

Background
Traumatic brain injury is a growing and under-recognized public health threat. The CDC estimates nearly 2.5 million people sustain a traumatic brain injury (TBI) each year with TBI-related healthcare expenditures near 80 billion dollars annually (1–4). The impact of TBI is highlighted not only by its high mortality rate but also by the significant long-term complications suffered by its survivors with the progessive development of motor, cognitive, and behavioral disorders (5–8). It is now recognized that even mild TBI, or concussions, may lead to significant long term morbidity and the insideous onset of neurodegenerative disease. Even subconcussive events, those resulting in subclinical brain dysfunction without the typical symptoms of concussion, may lead to long-term neurologic impairment. The immune response to TBI plays a fundamental role the development and progression of subseqent neurodegenerartive disease and represents a complex interplay between peripheral immunity and the resident immune system of the injured brain—microglia. Microglia are central to this process resulting in a microenvironment of simultaneous neuroprotection and neurotoxicity (9, 10). The nature of this bidirectional communication produces a local inflammatory microenvironment geared towards either repair and regeneration or a local milieu that propels and propagates the index injury (11–16).
Chronic traumatic encephalopathy (CTE) is a progressive neurodegenerative disease that occurs years to decades after prior episodes of TBI, often in the context of contact sports but also in military personal exposed to blast injury from explosive devices (17). In fact, over 20% of the two million soldiers deployed to Iraq and Afghanistan have sustained some degree of TBI and are at risk of developing CTE over the course of their lifetimes (18–20). Furthermore, it has also become evident that even a single episode of more severe TBI can result in development of the disease (21–23). Given the millions of children and young adults involved in contact sports or military service, the incidence of CTE will only increase over the ensuing years. One of the defining features of CTE is abnormal accumulation of phosphorylated tau (p-tau) proteins and amyloid-β peptide deposits within the brain— similar to other neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease (24). Indeed, several epidemiologic studies have suggested that moderate to severe TBI is a risk factor for subsequent development of the Alzheimer continuum of diseases (25, 26). The relationship between CTE and these neurodegenerative disease processes lies within the insidious, long-lasting, inflammation generated within the injured brain (27–29). This review will focus on the link between CTE and the resident innate immune system of the injured brain—microglia.
CTE—Historical Perspective
The first description of chronic traumatic encephalopathy in the medical literature is from 1927 when 100 clinical cases of concussion were examined. The authors concluded that concussion has the potential to result in chronic injury with secondary neurodegenerative changes seen histopathologically. They termed their pattern of findings “traumatic encephalopathy” (30, 31). The term chronic traumatic encephalopathy first appeared in 1940 and the syndrome continued to be described over the ensuing decades (32). The largest human histopathologic study of its time was carried out in 1973 and a re-examination of these brain sections in 1990 using modern day immunohistochemistry techniques was the first to reveal that almost all of these cases demonstrated extensive beta amyloid deposits similar to what is seen in Alzheimer’s disease (33). A connection between CTE and American football surfaced in 2005 when an autopsy of a retired National Football League (NFL) player, Mike Webster, demonstrated extensive changes consistent with CTE (34). In 2009, all cases of CTE in the world literature were examined and their clinical and pathologic characteristics were documented (35). In 2013 McKee and colleagues first introduced a staging system for CTE in which they reported that histopathologic findings of CTE can be observed in people who never demonstrated any clinical symptoms associated with the disease (36). Regardless of the stage, all confirmed cases of CTE involve a history TBI suggesting that TBI is required, although not necessarily sufficient for the development of CTE.
CTE—Neuropathology
CTE is an insidious disease process that progresses despite cessation of the initiating traumatic activity. It is most prominently associated with the formation and accumulation of phosphorylated tau protein into neurofibrillary tangles and amyloid beta deposits. While amyloid beta is a hallmark of Alzheimer disease, it is only found in 30% of acute TBI and 40% of CTE (37–39). The mechanism of amyloid beta accumulation in TBI is unknown, but may be associated with a more severe phenotype of CTE (39, 40). Tau is a microtubule binding protein that stabilizes microtubule fibrils in neurons. When tau is phosphorylated it binds other normal tau proteins leading to aggregation into neurofibrillary tangles and an inability to bind tubulin (41). This results in decreased neurite outgrowth, denervation, and neuron death (42). Normally, tau phosphatase and apolipoprotein E maintain tau in a unphosphorylated state (43). However, animal models of repetitive TBI have shown that brain trauma results in decreased levels of tau phosphatases and increased levels of tau kinases within activated glial cells tipping the balance towards tau hyperphosphorylation (44). Additionally, it has been shown that the apolipoprotein ε4 allele results in ineffective clearance of p-tau and that individuals possessing one or two copies of this allele are at increased risk for the development neurofibrillary tangles— demonstrating an important genetic component to the disease (45).
CTE is distinguishable from other tauopathies from the unique distribution of these p-tau neurofibrillary tangles. Indeed, region specific p-tau buildup is central to the diagnosis of CTE (18, 35). The largest study of neuropathologically confirmed CTE to date formed the foundation for the recent NIH Consensus criteria for the neuropathology of CTE: 1) macroscopic abnormalities of the septum pellucidum, 2) superficial location of neurofibrillary tangles at cortical layers II and III, 3) neurofibrillary tangles in the CA2 and CA4 regions of the hippocampus, 4) abnormal tau lesions in the subcortical nuclei, and 5) p-tau neurofibrillary tangles in sub-pial and perivascular locations (36, 46). The formation and accumulation of these misfolded p-tau aggregates also appears to progress in a very patterned manner following predictable anatomic connections. This spreading of misfolded protein appears to follow a prion-like mechanism and this process has been hypothesized to be one of the mechanisms behind the patterned anatomic progression of CTE and other tauopathies such as Alzheimer disease, Parkinson’s disease, and Huntington’s disease (47, 48).
The association between TBI and pathologic tau phosphorylation is a history of repeated head injuries. However, it is the cumulative exposure to brain trauma rather than the absolute number of concussions that best predicts the degree of tau phosphorylation. This finding suggests that clinically asymptomatic sub-concussive events play a much larger role in the process than has been previously recognized (49). In animal models of TBI a single mild injury was found to increase p-tau at one month, but these changes were resolved at six months post injury. However, when the single mild TBI was repeated daily for six days the animals developed marked neuroinflammation and p-tau aggregates at six months post injury (50, 51). On the other hand, there is emerging evidence that a single more severe TBI can also lead to significant accumulation of abnormal p-tau oligomers (52). Nonetheless, the central question that remains to be answered in this overarching process is what triggers the formation of misfolded p-tau in the first place.
The role of microglia in the development of CTE—
TBI triggers a robust inflammatory response within the injured brain. This acute neuroinflammation is initiated by several factors including necrotic debris at the site of injury, damage-associated molecular patterns, and infiltrating components of the peripheral innate immune response. The degree of this initial inflammatory response has significant value in predicting more long-term outcomes after TBI. A study of human patients showed that the level of IL-6 in the cerebrospinal fluid during the first week post injury correlated with neurologic recovery at 6 months (53). Even after the acute inflammatory response has resolved several studies have demonstrated residual long-lasting inflammation in both animal models and in human patients (54, 55). One of the main drivers of this continued low-level inflammation is the chronic activation of the resident innate immune system of the brain—microglia (55, 56). Microglia are embryologically distinct from peripheral bone marrow-derived macrophages, arising from the primitive yolk sac as opposed to the developing liver in the embryo (57). Additionally, microglia are self-renewing suggesting that bone marrow derived macrophages do not contribute to the maintenance of the mature microglia pool (58, 59). This is in keeping with the traditional view that the CNS is an immune privileged site (60). Microglia and peripheral bone marrow derived cells that infiltrate into the brain after TBI, therefore, represent distinct populations. In fact, each cell type relies on a distinctive set of transcription factors leading to separate patterns of gene expression (61, 62). Both cell types are responsible for different aspects of neural repair and potential neural toxicity after CNS injury (63). However, once the acute inflammatory process has resolved and peripheral bone marrow derived cells are no longer present, microglia have the potential to maintain a chronically activated state for years after the initial insult. Mouse models of single impact TBI have shown that microglial activation is maintained up to one-year post TBI (64). Likewise, human studies have demonstrated reactive microglia up to 18 years in survivors of TBI (65). In repetitive mild TBI, mouse models have shown reactive microglia 12 months post injury, but with a response greatly amplified as compared to animals receiving a single injury (66). This supports more recent studies suggesting that repeated mild injuries develop levels of neuroinflammation equivalent to a more severe single injury (67). Although all the above studies have relied on post-mortem analysis of brain tissue, recent advances in positron emission tomography (PET) technology has allowed for real time analysis of microglial activation. These studies have shown microglial activation persisting for up to 17 years post injury in human patients. Even mild TBI, especially repeated mild TBI, as seen in a cohort of retired NFL players has demonstrated persistent microglial activation years after retirement via advanced PET technology (68).
Over the course of injury these chronically activated microglia are responsible for increased levels of pro-inflammatory cytokines, chemokines, and reactive oxygen species thereby setting the stage for maintaining an insidious neurotoxic environment. This persistent low-level inflammation has the potential to generate an exaggerated inflammatory response to otherwise benign stimuli. This phenomenon is known as microglial priming (69). In fact, microglial priming may be the etiology behind the increased susceptibility to subsequent concussions once an index injury has been sustained. That is, primed microglia from an initial injury may respond in an amplified manner to subsequent insults, even if milder in nature (70, 71). This priming effect alters the ability of microglia to fully resolve the inflammatory process leading to a long-term, often exaggerated, neuroinflammatory response (Figure 1) (29). It also indicates that targeting the microglial response may be a novel therapeutic target for the treatment of TBI patients.
Figure 1. Schematic of microglial priming.
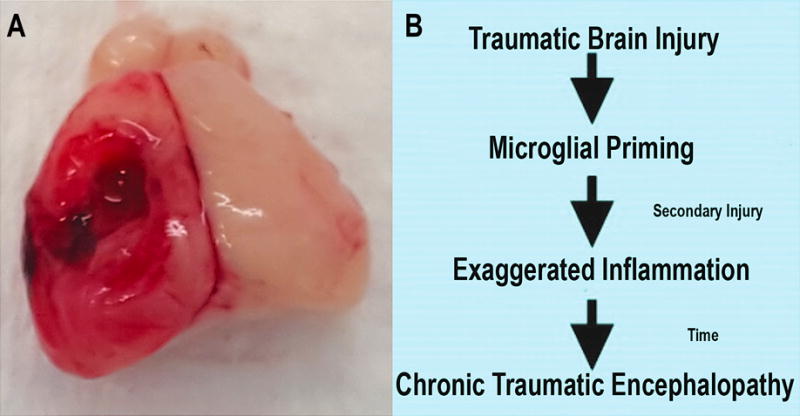
A) Standard rodent traumatic brain injury induced via controlled cortical impact. B) Traumatic brain injury generates a priming effect within microglia driving a chronic low-level inflammatory environment over time. This chronic inflammation leads to both an increased susceptibility to secondary injury as well as the long-term development of chronic traumatic encephalopathy.
Microglia and tau phosphorylation—
There is increasing acceptance that the abnormal accumulation and function of phosphorylated tau underlies the pathology of CTE. While the exact mechanisms of tau phosphorylation are yet to be fully determined, there is ample experimental evidence to suggest that activated microglia and their resultant neuroinflammation play a central role in the generation of p-tau after brain injury. For example, several investigative groups have shown that both early and late activation of microglia increases p-tau formation in mouse models of Alzheimer disease and other tauopathies (72–74). On the other hand, administration of the calcineurin inhibitor FK-506 in a transgenic mouse model of Alzheimer disease resulted in both improved survival and significantly reduced p-tau (75). This effect is likely secondary to the ability of FK-506 to suppress both NF-κB and MAPK in microglia resulting in lower levels of inflammatory gene expression (76). More direct evidence of microglial involvement comes from studies using CX3CR1 knockout mice. Microglia utilize fractalkine signaling via CX3CR1 to migrate to their synaptic targets and initiate phagocytosis and synaptic refinement (77). When mice deficient in CX3CR1 were crossed with a humanized tau (hTau) mouse model, there was significantly accelerated tau phosphorylation and aggregation into neurofibrillary tangles as well as enhanced activation of microglia resulting in neurocognitive impairment (78). Additional studies in these double transgenic animals found that microglial activation preceded the spread of p-tau. When microglia from these CX3CR1 deficient hTau mice were adoptively transferred into naïve mice, they were sufficient to induce tau phosphorylation (79). One of the more prominent components of microglia-induced neuroinflammation is IL-1β. IL-1β is produced by activated microglia and is involved in a variety of cellular activities. In wild type mice, implantation of a slow-release IL-1β pellet resulted in increased tau phosphorylation whereas chronic blockade of IL-1β via administration of IL-1R significantly reduced p-tau and improved cognition in a mouse model of Alzheimer disease (3xTg-AD) (80, 81). Administration of IL-1R also decreased NF-κB activation and reduced the levels of pro-inflammatory cytokines cumulatively suggesting that IL-1β mediates communication between glial cells and neurons (81). When the 3xTg-AD model was crossed with IL-1βXAT mice (allowing for hippocampus specific IL-1β expression) enhanced tau phosphorylation was noted throughout the hippocampus (82). While all of these findings have been made in animals genetically modeled for tauopathy, it should be noted that they are not present in naïve mice (83). This signals that there must be a pre-existing predilection for tau phosphorylation in addition to altered immune signaling. In TBI, it is postulated that the initial mechanical axonal injury and resultant neuroinflammation is what primes microglia and drives progression of the disease. Mechanistically, the force of injury causes tau to dissociate from the microtubule leading to phosphorylation and aggregation into NFTs (84, 85). The persistent neuroinflammation resulting from microglial priming then drives the progression of p-tau formation and propagates spread between neurons thereby slowly advancing the disease (Figure 2). Collectively, the bulk of the available literature indicates that chronically activated microglia within the traumatized brain are the key contributors to the pathological accumulation and spread of p-tau, both of which form the foundation and underlie the deficits seen in CTE.
Figure 2. Schematic of physiologic and pathophysiologic microglial activation.
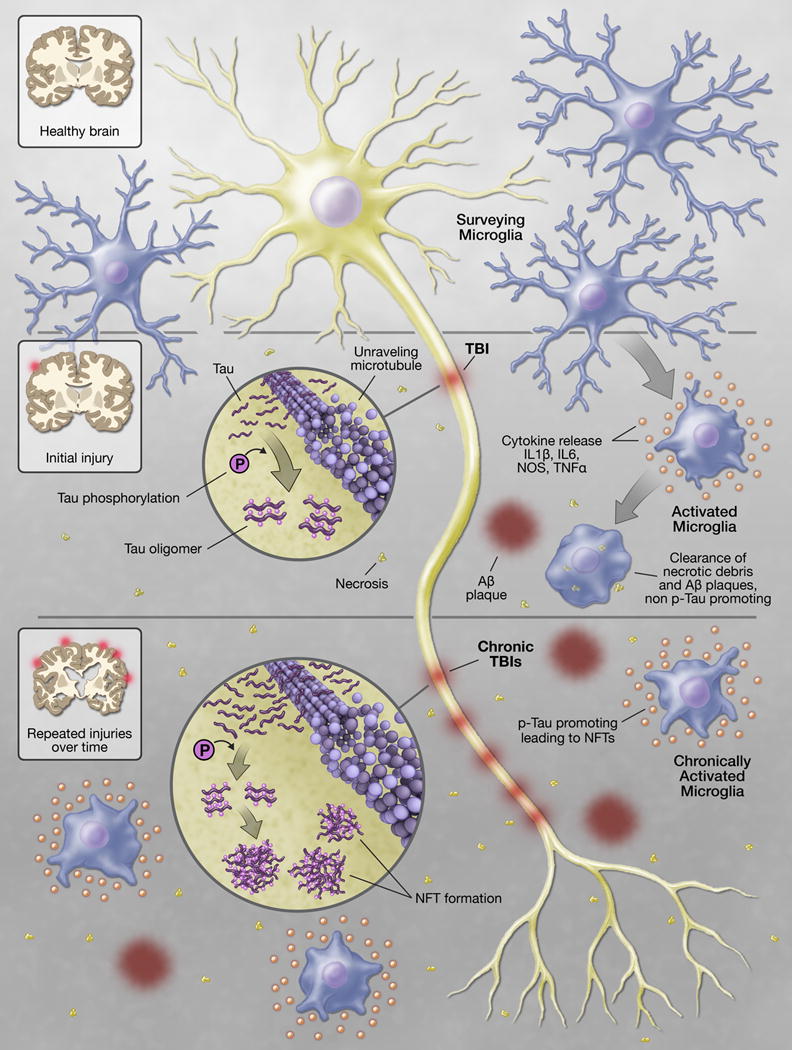
Under conditions of homeostasis microglia continually survey the local microenvironment for invading pathogens and cellular debris. Perturbations within this environment, such as traumatic injury, result in activation of microglia. Activated microglia release a myriad of inflammatory mediators to aid in the clearance of necrotic debris and amyloid beta plaques generated during the injury process. Activated microglia are also capable of limiting tau phosphorylation (p-tau) and the subsequent development of p-tau oligomers. If the inflammation fails to completely resolve, as is often the case with severe injury or repetitive injury, microglia can assume a pathophysiologic state of chronic activation. Chronically activated microglia are unable to clear amyloid beta, allow association of p-tau into oligomers and neurofibrillary tangles (NFT), and generate an exaggerated response to subsequent traumatic or inflammatory stimuli all of which lead to the subsequent development of chronic traumatic encephalopathy.
Potential therapeutic targets in the development and degree of CTE—
There are currently no effective treatments for patients with CTE. However, there are several potential therapeutic targets associated with the development and degree of CTE currently under investigation. Microglial activation and the ensuing neuroinflammatory response represent obvious areas for the development of therapeutic interventions. For example, targeting microglia-induced neuroinflammation has been extensively explored in both animal models and human trials of other neurodegenerative diseases. Non-steroidal anti-inflammatory drugs (NSAIDs) have been shown in human epidemiologic studies to reduce the incidence of Alzheimer disease and the administration of NSAIDs to Alzheimer disease mouse models have shown promise in reducing the amyloid beta plaque load (86, 87). Nonetheless, it should be noted that in a recent human clinical trial, NSAIDs failed to slow cognitive decline in patients who already have Alzheimer disease. However, given the latency between the initial traumatic insult and the development of CTE there may still be a role for NSAIDs as a preventative therapeutic prior to disease onset (88). Given these somewhat disappointing clinical results, there has been a shift in focus towards the prevention of p-tau formation and aggregation into p-tau oligomers. Given the prominent role of IL-1β in the generation of p-tau, IL-1 receptor antagonism has recently been assessed. In an animal model, delivery of an IL-1 receptor antagonist reduced p-tau formation, ameliorated cognitive dysfunction, and reversed spatial and contextual learning deficits (89). In addition, several trials are currently underway assessing the ability of compounds to inhibit p-tau aggregation. For example, a derivative of methylene blue (LMTX) was found to have a treatment benefit in a recent phase II clinical trail and is currently undergoing a phase III trial (90). A number of synthetic amino acids that sterically inhibit p-tau aggregation are under investigation as well (91). Microtubule stabilizers are also an active area of research given that microtubule stability deteriorates with tau pathology (92). The microtubule stabilizer epothilone D increased microtubule density, inhibited axonal loss, and reversed learning defects in an animal model of tauopathy (93). Alternatively, a number of pharmacologic and small chemical screens have identified certain drugs that may be useful in skewing the microglia polarization state for therapeutic purposes. For example, PPARγ agonists such as the glitazones, the tetracycline family of antibiotics, and the chemotherapeutic retinoic acid receptor agonists such as bexarotene have been shown to induce a more anti-inflammatory M2-like polarization state (94). In fact, minocycline was able to reduce levels of p-tau and insoluble tau in hTau mice (95). Perhaps the most exciting development in therapeutics targeted towards p-tau induced neurodegeneration is the recent identification of cis and trans isoforms of p-tau. It has recently been shown that trans p-tau is physiologic while cis p-tau is pathogenic promoting tauopathy after TBI. “Cistauosis” appears long before other tauopathies and treatment with anti cis p-tau antibody prevented the development of p-tau aggregates restoring several neuropathological and functional outcomes in a mouse model of TBI (96). Humanization of this anti cis p-tau antibody has the potential to become a revolutionary new treatment option for brain injured patients (97, 98). Lastly, it has also been recently shown that depletion of classical monocytes immediately after the induction of TBI results in skewing of the overall inflammatory microenvironment within the injured brain towards a more anti-inflammatory phenotype at 24 hours post injury. These changes correlated with significant reduction in cognitive dysfunction one-month post treatment (99). These data suggest that the infiltrating population of monocyte-derived cells may play a more significant role in the direction of the local inflammatory microenvironment than has been previously recognized. Indeed, the ability of infiltrating myeloid cells from the periphery to influence the inflammatory state of microglia at the site of neurologic injury has been previously reported in the stroke literature (100). Additional studies examining the infiltrating monocyte-microglia interaction have shown these infiltrating monocytic cells to play critical roles that the resident microglia are incapable of such as restricting amyloid beta plaque in Alzheimer disease and facilitating functional recovery in spinal cord injury (101–104). Both cell types are responsible for different aspects of neural repair and potential neural toxicity after CNS injury and how their interaction affects the long-term outcomes after TBI and subsequent development of CTE is poorly understood (63, 105). Understanding the nature of their crosstalk raises the possibility of therapeutically targeting their interaction as a novel target for the treatment of TBI.
Conclusions—
There is an abundance of evidence suggesting that the frequency and severity of TBI are key determinants in the development and degree of CTE. The precise mechanisms linking the two processes have yet to be fully elucidated. Given the long latency between the initial traumatic insult(s) and the later development of CTE, there is a large window of time in which potential intervention may have a clinically significant benefit. Navigating the complexities of the neuroinflammatory response to TBI and determining the precise mechanisms behind tau phosphorylation will be key steps towards combating this highly morbid injury process. Furthermore, the emerging role of microglia in this process provides an abundance of potential pathways for therapeutic intervention. The rapid advancement of next generation sequencing and its ability to identify pro-repair and pro-injury pathways within individual cell populations over the course of TBI will undoubtedly allow for rapid advancement on this front. CTE lies at the intersection between immunology and neuroscience. Advancements in our understanding of this complex injury process will challenge current dogmas and have the potential to be paradigm shifting as the field of neuroimmunology continues to establish itself within the scientific community.
Acknowledgments
Michael Gallagher created the artwork in figure 2.
Sources of Support: This study work was supported by NIH grant GM117341 and The American College of Surgeons C. James Carrico Research Fellowship to S.J.S. and NIH grant AR064313 to C.M.C. Further support from NIH grants AR064546, AR050250, AR054796, AI092490, HL108795, and funds provided by the Israel Binational Foundation and Mabel Greene Myers Professor Chair in Medicine to H.P.
References
- 1.Faul M. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- 2.Corso P, Finkelstein E, Miller T, Fiebelkorn I, Zaloshnja E. Incidence and lifetime costs of injuries in the United States. Inj Prev. 2006;12(4):212–218. doi: 10.1136/ip.2005.010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearson WS, Sugerman DE, McGuire LC, Coronado VG. Emergency department visits for traumatic brain injury in older adults in the United States: 2006–08. West J Emerg Med. 2012;13(3):289–293. doi: 10.5811/westjem.2012.3.11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitlock JA, Jr, Hamilton BB. Functional outcome after rehabilitation for severe traumatic brain injury. Arch Phys Med Rehabil. 1995;76(12):1103–1112. doi: 10.1016/s0003-9993(95)80117-0. [DOI] [PubMed] [Google Scholar]
- 5.Schwarzbold M, Diaz A, Martins ET, Rufino A, Amante LN, Thais ME, Quevedo J, Hohl A, Linhares MN, Walz R. Psychiatric disorders and traumatic brain injury. Neuropsychiatr Dis Treat. 2008;4(4):797–816. doi: 10.2147/ndt.s2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whelan-Goodinson R, Ponsford J, Johnston L, Grant F. Psychiatric disorders following traumatic brain injury: their nature and frequency. J Head Trauma Rehabil. 2009;24(5):324–332. doi: 10.1097/HTR.0b013e3181a712aa. [DOI] [PubMed] [Google Scholar]
- 7.Peskind ER, Brody D, Cernak I, McKee A, Ruff RL. Military- and sports-related mild traumatic brain injury: clinical presentation, management, and long-term consequences. J Clin Psychiatry. 74(2):180–188. doi: 10.4088/JCP.12011co1c. quiz 188, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin LA, Neighbors HW, Griffith DM. The experience of symptoms of depression in men vs women: analysis of the National Comorbidity Survey Replication. JAMA Psychiatry. 2013;70(10):1100–1106. doi: 10.1001/jamapsychiatry.2013.1985. [DOI] [PubMed] [Google Scholar]
- 9.Schwarzmaier SM, Plesnila N. Contributions of the immune system to the pathophysiology of traumatic brain injury - evidence by intravital microscopy. Front Cell Neurosci. 2014;8:358. doi: 10.3389/fncel.2014.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Rivero Vaccari JP, Dietrich WD, Keane RW. Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cereb Blood Flow Metab. 2014;34(3):369–375. doi: 10.1038/jcbfm.2013.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7(4):366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peruzzotti-Jametti L, Donega M, Giusto E, Mallucci G, Marchetti B, Pluchino S. The role of the immune system in central nervous system plasticity after acute injury. Neuroscience. 2014;283:210–221. doi: 10.1016/j.neuroscience.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6(10):775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- 14.Schwulst SJ, Trahanas DM, Saber R, Perlman H. Traumatic brain injury-induced alterations in peripheral immunity. J Trauma Acute Care Surg. 2013;75(5):780–788. doi: 10.1097/TA.0b013e318299616a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong XD, Bai S, Chen X, Wei HJ, Jin WN, Li MS, Yan Y, Shi FD. Alterations of natural killer cells in traumatic brain injury. Neurosci Bull. 2014;30(6):903–912. doi: 10.1007/s12264-014-1481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conway Morris A, Kefala K, Wilkinson TS, Dhaliwal K, Farrell L, Walsh T, Mackenzie SJ, Reid H, Davidson DJ, Haslett C, et al. C5a mediates peripheral blood neutrophil dysfunction in critically ill patients. Am J Respir Crit Care Med. 2009;180(1):19–28. doi: 10.1164/rccm.200812-1928OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stein TD, Alvarez VE, McKee AC. Concussion in Chronic Traumatic Encephalopathy. Curr Pain Headache Rep. 2015;19(10):47. doi: 10.1007/s11916-015-0522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKee AC, Robinson ME. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 2014;10(3 Suppl):S242–253. doi: 10.1016/j.jalz.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4(134):134ra160. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helmick KM, Spells CA, Malik SZ, Davies CA, Marion DW, Hinds SR. Traumatic brain injury in the US military: epidemiology and key clinical and research programs. Brain Imaging Behav. 2015;9(3):358–366. doi: 10.1007/s11682-015-9399-z. [DOI] [PubMed] [Google Scholar]
- 21.Omalu B. Chronic traumatic encephalopathy. Prog Neurol Surg. 2014;28:38–49. doi: 10.1159/000358761. [DOI] [PubMed] [Google Scholar]
- 22.Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, Fitzsimmons R. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. 2011;69(1):173–183. doi: 10.1227/NEU.0b013e318212bc7b. discussion 183. [DOI] [PubMed] [Google Scholar]
- 23.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22(2):142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755–766. doi: 10.1007/s00401-014-1349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heyman A, Wilkinson WE, Stafford JA, Helms MJ, Sigmon AH, Weinberg T. Alzheimer’s disease: a study of epidemiological aspects. Ann Neurol. 1984;15(4):335–341. doi: 10.1002/ana.410150406. [DOI] [PubMed] [Google Scholar]
- 26.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55(8):1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 27.Bouvier DS, Murai KK. Synergistic actions of microglia and astrocytes in the progression of Alzheimer’s disease. J Alzheimers Dis. 2015;45(4):1001–1014. doi: 10.3233/JAD-143156. [DOI] [PubMed] [Google Scholar]
- 28.Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 2015;96(Pt A):29–41. doi: 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andreasson KI, Bachstetter AD, Colonna M, Ginhoux F, Holmes C, Lamb B, Landreth G, Lee DC, Low D, Lynch MA, et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem. 2016;138(5):653–693. doi: 10.1111/jnc.13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osnato M, Gilliberti V. Postconcussion neurosis-traumatic encephalitis: a conception of postconcsussion phenomena. Arch Neurol Psychiatry. 1927;18:181–214. [Google Scholar]
- 31.Osnato M. The role of traumain various neruopsyciatric conditions. Am J Psychiatry. 1929;86:643–660. [Google Scholar]
- 32.Bowman KM, Blau A. In: Psychotic states following head and brain injury in adults and children. S B, editor. Baltimore, MD: Williams & Wilkins; 1940. pp. 309–359. [Google Scholar]
- 33.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3(3):270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 34.Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57(1):128–134. doi: 10.1227/01.neu.0000163407.92769.ed. discussion 128–134. [DOI] [PubMed] [Google Scholar]
- 35.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68(7):709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(Pt 1):43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 2010;11(5):361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sivanandam TM, Thakur MK. Traumatic brain injury: a risk factor for Alzheimer’s disease. Neurosci Biobehav Rev. 2012;36(5):1376–1381. doi: 10.1016/j.neubiorev.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Edwards G, 3rd, Moreno-Gonzalez I, Soto C. Amyloid-beta and tau pathology following repetitive mild traumatic brain injury. Biochem Biophys Res Commun. 2016 doi: 10.1016/j.bbrc.2016.07.123. [DOI] [PubMed] [Google Scholar]
- 40.Stone JR, Okonkwo DO, Singleton RH, Mutlu LK, Helm GA, Povlishock JT. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid Beta peptide in traumatic axonal injury. J Neurotrauma. 2002;19(5):601–614. doi: 10.1089/089771502753754073. [DOI] [PubMed] [Google Scholar]
- 41.Iqbal K, Gong CX, Liu F. Hyperphosphorylation-induced tau oligomers. Front Neurol. 2013;4:112. doi: 10.3389/fneur.2013.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cowan CM, Mudher A. Are tau aggregates toxic or protective in tauopathies? Front Neurol. 2013;4:114. doi: 10.3389/fneur.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lambrecht C, Haesen D, Sents W, Ivanova E, Janssens V. Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol Biol. 2013;1053:283–305. doi: 10.1007/978-1-62703-562-0_17. [DOI] [PubMed] [Google Scholar]
- 44.Kane MJ, Angoa-Perez M, Briggs DI, Viano DC, Kreipke CW, Kuhn DM. A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods. 2012;203(1):41–49. doi: 10.1016/j.jneumeth.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spillantini MG, Iovino M, Vuono R. Release of growth factors by neuronal precursor cells as a treatment for diseases with tau pathology. Arch Ital Biol. 2011;149(2):215–223. doi: 10.4449/aib.v149i1.1360. [DOI] [PubMed] [Google Scholar]
- 46.McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131(1):75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soto C. Transmissible proteins: expanding the prion heresy. Cell. 2012;149(5):968–977. doi: 10.1016/j.cell.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huber BR, Alosco ML, Stein TD, McKee AC. Potential Long-Term Consequences of Concussive and Subconcussive Injury. Phys Med Rehabil Clin N Am. 2016;27(2):503–511. doi: 10.1016/j.pmr.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petraglia AL, Plog BA, Dayawansa S, Dashnaw ML, Czerniecka K, Walker CT, Chen M, Hyrien O, Iliff JJ, Deane R, et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int. 2014;5:184. doi: 10.4103/2152-7806.147566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shultz SR, Wright DK, Zheng P, Stuchbery R, Liu SJ, Sashindranath M, Medcalf RL, Johnston LA, Hovens CM, Jones NC, et al. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain. 2015;138(Pt 5):1297–1313. doi: 10.1093/brain/awv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoshino S, Tamaoka A, Takahashi M, Kobayashi S, Furukawa T, Oaki Y, Mori O, Matsuno S, Shoji S, Inomata M, et al. Emergence of immunoreactivities for phosphorylated tau and amyloid-beta protein in chronic stage of fluid percussion injury in rat brain. Neuroreport. 1998;9(8):1879–1883. doi: 10.1097/00001756-199806010-00039. [DOI] [PubMed] [Google Scholar]
- 53.Kumar RG, Diamond ML, Boles JA, Berger RP, Tisherman SA, Kochanek PM, Wagner AK. Acute CSF interleukin-6 trajectories after TBI: associations with neuroinflammation, polytrauma, and outcome. Brain Behav Immun. 2015;45:253–262. doi: 10.1016/j.bbi.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 54.Kumar RG, Boles JA, Wagner AK. Chronic Inflammation After Severe Traumatic Brain Injury: Characterization and Associations With Outcome at 6 and 12 Months Postinjury. J Head Trauma Rehabil. 2015;30(6):369–381. doi: 10.1097/HTR.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 55.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol. 2013;9(4):211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat. 2015;11:97–106. doi: 10.2147/NDT.S65815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 59.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339(6116):156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12(9):623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 61.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 62.Gomez Perdiguero E, Schulz C, Geissmann F. Development and homeostasis of “resident” myeloid cells: the case of the microglia. Glia. 2013;61(1):112–120. doi: 10.1002/glia.22393. [DOI] [PubMed] [Google Scholar]
- 63.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14(9):1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 64.Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73(1):14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136(Pt 1):28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aungst SL, Kabadi SV, Thompson SM, Stoica BA, Faden AI. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab. 2014;34(7):1223–1232. doi: 10.1038/jcbfm.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol. 2016;173(4):681–691. doi: 10.1111/bph.13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coughlin JM, Wang Y, Munro CA, Ma S, Yue C, Chen S, Airan R, Kim PK, Adams AV, Garcia C, et al. Neuroinflammation and brain atrophy in former NFL players: An in vivo multimodal imaging pilot study. Neurobiol Dis. 2015;74:58–65. doi: 10.1016/j.nbd.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Witcher KG, Eiferman DS, Godbout JP. Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci. 2015;38(10):609–620. doi: 10.1016/j.tins.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muccigrosso MM, Ford J, Benner B, Moussa D, Burnsides C, Fenn AM, Popovich PG, Lifshitz J, Walker FR, Eiferman DS, et al. Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun. 2016;54:95–109. doi: 10.1016/j.bbi.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weil ZM, Gaier KR, Karelina K. Injury timing alters metabolic, inflammatory and functional outcomes following repeated mild traumatic brain injury. Neurobiol Dis. 2014;70:108–116. doi: 10.1016/j.nbd.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 72.Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25(39):8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;178(6):2811–2822. doi: 10.1016/j.ajpath.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 76.Yoshino T, Nakase H, Honzawa Y, Matsumura K, Yamamoto S, Takeda Y, Ueno S, Uza N, Masuda S, Inui K, et al. Immunosuppressive effects of tacrolimus on macrophages ameliorate experimental colitis. Inflamm Bowel Dis. 2010;16(12):2022–2033. doi: 10.1002/ibd.21318. [DOI] [PubMed] [Google Scholar]
- 77.Lyons A, Lynch AM, Downer EJ, Hanley R, O’Sullivan JB, Smith A, Lynch MA. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem. 2009;110(5):1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x. [DOI] [PubMed] [Google Scholar]
- 78.Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138(Pt 6):1738–1755. doi: 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sheng JG, Zhu SG, Jones RA, Griffin WS, Mrak RE. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp Neurol. 2000;163(2):388–391. doi: 10.1006/exnr.2000.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187(12):6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion MK. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33(11):5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Collins-Praino LE, Corrigan F. Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav Immun. 2016 doi: 10.1016/j.bbi.2016.09.027. [DOI] [PubMed] [Google Scholar]
- 84.Feuillette S, Miguel L, Frebourg T, Campion D, Lecourtois M. Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J Neurochem. 2010;113(4):895–903. doi: 10.1111/j.1471-4159.2010.06663.x. [DOI] [PubMed] [Google Scholar]
- 85.Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, Mandelkow E, Zweckstetter M. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48(42):10047–10055. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- 86.Deardorff WJ, Grossberg GT. Targeting neuroinflammation in Alzheimer’s disease: evidence for NSAIDs and novel therapeutics. Expert Rev Neurother. 2016:1–16. doi: 10.1080/14737175.2016.1200972. [DOI] [PubMed] [Google Scholar]
- 87.Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC, Cache County Study I Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. 2002;59(6):880–886. doi: 10.1212/wnl.59.6.880. [DOI] [PubMed] [Google Scholar]
- 88.Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH, Tarenflurbil Phase 3 Study G Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302(23):2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ben-Menachem-Zidon O, Ben-Menahem Y, Ben-Hur T, Yirmiya R. Intra-hippocampal transplantation of neural precursor cells with transgenic over-expression of IL-1 receptor antagonist rescues memory and neurogenesis impairments in an Alzheimer’s disease model. Neuropsychopharmacology. 2014;39(2):401–414. doi: 10.1038/npp.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, Kook KA, Harrington CR. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis. 2015;44(2):705–720. doi: 10.3233/JAD-142874. [DOI] [PubMed] [Google Scholar]
- 91.Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Munch J, Baker D, Eisenberg D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475(7354):96–100. doi: 10.1038/nature10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- 93.Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, Iba M, James MJ, Xie SX, Ballatore C, et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010;30(41):13861–13866. doi: 10.1523/JNEUROSCI.3059-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Amantea D, Bagetta G. Drug repurposing for immune modulation in acute ischemic stroke. Curr Opin Pharmacol. 2016;26:124–130. doi: 10.1016/j.coph.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 95.Noble W, Garwood C, Stephenson J, Kinsey AM, Hanger DP, Anderton BH. Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J. 2009;23(3):739–750. doi: 10.1096/fj.08-113795. [DOI] [PubMed] [Google Scholar]
- 96.Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523(7561):431–436. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, Zhou XZ. Potential of the Antibody Against cis-Phosphorylated Tau in the Early Diagnosis, Treatment, and Prevention of Alzheimer Disease and Brain Injury. JAMA Neurol. 2016;73(11):1356–1362. doi: 10.1001/jamaneurol.2016.2027. [DOI] [PubMed] [Google Scholar]
- 98.Albayram O, Herbert MK, Kondo A, Tsai CY, Baxley S, Lian X, Hansen M, Zhou XZ, Lu KP. Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci. 2016;6:59. doi: 10.1186/s13578-016-0124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, Ferguson AR, Rosi S. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci. 2015;35(2):748–760. doi: 10.1523/JNEUROSCI.2405-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43(11):3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 101.Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6(7):e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.London A, Itskovich E, Benhar I, Kalchenko V, Mack M, Jung S, Schwartz M. Neuroprotection and progenitor cell renewal in the injured adult murine retina requires healing monocyte-derived macrophages. J Exp Med. 2011;208(1):23–39. doi: 10.1084/jem.20101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.London A, Cohen M, Schwartz M. Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci. 2013;7:34. doi: 10.3389/fncel.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49(4):489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 105.Kokaia Z, Martino G, Schwartz M, Lindvall O. Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci. 2012;15(8):1078–1087. doi: 10.1038/nn.3163. [DOI] [PubMed] [Google Scholar]