarily as a means to improve colonization resistance.54 Considerable progress has been made in this area in the recent years in treatment of CDI. This includes development of protocols that allow cryopreservation of standardized healthy donor microbiota.62 These protocols can also be used to cryopreserve autologous microbiota that can be done before the initiation of myeloablative conditioning or significant exposure to antibiotics in HCT patients. The first clinical trial with autologous FMT in HCT patients has already been initiated in the Memorial Sloan-Kettering Cancer Center in New York City.
Administration of “probiotics” or defined one or several selected strains of microorganisms continues to be of interest to many but without substantial data to support its use in HCT. It is highly unlikely that any of the currently available probiotics, which were never developed as true therapeutics, could ever be clinically useful. However, it is conceivable that new probiotic drugs could emerge if their design is going to be mechanistically based. For example, microorganisms can be engineered to express growth factors to aid in restitution of a damaged intestinal epithelium, for example, epidermal growth factor and trefoil factor.63
However, there are multiple potential pitfalls associated with microbiota reconstitution, which need to be carefully considered. It may be challenging to administer complex microbial mixtures to highly immunosup-pressed patients. These patients may also develop spontaneous infections because of the underlying immunosuppression and barrier disruption, which could inevitably necessitate return to antibiotics and destruction of the microbiota graft. Therefore, an alternative or perhaps complimentary approach that needs to be tested may be administration of microbial products that have beneficial or protective properties. An example of such products is SCFAs, or their analogues, which may potentiate the gut barrier and promote the development of immunoregulatory T cells, as described previously.41 Polysaccharide A produced by B. fragilis is another microbial immunomodulatory molecule that might be developed as a therapeutic for GVHD prevention or treatment. Similarly, various microbiota-derived aryl hydrocarbon receptor ligands may be useful in stimulating mucosal immune cells that can enhance barrier immunity and mitigate GVHD. Indeed, it is likely that the human microbiome harbors many potential therapeutics that are yet to be discovered and developed.
Finally, antibiotic regimens that target pathogens, yet spare the indigenous microbiota in the distal gut, may be developed using innovative strategies of drug administration. For example, it may be possible to inactivate β-lactam antibiotics in the colon using orally administered β-lactamases.64,65 Another possible strategy is sequestering antibiotics away from indigenous gut microbiota using activated charcoal specifically formulated for site-specific release in the distal small intestine and proximal colon.66 This strategy has the advantage of being potentially applicable to a broad spectrum of antibiotics.
CONCLUSION
The traditional pathogen-centric framework in treating infections largely ignores the important roles that indigenous microbiota plays in protecting the host by providing colonization resistance and instructing the development and function of the immune system. This problem is especially germane and urgent in the context of HCT where broad-spectrum antibiotic treatments are routine and the host is especially vulnerable. Serious and often fatal infections remain common in HCT patients and can only be anticipated to rise with the steady emergence of increasingly virulent and multiple drug-resistant pathogens. In addition, microbiota altered by antibiotics may drive proinflammatory immune responses and worsen GVHD, which ultimately negatively affects long-term outcomes of HCT. Novel approaches that protect or restore the indigenous microbiota need to be developed and change the current standard care routines in administration of antimicrobial therapy to HCT patients.
Acknowledgments
This work was partially supported by a grant from the Office of Discovery and Translation at the University of Minnesota (grant number: ODAT2014-04-11) (A.K. and K.L.H.).
Abbreviations
- CDI
Clostridium difficile infection
- GVHD
graft-vs-host disease
- HCT
hematopoietic stem cell transplantation
- ILC
innate lymphoid cell
- iNKT
invariant natural killer T cell
- TLR
Toll-like receptor
Footnotes
Conflicts of Interest: A.K. has received research funding from CIPAC, Ld for development of standardized fecal microbiota preparations.
References
- 1.Freifeld AG, Bow EJ, Sepkowitz KA, et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2011;52:e56–93. doi: 10.1093/cid/cir073. [DOI] [PubMed] [Google Scholar]
- 2.Bruminhent J, Wang ZX, Hu C, et al. Clostridium difficile colonization and disease in patients undergoing hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2014;20:1329–34. doi: 10.1016/j.bbmt.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 3.Jain T, Croswell C, Urday-Cornejo V, et al. Clostridium Difficile Colonization in Hematopoietic Stem Cell Transplant Recipients: A Prospective Study of the Epidemiology and Outcomes Involving Toxigenic and Nontoxigenic Strains. Biol Blood Marrow Transplant. 2016;22:157–63. doi: 10.1016/j.bbmt.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 4.Huang AM, Marini BL, Frame D, Aronoff DM, Nagel JL. Risk factors for recurrent Clostridium difficile infection in hematopoietic stem cell transplant recipients. Transpl Infect Dis. 2014;16:744–50. doi: 10.1111/tid.12267. [DOI] [PubMed] [Google Scholar]
- 5.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–20. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 6.Cummings JH, Macfarlane GT. Role of intestinal bacteria in nutrient metabolism. JPEN J Parenter Enteral Nutr. 1997;21:357–65. doi: 10.1177/0148607197021006357. [DOI] [PubMed] [Google Scholar]
- 7.Stevens CE, Hume ID. Contributions of microbes in vertebrate gastrointestinal tract to production and conservation of nutrients. Physiol Rev. 1998;78:393–427. doi: 10.1152/physrev.1998.78.2.393. [DOI] [PubMed] [Google Scholar]
- 8.Ochman H, Worobey M, Kuo CH, et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 2010;8:e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van den Abbeele P, Van de Wiele T, Verstraete W, Possemiers S. The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol Rev. 2011;35:681–704. doi: 10.1111/j.1574-6976.2011.00270.x. [DOI] [PubMed] [Google Scholar]
- 10.Bloomfield SF, Stanwell-Smith R, Crevel RW, Pickup J. Too clean, or not too clean: the hygiene hypothesis and home hygiene. Clin Exp Allergy. 2006;36:402–25. doi: 10.1111/j.1365-2222.2006.02463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohnhoff M, Drake BL, Miller CP. Effect of streptomycin on susceptibility of intestinal tract to experimental Salmonella infection. Proc Soc Exp Biol Med. 1954;86:132–7. doi: 10.3181/00379727-86-21030. [DOI] [PubMed] [Google Scholar]
- 12.Miller CP, Bohnhoff M. Changes in the Mouse’s Enteric Microflora Associated with Enhanced Susceptibility to Salmonella Infection Following Streptomycin Treatment. J Infect Dis. 1963;113:59–66. doi: 10.1093/infdis/113.1.59. [DOI] [PubMed] [Google Scholar]
- 13.Ng KM, Ferreyra JA, Higginbottom SK, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502:96–9. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz-Ochoa VE, Jellbauer S, Klaus S, Raffatellu M. Transition metal ions at the crossroads of mucosal immunity and microbial pathogenesis. Front Cell Infect Microbiol. 2014;4:2. doi: 10.3389/fcimb.2014.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci USA. 2008;105:20858–63. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaishnava S, Yamamoto M, Severson KM, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–8. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bordin M, D’Atri F, Guillemot L, Citi S. Histone deacetylase inhibitors up-regulate the expression of tight junction proteins. Mol Cancer Res. 2004;2:692–701. [PubMed] [Google Scholar]
- 18.Al-Asmakh M, Hedin L. Microbiota and the control of blood-tissue barriers. Tissue Barriers. 2015;3:e1039691. doi: 10.1080/21688370.2015.1039691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nigro G, Rossi R, Commere PH, Jay P, Sansonetti PJ. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe. 2014;15:792–8. doi: 10.1016/j.chom.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Brandl K, Sun L, Neppl C, et al. MyD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc Natl Acad Sci U S A. 2010;107:19967–72. doi: 10.1073/pnas.1014669107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. 2015;33:747–85. doi: 10.1146/annurev-immunol-032414-112123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu J, Guo X, Chen ZM, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39:386–99. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasegawa M, Kamada N, Jiao Y, Liu MZ, Nunez G, Inohara N. Protective role of commensals against Clostridium difficile infection via an IL-lbeta-mediated positive-feedback loop. J Immunol. 2012;189:308591. doi: 10.4049/jimmunol.1200821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taur Y, Xavier JB, Lipuma L, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis. 2012;55:905–14. doi: 10.1093/cid/cis580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caballero S, Carter R, Ke X, et al. Distinct but Spatially Overlapping Intestinal Niches for Vancomycin-Resistant Enterococcus faecium and Carbapenem-Resistant Klebsiella pneumoniae. PLoS Pathog. 2015;11:e1005132. doi: 10.1371/journal.ppat.1005132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satlin MJ, Vardhana S, Soave R, et al. Impact of Prophylactic Levofloxacin on Rates of Bloodstream Infection and Fever in Neutropenic Patients with Multiple Myeloma Undergoing Autologous Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2015;21:1808–14. doi: 10.1016/j.bbmt.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–60. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dominguez-Bello MG, Blaser MJ. Asthma: Undoing millions of years of coevolution in early life? Sci Transl Med. 2015;7:307fs39. doi: 10.1126/scitranslmed.aad2741. [DOI] [PubMed] [Google Scholar]
- 32.Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7:887–94. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olszak T, An D, Zeissig S, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–93. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.An D, Oh SF, Olszak T, et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell. 2014;156:123–33. doi: 10.1016/j.cell.2013.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Round JL, Lee SM, Li J, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–7. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stefka AT, Feehley T, Tripathi P, et al. Commensal bacteria protect against food allergen sensitization. Proc Natl Acad Sci U S A. 2014;111:13145–50. doi: 10.1073/pnas.1412008111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–6. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 38.Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. 2008;19:587–93. doi: 10.1016/j.jnutbio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–5. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–73. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathewson ND, Jenq R, Mathew AV, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol. 2016;17:505–13. doi: 10.1038/ni.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khoruts A, Fraser JM. A causal link between lymphopenia and autoimmunity. Immunol Lett. 2005;98:23–31. doi: 10.1016/j.imlet.2004.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Bekkum DW, Roodenburg J, Heidt PJ, van der Waaij D. Mitigation of secondary disease of allogeneic mouse radiation chimeras by modification of the intestinal microflora. J Natl Cancer Inst. 1974;52:401–4. doi: 10.1093/jnci/52.2.401. [DOI] [PubMed] [Google Scholar]
- 44.Jones JM, Wilson R, Bealmear PM. Mortality and gross pathology of secondary disease in germfree mouse radiation chimeras. RadiatRes. 1971;45:577–88. [PubMed] [Google Scholar]
- 45.Vriesendorp HM, Heidt PJ, Zurcher C. Gastrointestinal decontamination of dogs treated with total body irradiation and bone marrow transplantation. Exp Hematol. 1981;9:904–16. [PubMed] [Google Scholar]
- 46.Beelen DW, Elmaagacli A, Muller KD, Hirche H, Schaefer UW. Influence of intestinal bacterial decontamination using metronidazole and ciprofloxacin or ciprofloxacin alone on the development of acute graft-versus-host disease after marrow transplantation in patients with hematologic malignancies: final results and longterm follow-up of an open-label prospective randomized trial. Blood. 1999;93:3267–75. [PubMed] [Google Scholar]
- 47.Vossen JM, Guiot HF, Lankester AC, et al. Complete suppression of the gut microbiome prevents acute graft-versus-host disease following allogeneic bone marrow transplantation. PLoS One. 2014;9:e105706. doi: 10.1371/journal.pone.0105706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vossen JM, Heidt PJ, van den Berg H, Gerritsen EJ, Hermans J, Dooren LJ. Prevention of infection and graft-versus-host disease by suppression of intestinal microflora in children treated with allogeneic bone marrow transplantation. Eur J Clin Microbiol Infect Dis. 1990;9:14–23. doi: 10.1007/BF01969527. [DOI] [PubMed] [Google Scholar]
- 49.Storb R, Thomas ED. Graft-versus-host disease in dog and man: the Seattle experience. Immunol Rev. 1985;88:215–38. doi: 10.1111/j.1600-065x.1985.tb01160.x. [DOI] [PubMed] [Google Scholar]
- 50.Shono Y, Docampo MD, Peled JU, et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci Transl Med. 2016;8:339ra71. doi: 10.1126/scitranslmed.aaf2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jenq RR, Ubeda C, Taur Y, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med. 2012;209:903–11. doi: 10.1084/jem.20112408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hippen KL, Bucher C, Schirm DK, et al. Blocking IL-21 signaling ameliorates xenogeneic GVHD induced by human lymphocytes. Blood. 2012;119:619–28. doi: 10.1182/blood-2011-07-368027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taur Y, Jenq RR, Perales MA, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood. 2014;124:1174–82. doi: 10.1182/blood-2014-02-554725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van der Waaij D, Vossen JM, Altes CK, Hartgrink C. Reconventionalization following antibiotic decontamination in man and animals. Am J Clin Nutr. 1977;30:1887–95. doi: 10.1093/ajcn/30.11.1887. [DOI] [PubMed] [Google Scholar]
- 55.Hardesty JS, Juang P. Fidaxomicin: a macrocyclic antibiotic for the treatment of Clostridium difficile infection. Pharmacotherapy. 2011;31:877–86. doi: 10.1592/phco.31.9.877. [DOI] [PubMed] [Google Scholar]
- 56.Louie TJ, Miller MA, Mullane KM, et al. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med. 2011;364:422–31. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 57.van Nood E, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–15. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 58.Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut microbes. 2013;4:125–35. doi: 10.4161/gmic.23571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weingarden A, Gonzalez A, Vazquez-Baeza Y, et al. Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome. 2015;3:10. doi: 10.1186/s40168-015-0070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seekatz AM, Aas J, Gessert CE, et al. Recovery of the gut microbiome following fecal microbiota transplantation. MBio. 2014;5:e00893–914. doi: 10.1128/mBio.00893-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shankar V, Hamilton MJ, Khoruts A, et al. Species and genus level resolution analysis of gut microbiota in Clostridium difficile patients following fecal microbiota transplantation. Microbiome. 2014;2:13. doi: 10.1186/2049-2618-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hamilton MJ, Weingarden AR, Sadowsky MJ, Khoruts A. Standardized frozen preparation for transplantation of fecal microbiota for recurrent Clostridium difficile infection. AmJ Gastroenterol. 2012;107:761–7. doi: 10.1038/ajg.2011.482. [DOI] [PubMed] [Google Scholar]
- 63.Huynh E, Li J. Generation of Lactococcus lactis capable of coexpressing epidermal growth factor and trefoil factor to enhance in vitro wound healing. Appl Microbiol Biotechnol. 2015;99:4667–77. doi: 10.1007/s00253-015-6542-0. [DOI] [PubMed] [Google Scholar]
- 64.Welling GW, Holtrop A, Slootmaker-van der Meulen C, et al. Inactivation of ceftriaxone by faecal enzyme preparations during ceftriaxone treatment. J Antimicrob Chemother. 1992;30:234–6. doi: 10.1093/jac/30.2.234. [DOI] [PubMed] [Google Scholar]
- 65.Pitout JD. IPSAT P1A, a class A beta-lactamase therapy for the prevention of penicillin-induced disruption to the intestinal microflora. Curr Opin Investig Drugs. 2009;10:838–44. [PubMed] [Google Scholar]
- 66.de Gunzburg J, Ducher A, Modess C, et al. Targeted adsorption of molecules in the colon with the novel adsorbent-based medicinal product, DAV132: A proof of concept study in healthy subjects. J Clin Pharmacol. 2015;55:10–6. doi: 10.1002/jcph.359. [DOI] [PubMed] [Google Scholar]