

Abstract
Aims
Human cytomegalovirus remains a significant issue for immunocompromised patients and existing viral polymerase targeting therapies are associated with significant toxicity. Accordingly, the viral terminase complex inhibitor, letermovir, is in development. We assessed letermovir pharmacokinetics in renal impairment.
Methods
This was a Phase 1, open‐label, nonrandomised trial. Estimated glomerular filtration rate based on the Modification of Diet Renal Disease equation was used to create three groups of eight subjects: healthy function (estimated glomerular filtration rate ≥ 90 ml min–1 1.73m–2), moderate (30–59 ml min–1 1.73m–2) and severe (<30 ml min–1 1.73m–2) impairment. Oral letermovir 120 mg was dosed once‐daily for 8 days and blood collected for pharmacokinetic analyses.
Results
All 24 subjects enrolled completed the trial. Moderate and severe renal impairment increased mean unbound letermovir fractions by 11% and 26%, respectively, vs. healthy subjects. Exposure (AUCτ,ss and Css,max) was increased with renal impairment [least square mean ratios (90% confidence intervals) total letermovir vs. healthy subjects, AUCτ,ss 192% (143–258%) and 142% (83–243%) for moderate and severe impairment, respectively; Css,max 125% (87–182%) and 106% (75–151%), respectively]. Clearance was decreased vs. healthy subjects. Correlation analyses indicated a correlation between decreasing renal function and increased unbound letermovir concentration (R2 = 0.5076, P < 0.0001). Correlations were identified between decreased clearance with both decreased renal function (R2 = 0.0662, P = 0.2249 and R2 = 0.1861, P = 0.0353 total and unbound clearance, respectively) and increased age (R2 = 0.3548, P = 0.0021 and R2 = 0.3166, P = 0.0042 total and unbound clearance, respectively). Multiple‐dose letermovir 120 mg was well tolerated across groups.
Conclusions
Renal impairment increased exposure to letermovir, although age was a confounding factor.
Keywords: antivirals, cytomegalovirus, letermovir, pharmacokinetics, renal insufficiency
What is Already Known about this Subject
Letermovir is a viral terminase inhibitor to prevent human cytomegalovirus viraemia and disease, successfully completing a Phase 3 trial.
Letermovir is primarily excreted unchanged via bile into faeces. Urine excretion is negligible.
Renal impairment can alter secretion/transport pathways. Therefore, drug pharmacokinetics and safety in renally impaired subjects must be assessed.
What this Study Adds
Letermovir 120 mg is well tolerated by subjects with moderate or severe renal impairment.
There is an increase in letermovir exposure associated with renal impairment and a weak correlation between estimated glomerular filtration rates and drug clearance.
Letermovir is suitable for further administration in patients with decreased renal function.
Introduction
Human cytomegalovirus (HCMV) remains a significant issue for immunocompromised individuals and those at particular risk include the recipients of bone marrow or solid organ transplants. HCMV infection or re‐activation of latent infection can be a life‐threatening disease 1. For example, following haematopoietic stem‐cell transplantation (HSCT) and in the absence of prophylaxis, 80% of seropositive patients become positive for infection, of whom 20–35% experience HCMV disease 2, 3. The most serious manifestation of HCMV disease in patients receiving HSCT is pneumonitis, which results in mortality rates of >50% 4, 5. Furthermore, approximately 50% of the US population is seropositive for HCMV, leading to this pathogen being a highly prevalent causative agent of viral infection associated with transplantations 6. Seropositivity can also reach close to 100% in patients of low socioeconomic backgrounds or in countries with a developing economy, such as India, Brazil, Chile and Turkey, putting these patients at high risk 7.
Due to the prevalence and potential severity of HCMV infections, antiviral prophylaxis can be indicated for allogeneic recipients of HSCT and other high‐risk patients. Patients requiring pre‐emptive treatment include those with graft‐vs.‐host disease or early cytomegalovirus infection 3. The current first‐line standard of care is (val)ganciclovir. However, (val)ganciclovir and other existing prophylactic/pre‐emptive therapies for HSCT or solid organ transplant recipients are associated with significant toxicity, including bone marrow suppression and nephrotoxicity 8, 9, 10. In an attempt to overcome these issues, brincidofovir (CMX001) was recently developed as a lipid acyclic nucleoside phosphonate, intended to supersede existing treatments; however, this, too, has demonstrated dose‐limiting adverse events (AEs) 11. More recently CMX001 failed to achieve its primary endpoint at Phase 3 12. Therefore, an urgent unmet medical need remains for new therapies that are effective without dose‐limiting toxicities.
Because existing nucleosidic drugs targeting viral polymerase induce significant toxicities due to interaction with human polymerase enzymes, non‐nucleosidic drugs inhibiting an alternative target (the viral terminase) have been identified and investigated 13, 14, 15. These studies ultimately led to the discovery of letermovir (AIC246/MK‐8228), a novel drug that has demonstrated efficacy in treating HCMV viraemia and disease 16, 17. By targeting the viral terminase complex, letermovir inhibits the formation and release of infectious virus particles, thereby contrasting with the mechanism of action of existing anti‐HCMV drugs that target the viral DNA polymerase 18, 19. Importantly, in contrast to nucleoside inhibitors, it does not require an activation step by a viral enzyme and is active in uninfected cells. To date, letermovir has demonstrated safety and efficacy as a prophylactic in recipients of HSCT and has met the primary endpoints of a Phase 3 trial 20, 21. However, as part of clinical development, the impact of renal impairment on letermovir pharmacokinetics should be investigated. Letermovir is primarily excreted unchanged via bile into the faeces, and excretion of dose‐related material into urine is negligible 22. Despite the primary secretion pathway of letermovir via the bile, renal function is known to have an unpredictable effect on drug secretion/transport pathways in the liver 23, 24. The recently finalized European Medicines Agency guideline also states that effects of severe renal impairment on nonrenal elimination mechanisms have been suggested to be attributed to accumulation of uraemic toxins that inhibit or suppress metabolising enzymes and transport proteins 25. Therefore, it cannot be excluded that the pharmacokinetic (PK) and safety profiles of letermovir may become compromised in the instance of renal impairment.
The present trial aimed to evaluate the effect of both moderate and severe renal impairment on letermovir PK. In addition, the safety and tolerability of letermovir in association with daily doses of 120 mg—corresponding with a dose previously shown to be safe and effective in Phase 2 trials 20—was further evaluated.
Methods
The present investigation was a Phase 1, open‐label, nonrandomized trial to assess the PK, safety and tolerability of multiple oral letermovir 120 mg once‐daily doses in subjects with differing degrees of renal impairment (EudraCT no.: 2012–003104‐12; AiCuris Protocol: AIC246–01‐I‐16; Merck protocol: 8228 P006). This single‐centre trial was performed at Clinical Research Services GmbH, Kiel, Germany. The trial population comprised three groups of eight subjects each, divided according to estimated glomerular filtration rate (eGFR) based on the Modification of Diet Renal Disease equation. Group 1 was comprised of healthy subjects (eGFR ≥ 90 ml min–1 1.73m–2), Group 2 of subjects with moderate renal impairment (eGFR: 30–59 ml min–1 1.73m–2), and Group 3 of subjects with severe renal impairment not requiring dialysis (eGFR < 30 ml min–1 1.73m–2). The trial was designed in accordance with the European Medicines Agency and the US Food and Drug Administration guidelines for evaluation of PK in subjects with renal impairment compared with healthy subjects. The primary objective of the trial was to investigate the effect of renal impairment on the PK of multiple oral once‐daily doses of letermovir 120 mg for 8 days (Days 1–8). The secondary objective of the trial was to investigate the safety and tolerability of letermovir. All participants gave written informed consent to participate in the trial. In addition, the trial was approved by the independent ethics committee of the Ärztekammer Schleswig‐Holstein and the German Federal Institute for Drugs and Devices and performed according to Good Clinical Practice Guidelines.
Trial population
Male and female Caucasian subjects aged 50–79 years were eligible for the trial. Other than renal impairment conforming to the trial groups, subjects were healthy, not smoking more than 10 cigarettes per day and not taking any drug that might interfere with letermovir, induce or inhibit xenobiotic metabolizing enzymes or transporters. In addition to being grouped according to renal function, the groups were also matched using the following parameters: arithmetic mean group age of Groups 2 and 3 should not differ by >10 years and the arithmetic mean body mass index should not differ by >15% compared with Group 1. On Days 1–8 of the trial, letermovir 120 mg was administered orally in the morning under fasted conditions.
Bioanalytical and PK evaluations
Blood was collected to determine total letermovir levels prior to dosing on Days 1, 5, 6 and 7 in addition to regular intervals until 144 h after the last dose. Unbound letermovir concentrations were calculated by correcting total letermovir for the fraction unbound, which was determined on Day 8, 1.5 h postdose. Bioanalytical analyses of total letermovir were conducted by A&M Labor für Analytik und Metabolismusforschung GmbH, Bergheim, Germany; the bioanalyses of the free fraction of letermovir was performed by Pharm‐Analyt Labor GmbH (Baden, Austria). PK analyses were performed by Kinesis Pharma BV (Breda, the Netherlands).
The total letermovir concentrations of the plasma samples were determined after protein precipitation by liquid chromatography–tandem mass spectrometry. For the fraction of unbound letermovir, analyte concentrations were determined before and after equilibrium dialysis by liquid chromatography–tandem mass spectrometry. Quantitation for both total and the fraction unbound letermovir were performed using the internal standard method. For determination of the curve fit for the assay, four validation batches were analysed, each consisting of one set of calibration standards (expected range: 0.1 ng ml–1–100 ng ml–1), a zero, a blank sample and five sets of quality control samples at three concentration levels. Assay precision ranged from 1.41–6.10% intrabatch and 5.98–7.11% interbatch. Because the free fraction of letermovir in human plasma is constant in the clinically relevant range of 0.5–50 mg l–1 26, there is no evident concentration dependency. Thus, the unbound letermovir concentrations could be calculated from a single time‐point per subject in accordance with guidelines 25.
The primary target characteristics for PK were both total and unbound plasma letermovir area under the curve over a dosing interval at steady‐state (AUCτ,ss) and maximal letermovir concentration observed at steady‐state (Css,max). Point estimates and 90% confidence intervals (CIs) as a percentage of healthy subject PK were used to assess the effect of renal impairment. The secondary variables analysed were time to maximum observed analyte concentration (Tmax), average steady‐state concentration over the dosing interval (Css,av), termination half‐life (t½,z), apparent clearance (CL/F) and apparent volume of drug distribution at steady state (Vz/F).
Safety assessments
AEs were assessed according to subject throughout the trial period. Vital sign and electrocardiogram measurements were performed at screening, predose on Days 1, 3, 5 and 8, and also up to 48 h after the last dose and at the final examination. Laboratory safety tests were performed at screening; at predose on Days 1 and 5, on Day 10; and also at the final examination. Physical examinations were performed at screening, on Day 10, and at the final examination.
Data analysis and PK statistics
Since this is an exploratory Phase 1 trial, no formal sample size estimation was performed. A total of 24 subjects, eight subjects per group, was considered to be sufficient to identify trends in PK relating to renal function. All subjects for whom bioanalytical data were available were included in the PK analysis. Subjects were eligible for safety evaluation if at least 1 dose of letermovir was administered to them. Safety data were listed individually and summarised as appropriate with descriptive statistics.
The PK characteristics of the drug were calculated from plasma concentrations of letermovir using noncompartmental procedures. Descriptive statistics were applied to plasma concentrations and derived PK parameters for total and unbound letermovir. The least square means (LSM) of the log‐transformed primary parameters, AUCτ,ss and Css,max, for each renal function group was estimated with a linear mixed‐effects model, controlling for renal function as fixed effect and subject as a random effect. A 90% CI was constructed around the difference between the LSMs of test (moderate or severe renal impairment subjects) and reference (healthy subjects). The available demographic variables, including eGFR, age, weight and body mass index, were plotted against letermovir PK parameters fraction unbound, CLss/F and Vz/F and the five most informative relationships were explored posthoc by correlation analysis after the results of the main analysis became available. To evaluate the effect of renal impairment on letermovir pharmacokinetics more closely, the following correlations were assessed: fraction unbound vs. ln eGFR, ln CL/F vs. age (for bound and unbound letermovir), and ln CL/F vs. ln eGFR (for total and unbound letermovir). The correlation analyses were performed in SAS (version 9.3, SAS Institute Inc., Cary, NC, USA) by Kinesis Pharma B.V. (Breda, NL) using the REG procedure (PROC REG). Correlation plots were produced showing the individual data points, line of best fit including 90% CI, and 90% prediction interval. The intercept and slope of the correlation lines were reported, as well as the r2 values and the P‐values.
Results
Trial population
Between October 2012 and May 2013, a total of 24 Caucasian subjects, 14 (58.3%) men and 10 (41.1%) women, aged 51.0–76.0 years, were enrolled and completed the trial (Table 1). The arithmetic mean age was highest in the group with moderate renal impairment (72.5 years) compared with the healthy subject and severe renal impairment groups (63.8 years and 63.9 years, respectively). The dispositions and eGFR characteristics of the grouped individuals can also be seen in Table 1. None of the subjects had hepatic impairment as determined by the Child–Pugh classification (no hepatic encephalopathy and ascites in medical history/physical examination, as well as normal bilirubin, albumin and partial thromboplastin time values throughout the trial). Concomitant medication was judged to have no relevant influence on the letermovir plasma concentrations.
Table 1.
Trial population and demographic characteristics
| Renal impairment | |||||
|---|---|---|---|---|---|
| Demographic parameter | Healthy n = 8 | Moderate n = 8 | Severe n = 8 | All n = 16 | Total N = 24 |
| Sex, n (%) | |||||
| Male | 4 (50.0) | 5 (62.5) | 5 (62.5) | 10 (62.5) | 14 (58.3) |
| Female | 4 (50.0) | 3 (37.5) | 3 (37.5) | 6 (37.5) | 10 (41.7) |
| Ethnic origin, n (%) | |||||
| Caucasian | 8 (100) | 8 (100) | 8 (100) | 16 (100) | 24 (100) |
| Age, years | |||||
| Mean (SD) | 63.8 (3.0) | 72.5 (4.2) | 63.9 (8.5) | 68.2 (7.9) | 66.7 (6.9) |
| Median | 64.0 | 73.5 | 64.0 | 70.5 | 67.0 |
| Range | 59.0–68.0 | 64.0–76.0 | 51.0–75.0 | 51.0–76.0 | 51.0–76.0 |
| Body weight, kg | |||||
| Mean (SD) | 75.8 (8.2) | 82.5 (13.4) | 77.4 (6.1) | 79.9 (10.4) | 78.6 (9.7) |
| Median | 75.1 | 81.5 | 77.1 | 79.0 | 77.1 |
| Range (min–max) | 61.4–90.4 | 65.7–105.3 | 66.4–86.1 | 65.7–105.3 | 61.4–105.3 |
| BMI, kg/m2 | |||||
| Mean (SD) | 26.3 (3.0) | 27.2 (4.0) | 26.4 (3.1) | 26.8 (3.5) | 26.6 (3.3) |
| Median | 25.1 | 27.4 | 26.6 | 26.7 | 26.1 |
| Range (min–max) | 24.6–33.2 | 22.2–32.5 | 19.8–29.8 | 19.8–32.5 | 19.8–33.2 |
| eGFR, ml min–1 1.73m–2 | |||||
| Mean (SD) | 98.30 (6.66) | 39.85 (8.78) | 20.66 (5.11) | 30.25 (12.10) | 59.94 (34.39) |
| Median | 96.59 | 36.75 | 21.02 | 29.59 | 36.75 |
| Range (min–max) | 92.90–112.27 | 31.03–56.83 | 11.86–28.14 | 11.86–56.83 | 11.86–112.27 |
BMI, body mass index; eGFR, estimated glomerular filtration rate; max, maximum; min, minimum; SD, standard deviation.
Plasma PK of letermovir
Analysis of trough samples from Days 5–8 demonstrated that steady‐state was reached in healthy subjects and in both the moderate and severe renal impairment groups. The arithmetic mean concentration–time profiles at Day 8 are shown in Figure 1. In healthy subjects, as well as in patients with renal impairment, mean average total letermovir plasma concentrations started to increase immediately after dosing, reaching a maximal plasma concentration after approximately 1.5–2.0 h and decreasing thereafter. The arithmetic mean (± standard deviation) unbound fractions were increased from 0.00945 ± 0.00179 in the normal renal function group to 0.0105 ± 0.00101 and 0.0119 ± 0.00133 in subjects with moderate renal impairment and severe renal impairment, respectively. Thus, the mean unbound fractions in the moderate and severe impairment groups were approximately 11% and 26%, respectively, higher compared with mean unbound fraction in healthy subjects. PK parameters for total and unbound letermovir, as well as statistical comparison of each of the renal impairment groups compared with the healthy volunteer group are shown in Table 2. The exposure of renally impaired patients to letermovir was increased as indicated by the AUCτ,ss and Css,max values. This was also reflected in the LSM ratios that were increased for subjects with renal impairment. For total letermovir, Css,max was increased in the moderate impairment group but not the severe impairment group, whilst unbound letermovir was increased for both. AUCτ,ss was increased in both groups of subjects with renal impairment and for both total and unbound letermovir. In addition, arithmetic mean Css,av and t1/2,z were increased in the renal impairment groups, whilst clearance was decreased in comparison with the healthy subjects. These differences were greatest in the moderate renal impairment group, while the more variable data for the severe impairment group were, in general, not as altered in comparison with the healthy group. It is of note that no relevant change was detected for total protein and albumin after dosing compared with baseline in any of the trial participants.
Figure 1.
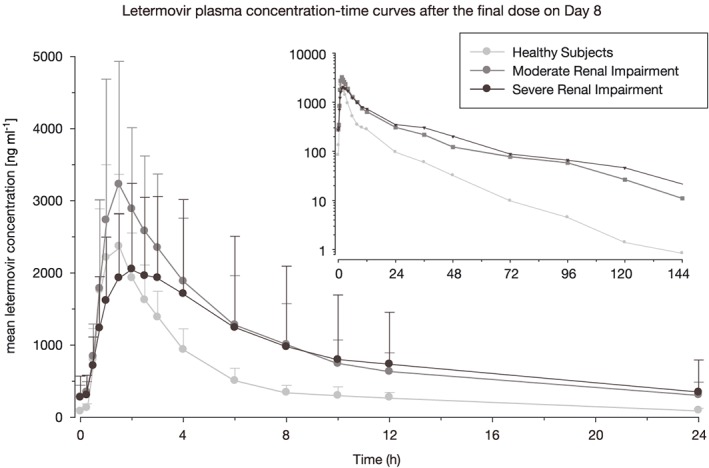
Letermovir total plasma concentration‐time curves after the final dose on Day 8
Table 2.
Plasma PK of letermovir in healthy and renally impaired subjects
| PK parameter | Renal impairment | ||
|---|---|---|---|
| Healthy | Moderate | Severe | |
| n = 8 | n = 8 | n = 8 | |
| Mean (SD), T max : median [range] ( total letermovir) | |||
| Css,max, ng ml–1 | 2614 (1042) | 3301 (1670) | 2714 (929.9) |
| tmax, h | 1.50 (1.00–2.50) | 1.51 (1.00–2.00) | 1.75 (1.00–4.00) |
| AUCτ,ss, ng.h ml–1 | 11 413 (3194) | 22 694 (9944) | 21 013 (17 919) |
| Css,av, ng ml–1 | 475.5 (133.1) | 945.6 (414.3) | 875.4 (746.7) |
| t½,z, h | 16.21 (7.705) | 25.95 (15.83) | 21.69 (9.295) |
| CL/F, L h–1 | 11.25 (3.090) | 6.024 (2.119) | 9.929 (7.124) |
| Vz/F, L | 261.4 (148.2) | 222.8 (142.4) | 314.3 (308.9) |
| Mean (SD) ( unbound letermovir) | |||
| Css,max, ng ml–1 | 24.20 (8.65) | 34.01 (15.01) | 32.07 (11.34) |
| AUCτ,ss, ng.h ml–1 | 107.8 (36.4) | 234.3 (93.3) | 253.2 (238.4) |
| Css,av, ng ml–1 | 4.492 (1.516) | 9.764 (3.888) | 10.55 (9.934) |
| LSM ratio (90% CI), % ( total letermovir) | |||
| Test 1 vs. reference 8 vs.8 | Test 2 vs. reference 8 vs. 8 | ||
| Css,max, ng ml–1 | 125.33 (86.54–181.50) | 106.11 (74.71–150.50) | |
| AUCτ,ss, ng.h ml–1 | 191.79 (142.58–257.98) | 142.02 (83.10–242.71) | |
| LSM ratio (90% CI), % ( unbound letermovir) | |||
| Css,max, ng ml–1 | 140.50 (98.41–200.60) | 134.90 (95.85–189.86) | |
| AUCτ,ss, ng.h ml–1 | 215.00 (158.77–291.14) | 180.56 (104.48–312.06) | |
AUCτ,ss, area under the curve over the dosing interval at steady state; CI, confidence interval; CL/F, apparent clearance; Css,av, average steady‐state concentration over the dosing interval; Css,max, maximal concentration observed at steady state; LSM, least squares mean; t½,z, termination half‐life; Tmax, time at which maximal concentration is observed; Vz/F, apparent volume of drug distribution at steady state.
The available demographic variables of eGFR, age, weight and body mass index were plotted against the following letermovir PK parameters: fraction unbound, apparent clearance at steady state and Vz/F. From these plots, significant relationships were described associated with ln eGFR vs. fraction unbound, ln clearance vs. age and ln clearance vs. ln eGFR. Decreasing ln eGFR correlated with increasing unbound fraction of letermovir (R2 = 0.5076, P < 0.0001). While the correlation analysis of ln clearance vs. ln eGFR (letermovir unbound; Figure 2) suggested a correlation between decreasing clearance and decreasing renal function determined by eGFR, this association was ultimately weak [R2 = 0.0662 (P = 0.2249) and 0.1861 (0.0353) for total and unbound clearance, respectively]. In addition, advanced age may be associated with the observed increases in exposure as shown by a weak correlation reported by the correlation analysis of ln clearance vs. age [letermovir unbound in Figure 3; R2 = 0.3548 (P = 0.0021) and 0.3166 (P = 0.0042) for total and unbound clearance, respectively].
Figure 2.
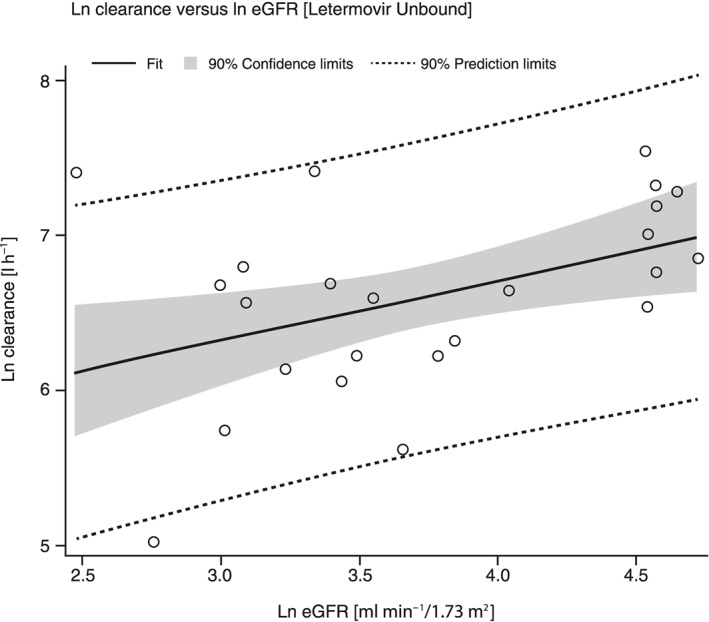
Correlation plot of Ln clearance of unbound letermovir vs. the ln eGFR of each patient
Figure 3.
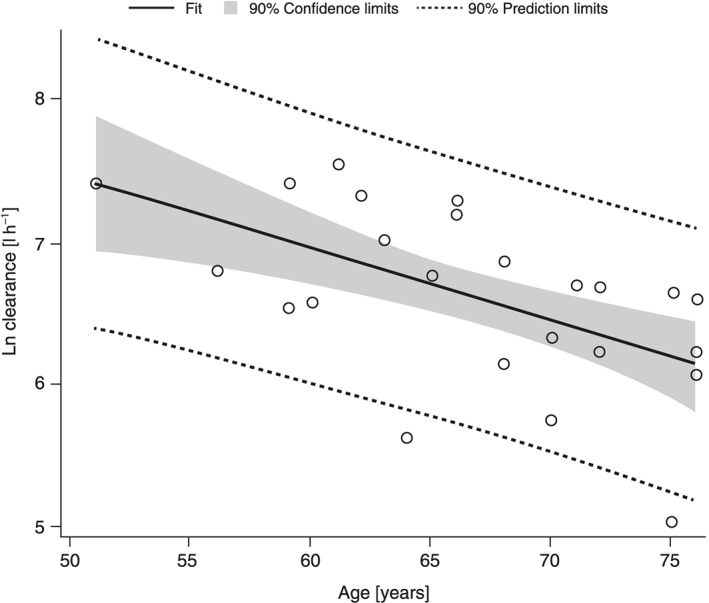
Correlation plot for Ln clearance of unbound letermovir vs. the age of each patient
Safety
All groups tolerated the multiple doses of letermovir 120 mg. Treatment‐emergent AEs (TEAEs) were reported in two subjects. The first subject from the moderate renal impairment group had a mild oedema of both lower legs. The TEAE started 2 days after the first dose of letermovir, lasted for 7 days and was not considered to be related to letermovir. The second subject from the severe renal impairment group had a severe acute ischaemia of the left lower leg. The TEAE started 7 days after the last dose of letermovir, and the subject was hospitalised 2 days later. Eight days after initial onset the event was resolved and not considered related to letermovir. The subject was known to have a vascular sclerosis and already received earlier aortofemoral stenting.
Clear baseline differences in mean haematology and clinical chemistry parameters were apparent between subject groups and consistent with underlying differences in renal insufficiency and chronic disease. Within each group of subjects, no relevant change of any of the parameters occurred.
Differences between subject groups were also apparent in baseline cardiac parameters (PR, QRS and QTcF) and were consistent with underlying differences in renal insufficiency. There were no apparent trends in changes from baseline in electrocardiogram parameters over time in any subject group.
Baseline vital sign parameters were comparable between subject groups, and there were no apparent trends in absolute values or changes from baseline in vital sign parameters in any subject group during the treatment period.
Discussion
In this Phase 1 trial, letermovir PK and safety profiles in both healthy and renally impaired subjects were compared. Overall, the results suggest that the administration of letermovir is suitable for patients with moderate or severe renal impairment. Throughout the trial, the investigated drug was well tolerated, although many PK parameters were mildly affected, as was exposure as identified by AUCτ,ss and Css,max. In the renally impaired groups, total exposure to the drug was increased by up to 92% for AUCτ,ss and 25% for Cmax. Following the findings of the study, the relationships between letermovir exposure and subject characteristics were further explored using correlation analyses. The correlation analyses performed suggested a weak correlation between decreased clearance and increased renal impairment, possibly contributing to the observation of greater exposure. However, the study was powered for the current statistical analysis and lacks sufficient power for further assessment using a covariate analysis.
Previously, letermovir was shown to be excreted primarily unchanged into the faeces and excretion into urine was negligible 22. Despite the apparent lack of excretion by the kidneys, renal impairment is known to potentially alter the PK profiles of nonrenally cleared drugs 23, 25, 27, 28. For many hepatically cleared drugs, the increase in drug exposure due to renal impairment is 1.5–3.0‐fold 23. The results of the current trial fall at the lower end of this range, suggesting that the effects of renal impairment on letermovir exposure are marginal. The role of renal impairment on letermovir PK, particularly the elimination half‐life, was very modest in comparison with the current standard of care: valganciclovir; the elimination half‐life of which has been shown to increase from 3.5 in subjects with normal renal function to 68.1 h in patients with severe renal impairment 29. The difference between these drugs is probably due to the elimination of valganciclovir via the kidneys in contrast to letermovir, which is primarily excreted via bile. In addition, the effect of severe renal impairment vs. healthy subjects on valganciclovir increased Cmax from 5800 ng ml–1 to 8500 ng ml–1, AUC0‐∞ from 28 100 ng.h ml–1 to 252 000 ng.h ml–1, and Tmax from 2 h to 4.3 h 29. In the present trial, Cmax, Tmax and AUC were less affected by severe renal impairment—total plasma letermovir geometric mean Cmax increasing from 2423 ng ml–1 to 2571 ng ml–1, median tmax from 1.50 h to 1.75 h, and geometric mean AUCτ,ss from 11 032 ng.h ml–1 to 15 668 ng.h ml–1 in healthy and severe impairment, respectively.
In addition to the weak correlation between increased renal impairment and decreased clearance, increasing age also demonstrated a weak correlation with decreased clearance. This feature has been previously described as a general effect of age due to decreased blood flow to the liver and, therefore, was to be expected 30. Interestingly, throughout the PK data, a trend towards the group with moderate impairment deviating further from the healthy group in comparison with the group with severe impairment was observed. Furthermore, greater variability in the data was associated with the severe impairment group in comparison with the other groups. Given this weak correlation between age and clearance, it may be that the effect of renal impairment is potentially confounded by the increased age of the moderately impairment group in comparison with the others. Furthermore, the greatest variability in age was in the severe impairment group, potentially contributing to the variability of results in this dataset. However, it must be noted that no liver insufficiency was determined for the subjects according to Child–Pugh scoring, which may suggest that the age‐related decreased hepatic blood flow may not entirely be responsible for the trends observed. Therefore, this potentially warrants further evaluation in greater detail using Phase 3 data.
In addition to the correlations between clearance and both renal function and age, there was also a correlation between decreased renal function and increased unbound letermovir. Letermovir binds to human serum albumin and human α1‐acid glycoprotein. The correlation between decreased renal function and increased unbound treatment has previously been observed with other drugs. This feature has been attributed to changes in plasma albumin and competition for protein binding by uraemic toxins present in subjects with renal impairment 31, 32, 33, 34.
Despite the observed increase in exposure and decrease in clearance associated with impaired kidney function, no AEs were attributed to increased drug exposure. Previously, higher doses of letermovir 240 mg were demonstrated to induce fewer AEs in a group of 34 patients compared with a placebo in patients receiving HSCT 20. Although the PK was not explored in this earlier trial, it appears that higher exposure due to either an initial 240 mg dose or 120 mg in renally impaired patients does not increase the incidence of AEs amongst subjects. The current trial demonstrated that multiple oral doses of once‐daily letermovir 120 mg are generally safe and well tolerated by both healthy subjects and subjects with moderate or severe renal impairment. This provides further reassurance of the suitability of this agent for use prophylactically or pre‐emptively amongst transplant patients. However, caution should be taken when applying doses > 120 mg in patients with renal impairment, such as the 240 mg being used in further clinical trials, as the subsequent exposure is unknown. In addition, comorbidities such as decreased hepatic function and drug–drug interactions should also be considered.
In this trial, a multiple‐dose design was used. According to current guidelines 25, a single‐dose design can be chosen when the investigated treatment is expected to exhibit dose‐linear and time‐independent PK in subjects with renal impairment and, thus, steady‐state PK can be predicted from single‐dose data. As letermovir demonstrates nonlinear PK following single doses in earlier trials, a multiple‐dose design was deemed suitable for the current trial.
Conclusions
In this Phase 1 trial, renal impairment was shown to mildly increase subject exposure to letermovir. Reduced clearance may be a factor; however, the correlation between eGFR and clearance was weak. Despite this observed increase in exposure, daily doses of letermovir 120 mg were well tolerated by subjects with a range of renal impairment, and no AEs were considered to be related to the drug. This preliminary evidence suggests that letermovir may be suitable for patients with impaired renal function in the future.
Competing Interests
Funding for this research was provided by AiCuris Anti‐infective Cures GmbH.
D.K., J.S., H.P.S., H.Z. and H.R.S. are current or former employees of AiCuris Anti‐infective Cures GmbH. E.H. is a former employee of Merck Sharp & Dohme, the Netherlands. A.v.S. is a former employee of, and currently working as a consultant for, Merck, Sharp & Dohme, The Netherlands. K.E. was a consultant for AiCuris Anti‐infective Cures GmbH. A.H. is an employee of CRS Clinical Research Services Kiel GmbH, Kiel, Germany.
Medical writing assistance was provided by Edward Rochford, PhD, of Complete Medical Communications, Hackensack, NJ, USA. This assistance was funded by Merck & Co., Inc., Kenilworth, NJ, USA.
Contributors
D.K. contributed to study design, data acquisition and analysis, interpretation of the results, drafting of the manuscript, and critical review of the manuscript. J.S. contributed to study design, interpretation of the results, drafting of the manuscript, and critical review of the manuscript. K.E. contributed to study design, interpretation of the results, and critical review of the manuscript. A.H. contributed to study design, data acquisition and analysis, interpretation of the results, and critical review of the manuscript. H.P.S. contributed to study design, data analysis, interpretation of the results, and critical review of the manuscript. E.H. contributed to interpretation of the results and critical review of the manuscript. A.v.S. contributed to interpretation of the results and critical review of the manuscript. H.Z. contributed to study design, data analysis, interpretation of the results, and critical review of the manuscript. H.R.S. contributed to study design, data analysis, interpretation of the results, drafting of the manuscript, and critical review of the manuscript.
Kropeit, D. , Scheuenpflug, J. , Erb‐Zohar, K. , Halabi, A. , Stobernack, H.‐P. , Hulskotte, E. G. J. , van Schanke, A. , Zimmermann, H. , and Rübsamen‐Schaeff, H. (2017) Pharmacokinetics and safety of letermovir, a novel anti‐human cytomegalovirus drug, in patients with renal impairment. Br J Clin Pharmacol, 83: 1944–1953. doi: 10.1111/bcp.13292.
European Clinical Trial Registry no.: 2012‐003104‐12
References
- 1. Beam E, Razonable RR. Cytomegalovirus in solid organ transplantation: epidemiology, prevention, and treatment. Curr Infect Dis Rep 2012; 14: 633–641. [DOI] [PubMed] [Google Scholar]
- 2. Boeckh M. Current antiviral strategies for controlling cytomegalovirus in hematopoietic stem cell transplant recipients: prevention and therapy. Transpl Infect Dis 1999; 1: 165–178. [DOI] [PubMed] [Google Scholar]
- 3. Ljungman P, Hakki M, Boeckh M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Infect Dis Clin North Am 2010; 24: 319–337. [DOI] [PubMed] [Google Scholar]
- 4. Konoplev S, Champlin RE, Giralt S, Ueno NT, Khouri I, Raad I, et al. Cytomegalovirus pneumonia in adult autologous blood and marrow transplant recipients. Bone Marrow Transplant 2001; 27: 877–881. [DOI] [PubMed] [Google Scholar]
- 5. Ljungman P. Cytomegalovirus pneumonia: presentation, diagnosis, and treatment. Semin Respir Infect 1995; 10: 209–215. [PubMed] [Google Scholar]
- 6. Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: The national health and nutrition examination surveys, 1988‐2004. Clin Infect Dis 2010; 50: 1439–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol 2010; 20: 202–213. [DOI] [PubMed] [Google Scholar]
- 8. Melendez DP, Razonable RR. Letermovir and inhibitors of the terminase complex: A promising new class of investigational antiviral drugs against human cytomegalovirus. Infect Drug Resist 2015; 8: 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jacobsen T, Sifontis N. Drug interactions and toxicities associated with the antiviral management of cytomegalovirus infection. Am J Health Syst Pharm 2010; 67: 1417–1425. [DOI] [PubMed] [Google Scholar]
- 10. Salzberger B, Bowden RA, Hackman RC, Davis C, Boeckh M. Neutropenia in allogeneic marrow transplant recipients receiving ganciclovir for prevention of cytomegalovirus disease: Risk factors and outcome. Blood 1997; 90: 2502–2508. [PubMed] [Google Scholar]
- 11. Marty FM, Winston D, Rowley SD, Boeckh M, Vance E, Papanicolaou G, et al. CMX001 for prevention and control of CMV infection in CMV‐seropositive allogeneic stem‐cell transplant recipients: A phase 2 randomized, double‐blind, placebo‐controlled, dose‐escalation trial of safety, tolerability and antiviral activity. Biol Blood Marrow Transplant 2012; 18: S203–S204. [Google Scholar]
- 12. Chimerix . Chimerix announces top‐line results from phase 3 SUPPRESS trial of Brincidofovir. Available at http://ir.chimerix.com/releasedetail.cfm?releaseid=948172. Updated 2015 (last accessed 18 February 2016).
- 13. Buerger I, Reefschlaeger J, Bender W, Eckenberg P, Popp A, Weber O, et al. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J Virol 2001; 75: 9077–9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reefschlaeger J, Bender W, Hallenberger S, Weber O, Eckenberg P, Goldmann S, et al. Novel non‐nucleoside inhibitors of cytomegaloviruses (BAY 38‐4766): in vitro and in vivo antiviral activity and mechanism of action. J Antimicrob Chemother 2001; 48: 757–767. [DOI] [PubMed] [Google Scholar]
- 15. Weber O, Bender W, Eckenberg P, Goldmann S, Haerter M, Hallenberger S, et al. Inhibition of murine cytomegalovirus and human cytomegalovirus by a novel non‐nucleosidic compound in vivo . Antiviral Res 2001; 49: 179–189. [DOI] [PubMed] [Google Scholar]
- 16. Kaul DR, Stoelben S, Cober E, Ojo T, Sandusky E, Lischka P, et al. First report of successful treatment of multidrug‐resistant cytomegalovirus disease with the novel anti‐CMV compound AIC246. Am J Transplant 2011; 11: 1079–1084. [DOI] [PubMed] [Google Scholar]
- 17. Stoelben S, Arns W, Renders L, Hummel J, Muhlfeld A, Stangl M, et al. Preemptive treatment of cytomegalovirus infection in kidney transplant recipients with letermovir: results of a phase 2a study. Transpl Int 2014; 27: 77–86. [DOI] [PubMed] [Google Scholar]
- 18. Goldner T, Hewlett G, Ettischer N, Ruebsamen‐Schaeff H, Zimmermann H, Lischka P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol 2011; 85: 10884–10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lischka P, Hewlett G, Wunberg T, Baumeister J, Paulsen D, Goldner T, et al. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob Agents Chemother 2010; 54: 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chemaly RF, Ullmann AJ, Stoelben S, Richard MP, Bornhäuser M, Groth C, et al. Letermovir for cytomegalovirus prophylaxis in hematopoietic‐cell transplantation. N Engl J Med 2014; 370: 1781–1789. [DOI] [PubMed] [Google Scholar]
- 21. Marty FM, Ljungman PT, Chemaly RF, Maertens JA, Snydman DR, Duarte RF, et al. A phase III randomized, double‐blind, placebo‐controlled trial of Letermovir (LET) for prevention of cytomegalovirus (CMV) infection in adult CMV‐seropositive recipients of allogeneic hematopoietic cell transplantation (HCT). Paper presented at the BMT Tandem Meetings 2017, 2017.
- 22. AiCuris Anti‐infective Cures GmbH . Unpublished data on file. 2009.
- 23. Yeung CK, Shen DD, Thummel KE, Himmelfarb J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int 2014; 85: 522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. FDA . Guidance for industry: pharmacokinetics in patients with impaired renal function — Study design, data analysis, and impact on dosing and labeling. 2010.
- 25. European Medicines Agency . Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/02/WC500200841.pdf. Updated 2015 (last accessed 16 August 2016).
- 26. AiCuris Anti‐infective Cures GmbH . Unpublished data on file. 2005.
- 27. Naud J, Michaud J, Leblond FA, Lefrancois S, Bonnardeaux A, Pichette V. Effects of chronic renal failure on liver drug transporters. Drug Metab Dispos 2008; 36: 124–128. [DOI] [PubMed] [Google Scholar]
- 28. Touchette MA, Slaughter RL. The effect of renal failure on hepatic drug clearance. DICP 1991; 25: 1214–1224. [DOI] [PubMed] [Google Scholar]
- 29. Czock D, Scholle C, Rasche FM, Schaarschmidt D, Keller F. Pharmacokinetics of valganciclovir and ganciclovir in renal impairment. Clin Pharmacol Ther 2002; 72: 142–150. [DOI] [PubMed] [Google Scholar]
- 30. Wynne HA, Goudevenos J, Rawlins MD, James OF, Adams PC, Woodhouse KW. Hepatic drug clearance: The effect of age using indocyanine green as a model compound. Br J Clin Pharmacol 1990; 30: 634–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fujita KI, Masuo Y, Okumura H, Watanabe Y, Suzuki H, Sunakawa Y, et al. Increased plasma concentrations of unbound SN‐38, the active metabolite of irinotecan, in cancer patients with severe renal failure. Pharm Res 2016; 33: 269–282. [DOI] [PubMed] [Google Scholar]
- 32. Hooper WD, Bochner F, Eadie MJ, Tyrer JH. Plasma protein binding of diphenylhydantoin. Effects of sex hormones, renal and hepatic disease. Clin Pharmacol Ther 1974; 15: 276–282. [DOI] [PubMed] [Google Scholar]
- 33. Roberts JA, Pea F, Lipman J. The clinical relevance of plasma protein binding changes. Clin Pharmacokinet 2013; 52: 1–8. [DOI] [PubMed] [Google Scholar]
- 34. Lalande L, Charpiat B, Leboucher G, Tod M. Consequences of renal failure on non‐renal clearance of drugs. Clin Pharmacokinet 2014; 53: 521–532. [DOI] [PubMed] [Google Scholar]