

Abstract
Although the BTK inhibitor ibrutinib has transformed the management of patients with CLL, it does not induce substantial apoptosis in vitro, and as such the mechanisms underlying its ability to kill CLL cells are not well understood. Acalabrutinib, a more specific BTK inhibitor now in development, also appears to be highly effective in CLL, but the connection of its mechanism with CLL cell death is also unclear. Using dynamic BH3 profiling, we analyzed alterations in the function of the mitochondrial apoptotic pathway induced by ibrutinib and acalabrutinib. We studied CLL patient samples treated ex vivo with both drugs, as well as primary samples from CLL patients on clinical trials of both drugs. We found that BTK inhibition enhances mitochondrial BCL-2 dependence without significantly altering overall mitochondrial priming. Enhancement of BCL-2 dependence was accompanied by an increase in the pro-apoptotic protein BIM. In contrast, treatment with the selective BCL-2 inhibitor venetoclax enhanced overall mitochondrial priming without increasing BCL-2 dependence. Pre-treatment of CLL cells with either BTK inhibitor, whether ex vivo or in vivo in patients, enhanced killing by venetoclax. Our data suggest that BTK inhibition enhances mitochondrial BCL2 dependence, supporting the ongoing development of clinical trials combining BTK and BCL-2 inhibition.
Keywords: BTK, BCL-2, ibrutinib, acalabrutinib (ACP-196), venetoclax, BH3 profiling, mitochondrial priming
Introduction
The treatment of chronic lymphocytic leukemia (CLL) has recently been transformed by novel oral agents targeting B cell receptor (BCR) pathway kinases that are critical to malignant B cell survival.1 Ibrutinib is a covalent, irreversible inhibitor of Bruton’s tyrosine kinase, a key BCR protein.2 It was approved for the treatment of CLL based on its excellent efficacy and tolerability, even in patients with high risk disease.3 Interestingly, although ibrutinib inhibits malignant B cell proliferation, survival, and migration, resulting in decreased tumor burden in patients, it typically does not induce a substantial amount of apoptosis in vitro4, and complete responses occur in only 2–4% of patients.3, 5 However, the progression free survival benefits of ibrutinib are substantial, and the complete response rate is likely to rise as patients stay on therapy longer.6, 7 Despite these clear clinical observations, the mechanisms by which ibrutinib interacts with the apoptotic cascade are incompletely understood.
In addition to BTK, ibrutinib targets several other kinases such as ITK and TEC, and it is likely that some of its toxicities are related to these off-target effects. A more specific BTK inhibitor, acalabrutinib (ACP-196) recently entered the clinic and was also found to be highly effective in relapsed/refractory CLL with a favorable toxicity profile in early phase clinical trials.8 As with ibrutinib, the effect of BTK inhibition with acalabrutinib on the mitochondrial pathway of apoptosis has not been fully explored.
Dysregulation of the mitochondrial pathway of apoptosis is one of the hallmarks of CLL cell pathophysiology. Indeed, CLL cells rely heavily on the anti-apoptotic protein BCL-2 for survival,9 and thus targeting this protein is another promising new therapeutic strategy. Venetoclax (ABT-199/GDC-0199) is a highly selective oral BCL-2 antagonist10 that induces rapid and deep responses in CLL,11 even in patients with high risk TP53 deficient disease.12, 13 The mechanism of action and toxicities of venetoclax are distinct from BTK inhibitors, suggesting that the combination of these agents would be feasible in the clinic. Recent work has provided evidence for the preclinical efficacy of the ibrutinib plus venetoclax combination in CLL.14 In that study, levels of the anti-apoptotic proteins MCL-1 and BCL-XL were found to decrease after ibrutinib therapy. Whether this effect is a property of ibrutinib specifically, or of BTK inhibition more generally, is not known. We hypothesized that if we found the same effects with ibrutinib and the more BTK-specific inhibitor acalabrutinib that this would support the idea that BTK inhibition is the critical factor that enhances the sensitivity of malignant B cells to BCL-2 antagonism by venetoclax.
Our group previously developed BH3 profiling, a functional assay designed to interrogate mitochondria to determine their proximity to the threshold of apoptosis, a property called “mitochondrial priming”, which can be measured using a BH3 peptide derived from the pro-apoptotic protein BIM, which interacts with all major anti-apoptotic proteins.15 BH3 profiling is also able to determine BCL-2 dependence of a cell compared to related anti-apoptotic proteins such as MCL-1 or BCL-XL, as measured using a BH3 peptide derived from the pro-apoptotic protein BAD, which interacts selectively with BCL-2 in CLL cells. Dynamic BH3 profiling (DBP) is a new variation of this technique that measures early changes in net pro-apoptotic signaling at the mitochondrion that are induced in cancer cells treated with anti-cancer agents.16 We hypothesized that DBP would allow us to determine how BTK inhibitors and venetoclax influence mitochondrial priming and anti-apoptotic dependence in primary CLL cells.
Here, we utilize DBP to show that venetoclax increases the overall level of mitochondrial priming of CLL cells ex vivo. In contrast, ibrutinib and acalabrutinib selectively increase the dependence of mitochondria on BCL-2, and hence increase the sensitivity of CLL cells to venetoclax. Importantly, we confirmed these findings in vivo in primary CLL cells isolated from patients treated with ibrutinib on the phase 3 PCYC-1112 (RESONATE) trial17 and with acalabrutinib on its phase I first-in-human trial.8 These data help elucidate the interactions of BTK inhibitors with the mitochondrial pathway of apoptosis, and provide further preclinical rationale for clinical trials combining these novel agents in the clinic.
Methods
CLL patient samples and cell purification
After obtaining informed consent, peripheral blood was obtained from patients fulfilling diagnostic criteria for CLL. Consent was obtained in accordance with the Declaration of Helsinki on protocols reviewed and approved by the Dana-Farber / Harvard Cancer Center Institutional Review Board.
Mononuclear cells were isolated from blood and tissue samples by Ficoll-Paque (GE Healthcare, Waukesha, WI) density gradient centrifugation. Samples were viably frozen in 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO) in fetal bovine serum (FBS, Sigma-Aldrich), stored in liquid nitrogen, and later thawed for analysis. Single cell suspensions were prepared, and CD19+CD5+ CLL cells generally accounted for >85% of analyzed cells.
CLL cell and stromal cell co-cultures
The stromal NKTert cell line was purchased from the Riken cell bank (Tsukuba, Japan) and maintained in RPMI 1640 medium supplemented with 10% FBS, 2.05mM L-glutamine, and penicillin-streptomycin (Life Technologies, Grand Island, NY). Primary CLL cells were cultured with the same complete RPMI media. For co-culture experiments, CLL cells were seeded with NKTert as previously described.18 Cells were then treated with drugs for the specified time periods (see Supplemental Methods) and analyzed.
Cell viability testing, reagents
CLL cell viability was determined by flow cytometric analysis using surface marker staining antibodies, Annexin V-FITC (BD Biosciences, San Diego, CA) and Propidium Iodide (PI) (Sigma). Analysis was performed with a BD FACS Fortessa or Fortessa × 20 machine.
Dynamic BH3 profiling
BH3 profiling was performed by flow cytometry, as previously described.18, 19 Briefly, CLL cells were thawed from viably-frozen vials or harvested from ex vivo treatments, washed, and stained with fluorescent antibodies. Single cell suspensions were exposed to 0.002% digitonin and then BH3-only peptides for 60 minutes. After formaldehyde fixation, anti-cytochrome C-Alexa 488 (BD Pharmingen) was added, and to analyze the plate flow cytometry was performed.
Western blot analysis
Protein lysates were obtained by cell lysis, electrophoretically separated on NuPAGE 10% Bis-Tris polyacrylamide gels (Life Technologies) and transferred to PVDF membrane (EMD Millipore, MA). Images were obtained by exposing membranes to ECL solution and the signals were captured by an LAS 4000 imager (Fuji Film). Densitometry was done with ImageQuant software equipped in the imager.
Data analysis and statistics
Flow cytometry data were analyzed using FACS Diva version 6.1.1 (BD Pharmingen). Delta priming was generated by subtracting the values of loss of cytochrome C in DMSO- or pre- treated samples from drug treated samples; and maximal viability decrease was the biggest difference in viability calculated from dose curve experiments. Statistical analysis was done by GraphPad Prism 6 software for PC (GraphPad Software, San Diego, CA). After assessing the data for normality by the Shapiro-Wilk normality test, a one sample t test was used to validate if delta priming was significantly different from 0. In cases where data sets did not pass the normality test, a sensitivity analysis using a non-parametric one sample Wilcoxon was also performed. Student’s paired t-tests were performed to compare two different drug treatments, and one-way ANOVA was used for > two group comparisons. P value ≤ 0.05 was considered statistically significant.
Results
Ex vivo BTK inhibition increases BCL-2 dependence in primary CLL cells
We initially compared the ability of venetoclax and ibrutinib to induce apoptosis in primary patient CLL cells ex vivo. Consistent with prior studies, venetoclax triggered apoptosis rapidly, with an EC50 below 10 nM, whereas ibrutinib did not elicit significant apoptotic cell death even at 1 uM (Supplemental Figure 1).
Apoptosis is a threshold phenomenon, and we utilize BH3 profiling to assess mitochondrial priming, a cell’s proximity to the apoptotic threshold. We next asked whether even if ibrutinib was not inducing frank cell death, could it nonetheless be inducing sub-lethal pro-apoptotic signaling? We assessed for changes in pro-apoptotic signaling by dynamic BH3 profiling (DBP), a technique that allows us to measure “delta priming”, defined as the difference in mitochondrial response to a BH3 peptide in a drug-treated sample versus an untreated control (Supplemental Figure 2A).16 The BIM BH3 peptide interacts promiscuously with all of the anti-apoptotic proteins (Supplemental Figure 2B), as well as with the pro-apoptotic effector proteins BAX and BAK. As such, mitochondrial sensitivity to BIM is a measure of the overall proximity of the cell to the apoptotic threshold.
Primary CLL cells were prepared from a panel of CLL patients and cultured ex vivo with or without drugs for 72 hours in the presence of the stroma to facilitate CLL cell survival for BH3 profiling analysis. We assessed whether delta priming after drug treatment was significantly different from 0 (i.e. no effect). We chose to study previously untreated CLL patients with uniform prognostic markers for these experiments to avoid potential confounding by biological heterogeneity in the CLL cells themselves (Supplemental Table 1A). We found that ibrutinib did not alter mitochondrial sensitivity to the BIM peptide (Figure 1A) suggesting that the drug had little effect on overall mitochondrial priming of the CLL cells. The BAD BH3 peptide interacts more selectively with BCL-2, BCL-XL, and BCL-w (Supplemental Figure 2B). BCL-XL and BCL-w are expressed at much lower levels than BCL-2 in CLL.9 Therefore, in CLL, mitochondrial sensitivity to the BAD BH3 peptide can be considered a measure of the cell’s BCL-2 dependence. In contrast to the results with BIM peptide, ibrutinib consistently increased the response to BAD peptide (20–50% positive delta priming, Figure 1A). Ibrutinib treatment did not yield significant priming changes to MS1 (specific for dependence on MCL-1)20 or HRK (specific for dependence on BCL-XL)15 peptide (Supplemental Figure 3). Taken together, these results suggest that ibrutinib selectively enhances CLL cell dependence on BCL-2.
Figure 1. Ex vivo BTK inhibition increases CLL cell BCL-2 dependence as shown by dynamic BH3 profiling.
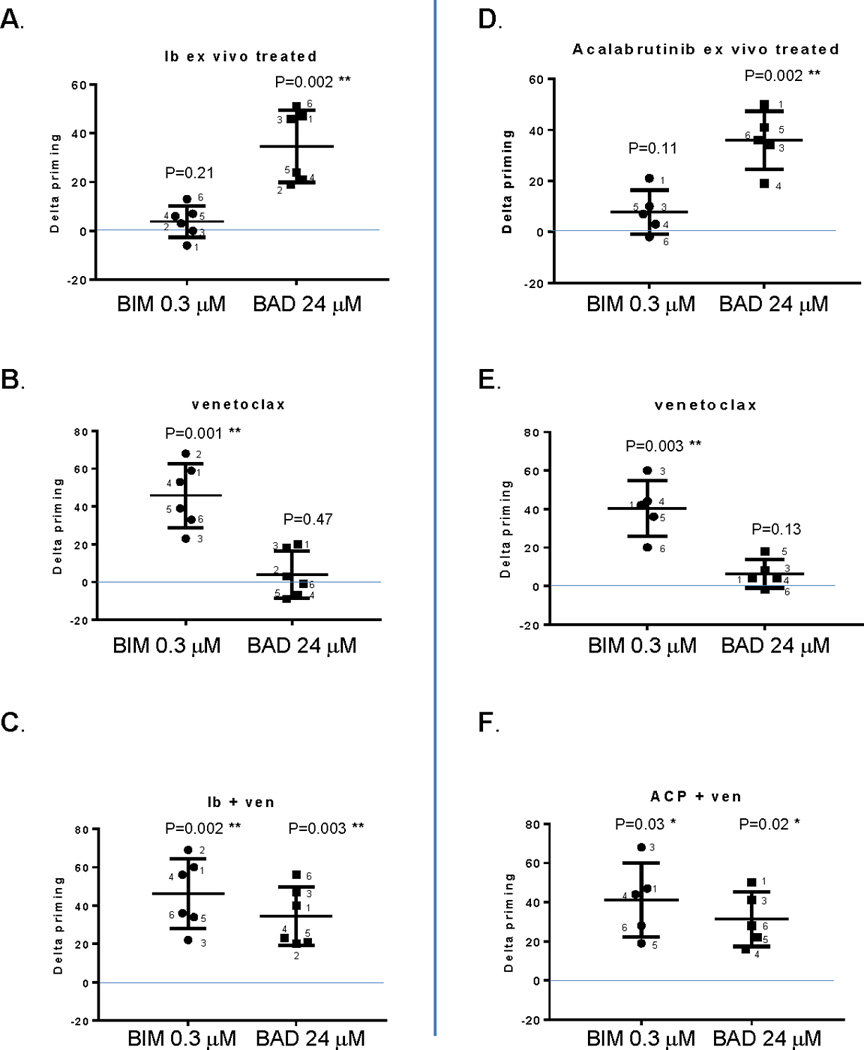
Primary CLL cells were co-cultured with the NKTert stromal cell line, and subjected to single drug or combination treatment with ibrutinib alone (panel A), venetoclax alone (panel B), or the ibrutinib + venetoclax combination (panel C). Analogous experiments were performed with acalabrutinib alone (panel D), venetoclax alone (panel E), or the acalabrutinib + venetoclax combination (panel F). Ibrutinib and acalabrutinib were both used at 1 uM for 72 hours, while venetoclax at 1.5 nM was added only for the last 20 hours. Cells were then harvested for BH3 profiling. Delta priming is the difference in mitochondrial response to a BH3 peptide between DMSO- and drug treated samples (Y-axis). The response from BAD peptide, a specific indicator for BCL2 dependence, is shown side-by-side with BIM peptide response. Means are depicted as horizontal bars +/− SD error bars. p values were calculated using a one sample t test to validate if delta priming is different from 0. Ib, ibrutinib; ACP, acalabrutinib. * and ** indicate statistical significance.
When CLL cells were treated with venetoclax, the profiles revealed a pattern of BIM-BAD peptide response complementary to that observed with ibrutinib. Venetoclax elicited a 20–70% positive delta-priming in response to BIM BH3 peptide, whereas the delta-priming in response to the BAD peptide was minimal (Figure 1B). These results suggest that venetoclax moves the cell closer to the threshold of apoptosis, but it does not render the mitochondrion more sensitive to the BAD BH3 peptide. In other words, it does not selectively enhance BCL-2 dependence. The combination of ibrutinib and venetoclax increased both BIM and BAD BH3 peptide response (Figure 1C), suggesting that this drug combination increases both CLL cell mitochondrial priming and BCL-2 dependence.
We next sought to evaluate whether the selective increase in BCL-2 dependence observed with ibrutinib in the ex vivo treated CLL patient samples was a BTK-specific effect. To assess this, we repeated the same set of experiments on samples from the same patients, this time treated ex vivo with a more BTK-specific inhibitor acalabrutinib. As with ibrutinib, acalabrutinib consistently induces positive delta-priming (20–50%) in response to BAD but not BIM BH3 peptide (Figure 1D), suggesting that it also increases BCL-2 dependence without altering overall mitochondrial priming. The specific BCL-2 dependence was again confirmed by the minimal effects of MS1 or HRK BH3 peptides, suggesting that acalabrutinib did not alter dependency on MCL-1 or BCL-XL, respectively (Supplemental Figure 4). When these CLL cells were treated with venetoclax, BH3 profiling again demonstrated an increase in delta-priming in response to BIM BH3 peptide but not to BAD BH3 peptide (Figure 1E), and the combination of acalabrutinib and venetoclax increased both priming and BCL-2 dependence (Figure 1F). These data suggest that the increase in BCL-2 dependence in CLL cells observed with ex vivo ibrutinib treatment is likely due to a BTK-specific effect, rather than to an off-target effect of the drug.
Ex vivo BTK inhibition increases CLL cell sensitivity to BCL-2 inhibition
Our DBP results led us to hypothesize that pre-treatment with a BTK inhibitor would sensitize CLL cells to BCL-2 inhibition and would therefore be highly effective at killing CLL cells. To test this hypothesis, we pre-treated primary CLL cells with ibrutinib or acalabrutinib and then subsequently added venetoclax. As predicted, pre-treatment with ibrutinib or acalabrutinib increased CLL cell sensitivity to venetoclax (supplemental Figures 5 and 6). The maximal decrease in viability for each combination was calculated from the dose response curves and is summarized in Figure 2 (panel A for pretreatment with ibrutinib, panel B for pretreatment with acalabrutinib). Both drugs significantly enhanced killing by venetoclax.
Figure 2. Pre-treatment with ibrutinib or acalabrutinib increases CLL cell sensitivity to venetoclax.
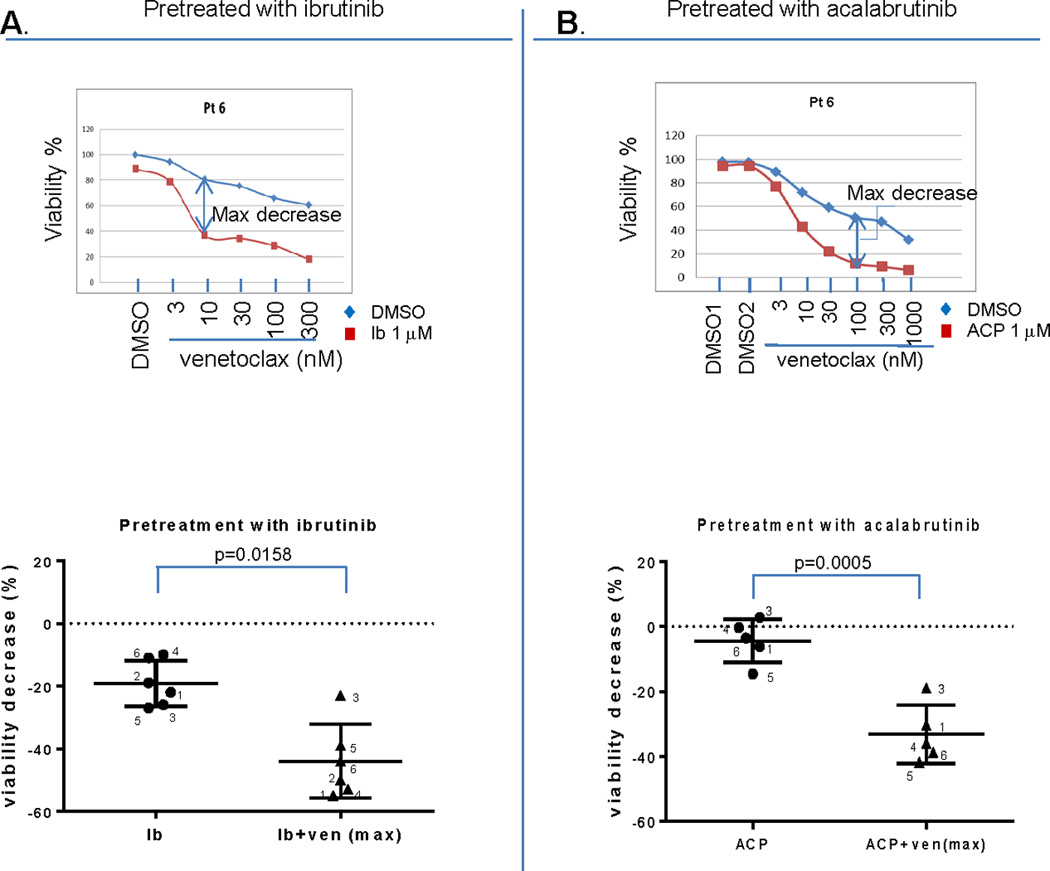
Primary CLL cells were seeded on top of the NKTert stromal cells and treated with 1 uM ibrutinib or acalabrutinib for 72 hours. Venetoclax was added for the last 1 hour, and viability was measured by flow cytometry analysis using FITC conjugated Annexin V and propidium iodide staining. Panel A and B top panels show representative dose curves for ibrutinib and acalabrutinib experiments, respectively. Panel A and B bottom panels show the maximal decrease of viability from each CLL primary sample calculated from dose curve experiments shown in the supplemental data. The statistical analysis used paired t-test (two-tailed) for a BTK inhibitor alone or in combination with venetoclax was shown in the bottom panels. Means are depicted as horizontal bars +/− SD error bars.
To assess whether the effects we observed were specific to venetoclax or whether they may also be observed with chemotherapy, we repeated our viability experiments by pre-treating primary CLL cells with either ibrutinib or a vehicle treated control, and then adding the purine analogue fludarabine. While there was some increased killing observed with ibrutinib, the effects were less consistent than with venetoclax (Supplemental Figure 5B). This suggests that the increase in BCL-2 dependence caused by BTK inhibition enhances CLL cell killing by venetoclax more than by fludarabine.
Ex vivo BTK inhibition increases expression of the BH3-only protein BIM
We next assessed whether changes in BCL-2 family protein expression in response to BTK and BCL-2 inhibition corresponded to the changes we observed in our BH3 profiling and viability assays. To test this, we prepared cell extracts from control and drug treated CLL primary cells from our earlier experiments and performed Western blot analysis. Consistent with our results above, we found that expression of the BH3-only protein BIM increased when cells were treated with ibrutinib (Figure 3A) or acalabrutinib (Figure 3B). Densitometry analysis of BIM-EL protein expression confirmed this increased expression with both ibrutinib and acalabrutinib (Figure 3C). BAD protein was increased by ibrutinib but not acalabrutinib (Figure 3C). Venetoclax treatment did not significantly alter BIM or BAD protein expression. No consistent changes were observed in protein expression of other BCL-2 family members in response to any of the drug treatments, including anti-apoptotic proteins MCL-1 and BCL-2 or the pro-apoptotic multi-domain protein BAX (Supplemental Figure 7).
Figure 3. Ex vivo BTK inhibition increases expression of the pro-apoptotic BH3-only proteins BIM.
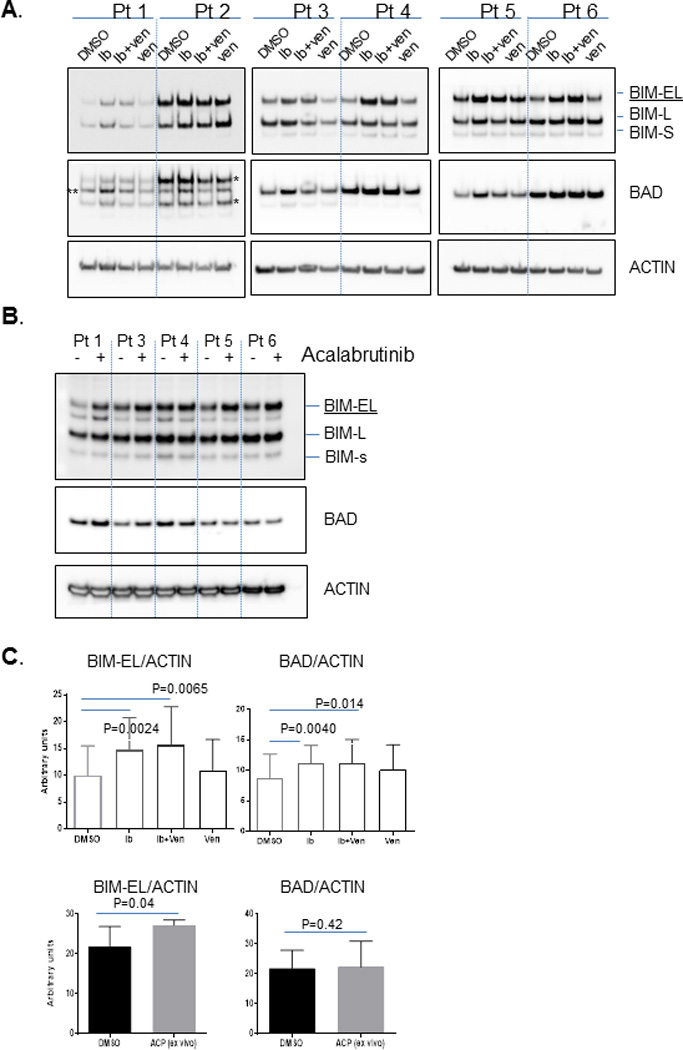
Primary CLL cells were treated with ibrutinib or acalabrutinib alone or in combination with venetoclax under identical conditions as in Figure 2. Cells were collected at the end of treatment to prepare protein lysates for SDS-PAGE electrophoresis. Specific proteins were detected by the antibodies indicated in the figure. Densitometry analysis was performed for images shown in panel A and B. Ratio of BIM-EL or BAD protein to ACTIN was compared within different treatments and shown in panel C. The concentrations used in the experiments were 1 uM for ibrutinib and acalabrutinib; 1.5 nM for venetoclax. In patients 1 and 2 in panel A, the BIM antibody was run before the BAD antibody, resulting in multiple bands in the box labeled BAD. For all other patients BAD antibody was run after BIM antibody. * leftover BIM protein, ** BAD protein.
In vivo BTK inhibition increases BCL-2 dependence in CLL patients
We next asked whether the increased BCL-2 dependence seen with BTK inhibition ex vivo could be observed in vivo in primary CLL cells obtained from patients treated on ibrutinib and acalabrutinib clinical trials (Supplemental Table 1B). We utilized samples that were both pre-treatment and from multiple time points after dosing, and conducted experiments in a manner similar to the ex vivo work described above. These samples came from patients who were a mix of previously treated and untreated so that we could assess whether the biology we observed in CLL cells from untreated patients was retained in previously treated patients.
Consistent with our ex vivo findings, ibrutinib therapy led to increased mitochondrial BCL-2 dependence in vivo, as indicated by the positive delta-priming observed in response to the BAD BH3 peptide in all five patients tested (Figure 4A, B). “Delta-priming” here describes the difference in mitochondrial response to a BH3 peptide in a clinical sample obtained on treatment compared to a pre-treatment control sample. The positive delta-priming was observed as early as 6 hours after the first dose of ibrutinib (Figure 4A), and by day 15 on trial all five patients had at least a +20% delta-priming (Figure 4B). As an internal control, we utilized venetoclax applied directly to mitochondria like a BH3 peptide and obtained the same result (Figure 4A, B). Similar to the ex vivo treatment, the delta-priming observed with the BIM peptide was minimal after in vivo ibrutinib therapy (Figure 4A, B), and was significantly less than that observed with BAD peptide. No changes in MCL-1 or BCL-XL dependence were observed in vivo, as indicated by the lack of delta-priming after treatment with the MS-1 or HRK peptide, respectively (Supplemental Figure 8).
Figure 4. In vivo BTK inhibition increases BCL-2 dependence in cells from CLL patients.
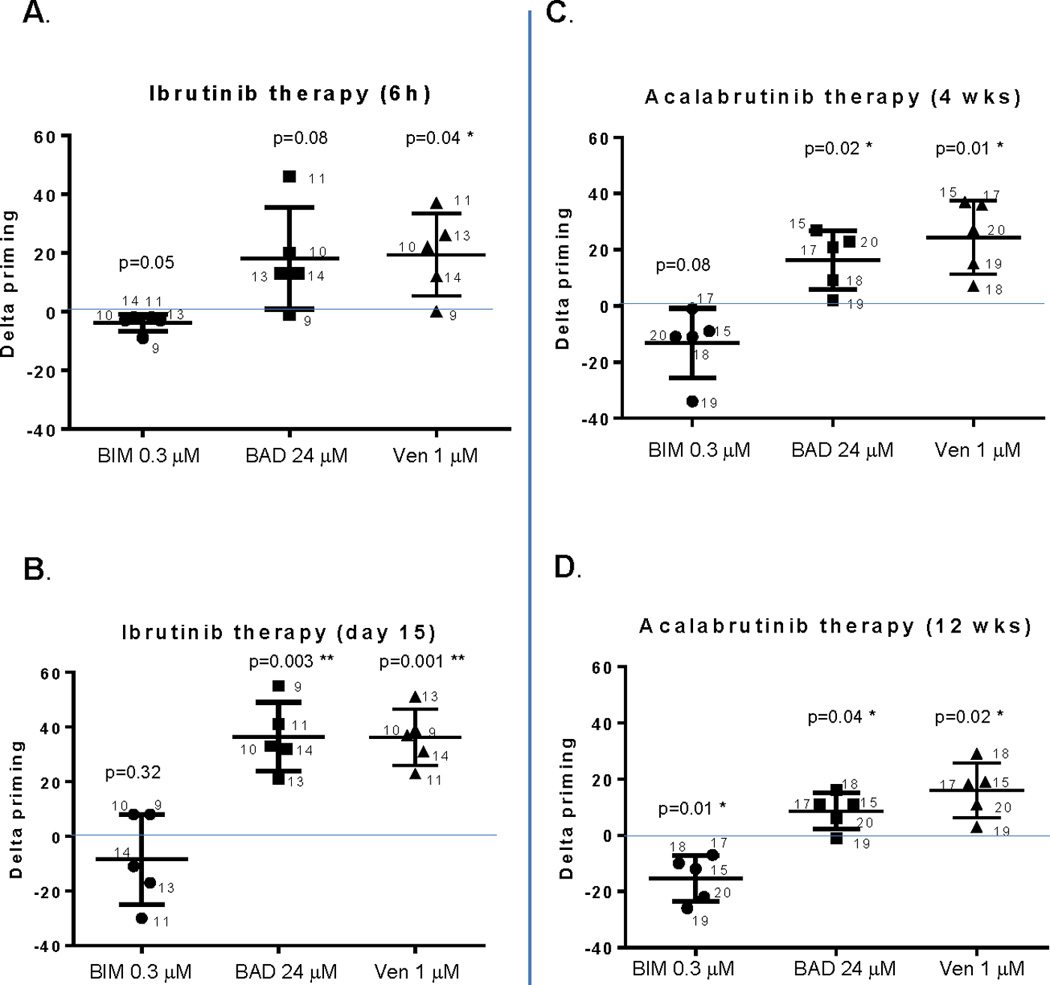
CLL patient samples were collected before and after ibrutinib or acalabrutinib therapy in the respective clinical trials. BH3 profiling was performed under identical conditions as the ex vivo samples shown in Figure 1. Delta priming was calculated from pre- and post-treatment mitochondrial responses to BIM or BAD peptides and low-dose venetoclax (used like a peptide in the BH3 profiling assay). Ibrutinib samples were obtained 6 hours (panel A) and 15 days (panel B) after the first dose. Acalabrutinib samples were obtained 4 weeks (panel C) and 12 weeks (panel D) after the first dose. Means are depicted as horizontal bars +/− SD error bars. One sample t test was used to validate if the delta priming is different from 0 and p values are indicated with * and ** indicating statistical significance. Normality was confirmed in all experiments with the exception of BIM 0.3 uM in panel (A), which was also subjected to a Wilcoxon rank test and was not significant (0.0625), suggesting that delta-priming for this peptide is not significantly changed.
We next tested whether increased BCL-2 dependence could also be observed in CLL cells from patients treated with acalabrutinib. Of note, due to sample availability, the correlative acalabrutinib trial samples were from later time points than our samples from ibrutinib-treated patients (week 4 and week 12 vs. 6 hours, day 8, and day 15 post-dosing, respectively). Nonetheless, we observed that acalabrutinib therapy also enhances mitochondrial BCL-2 dependence in vivo. Both BAD peptide and venetoclax used like a peptide in the BH3 profiling assay resulted in positive delta-priming at both 4 weeks and 12 weeks on acalabrutinib therapy, and in both cases the delta-priming was significantly greater for BAD peptide than BIM peptide, which was negative in some cases, suggesting decreased priming (Figure 4C, D). This negative delta-priming in the less primed in vivo samples may reflect that the cells recently exited the protective stromal microenvironment, in contrast to the ex vivo treated samples that become more primed in culture. The delta-priming in response to MS1 and HRK was minimal (Supplemental Figure 9), suggesting again that BTK inhibition enhances BCL-2 dependence specifically, without affecting dependence on other anti-apoptotic proteins or the overall level of mitochondrial priming in the cell. As in the ex vivo setting, this enhanced BCL-2 dependence likely contributes to increased CLL cell apoptosis in patients, as has previously been demonstrated by other groups.21
In vivo BTK inhibition sensitizes CLL cells to venetoclax
We next tested whether CLL cells from patients treated with BTK inhibitors in vivo demonstrated increased ex vivo sensitivity to venetoclax in a standard viability assay. We exposed CLL cells from these patients to venetoclax ex vivo for 1 hour and measured viability by Annexin/PI. Using dose response curves, we found that venetoclax enhanced CLL cell killing at a variety of concentrations (supplemental Figures 10 and 11). The maximal viability decrease with the combination of a BTK inhibitor plus venetoclax is depicted in Figure 5. Compared to the viability decrease by ibrutinib alone this cell killing by ibrutinib plus venetoclax was significant (Figure 5A). There was a trend toward increased killing by acalabrutinib, but this did not achieve significance (Figure 5B). Overall, venetoclax did induce more ex vivo cell death in CLL cells from ibrutinib-trial patients compared to acalabrutinib-trial patients (supplemental Figure 12A), which is consistent with our results above showing that ibrutinib induced a greater change in delta-priming than did acalabrutinib (Supplemental Figure 12B, C).
Figure 5. In vivo BTK inhibition sensitizes CLL cells to venetoclax.
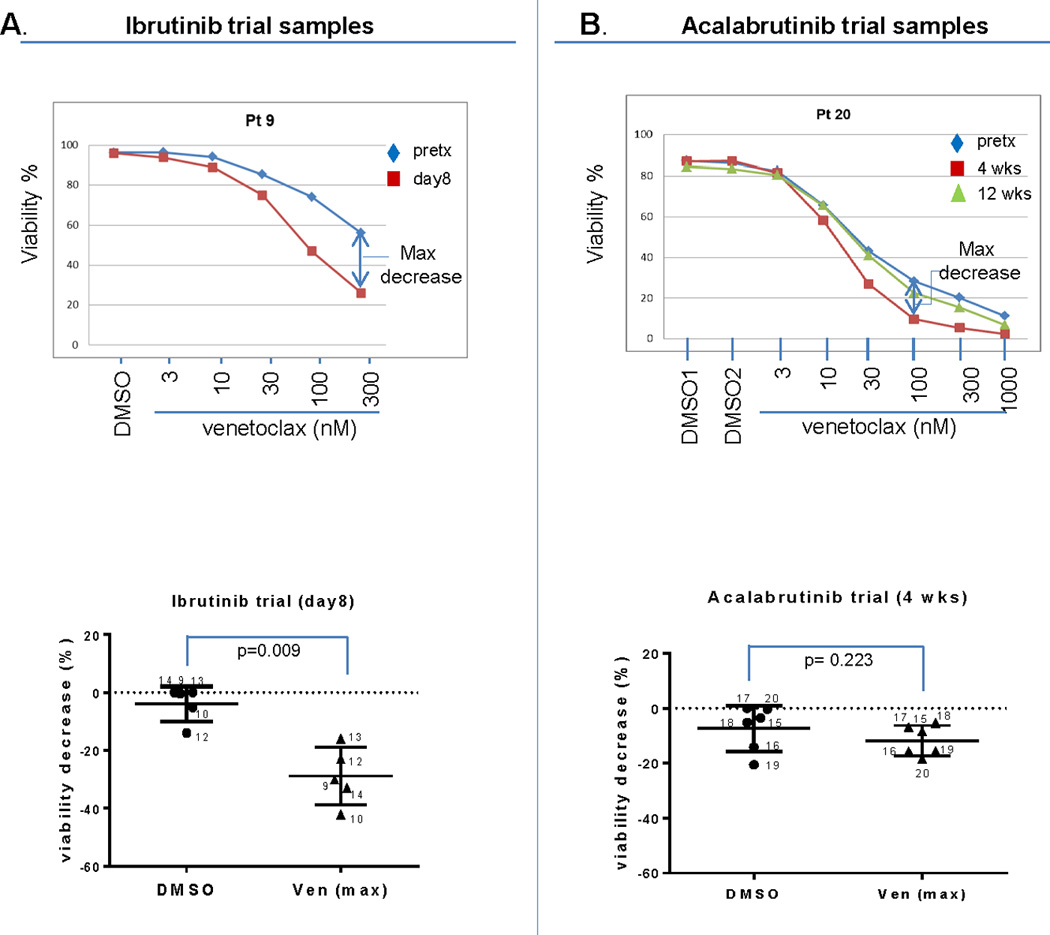
CLL patient samples obtained from the BTK inhibitor clinical trials for the BH3 profiling assays were also aliquoted for viability assessment. Cells were subjected to ex vivo venetoclax treatment for 1 hour and viability was measured by flow cytometry analysis using FITC conjugated AnnexinV and PI staining. Panel A and B top panels show representative dose curves for ibrutinib and acalabrutinib treated patients, respectively. Panel A and B bottom panels show the maximal decrease of viability from each CLL patient sample calculated from dose curve experiments shown in the supplemental data. The statistical analysis used paired t-test (two-tailed) for viability decrease resulting from a BTK inhibitor alone or in combination with venetoclax is shown in bottom panels. Means are depicted as horizontal bars +/− SD error bars. Normality was confirmed in all experiments with the exception of the data for ibrutinib at day 15 and acalabrutinib at week 12. N/A: sample not available.
In vivo BTK inhibition upregulates the pro-apoptotic protein BIM
To assess whether there is evidence at the protein level to support our findings with BH3 profiling and cell viability assays, we performed Western blot analysis with proteins lysates prepared from the same patient samples. As with our ex vivo experiments, we again found that BIM protein generally increased in CLL cells from patients treated with either ibrutinib (Figure 6A) or acalabrutinib (Figure 6B). For acalabrutinib, the increase appeared greater at 4 weeks than 12 weeks (Figure 6C), perhaps reflecting selection against high BIM expressing cells at the later time point. BAD protein expression also increased in some ibrutinib treated patient samples. Other multi-domain BCL-2 family member proteins, including BCL-2, MCL-1, and BAX did not change consistently (supplemental Figure 13). These findings at the protein level in our in vivo samples from patients treated with BTK inhibitors are consistent with our observations in the primary CLL samples subjected to ex vivo BTK inhibition.
Figure 6. BIM expression is increased in CLL cells treated in vivo with BTK inhibition.
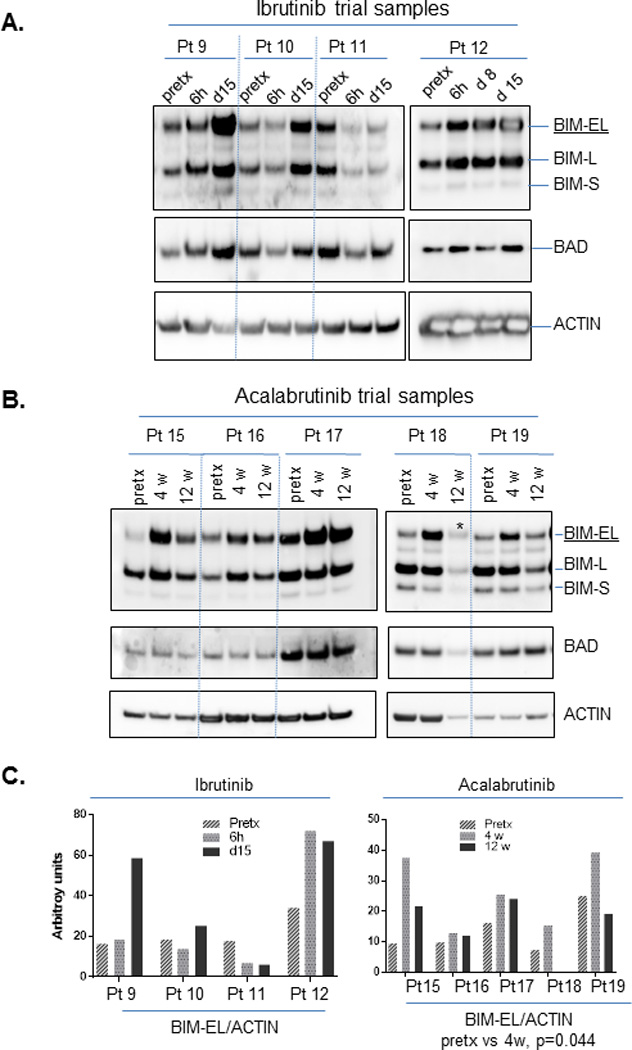
Protein lysates were prepared using aliquots of primary CLL cells obtained from clinical trials of ibrutinib (panel A) or acalabrutinib (panel B), and analyzed by SDS-PAGE electrophoresis. Antibodies were used to detect BCL-2 family proteins as indicated in the figure. Densitometry analysis was performed for BIM-EL and ACTIN, and their ratio is shown in panel C. One-way ANOVA for BIM-EL/ACTIN ratios in acalabrutinib pretreated and week 4 samples showed a p<0.05. Pretx=pre-treatment samples. Other specified time points refer to the time after initial dosing of the respective drug. * underloaded samples not calculated for BIM-EL/ACTIN ratio.
Discussion
Although ibrutinib monotherapy is highly effective for patients with CLL, it rarely induces complete remission, and the durability of response is limited in patients with high risk CLL such as those with del(17p) and/or complex karyotype.22 In addition, the acquisition of somatic resistance mutations has been described, such as the BTK C481S identified in several patients who progressed on ibrutinib.23 Given the plethora of novel agents now available for CLL, it is not feasible to examine every ibrutinib combination strategy in a clinical trial. Therefore, sound preclinical data allow for the rational design of CLL trials to explore optimal combination partners and drug sequences.
BTK inhibition leads to disruption of tonic BCR signaling, migration, and proliferation of CLL cells; however, the details of how BTK inhibition interacts with the intrinsic pathway of mitochondrial apoptosis are not fully understood. The preclinical efficacy of the ibrutinib/venetoclax combination in killing CLL cells has recently been demonstrated using conventional cell death testing in both ex vivo treated CLL cells and in vivo samples from patients on ibrutinib therapy.14 Although this important work is suggestive of a mechanistic connection between ibrutinib and BCL-2, it does not address the fundamental question of how this interaction occurs on a functional level, nor does it shed light on whether this interaction is due to BTK inhibition or off target effects of ibrutinib.
In our ex vivo studies on CLL cells from patients, we found through dynamic BH3 profiling (DBP) that ibrutinib causes a substantial increase in BCL-2 dependence with minimal change in the overall level of mitochondrial priming, whereas venetoclax had the opposite effect. DBP provides the first direct evidence of the uniquely complementary nature of the effects of these two drugs on mitochondria. Importantly, we confirmed our ex vivo findings in samples from patients on ibrutinib, showing that these effects also apply in vivo. By increasing mitochondrial dependence on BCL-2, CLL cells are sensitized to BCL-2 antagonism with venetoclax, which helps to explain why this combination is particularly effective at killing CLL cells (Supplemental figure 14). A possible model of how BTK inhibition primes CLL cells for apoptosis through BCL-2 inhibition is presented in Figure 7.
Figure 7. Schema summarizing the complementary effects of BTK and BCL-2 inhibition on CLL cell mitochondria.
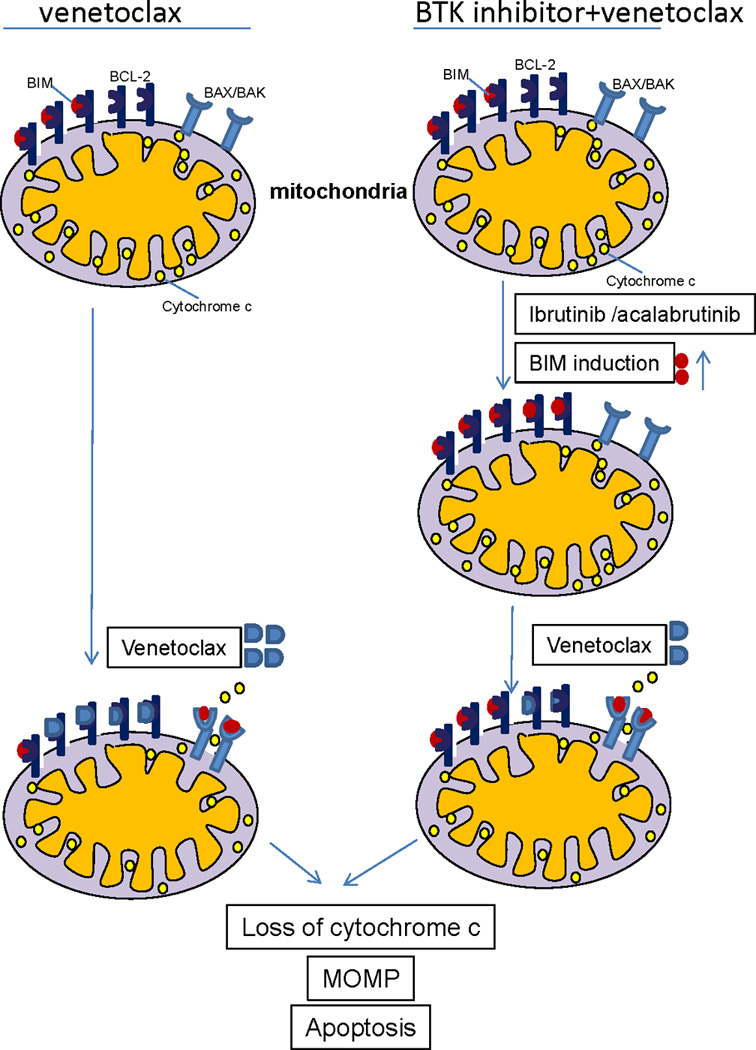
At baseline (top left), CLL cells are BCL-2 dependent and primed for apoptosis, with the pro-apoptotic protein BIM occupying some, but not all of the binding sites on BCL-2. Treatment with venetoclax (bottom left) leads to apoptosis in most CLL cells by displacing BIM from BCL-2, allowing it to bind to the pro-apoptotic effector proteins BAX/BAK, which can then homo-oligomerize leading to mitochondrial outer membrane permeabilization (MOMP), cytochrome C release, and subsequent caspase-mediated cell death. BTK inhibition with ibrutinib or acalabrutinib (right side) can increase BIM protein thereby saturating essentially all of the binding sites on BCL-2 and increasing the BCL-2 dependence of the CLL cell even further. Subsequently adding venetoclax (lower right) rapidly triggers MOMP and subsequent CLL cell death.
Our discovery of the complementary mechanisms of action of ibrutinib and venetoclax on mitochondria prompted the question of whether this interaction is a BTK specific phenomenon or whether it may be due to off target effects of ibrutinib. Mirroring our ibrutinib results, the highly selective BTK inhibitor acalabrutinib induced a significant increase in BCL-2 dependence in CLL cells both ex vivo and in vivo. This suggests that BTK inhibition itself may be sufficient to enhance BCL-2 dependence in CLL cells, although differences in the ex vivo and in vivo kinase inhibitor profiles of these drugs makes this challenging to prove conclusively.
Our results suggest that BTK inhibition perturbs the intrinsic apoptotic pathway in CLL cells by increasing BCL-2 dependence directly at the level of the mitochondria. Although there are several potential mechanisms, one possibility is that BTK inhibition, by increasing BIM levels, increases the occupancy of BCL-2. Thus, when venetoclax competes for the BH3 binding site on BCL-2, more BIM is displaced, resulting in more efficient mitochondrial outer membrane permeabilization (MOMP)9, the point of no return in apoptotic cell death. Alternatively, BTK inhibition might cause a decrease in the abundance or function of another key anti-apoptotic protein, MCL-1.14 In our experiments, although MCL-1 protein levels did appear to decrease in some samples (Supplemental Figure 13), other patients had increased BCL-2 dependence without a significant change in MCL-1 protein expression. This is consistent with recent in vitro observations by another group,24 and suggests that ibrutinib may inhibit MCL-1 function without necessarily decreasing its levels. A previous study found that phosphorylation events at serine-64 on MCL-1 may affect the function of the protein.25 Given that ibrutinib inhibits over a dozen other kinases besides BTK, it may be informative to determine whether one of these kinases normally phosphorylates MCL-1 under physiologic conditions, and whether ibrutinib treatment can abrogate this phosphorylation and thereby impair MCL-1 function, although this is beyond the scope of our current study.
An important advantage of DBP over prior studies that relied on conventional cell death assays is that it allows us to identify these pro-apoptotic signals even in the absence of frank cell death. Thus, we could study living tumor cells that had survived contact with ibrutinib in the patient. This approach avoids the potential confounding factors that inevitably arise when CLL cells are cultured over time. Of note, although significant changes were seen in BCL-2 dependence through DBP, our western blot analyses demonstrated minimal changes in protein expression for most of the Bcl-2 family members. This highlights that examining protein expression levels alone fails to adequately assess the complexity of the Bcl-2 family, which involves protein-protein interactions, post-translational modifications, and other unmeasured factors which are best captured by a functional assay. This is particularly important in CLL where, unlike in other types of cancer, novel agents do not target specific somatic mutations, but rather the pathophysiology of the disease itself.26
Our study is the first demonstration of DBP using in vivo treated patient samples. By observing changes in circulating patient cells, we could predict the efficacy of a new therapeutic combination. This approach offers promise for a paradigm in which DBP can be used for serial tailoring of therapy on patients with accessible tumor tissue. While we focused here on BTK and BCL-2 inhibition, DBP can be used to examine the effects of many therapies with different mechanisms of action in parallel, offering the opportunity for a real-time approach to precision medicine.
Our finding of the complementary effects of BTK inhibitors and venetoclax on CLL mitochondria strongly supports the exploration of these combinations in the clinic. One of these trials involving the ibrutinib/venetoclax combination in CLL (NCT02427451) recently opened, and future studies may utilize this backbone as a highly effective combination regimen in CLL. Such approaches hold promise for a well-tolerated, time-limited therapy for CLL that can achieve deep and durable remissions.
Supplementary Material
Acknowledgments
J.R.B. has received consulting fees from Abbvie, Genentech, Pharmacyclics, and Janssen. A.L. is a paid advisor to and his laboratory receives research sponsorship from AbbVie, Astra-Zeneca, and Tetralogic. M.S.D. has received research sponsorship from Pharmacyclics and Genentech and has received consulting fees from Genentech, Abbvie, Pharmacyclics, and Janssen.
The authors thank the patients who generously donated their samples for this study; John F. Daley II and Suzan Lazo-Kallanian for technical support with flow cytometry; Abbvie for providing venetoclax, and Acerta for providing acalabrutinib. This work was funded by the National Institutes of Health (R01CA129974) and a Dana-Farber Gloria Spivak Faculty Advancement Award. M.S.D. is an American Society of Clinical Oncology Career Development Award recipient and a recipient of a National Institutes of Health Loan Repayment Program award.
Footnotes
Conflicts of Interest
J.D., E.I, and S.F. have no relevant conflicts of interest.
Note: Supplementary information is available at Leukemia's website
References
- 1.Wiestner A. BCR pathway inhibition as therapy for chronic lymphocytic leukemia and lymphoplasmacytic lymphoma. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2014 Dec 5;2014(1):125–134. doi: 10.1182/asheducation-2014.1.125. [DOI] [PubMed] [Google Scholar]
- 2.Davids MS, Brown JR. Ibrutinib: a first in class covalent inhibitor of Bruton's tyrosine kinase. Future oncology (London, England) 2014 May;10(6):957–967. doi: 10.2217/fon.14.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013 Jul 4;369(1):32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012 Feb 2;119(5):1182–1189. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burger JA, Styles L, Kipps TJ. Ibrutinib for Chronic Lymphocytic Leukemia. N Engl J Med. 2016 Apr 21;374(16):1594–1595. doi: 10.1056/NEJMc1600328. [DOI] [PubMed] [Google Scholar]
- 6.O'Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014 Jan;15(1):48–58. doi: 10.1016/S1470-2045(13)70513-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015 Apr 16;125(16):2497–2506. doi: 10.1182/blood-2014-10-606038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrd JC, Harrington B, O'Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016 Jan 28;374(4):323–332. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. The Journal of clinical investigation. 2007 Jan;117(1):112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013 Feb;19(2):202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 11.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016 Jan 28;374(4):311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson MA, Deng J, Seymour JF, Tam C, Kim SY, Fein J, et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood. 2016 Jun 23;127(25):3215–3224. doi: 10.1182/blood-2016-01-688796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stilgenbauer S, Eichhorst B, Schetelig J, Coutre S, Seymour JF, Munir T, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016 Jun;17(6):768–778. doi: 10.1016/S1470-2045(16)30019-5. [DOI] [PubMed] [Google Scholar]
- 14.Cervantes-Gomez F, Lamothe B, Woyach JA, Wierda WG, Keating MJ, Balakrishnan K, et al. Pharmacological and Protein Profiling Suggests Venetoclax (ABT-199) as Optimal Partner with Ibrutinib in Chronic Lymphocytic Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015 Aug 15;21(16):3705–3715. doi: 10.1158/1078-0432.CCR-14-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer cell. 2006 May;9(5):351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 16.Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, Ercan D, et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell. 2015 Feb 26;160(5):977–989. doi: 10.1016/j.cell.2015.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrd JC, Brown JR, O'Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus of atumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014 Jul 17;371(3):213–223. doi: 10.1056/NEJMoa1400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davids MS, Deng J, Wiestner A, Lannutti BJ, Wang L, Wu CJ, et al. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood. 2012 Sep 5; doi: 10.1182/blood-2012-02-414060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proceedings of the National Academy of Sciences of the United States of America. 2010 Jul 20;107(29):12895–12900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foight GW, Ryan JA, Gulla SV, Letai A, Keating AE. Designed BH3 peptides with high affinity and specificity for targeting Mcl-1 in cells. ACS chemical biology. 2014 Sep 19;9(9):1962–1968. doi: 10.1021/cb500340w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wodarz D, Garg N, Komarova NL, Benjamini O, Keating MJ, Wierda WG, et al. Kinetics of CLL cells in tissues and blood during therapy with the BTK inhibitor ibrutinib. Blood. 2014 Jun 26;123(26):4132–4135. doi: 10.1182/blood-2014-02-554220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson PA, O'Brien SM, Wierda WG, Ferrajoli A, Stingo F, Smith SC, et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer. 2015 Oct 15;121(20):3612–3621. doi: 10.1002/cncr.29566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance Mechanisms for the Bruton's Tyrosine Kinase Inhibitor Ibrutinib. N Engl J Med. 2014 May 28; doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bojarczuk K, Sasi BK, Gobessi S, Innocenti I, Pozzato G, Laurenti L, et al. BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood. 2016 Jun 23;127(25):3192–3201. doi: 10.1182/blood-2015-10-675009. [DOI] [PubMed] [Google Scholar]
- 25.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS letters. 2010 Jul 16;584(14):2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 26.Davids MS. Boldly Targeting Kinases without mutations. Blood. 2014 Feb 20;123(8):1119–1121. doi: 10.1182/blood-2013-12-543322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.