

Abstract
Investigations of teriparatide (rPTH) as a potential treatment for critical defects have demonstrated the predicted anabolic effects on bone formation, and significant non-anabolic effects on healing via undefined mechanisms. Specifically, studies in murine models of structural allograft healing demonstrated that rPTH treatment increased angiogenesis (vessels <30μm), and decreased arteriogenesis (>30 μm) and mast cell numbers, which lead to decreased fibrosis and accelerated healing. To better understand these non-anabolic effects, we interrogated osteogenesis, vasculogenesis and mast cell accumulation in mice randomized to placebo (saline), rPTH (20μg/kg/2days), or the mast cell inhibitor sodium cromolyn (SC) (24μg/kg/2days), via longitudinal micro-CT and multiphoton laser scanning microscopy (MPLSM), in a critical calvaria defect model. Micro-CT demonstrated that SC significantly increased defect window closure and new bone volume versus placebo (p<0.05), although these effects were not as great as rPTH. Interestingly, both rPTH and SC has similar inhibitory effects on arteriogenesis versus placebo (p<0.05) without affecting total vascular volume. MPLSM time course studies in untreated mice revealed that large numbers of mast cells were detected 1 day post-op (43 +/− 17), peaked at 6 days (76 +/− 6), and were still present in the critical defect at the end of the experiment on day 30 (20 +/− 12). In contrast, angiogenesis was not observed until day 4, and functional vessels were first observed on 6 days, demonstrating that mast cell accumulation precedes vasculogenesis. To confirm a direct role of mast cells on osteogenesis and vasculogenesis, we demonstrated that specific diphtheria toxin-α deletion in Mcpt5-Cre-iDTR mice results in similar affects as SC treatment in WT mice. Collectively, these findings demonstrate that mast cells inhibit bone defect healing by stimulating arteriogenesis associated with fibrotic scaring, and that an efficacious non-anabolic effect of rPTH therapy on bone repair is suppression of arteriogenesis and fibrosis secondary to mast cell inhibition.
Introduction
Critical bone defects caused by birth defects, traumatic injuries, infection or cancer remain a great clinical challenge.(1) One of the approaches that has been investigated to address this problem is the use of recombinant parathyroid hormone (rPTH, teriparatide) adjuvant therapy,(2) which was based on its well-established anabolic effects as a FDA-approved treatment for osteoporosis,(3) and positive findings in phase 2 clinical trials on adult fractures.(4–6) Moreover, data from pre-clinical studies(7–9) and clinical case reports(10–12) have demonstrated that rPTH treatment during bone repair has additional non-anabolic effects that alter vascularity, and inhibits fibrosis to accelerate healing and bony union.
Mechanistic studies in murine models of structural bone grafting have shown that efficient live autograft healing is characterized by angiopoietin-1 mediated angiogenesis (blood vessels <30μm in diameter) with a paucity of arteriogenesis (blood vessels >30 μm in diameter), while defective allograft healing occurs in the presence of high levels of angiopoietin-2 that promotes arteriogenesis and fibrosis.(13) Furthermore, it was shown that rPTH treatment induced angiopoietin-1 (8-fold), while dramatically decreasing angiopoietin-2 (70-fold) at day 7 of allograft healing, which significantly reduced arteriogenesis and fibrosis.(13) These rPTH inhibitory effects on vasculogenesis and fibrosis were largely recapitulated with anti-angiopoietin-2 peptibody treatment,(13) formally demonstrating the adverse effects of this factor and arteriogenesis in the setting of bone regeneration.
Another surprising effect of rPTH treatment on both femoral and calvarial allograft healing in mice was the finding that the drug eliminates large numbers of mast cells that accumulate around large vessels in the transitional tissue at the graft-host junction.(8,13) Interestingly, it has long been recognized that mast cells may play a role in fracture healing.(14) Histology studies of fractures in rats revealed that in the first two weeks, mast cells are found either in the vicinity of blood vessels or in the vascularized tissue proliferating into the cartilaginous portion of subperiosteal callus.(15) This finding led to the view that mast cells are involved in digestion of extracellular matrix and angiogenesis in the early stages of fracture healing. However, mast cells are also known to be central mediators of chronic fibrosis via degranulation and release of fibroblast growth factors (FGF), tumor growth factors (TGF), platelet derived growth factor (PDGF), granulocyte macrophage colony-stimulating factor (GM-CSF), and other factors that promote progressive sclerosis,(16) and several chronic fibrotic conditions (i.e. pulmonary fibrosis,(17) renal fibrosis,(18) and scleroderma (19)). Moreover, the recent studies identifying mast cells as potential mediators in musculoskeletal diseases (i.e. tendinopath,(20) inflammatory myopathy(21)), via their deregulation and TGFβ1-induced fibrosis, suggests a role for mast cells in failed tissue healing.(22)
Based on the aforementioned data, we proposed that fundamental differences between the “scarless” healing observed with live autografts, versus the “scarful” healing observed with structural allografts, is the accumulation of mast cells around large vessels in the transitional tissue at the graft-host junction, and that the non-anabolic efficacy observed with rPTH treatment is due to the inhibition of these pathologic factors.(23) However, formal hypothesis testing of the cause and effect relationships between arteriogenesis, mast cells and critical defects were limited by the absence of an in vivo model with sufficient spatiotemporal resolution and genetic functionality. To address this, we developed a chronic cranial defect window model for in vivo multiphoton laser scanning microscopy (MPLSM) with quantitative outcomes, to interrogate the natural history of osteogenesis and vasculogenesis during bone repair.(24) Additionally, this MPLSM approach allows for the use of genetically modified strains for lineage tracing and loss of function studies. For our studies, we chose the Mcpt5-Cre+iDTR+ transgenic mice,(25) which express Cre recombinase from the mast cell protease (Mcpt) 5 promoter (Mcpt5-Cre),(26) and allows for the expression of a simian diphtheria toxin receptor (DTR) only in mast cells. These Mcpt5-Cre+iDTR+ mice are phenotypically WT until they receive an intraperitoneal injection of Corynebacterium diphtheria toxin-alpha (DTA), which results in an almost complete loss (98.7%) of mast cells.(25) Utilizing these technical advances, here we prospectively tested the hypotheses that: 1) arteriogenesis in critical bone defects allows for mast cell accumulation around large vessels; 2) mast cells inhibit bone formation; and 3) rPTH efficacy is derived from both the direct anabolic effects on bone formation and its inhibitory effects on arteriogenesis and mast cells.
Materials and Methods
Animals and in vivo experiments
All experiments with live animals were performed on University of Rochester Committee for Animal Resources approved protocols. All mouse strains were obtained from the Jackson laboratory (Bar Harbor, ME). C57BL/6 mice (The Jackson laboratory, 000664 – C57BL/6J) were used as the wild type (WT) strain. B6.FVB-Tg(CMA1-cre)6Thhe/J (The Jackson laboratory, stock # 014643) and B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J) (The Jackson laboratory, stock # 006148) were crossed to generate the experimental Mcpt5-Cre-YFP mice as previously described(26). B6.FVB-Tg(CMA1-cre)6Thhe/J (The Jackson laboratory, stock # 014643) and C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J) (The Jackson laboratory, stock # 007900) were crossed to generate the experimental Mcpt5-Cre-iDTR mice as previously described(27). All experimental mice were ~2-month of age weighing 20–24g at the beginning of the study. The mice were randomly assigned to treatment groups such that there were no significant differences in age, weight or sex. The animals were used in 4 independent in vivo experiments as follows.
Experiment 1
To assess the differential effect of rPTH and SC on blood vessels and bone formation versus placebo, female C57BL/6 mice (n≥4) underwent cranial defect window surgeries, and were subsequently randomized to: 1) placebo (saline), 2) rPTH (20μg/kg/2days, teriparatide obtained from Lilly Inc., Indianapolis, IN), or 3) SC (24μg/kg/2days, catalog number: C0399, Sigma Inc., St. Louis, MO). The treatments were administered by sub-cutaneous injection every other day until euthanasia on day 28 post-op. In vivo micro-CT was performed on days 1, 14 and 28 post-op. In vivo MPLSM was performed on days 15 and 29 post-op.
Experiment 2
To assess the spatiotemporal relationships between mast cell accumulation and vasculogenesis in critical bone defects, female C57BL/6 mice (n=3) underwent cranial defect window surgeries, and in vivo MPLSM was performed on days 1, 4, 6, 10, 17 and 30 post-op.
Experiment 3
To confirm rPTH effects on mast cell numbers via direct fluorescent MPLSM, male and female Mcpt5-Cre-YFP mice (n=4) underwent cranial defect window surgeries, and were subsequently randomized to: 1) placebo (saline), or 2) rPTH (20μg/kg/2days). The treatments were administered by sub-cutaneous injection every other day until euthanasia on day 28 post-op. In vivo MPLSM was performed on days 15 and 29 post-op.
Experiment 4
To confirm mast cell loss of function effects on osteogenesis and vasculogenesis during critical bone defect healing, male and female Mcpt5-Cre-iDTR mice (n=4) underwent cranial defect window surgeries, and were subsequently randomized to: 1) placebo (saline), or 2) DTA (50μg/kg/day, catalog number D0564, Sigma Inc. St. Louis, MO)(27), which were injected intraperoneally every day until euthanasia on day 28 post-op. In vivo micro-CT was performed on days 1 and 28 post-op. In vivo MPLSM was performed on day 29 post-op.
Murine cranial defect window model
The operation was carried out under a ketamine anesthetic and a buprenorphine painkiller. Cranial defect window surgeries were performed on 2 months old mice, as previously detailed(24). Briefly, the mouse under anesthesia was weighed and hair was removed from surgery site of the skull. A motorized stereotaxic instrument (Stoelting Inc., Wood Dale, IL) was used to stabilize mouse head for surgery under dissection microscope. A custom-made 0.5-mm-thick spacer made of poly (aryl-ether-ether-ketone) (PEEK) was glued onto the skull using cyanoacrylate glue (Loctite; Cat #45404, Düsseldorf, Germany). Using a micro drill (Stoelting Inc., Wood Dale, IL), a 1.75mm circular full thickness bone defect was created. The wound was filled with DMEM (with 1% P/S) and sealed with an ultra-thin cover glass using Denture Material Methyl Methacrylate (Co-Oral-Ite Dental MFG. Co., New Franken, WI) to create a window for high resolution imaging.
Longitudinal micro-CT scanning
Mice were sedated with isoflurane for 15min, and then transferred and restrained in a custom-made chamber with a constant flow of isoflurane. The mouse skull was scanned by a Scanco Viva CT40 system (Scanco Medical, Brüttisellen, Switzerland) at a voxel size of 30 microns.
Longitudinal Multiphoton-Laser-Scanning Microscopy (MPLSM)
Blood vessels were visualized with Texas red dextran (Dextran, Texas Red®, 70,000 MW, Neutral, Catalog number: D1830, Thermo Fisher Scientific Inc., Waltham, MA) via retro-orbital injection (2.5mg/ml in PBS, 100μl) administered immediately prior to MPLSM. Indirect immunofluorescent imaging of mast cells was performed 3 hrs after retro-orbital injection of FITC-conjugated anti-Mcpt5 antibody (0.5mg/ml in PBS, 150μl catalog number: bs-2353R-FITC, Bioss Inc., Woburn, MA). Images of Second Harmonic generation (SHG), blood vessels and mast cells were collected on an Olympus FV1000-AOM multiphoton imaging system (Olympus, Tokyo, Japan), equipped with a Titanium:Sapphire laser (Spectra-Physics, Santa Clara, CA), a C-Apochromat 10X/0.45 (Zeiss, Oberkochen, Germany), and a 25X/1.05 (Olympus, Tokyo, Japan), water immersion objective. Images were acquired at 512×512 pixels, 0.2 μs pixel dwell with the laser tuned to 780nm. SHG signals and blood vessel signals were collected using the 4× objective lens (Zeiss, Oberkochen, Germany), and mast cell signals were collected using the 4× lens for number counting and 10× and 25× water immersion objective lens (Zeiss, Oberkochen, Germany) for mast cell identification, a 1.3-mm × 1.3-mm multichannel Z-series stack with 2-μm steps was obtained. The fluorescence of SHG, YFP and Texas Red was collected with a 370/410 nm, a 520/560 nm filter and near red 575/630 nm band pass filter respectively.
3D-reconstruction of SHG, blood vessel images and mast cell images from MPLSM and the images from micro-CT scanning
Amira 6.0 software developed by Visualization Sciences Group (VSG, Burlington, MA), was used for editing and visualization of volumetric data. The z-stack images of SHG, blood vessels and mast cells from MPLSM or the images from micro-CT scanning were visualized by 3D-reconstruction with Amira 6.0 software developed by Visualization Sciences Group (VSG, Burlington MA). The data were imported with resolution 1.2 in voxel size for blood vessel images from MPLSM and 1.0 for bone signal from micro-CT. Blood vessels were segmented using “Label field” in Amira and classified using the “brush” tool in the segmentation editor.
Quantification of blood vessels, bone formation, fibrosis and mast cells
The length and diameter of the blood vessels were quantified based on the two-dimensional images collected following 3D-reconstruction with Amira. The region of interest was drawn based on the size of the defect visualized on the first day of operation. The further segmentation and quantification were performed on 2D images according to the protocols developed by the first author of this work with Visiopharm software 6.0 (Visiopharm Inc., Hoersholm, Denmark). The average blood vessel diameter and blood vessel diameter distribution were generated by drawing cross lines on the segmented blood vessels, while the total length of the blood vessels were measured via skeletonization of the blood vessel. Mast cell number was counted based on the MPLSM signal. The total volume of the defect in which blood vessels and mast cells were quantified was determined by the initial cranial defect (i.e. area A= (PI*(1.75/2)ˆ2) =2.4 mm2, diameter of initial defect = 1.75mm, thickness = 0.5mm). To measure the volume of bone formation, DICOM files from micro-CT were imported into Amira software. The region of interest is defined based on the initial defect size. The new bone volume over a period of 28 days was segmented, classified and quantified. – using a Cylinder transformer in “Volume Edit” in Amira software. The exact same cylinder transformer was used for bone volume evaluation. To calculate the closure (%) of the cranial defect window at day X, closure (%) at day X = (Aday 1 − Aday x)/ Aday 1*100, in which Aday 1 is the initial defect area, and Aday x is the defect area of the same mouse at day X. In a similar way, but using images from the second harmonic generation (SHG) channel of MPLSM at day 28, 3D-reconstruction was performed to quantify fibrosis volume using the initial defect (at day 1) as a reference. The cylinder transformer, which includes both bone and fibrosis defined by SHG signal collected on type-1 collagen, was used to calculate Vf = Vt – Vn, where Vf is volume of fibrosis, Vt is the total volume, and Vn is the new bone volume.
Histology and immunofluorescent imaging of mast cells
Skulls containing the cranial defect window were harvested from the Mcpt5-Cre-YFP mice, and fixed in 4% paraformaldehyde in neutral buffered saline, and processed for decalcified histology on frozen slides. The skull piece with cranial defect window was laid flat and embedded in O.C.T. using standard protocol. The protocol is publicly available on the website of the Center for Musculoskeletal Research, URMC, University of Rochester (https://www.urmc.rochester.edu/MediaLibraries/URMCMedia/musculoskeletal-research/core-services/histology/documents/Preparation_of_Frozen_Specimens.pdf). Cross sections were cut and the frozen slides were stained with Toluidine Blue/Fast Green Stain, protocol available online as well (https://www.urmc.rochester.edu/MediaLibraries/URMCMedia/musculoskeletal-research/core-services/histology/documents/ToluidineBlueStain.pdf). Direct fluorescence and immunofluorescent imaging of the cranial defect window histology was performed with an Olympus IX81 confocal microscope and the bright fields were imaged with a Zeiss axioskop 40 microscope.
Osteoblast and mast cell co-culture, in vitro rPTH stimulation and RNAseq
The osteoblast cell line 7F2(ATCC® CRL-12557™) and the mast cell line MC/9(ATCC® CRL-8306™) were purchased from the American Type Culture Collection (ATCC®, Minneapolis, MN), and grown in vitro according to the manufacture’s protocol. To assess 7F2 and MC/9 cell responses to rPTH in vitro, we assessed cAMP levels and viability as follows. For cAMP assays, 7F2 or MC/9 cells were added to 96-well plates at a density of 104 cells/well (n=4), and cultured overnight in Dulbecco’s Modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO). The media was replaced with DMEM without FBS and incubated for 1hr at 37°C prior to treatment with rPTH at 1, 10, 100nM in DMEM for 10 minutes, and cells were harvested with lysis buffer. cAMP concentration was determined by EIA assay (Cyclic AMP XP™ Assay Kit #4339, Cell Signaling Technology), which was normalized to total protein concentration in cell lysates determined with Pierce™ BCA Protein Assay Kit (Cat. # 23225, Thermo Scientific). To determine mast cell viability in co-culture, 104 7F2 cells were plated in 6-well dishes and incubated for 24hrs at 37°C to establish an osteoblast monolayer. Then 104 MC/9 cells were added to the 7F2 cell monolayer. Then cultures were stimulated with varying doses of rPTH or 100nM SC, incubated for 24hrs at 37°C, and trypan blue staining was performed to quantify the % viable cells via dye exclusion (n=3). To identify rPTH induced genes in co-culture, 6-well co-cultures of 7F2 and MC/9 cells were established and stimulated with or without 10nM rPTH (n=3) for 24hrs as described above. Afterwards, total RNA was extracted, and RNAseq was performed by the University of Rochester Genomic Research Center (Rochester, NY). The genes with p < 0.05 compared with the average factor of all groups were screened out as candidate rPTH-induced genes.
Statistical analyses
Statistical analyses were performed with Prism 7.0 to assess significant differences between groups using one-way ANOVA and Tukey’s multiple comparisons test using two way ANOVA. Pairwise comparisons were performed with Prism 7.0 using a t-test. The data are expressed as the mean ± SEM in which p <0.05 was considered statistically significant.
Results
Pharmacological inhibition of mast cells enhances cranial defect healing and inhibits arteriogenesis
Sodium cromolyn (SC) is a standard of care anti-inflammatory drug commonly used to treat allergic reactions based on its selective inhibitory effects on mast cells.(28,29) Thus, we utilized SC for an initial assessment of mast cell effects on bone healing in the cranial defect window model, compared to placebo and rPTH controls. Longitudinal micro-CT demonstrated that SC significantly increased defect healing 1.4-fold, and new bone volume 1.4-fold, versus placebo by day 28 post-op (Figure 1). However, rPTH treatment was superior, demonstrating significant increases in defect closure and new bone formation versus placebo by day 14, and greater effects versus SC at day 28 post-op (Figure 1). Interestingly, longitudinal assessment of vasculogenesis by MPLSM revealed similar differential effects of rPTH and SC treatment on angiogenesis and arteriogenesis during cranial defect healing (Figure 2). While no differences between groups were observed on day 15 (Fig. 2G), SC significantly decreased small blood vessel numbers, and increased large vessel numbers in the defect versus placebo on day 29 post-op, and greater effects were observed in mice treated with rPTH (Fig. 2H). Remarkably, these changes in small versus large vessel numbers were commensurate in the rPTH treatment group, as no significant differences in total vascular volume were observed versus placebo at any time point. Also of note is that there were no differences in total blood vessel length between the groups at either time point, and that SC significantly decreased total blood vessel volume on day 29 versus placebo. We also assessed rPTH and SC effects on fibrosis within the defect, via MPLSM of images in the SHG channel. While fibrotic tissues could not be clearly discerned at day 15, quantification of the fibrosis volume at day 29 demonstrated that both rPTH and SC significantly decreased this tissue formation (Fig. 1L).
Figure 1. rPTH and SC treatment accelerate cranial defect healing.
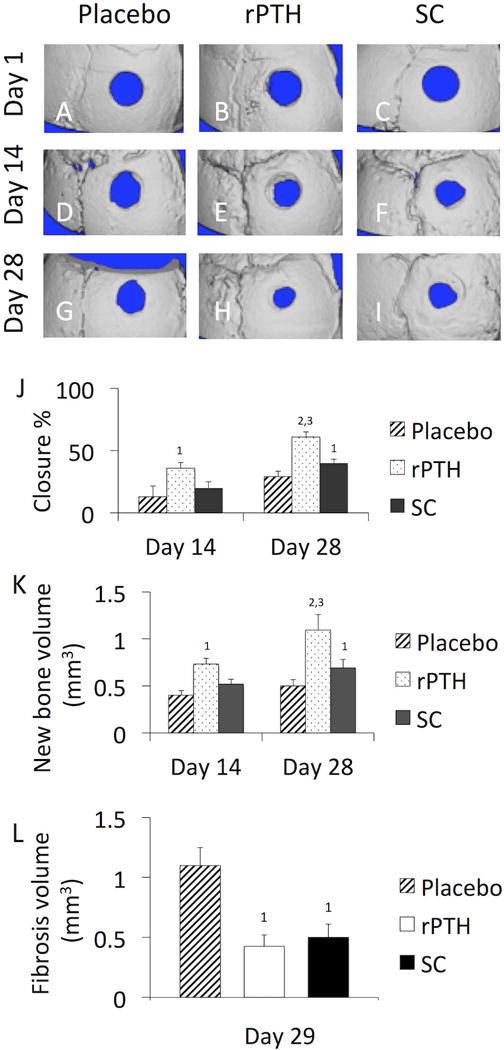
WT female mice (n≥4) underwent cranial window defect surgery, were randomized to Placebo, PTH and SC treatment groups, and micro-CT scans were performed on days 1, 14 and 28 post-op. 3D renderings of the calvariae from a representative mouse in each group are shown to illustrate the relative defect closure rates for the Placebo (A, D, G), rPTH (B, E, H) and SC (C,F,I) treatment groups. The % defect closure (J), new bone volume (K), and the fibrosis volume (L) were quantified via volumetric registration of the 3D images as described in Materials and Methods, and the data are presented as the mean +/− SEM (1p<0.05 vs. Placebo; 2p<0.01 vs. Placebo; 3p<0.05 vs. SC at the same time point).
Figure 2. rPTH and SC treatment inhibits arteriogenesis during cranial defect healing.
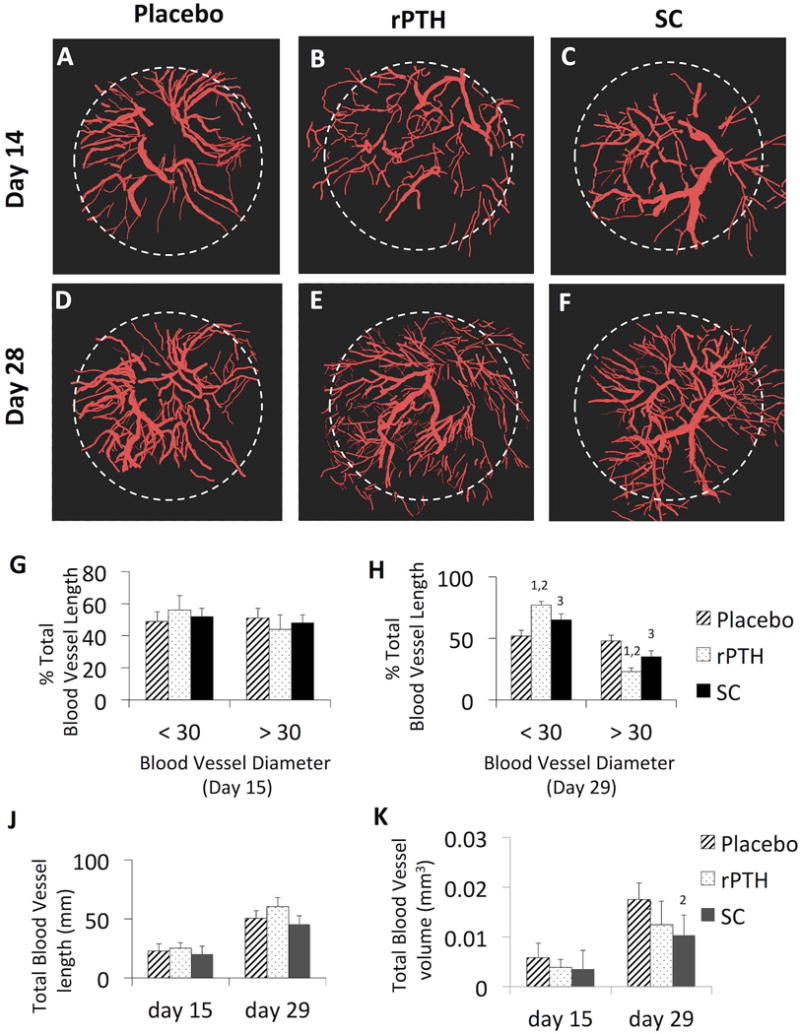
MPLSM was performed on the mice described in Figure 1 immediately after intravenous injection of Texas red dextran on days 15 and 29 post-op. 3D rendered images were generated with Visiopharm software, and representative images of Placebo (A, D), rPTH (B, E) and SC (C, F) treated mice are shown to illustrate the extent of angiogenesis (blood vessels <30μm in diameter) versus arteriogenesis (blood vessels >30μm in diameter) within the defect (white dashed circle). This threshold was used to assess drug effects on arteriogenesis versus arteritogenesis as quantified by the percentage of total blood vessel length on day 15 (G) and day 29 (H) post-op, and total blood vessel length (J) and volume (K) at these time points (data are presented as the mean +/− SEM for the group, 1p<0.01 vs. Placebo; 2p<0.05 vs. Placebo; 3p<0.05 vs. Placebo at the same time point).
Mast cell accumulation at the edge of cranial defects occurs within 24hrs of injury prior to marked angiogenesis at 1 week
To test our hypothesis that large vessel-derived factors recruit, differentiate and/or sustain mast cells within critical defects,(23) we performed longitudinal indirect immunofluorescence MPLSM with FITC-conjugated anti-MCPT5 antibodies (Figure 3). In contrast to our theory, we observed large numbers of mast cells around the edge of the defect within 24hrs of injury (Fig. 3A). We also found that mast cell numbers peaked around 1-week post-op, which was at the commencement of detectable angiogenesis in the defect (Figs. 3C & C’). Arteriogenesis commenced around day 17 post-op (Fig. 3E), and continued throughout the study (Figs. 3 E & F). Thus, mast cell accumulation occurs independent of arteriogenesis, and these results suggest that mast cell may contribute to arteriogenesis during critical defect healing.
Figure 3. Mast cells accumulate at the edge of cranial defects within 24hrs of injury prior to marked angiogenesis at 1 week.
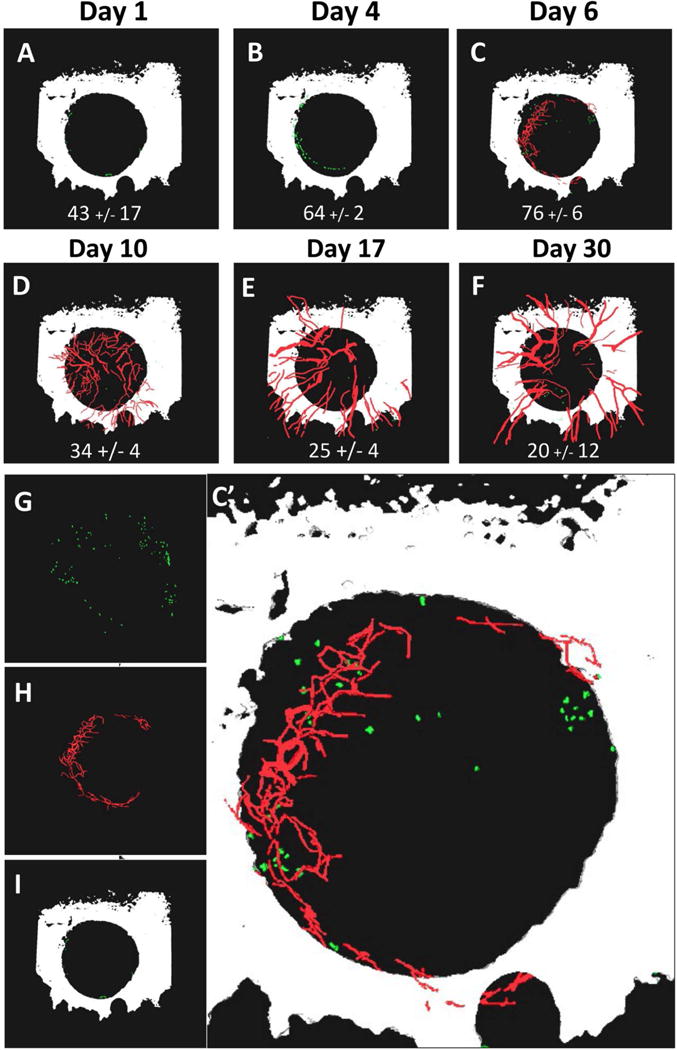
WT female C57BL/6 mice (n=3) underwent cranial defect window surgery, and MPLSM was performed after injection of Texas red dextran and FITC conjugated anti-MCPT5 antibody on days 1, 4, 6, 10, 17 and 30 post-op as described in Materials and Methods. 3D reconstruction of the multiphoton images acquired at 4× was performed with Amria, and images from a representative mouse with the number of mast cells (mean +/− SEM for the cohort) within the defect at each time point is shown (A–F) to illustrate the rapid accumulation of mast cells around the edge of the defect relative to angiogenesis over the course of bone defect healing. Individual renderings of the day 6 image (C) are shown to better illustrate the FITC-labeled mast cells (green in G), Texas red dextran labeled vasculature (red in H), and bone from second harmonic generation (white in I), and combined in a enlarged 3D rendering (C’).
rPTH reduces mast cell numbers within critical defects during bone healing
Previous cross-sectional histology studies have demonstrated that rPTH treatment reduces numbers of mast cells in transitional tissue during the late stages of femoral and calvaria allograft healing, concomitant with decreased arteriogenesis.(8,13) Thus, to test if rPTH inhibition of mast cells occurs prior to arteriogenesis, we aimed to performed longitudinal MPLSM on cranial defect windows generated in mice treated with placebo or rPTH. However, to eliminate potentially confounding effects of anti-Mcpt5 antibodies on mast cells during the experiment, we utilized Mcpt5-Cre-YFP transgenic mice that have restricted yellow fluorescent protein (YFP) expression in their mast cells.(26) To validate the YFP signal as a marker of mast cells in our model, we performed confocal immunohistochemistry on calvaria tissue section from Mcpt5-Cre-YFP mice harvested on day 7 after cranial defect window surgery (Figure 4). The result demonstrated obvious co-localization of cytoplasmic direct fluorescence (YFP) and indirect fluorescence (FITC-conjugated anti-Mcpt5 immunoreactivity) around the nuclei of mast cells. We further validated the YFP signal as a marker of mast cells via bright and dark field histology (Figure 5A–F).
Figure 4. Validation of direct and indirect fluorescent imaging of mast cells in cranial defects.
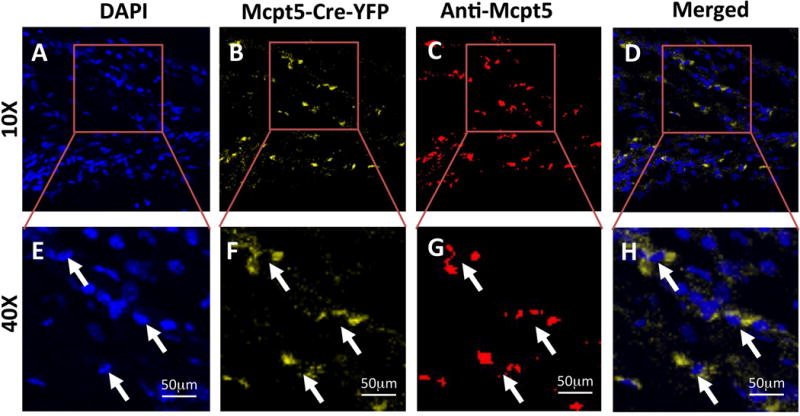
Mcpt5-Cre-YFP mice (n=3) underwent cranial defect window surgery, and the calvariae were harvested on day 7 post-op for immunohistochemistry with FITC-conjugated anti-Mcpt5 antibody, countersated with DAPI. Confocal fluorescent images of a representative tissue section within the defect obtain at 10× (A–D) and 40× (E–H) are shown. The authenticity of the direct fluorescent YFP signal (B,F) and indirect FITC signal (C,G) in the cytoplasm of mast cells (arrows) is apparent based on its localization around the DAPI stained nuclei (A,E), which is further illustrated in merged images of the DAPI and YFP fluorescent signals (D,H).
Figure 5. rPTH treatment decreases mast cell numbers within cranial defects.
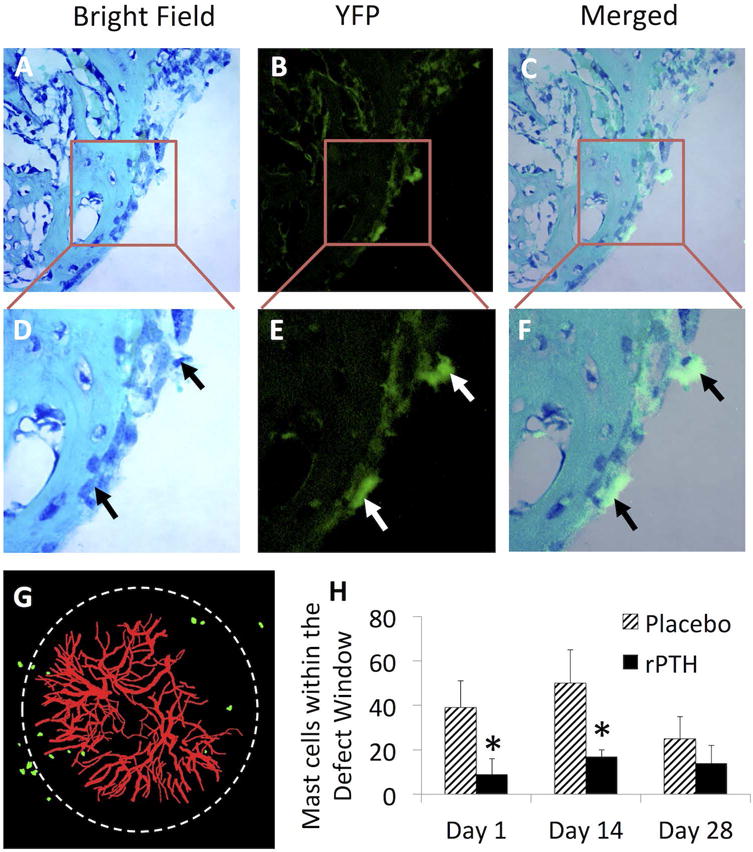
Histology was performed on calvariae from Mcpt5-Cre-YFP transgenic mice (n=4) harvested on day 28 following cranial window surgery to assess co-localization of the YFP signal with mast cells in our model. A representative 40× image of a toluidine blue & fast green stained section of the defect is shown as the bright field (A), dark field fluorescence (B), and merged image (C). The boxed regions in A–D are also shown magnified (D–F) to highlight the two YFP+ mast cells in this field at the edge of the defect (arrows). Mcpt5-Cre-YFP transgenic mice randomized to Placebo and rPTH treatment after cranial window defect surgery, and MPLSM was performed as described in Figure 2. A representative 3D-rendered image from a Placebo treated mouse on day 28 is shown to illustrate the YFP-signal (mast cells), and Texas red dextran labeled blood vessels within the defect (white circle) (G). The number of YFP+ mast cells within the defect window were quantified from the MPLSM images obtained on days 1, 14 and 28 post-op (H), and the data are presented as the mean +/− SEM (*p < 0.05 vs. Placebo at the same time point).
Based on these findings, we utilized direct fluorescence MPLSM to quantify mast cell number in Mcpt5-Cre-YFP mice treated with Placebo or rPTH following cranial defect window surgery (Figs. 5G–H). 3D rendering of the MPLSM images showed a similar number of YFP+ mast cells around the edge of the defect (Fig. 5G) as we observed with indirect MPLSM using anti-Mcpt5-FITC antibodies (Figure 3). This finding validated our indirect fluorescence MPLSM studies, and confirmed that absence of confounding effects of the anti-mast cell antibodies. Additionally, quantitation of mast cell numbers in the defect revealed that rPTH treatment reduced mast cell numbers significantly on days 1 and 14 post-op versus placebo (p < 0.05). However, no difference in mast cell numbers were observed on day 28 post-op, demonstrating that this immunomodulatory effect of rPTH is restricted to the inflammatory phase of bone healing.
Deletion of mast cells enhances critical defect healing and inhibits arteriogenesis
In order to directly demonstrate mast cell loss of function effects on bone healing and vasculogenesis in our cranial defect window model, we utilized Mcpt5-iDTR transgenic mice. DTA or placebo was administered to mice immediately after surgery, and DTA-mediated deletion of mast cells was confirmed by indirect MPLSM using anti-Mcpt5-FITC antibodies (data not shown). Longitudinal micro-CT assessment of bone healing revealed that DTA-deletion of mast cells significantly enhanced defect closure and new bone formation compared to placebo (Figs 6A–F). Additionally, MPLSM of Texas red-dextran labeled blood vessels demonstrated that DTA-deletion of mast cells significantly increased angiogenesis and decreased arteriogenesis and fibrosis versus placebo (Figs. 6G–F). Of note is that these DTA effects on bone formation (Figs. 6E, F), vasculogenesis (Fig. 6I) and fibrosis (Fig. 6J) observed in Mcpt5-iDTR transgenic mice were similar to the SC effects observed in WT mice (Figs 1 & 2).
Figure 6. Deletion of mast cells enhances critical defect healing and inhibits arteriogenesis.
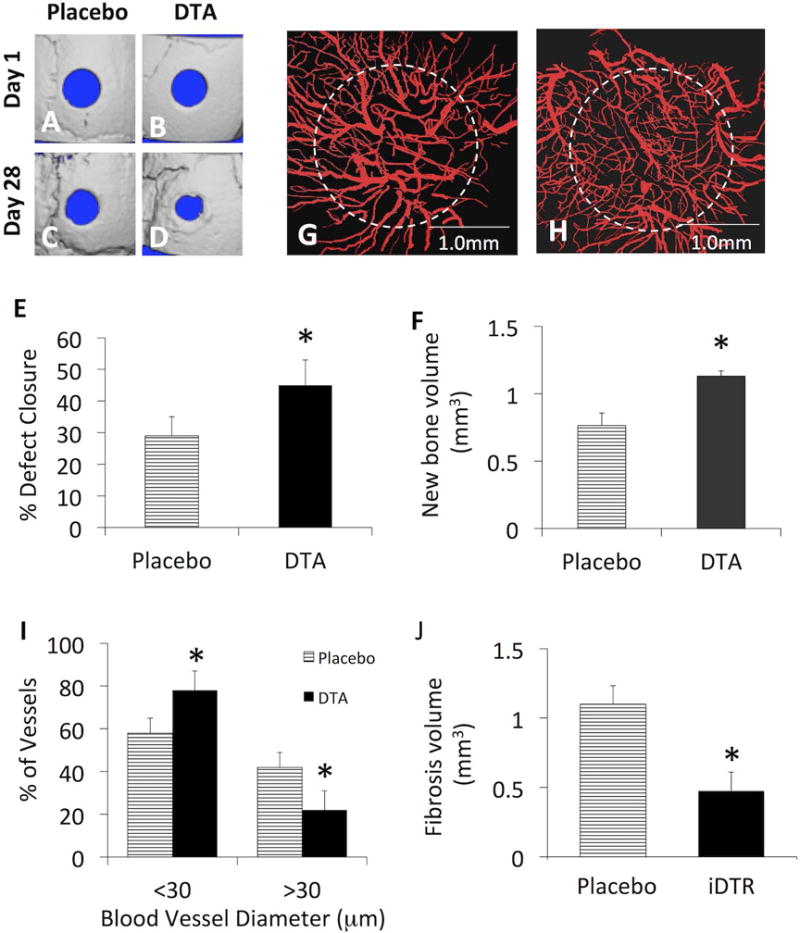
Mcpt5-Cre-YFP mice (n=4) underwent cranial window defect surgery, randomized to Placebo and DTA treatment groups. Micro-CT was performed on days 1 and 28 post-op, and MPLSM was performed on day 29 post-op, as described in Materials and Methods. 3D renderings of the micro-CT scans (A–D) and confocal fluorescent images obtained at 4× (G & H) from representative mice in each group are shown to illustrate the effects of mast cell depletion on bone healing and arteriogenesis respectively. The % defect closure (E), new bone volume (F), % of small vs. large blood vessels in the defect (I), and the fibrosis volume (J), were measured, and the data are presented as the mean +/− SEM (*p < 0.05 vs. Placebo).
rPTH inhibits mast cells indirectly through osteoblasts
To the end of elucidating the mechanism by which rPTH inhibits mast cell-mediated arteriogenesis and fibrosis during critical bone defect healing, we preformed studies to assess PTH1 receptor (PTH1R) expression in mast cells, to determine if rPTH action is direct or indirect through accessory cells. Initially, we performed quantitative real time RT-PCR studies on 7F2 and MC/9 cells, which demonstrated pth1r expression in osteoblasts and undetectable mRNA levels in mast cells (data not shown). To confirm this, we also performed an in silico analysis of transcriptome arrays in the NCBI database, which also demonstrated the absence of detectable pth1r transcripts in mast cells (data not shown). Next, we performed immunohistochemistry on murine calvaria cranial window tissues to assess PTH1R protein expression on osteoblasts and mast cells in our model. The results showed positive staining of bone lining osteoblasts, and no immunoreactivity against toluidine blue staining mast cells (Figs 7 A–F).
Figure 7. rPTH inhibits mast cells indirectly via osteoblasts.
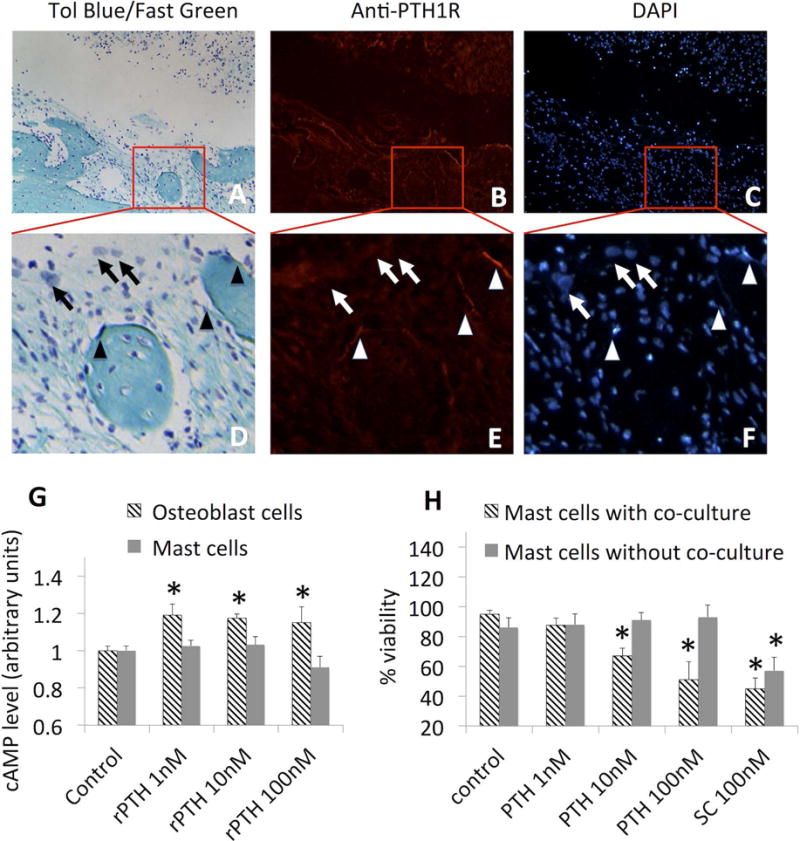
Cranial window surgery was performed on WT mice (n=4), and the calvariae were harvested on day (28) for histology and immunohistochemistry. Representative micrographs obtained at 20× (A–C); and 40× (D–F) of Toluidine Blue/Fast Green (A, D), immunohistochemistry with FITC-conjugated anti-PTH1R antibody (B,E), and DAPI (C, F) staining histology are shown to illustrate that osteoblasts (arrowheads) and not mast cells (arrows) express PTH1R. Individual cultures of 7F2 (osteoblast cells) or MC/9 (mast cells) were stimulated with the indicated dose of rPTH for 10min, then harvested for cAMP ELISA as described in methods. Data are presented as the mean+/− SEM normalized to the no-rPTH control, whose values are arbitrarily set to 1.0 (G; *p<0.05 increase vs control). MC/9 mast cells were cultured in the presence or absence of 7F2 osteoblasts, stimulated with the indicated dose of rPTH or SC for 24 hours, and cell viability was determined by trypan blue staining (H). Data are presented as mean +/− SEM (*p<0.05 vs control).
We also tested direct and indirect effects of rPTH treatment of mast cells using in vitro functional studies. Initially, we assessed rPTH dose-dependent induction of cAMP levels in 7F2 and MC/9 cells via EIA. While the results demonstrated that all doses of rPTH induced cAMP levels in osteoblastic 7F2 cells, cAMP induction was not observed in the mast cell line at any dose (Fig 7G). To assess direct and indirect effects of rPTH on mast cell viability, we treated MC/9 cells alone, or in co-cultures with 7F2 cells, and performed live/dead trypan blue staining at 24hrs compared to SC control. The results showed that decreased mast cell viability was only observed in co-cultures stimulated with ≥10nM rPTH, while SC treatment directly reduced mast cell viability (Fig 7H). Taken together, these findings demonstrate that rPTH effects on mast cells are indirect, and rPTH stimulation of osteoblastic cells induces anti-mast cell factor(s).
As a critical future direction is elucidation of the indirect anti-mast cell effects of rPTH treatment, we performed a preliminary RNAseq analysis on our 7F2 and MC/9 co-cultures. As there are already several published rPTH-induced osteoblast transciptomes in the literature, we focused our analysis on RNA isolated primarily from the non-adherent MC/9 cells in the co-culture. The initial results identified 11 candidate mast cell genes that were expressed at significantly different levels in rPTH treated co-cultures (Table 1). These genes could be divided into four groups: 1) genes related to fibrosis, including Igsf10, Ifi44, Pde8a; 2) genes related to apoptosis, including Cecr2; 3) genes related to blood vessel formation, including Thbs2 and Gm8692; and 4) the remaining genes whose functions are currently unknown. Once the induction of these genes are confirm, functional studies can be performed to assess their roles in bone repair.
Table 1.
rPTH-induced genes in mast cell and osteoblast co-cultures.
| Gene name | Description |
|---|---|
| Igsf10a | Immunoglobulin superfamily, member 10. Fibrosis related(58). |
| Ifi44a | Interferon-induced protein 4, a putative biomarker of fibrosis(59,60). |
| Cecr2b | Histone acetyl-lysine reader, apoptosis and cellular component disassembly(61). |
| Pde8aa | Phosphodiesterase 8A, physical interaction with cystic fibrosis (CF) transmembrane conductance regulator (CFTR)(62). |
| Olfr56a | Olfactory receptor 56. |
| Thbs2a | Thrombospondin 2, regulation of bone collagen ultrastructure (63,64), regulation on angiogenic cell function(65). |
| Gm7079b | Predicted pseudogene 7079. |
| Gm11694b | Predicted gene 11694. |
| Gm8692b | Predicted gene 8692, may be related to microvasculature(66). |
| Gm20302b | Predicted gene 20302. |
| Gm16106b | Predicted gene 16106. |
The 11 mast cell genes that were significantly up-regulated
or down regulated
>2-fold in the RNAseq analysis of rPTH treated vs. untreated MC/9 mast cell and 7F2 osteoblast co-cultures are presented. The GEO accession numbers for the primary data sets (GSE98510 RNAseq in mast cells) are: GSM2597688, GSM2597689, GSM2597690, GSM2597691, GSM2597692 and GSM2597693.
Discussion
In contrast to epimorphic regeneration and scar-free healing that only occurs in special tissues (e.g. reindeer antlers) and settings (skin injury in utero), tissue repair following traumatic injury in adult mammals occurs following a robust inflammatory cascade that leads to imperfect healing as a result of fibrotic scaring.(30,31) Ironically, in the case of bone repair, the inflammatory phase that commences immediately after fracture, which leads to fibrosis in non-unions and critical defects, is required for the proliferative and remodeling phases of normal healing.(32) Thus, elucidation of targetable factors uniquely involved in fibrotic scaring is critical towards development of interventions for challenging bone injuries, and reconstruction of large segmental defects. To this end, we have evaluated the biomechanical, immunological, and vascular differences between the highly efficient “scarless” healing observed in live structural autografts, versus the “scarful” poor/defective healing observed with necrotic intercalary allografts.(8,10,13,23,33–40) The results from these studies displayed several global differences that remain a focus of current research. At the skeletal level, it is now clear that the remarkable bridging bone that spans the entire length of autografts and live isografts is driven by donor osteoprogenitor cells on the surface of the graft, which directly contribute to intramembranous and endochondral ossification, and that their absence in allografts leads to fibrosis.(34–37) Immunologically, it was found that the extensive transitional-inflammatory tissue that surrounds allografts in the proliferation phase, which remodels into fibrous non-unions at the graft-host junctions, is primarily due to an innate immune response, as processed isografts from genetically identical littermates provokes the same response as allografts, as does allograft bone implanted into immunocompromised mice that lack B-cells and T-cells.(33,39,40) Vasculogenesis is also remarkably distinct, as autografts heal via perpetuating angiogenesis of small vessels, while allografts induce angiopoietin-2 and arteriogenesis of large vessels.(13,23,34) Although the mechanism by which these large vessels contribute to scarful healing of allografts is unknown, the finding that they are elaborated with mast cells suggested that they trigger the pro-inflammatory cascade that leads to fibrotic non-unions.(8,13,23)
Two parallel fields of tissue regeneration research that have also identified critical roles of inflammation, hypoxia, angiogenesis and arteriogenesis in the repair process are hind limb ischemia(41) and distraction osteogenesis.(42) Data from animal models have shown that the response to hind limb ischemia is a complex process regulated by inflammatory cells and chemokines that induce arteriogenesis in tissues with normal perfusion, and angiogenesis in necrotic areas undergo skeletal muscle regeneration.(43–45) In a model of distraction osteogenesis, Morgan et al demonstrated that intense osteogenesis is concurrent with angiogenesis in the intraosteal region during consolidation.(46) In contrast, the period of active distraction is characterized primarily by arteriogenesis in the surrounding muscle.(46) Most recently, Bragdon et al used an ectopic bone formation model to demonstrate that angiogenesis plays a developmental role in endochondral bone formation prior to chondrogenesis.(47) Collectively, these findings are consistent with our model of angiogenesis in scarless healing of non-critical defects, and arteriogenesis in adjacent tissue to support the new vasculature in the regenerate. However, in the setting of a critical defect where the regenerate cannot keep pace with vasculogenesis, mast cells promote arteriogenesis and scarful healing.
Given the major challenges in treating fracture non-unions and critical bone defects, the experimental and clinical evidence supporting the use of rPTH as a potential adjuvant therapy has been met with great enthusiasm.(4–12) While the original rationale for this treatment was based on its well-established effects on bone formation, it is now clear that rPTH has several non-anabolic effects that can transition scarful allograft healing towards the scarless healing observed in autografts.(2,23) Most notably, rPTH inhibits arteriogenesis, mast cells and fibrosis during structural allograft healing.(8,13) However, the functional significance of these rPTH non-anabolic effects on bone healing, and the spatiotemporal relationship between mast cells and arteriogenesis during critical bone defect healing are poorly understood. To address this, we adopted the murine cranial defect window model, which allows for longitudinal micro-CT and intravital microscopy, to assess osteogenesis and vasculogenesis during bone healing.(24) Consistent with a functional role of mast cells in the inhibition of bone formation and the promotion of arteriogenesis, we found that SC treatment significantly increased the bone defect closure rate and new bone formation, and inhibited arteriogenesis and fibrosis (Figures 1 & 2). We also confirmed these findings via direct ablation of mast cells (Figure 6). Thus, we conclude that mast cells are initiators of the pro-fibrotic cascade in critical defects. Interestingly, and in contrast to our initial hypothesis, mast cell accumulation occurs immediately after injury, and at least two weeks before arteriogenesis (Figure 3). Therefore, large vascular smooth muscle containing vessels are not required for recruitment, differentiation and/or retention of mast cells in critical bone defects as we previously proposed.(23) Moreover, these finding suggest that mast cells may be a source of the vascular endothelial growth factors and angiopoietin-2 that drives angiogenesis and subsequent arteriogenesis, as has been reported in other models.(48,49) Thus, formally testing this, and elucidating the mechanisms responsible for mast cell accumulation in critical defects remain important areas for future investigation.
By comparing the efficacy of SC versus rPTH treatment in the cranial defect model we also confirmed that teriparatide has both anabolic and non-anabolic effects on bone healing. We interpret the increased bone healing above that observed with SC (Figure 1) to be the anabolic drug effects on both mesenchymal progenitors and bone forming osteoblasts.(50–53) Also of note is that preliminary results with combination rPTH and SC treatment failed to demonstrate any additional effects over rPTH alone on bone healing in the cranial defect window model (P = 0.072 for % closure and P = 0.13 for bone volume on day 28; n=5; data not shown). Thus, although SC had modest inhibitory effects on arteriogenesis over rPTH (Fig. 2K), presumably due to its specific effects on mast cell granule stabilization, the findings that rPTH directly reduces mast cell numbers (Figure 5), suggests that its major non-anabolic effects are secondary to the absence of these potent mediators of inflammation and fibrosis.
Based on this new information, we propose a revised model of scarful versus rPTH-mediated scarless healing of critical bone defects (Figure 8). In the native host response to injury, mast cells immediately accumulate around the edge of the defect and release pro-inflammatory and pro-fibrotic factors (i.e. TGF-beta, FGF, PDGF and GM-CSF) that stimulate the differentiation of mesenchymal progenitor cells into fibroblasts. The mast cells also produce chymases (i.e. Mcpt5), which cleave the latent form of TGF-beta from cell membranes,(54) and drives collagen production in the fibroblasts. This also stimulates fibroblast differentiation into myofibroblasts.(54–57) Finally, the extended hypoxia in the center of the defect leads to angiopoietin-2 expression, from a yet to be identified source, which remodels newly formed capillaries into large vascular smooth muscle containing blood vessels. Although we show here that these large vessels are not required for initial mast cell accumulation following injury (Figure 3), our prior observation that mast cells reside in immediate proximity to large vessels during the late stages of critical defect healing suggests that this complex perpetuates fibrosis in the center of the defect,(23) and warrants further investigation. In contrast to scarful healing, the inflammatory phase of rPTH-induced scarless healing is characterized by small numbers of mast cells (Figure 5), and reciprocal expression of angiopoietins 1 & 2.(13) Consequently, rPTH significantly increases angiogenesis (Figure 2), which would improve new bone formation (46). In contrast, perivascular mast cells play a central role in regulating the shear stress-induced arteriogenesis by orchestrating leukocyte function and growth factor/cytokine release. This leads to a vicious cycle of mast cell recruitment and activation, and stimulation of arteriogenesis via recruitment of neutrophils, monocytes and T cells.(49)
Figure 8. Working model of native Scarful Healing versus rPTH-induced Scareless Healing of bone defects.
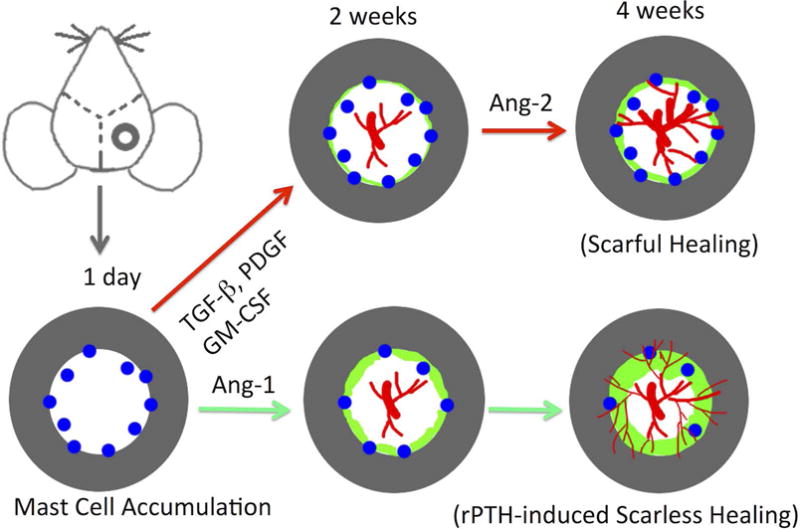
The schematic illustration of the cranial defect in a mouse is shown, highlighting the mast cells (blue) that accumulate proximal to the edge of bone defects immediately after injury, reaching peak numbers within 24hrs such that angiogenesis (red) within the defect occurs over the next two weeks in the presence of large numbers of these pro-inflammatory/pro-fibrotic cells. The model proposes that this accumulation and activation of mast cells consequently promotes arteriogenesis and fibrosis, and concomitantly inhibits new bone formation (green) in a “scarful healing” process. Intermittent rPTH treatment enhances defect healing via its established direct anabolic effects on osteoprogenitor cells and osteoblasts; and via inhibition of mast cells, which results in increased angiogenesis and decreased arteriogenesis and fibrosis, to achieve a “scarless healing” process.
Finally, although SC is broadly used in a variety of patients, we are unaware of any clinical studies that have specifically evaluated the effects of mast cell inhibition bone healing. Thus, future pre-clinical and clinical studies are needed to assess the translational potential of targeting mast cells as an intervention for non-unions and critical bone defect.
Acknowledgments
The authors would like to thank Michael Thullen for technical assistance with micro-CT, and Sarah Mack and Karen de Mesy Bentley for technical assistance with histology. This research was funded by grants (R01 DE019902, R01AR067859, R21DE026256, P30 AR069655, P50AR054041, 1S10RR027340, and T32AR53459) from the National Institutes of Health.
Grant sponsors: National Institutes of Health, Grant numbers: R01 DE019902, R01AR067859 R21DE026256, P30 AR069655, P50AR054041, 1S10RR027340, and T32AR53459.
Footnotes
Conflict of interest: The teriparatide used in this study was a gift from Lilly Inc.
References
- 1.Molina CS, Stinner DJ, Obremskey WT. Treatment of Traumatic Segmental Long-Bone Defects: A Critical Analysis Review. JBJS Rev. 2014;2(4) doi: 10.2106/JBJS.RVW.M.00062. [DOI] [PubMed] [Google Scholar]
- 2.Dhillon RS, Schwarz EM. Teriparatide Therapy as an Adjuvant for Tissue Engineering and Integration of Biomaterials. J Mater Res. 2011;4(6):1117–31. doi: 10.3390/ma4061117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434–41. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 4.Aspenberg P, Genant HK, Johansson T, et al. Teriparatide for acceleration of fracture repair in humans: a prospective, randomized, double-blind study of 102 postmenopausal women with distal radial fractures. J Bone Miner Res. 2009;25(2):404–14. doi: 10.1359/jbmr.090731. [DOI] [PubMed] [Google Scholar]
- 5.Aspenberg P, Johansson T. Teriparatide improves early callus formation in distal radial fractures. Acta Orthop. 2010;81(2):234–6. doi: 10.3109/17453671003761946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peichl P, Holzer LA, Maier R, Holzer G. Parathyroid hormone 1–84 accelerates fracture-healing in pubic bones of elderly osteoporotic women. J Bone Joint Surg Am. 2011;93(17):1583–7. doi: 10.2106/JBJS.J.01379. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds DG, Takahata M, Lerner AL, O’Keefe RJ, Schwarz EM, Awad HA. Teriparatide therapy enhances devitalized femoral allograft osseointegration and biomechanics in a murine model. Bone. 2010;48(3):562–70. doi: 10.1016/j.bone.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheyn D, Cohn Yakubovich D, Kallai I, et al. PTH Promotes Allograft Integration in a Calvarial Bone Defect. Mol Pharm. 2013;10(12):4462–71. doi: 10.1021/mp400292p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheyn D, Shapiro G, Tawackoli W, et al. PTH Induces Systemically Administered Mesenchymal Stem Cells to Migrate to and Regenerate Spine Injuries. Mol Ther. 2016;24(2):318–30. doi: 10.1038/mt.2015.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynolds DG, Shaikh S, Papuga MO, et al. muCT-based measurement of cortical bone graft-to-host union. J Bone Miner Res. 2009;24(5):899–907. doi: 10.1359/JBMR.081232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bashutski JD, Eber RM, Kinney JS, et al. Teriparatide and osseous regeneration in the oral cavity. N Engl J Med. 2010;363(25):2396–405. doi: 10.1056/NEJMoa1005361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubery PT, Bukata SV. Teriparatide may accelerate healing in delayed unions of type III odontoid fractures: a report of 3 cases. J Spinal Disord Tech. 2010;23(2):151–5. doi: 10.1097/BSD.0b013e31819a8b7a. [DOI] [PubMed] [Google Scholar]
- 13.Dhillon RS, Xie C, Tyler W, et al. PTH-enhanced structural allograft healing is associated with decreased angiopoietin-2-mediated arteriogenesis, mast cell accumulation, and fibrosis. J Bone Miner Res. 2012;28(3):586–97. doi: 10.1002/jbmr.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindholm R, Lindholm S, Liukko P. Fracture healing and mast cells. I. The periosteal callus in rats. Acta Orthop Scand. 1967;38(2):115–22. doi: 10.3109/17453676708989624. [DOI] [PubMed] [Google Scholar]
- 15.Banovac K, Renfree K, Makowski AL, Latta LL, Altman RD. Fracture healing and mast cells. J Orthop Trauma. 1995;9(6):482–90. doi: 10.1097/00005131-199509060-00005. [DOI] [PubMed] [Google Scholar]
- 16.Puxeddu I, Piliponsky AM, Bachelet I, Levi-Schaffer F. Mast cells in allergy and beyond. Int J Biochem Cell Biol. 2003;35(12):1601–7. doi: 10.1016/s1357-2725(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 17.Wygrecka M, Dahal BK, Kosanovic D, et al. Mast cells and fibroblasts work in concert to aggravate pulmonary fibrosis: role of transmembrane SCF and the PAR-2/PKC-alpha/Raf-1/p44/42 signaling pathway. Am J Pathol. 2013;182(6):2094–108. doi: 10.1016/j.ajpath.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Holdsworth SR, Summers SA. Role of mast cells in progressive renal diseases. J Am Soc Nephrol. 2008;19(12):2254–61. doi: 10.1681/ASN.2008010015. [DOI] [PubMed] [Google Scholar]
- 19.Pearson ME, Huff JC, Giorno RC, Panicheewa S, Claman HN, Steigerwald JC. Immunologic dysfunction in scleroderma: evidence for increased mast cell releasability and HLA-DR positivity in the dermis. Arthritis Rheum. 1988;31(5):672–7. doi: 10.1002/art.1780310514. [DOI] [PubMed] [Google Scholar]
- 20.Scott A, Lian O, Bahr R, Hart DA, Duronio V, Khan KM. Increased mast cell numbers in human patellar tendinosis: correlation with symptom duration and vascular hyperplasia. Br J Sports Med. 2008;42(9):753–7. doi: 10.1136/bjsm.2007.040212. [DOI] [PubMed] [Google Scholar]
- 21.Yokota M, Suzuki K, Tokoyoda K, et al. Roles of mast cells in the pathogenesis of inflammatory myopathy. Arthritis Res Ther. 2014;16(2):R72. doi: 10.1186/ar4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behzad H, Sharma A, Mousavizadeh R, Lu A, Scott A. Mast cells exert pro-inflammatory effects of relevance to the pathophyisology of tendinopathy. Arthritis Res Ther. 2014;15(6):R184. doi: 10.1186/ar4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antebi B, Zhang L, Sheyn D, et al. Controlling Arteriogenesis and Mast Cells Are Central to Bioengineering Solutions for Critical Bone Defect Repair Using Allografts. Bioengineering (Basel) 2016;3(1) doi: 10.3390/bioengineering3010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang C, Ness VP, Yang X, et al. Spatiotemporal Analyses of Osteogenesis and Angiogenesis via Intravital Imaging in Cranial Bone Defect Repair. J Bone Miner Res. 2015;30(7):1217–30. doi: 10.1002/jbmr.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ribatti D, Crivellato E. Mast cells, angiogenesis, and tumour growth. Biochim Biophys Acta. 2012;1822(1):2–8. doi: 10.1016/j.bbadis.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 26.Scholten J, Hartmann K, Gerbaulet A, et al. Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res. 2008;17(2):307–15. doi: 10.1007/s11248-007-9153-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saito M, Iwawaki T, Taya C, et al. Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat Biotechnol. 2001;19(8):746–50. doi: 10.1038/90795. [DOI] [PubMed] [Google Scholar]
- 28.Ashmole I, Bradding P. Ion channels regulating mast cell biology. Clin Exp Allergy. 2013;43(5):491–502. doi: 10.1111/cea.12043. [DOI] [PubMed] [Google Scholar]
- 29.Kennedy LL, Hargrove LA, Graf AB, et al. Inhibition of mast cell-derived histamine secretion by cromolyn sodium treatment decreases biliary hyperplasia in cholestatic rodents. Lab Invest. 2014;94(12):1406–18. doi: 10.1038/labinvest.2014.129. [DOI] [PubMed] [Google Scholar]
- 30.Ferguson MW, O’Kane S. Scar-free healing: from embryonic mechanisms to adult therapeutic intervention. Philos Trans R Soc Lond B Biol Sci. 2004;359(1445):839–50. doi: 10.1098/rstb.2004.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ud-Din S, Volk SW, Bayat A. Regenerative healing, scar-free healing and scar formation across the species: current concepts and future perspectives. Exp Dermatol. 2014;23(9):615–9. doi: 10.1111/exd.12457. [DOI] [PubMed] [Google Scholar]
- 32.Einhorn TA, Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nature reviews Rheumatology. 2015;11(1):45–54. doi: 10.1038/nrrheum.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tiyapatanaputi P, Rubery PT, Carmouche J, Schwarz EM, O’Keefe RJ, Zhang X. A novel murine segmental femoral graft model. J Orthop Res. 2004;22(6):1254–60. doi: 10.1016/j.orthres.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Xie C, Lin AS, et al. Periosteal progenitor cell fate in segmental cortical bone graft transplantations: implications for functional tissue engineering. J Bone Miner Res. 2005;20(12):2124–37. doi: 10.1359/JBMR.050806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Long T, Zhu Z, Awad HA, Schwarz EM, Hilton MJ, Dong Y. The effect of mesenchymal stem cell sheets on structural allograft healing of critical sized femoral defects in mice. Biomaterials. 2014;35(9):2752–9. doi: 10.1016/j.biomaterials.2013.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie C, Reynolds D, Awad H, et al. Structural bone allograft combined with genetically engineered mesenchymal stem cells as a novel platform for bone tissue engineering. Tissue Eng. 2007;13(3):435–45. doi: 10.1089/ten.2006.0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie C, Xue M, Wang Q, Schwarz EM, O’Keefe RJ, Zhang X. Tamoxifen-inducible CreER-mediated gene targeting in periosteum via bone-graft transplantation. J Bone Joint Surg Am. 2008;90(Suppl 1):9–13. doi: 10.2106/JBJS.G.01212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reynolds DG, Hock C, Shaikh S, et al. Micro-computed tomography prediction of biomechanical strength in murine structural bone grafts. J Biomech. 2007;40(14):3178–86. doi: 10.1016/j.jbiomech.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Ito H, Koefoed M, Tiyapatanaputi P, et al. Remodeling of cortical bone allografts mediated by adherent rAAV-RANKL and VEGF gene therapy. Nat Med. 2005;11(3):291–7. doi: 10.1038/nm1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Awad HA, O’Keefe RJ, Guldberg RE, Schwarz EM. A perspective: engineering periosteum for structural bone graft healing. Clin Orthop Relat Res. 2008;466(8):1777–87. doi: 10.1007/s11999-008-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shireman PK. The chemokine system in arteriogenesis and hind limb ischemia. J Vasc Surg. 2007;45(Suppl A):A48–56. doi: 10.1016/j.jvs.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carvalho RS, Einhorn TA, Lehmann W, et al. The role of angiogenesis in a murine tibial model of distraction osteogenesis. Bone. 2004;34(5):849–61. doi: 10.1016/j.bone.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 43.Tang GL, Chang DS, Sarkar R, Wang R, Messina LM. The effect of gradual or acute arterial occlusion on skeletal muscle blood flow, arteriogenesis, and inflammation in rat hindlimb ischemia. J Vasc Surg. 2005;41(2):312–20. doi: 10.1016/j.jvs.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 44.Scholz D, Ziegelhoeffer T, Helisch A, et al. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol. 2002;34(7):775–87. doi: 10.1006/jmcc.2002.2013. [DOI] [PubMed] [Google Scholar]
- 45.Heil M, Eitenmuller I, Schmitz-Rixen T, Schaper W. Arteriogenesis versus angiogenesis: similarities and differences. J Cell Mol Med. 2006;10(1):45–55. doi: 10.1111/j.1582-4934.2006.tb00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morgan EF, Hussein AI, Al-Awadhi BA, et al. Vascular development during distraction osteogenesis proceeds by sequential intramuscular arteriogenesis followed by intraosteal angiogenesis. Bone. 2012;51(3):535–45. doi: 10.1016/j.bone.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bragdon B, Lam S, Aly S, et al. Earliest phases of chondrogenesis are dependent upon angiogenesis during ectopic bone formation in mice. Bone. 2017 doi: 10.1016/j.bone.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Norrby K. Mast cells and angiogenesis. APMIS. 2002;110(5):355–71. doi: 10.1034/j.1600-0463.2002.100501.x. [DOI] [PubMed] [Google Scholar]
- 49.Chillo O, Kleinert EC, Lautz T, et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016;16(8):2197–207. doi: 10.1016/j.celrep.2016.07.040. [DOI] [PubMed] [Google Scholar]
- 50.Ohishi M, Schipani E. PTH and stem cells. J Endocrinol Invest. 2011;34(7):552–6. doi: 10.3275/7620. [DOI] [PubMed] [Google Scholar]
- 51.Choudhary S, Huang H, Raisz L, Pilbeam C. Anabolic effects of PTH in cyclooxygenase-2 knockout osteoblasts in vitro. Biochem Biophys Res Commun. 2008;372(4):536–41. doi: 10.1016/j.bbrc.2008.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu B, Zhao X, Yang C, et al. Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. J Bone Miner Res. 2012;27(9):2001–14. doi: 10.1002/jbmr.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calvi LM, Sims NA, Hunzelman JL, et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107(3):277–86. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem. 1995;270(9):4689–96. doi: 10.1074/jbc.270.9.4689. [DOI] [PubMed] [Google Scholar]
- 55.Hugle T. Beyond allergy: the role of mast cells in fibrosis. Swiss Med Wkly. 2014;144:w13999. doi: 10.4414/smw.2014.13999. [DOI] [PubMed] [Google Scholar]
- 56.Gailit J, Marchese MJ, Kew RR, Gruber BL. The differentiation and function of myofibroblasts is regulated by mast cell mediators. J Invest Dermatol. 2001;117(5):1113–9. doi: 10.1046/j.1523-1747.2001.15211.x. [DOI] [PubMed] [Google Scholar]
- 57.Veerappan A, O’Connor NJ, Brazin J, et al. Mast cells: a pivotal role in pulmonary fibrosis. DNA Cell Biol. 2013;32(4):206–18. doi: 10.1089/dna.2013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Segev O, Samach A, Faerman A, et al. CMF608-a novel mechanical strain-induced bone-specific protein expressed in early osteochondroprogenitor cells. Bone. 2004;34(2):246–60. doi: 10.1016/j.bone.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 59.Farina G, Lafyatis D, Lemaire R, Lafyatis R. A four-gene biomarker predicts skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2010;62(2):580–8. doi: 10.1002/art.27220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mahoney JM, Taroni J, Martyanov V, et al. Systems Level Analysis of Systemic Sclerosis Shows a Network of Immune and Profibrotic Pathways Connected with Genetic Polymorphisms. Plos Comput Biol. 2015;11(1) doi: 10.1371/journal.pcbi.1004005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu M, Xiang F, Beyer RP, Farin FM, Bammler TK, Chin MT. Transcription Factor CHF1/Hey2 Regulates Specific Pathways in Serum Stimulated Primary Cardiac Myocytes: Implications for Cardiac Hypertrophy. Curr Genomics. 2010;11(4):287–96. doi: 10.2174/138920210791233117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang X, Venable J, LaPointe P, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127(4):803–15. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 63.Manley E, Jr, Perosky JE, Khoury BM, Reddy AB, Kozloff KM, Alford AI. Thrombospondin-2 deficiency in growing mice alters bone collagen ultrastructure and leads to a brittle bone phenotype. J Appl Physiol (1985) 2015;119(8):872–81. doi: 10.1152/japplphysiol.00340.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alford AI, Golicz AZ, Cathey AL, Reddy AB. Thrombospondin-2 facilitates assembly of a type-I collagen-rich matrix in marrow stromal cells undergoing osteoblastic differentiation. Connect Tissue Res. 2013;54(4–5):275–82. doi: 10.3109/03008207.2013.811236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bae ON, Wang JM, Baek SH, Wang Q, Yuan H, Chen AF. Oxidative stress-mediated thrombospondin-2 upregulation impairs bone marrow-derived angiogenic cell function in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2013;33(8):1920–7. doi: 10.1161/ATVBAHA.113.301609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yan L, Chen X, Talati M, et al. Bone Marrow-derived Cells Contribute to the Pathogenesis of Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2016;193(8):898–909. doi: 10.1164/rccm.201502-0407OC. [DOI] [PMC free article] [PubMed] [Google Scholar]