

Abstract
Aims
The currently licensed seasonal trivalent influenza vaccines contain 15 μg haemagglutinin per strain for adult, and up to 60 μg for elderly patients. However, due to recent shortages, dose sparing to increase production capacity would be highly desirable. In the present study, we attempted to find a dose–response relationship for immunogenicity and, thus, the optimal dose for seasonal influenza vaccines in adult and elderly patients.
Methods
A total of 256 subjects, including adult (aged 18–60 years) and elderly (aged over 60 years) individuals, were enrolled. Subjects were randomly assigned in a 1:1:1:1 ratio to receive a whole‐virion, aluminium‐adjuvanted trivalent influenza vaccine containing 3.5, 6, 9 or 15 μg haemagglutinin of seasonal A/H1N1, A/H3N2 and B influenza antigens manufactured by Omninvest Ltd., Hungary. Serum antibody titres against the vaccine virus strains were measured by haemagglutination inhibition.
Result
All vaccines were well tolerated. All four vaccines fulfilled all three immunogenicity licensing criteria, as determined by the European Committee for Proprietary Medicinal Products (CPMP)/Biotechnology Working Party (BWP)/214/96 guideline for all three virus strains and both age groups. The 3.5 μg vaccine showed 28% less seroconversion compared to the 15 μg dose in terms of influenza AH3N2 in the adult group (95% confidence interval –51, −3; P < 0.05). All other doses showed no significant difference in immunogenicity compared with the licensed vaccine containing 15 μg haemagglutinin.
Conclusions
Our data suggested that significant dose sparing is possible with the use of whole‐virion vaccines and aluminium adjuvants, without compromising safety. This could have significant economic and public health impacts.
Keywords: adverse events, dose response, economics, immunology, safety, vaccine
What is Already Known about this Subject
The currently licensed seasonal trivalent influenza vaccines are split‐virion or subunit vaccines containing only viral surface glycoproteins, and include 15 μg haemagglutinin (HA) per virus strain for adult and up to 60 μg for elderly patients.
What this Study Adds
The study addressed the possibility of a dose reduction in adult and elderly patients, and attempted to establish the optimal dose.
The study showed that by using whole‐virion vaccines and alum adjuvants, a significant dose reduction is possible in adult and even elderly patients.
Based on our results, a new, reduced‐dose seasonal vaccine is being licensed in the European Union member state of Hungary, which will have an impact on production capacities and, ultimately, vaccine availability to patients.
Introduction
The currently licensed seasonal trivalent influenza vaccines are split‐virion or subunit vaccines containing only viral surface glycoproteins, and include 15 μg haemagglutinin (HA) per virus strain for adult and up to 60 μg for elderly patients 1, 2, 3. However, due to recurring vaccine shortages, such as during the 2009–2010 influenza pandemic and previous shortages of seasonal influenza vaccines, dose sparing to increase production capacity would be highly desirable 4.
During the 2009–2010 influenza pandemic, our group demonstrated that when using a monovalent, aluminium‐adjuvanted whole‐virion vaccine, just 6 μg HA is sufficient to produce an immunogenic vaccine 5. Furthermore, we have recently shown that a whole‐virion, aluminium phosphate‐adjuvanted seasonal trivalent influenza vaccine is safe and immunogenic at a dose of only 6 μg HA per strain – not only adult, but also in elderly patients 6. In the present study, we attempted to find a dose–response relationship for immunogenicity and, thus, to establish the optimal dose for seasonal trivalent influenza vaccines in adult and elderly patients.
Methods
Vaccines
The vaccine strains were influenza A/California/7/2009(H1N1)‐derived NYMC X‐179A reassortant, A/Perth/16/2009(H3N2)‐like A/Victoria/210/2009(H3N2)‐derived NYMC X‐187 reassortant and B/Brisbane/60/2008‐derived NYMC BX‐35 reassortant, chosen according to the European Union (EU) recommendations for the seasonal influenza vaccine composition for the season 2011–2012 7. The strains were grown in embryonated hen eggs, inactivated by formaldehyde, purified and concentrated, and absorbed on aluminium phosphate gel. All vaccines were manufactured according to good manufacturing practice requirements by Omninvest Ltd., Hungary, as described in detail previously 8. Four different concentrations were prepared as follows: 0.5 ml of the vaccine contained 3.5 μg (Lot number: FL‐K‐01/11), 6 μg (Lot number: FL‐K‐02/11), 9 μg (Lot number:FL‐K‐03/11) or 15 μg (Lot number: FL‐K‐04/11) HA per strain. The 15 μg dose met the requirements of the European Agency for the Evaluation of Medicinal Products for seasonal influenza vaccines, and was licensed for the season 2011–2012 in Hungary under licence number OGYI‐T‐8998 by the National Institute of Pharmacy, and administered safely in 1.3 million cases 9.
Prior to the addition of the aluminium phosphate adjuvant, the HA content of the vaccines was measured using the single radial immunodiffusion test, using reagents provided by the National Institute for Biological Standards and Control (NIBSC), UK, as described previously 10. Purity was assessed by measuring the endotoxin content, which was <0.05 IU per dose, and the amount of ovalbumin, which was <5 ng per dose. Both values are considerably lower than the concentrations considered acceptable (100 IU and 1000 ng per dose, respectively) by the European Pharmacopoeia 11. Aluminium phosphate was used as an adjuvant at a quantity of 0.33 mg per ampoule, and merthiolate was added as preservative (0.1 mg ml–1), meeting the requirements of the European Pharmacopoeia 11.
Study settings and trial design
Between 5 September and 17 October 2011, we completed a prospective, multicentre, randomized, double‐blind clinical trial at four state‐run primary care centres in Budapest, Hungary, following a multi‐arm parallel design.
Outcomes
The first of the two prespecified immunogenicity objectives was to assess the immunogenicity of one 0.5 ml intramuscular injection of four trivalent seasonal influenza vaccines containing either 3.5, 6, 9 or 15 μg HA of seasonal A/H1N1, A/H3N2 and B influenza antigens, as measured by haemagglutination inhibition (HI) test 21 days after vaccination, in compliance with the requirements of the current EU and US Food and Drug Administration (FDA) recommendations (objective A) 9, 12. The second objective was to determine the dose–effect relationship between the above four vaccines by means of the immune response provoked 21 days after vaccination, in terms of pre‐ and post‐immunization HA titres, as measured by HI (objective B).
The safety objective was to evaluate the tolerability and safety of the administration of one 0.5 ml intramuscular injection of four trivalent influenza vaccines containing either 3.5, 6, 9 or 15 μg HA of seasonal A/H1N1, A/H3N2 and B influenza antigens, as determined by the licensing requirements of the EU 9.
Participants
Participants were recruited by their primary care physicians. The planned enrolment was 256 subjects overall. A total of 256 healthy volunteers were screened, all of whom were found to be eligible, and were enrolled to be vaccinated, with 127 adult and 129 elderly subjects. A written informed consent was obtained from all participants. A negative pregnancy test on day 0 was required for all females of childbearing age, and the use of an acceptable contraception method was required for the duration of the study. Exclusion criteria included immunodeficiency, a history of Guillain–Barré syndrome, disease states that might have affected immune reactivity [e.g. malignancies, chronic infections (HIV or hepatitis B or C), uncontrolled diabetes mellitus or autoimmune diseases], use of immunosuppressive medications, conditions precluding compliance, receipt of any vaccines 28 days prior to the study, use of influenza vaccines within the previous 6 months, use of investigational agents within the previous 30 days, having received any blood products or immunoglobulins in the past 6 months, acute febrile illness 1 week prior to vaccination, breast feeding and hypersensitivity to any of the vaccine components. All subjects were of Caucasian ethnicity.
Randomization
Based on the stratified randomization method, subjects were stratified according to age: adult (aged 18–60 years) and elderly (aged >60 years) individuals. The subjects were randomly assigned to one of the groups in a 1:1:1:1 ratio between the centres using http://randomization.com. The groups were as follows: Group 1 received the trivalent influenza vaccine containing 3.5 μg of HA per strain; Group 2 received the trivalent influenza vaccine containing 6 μg HA per strain; Group 3 received the trivalent influenza vaccine containing 9 μg HA per strain; and Group 4 received the trivalent influenza vaccine containing 15 μg HA per strain. All four vaccine groups had two subgroups, for adult (aged 18–60 years) and elderly (aged over 60 years) subjects.
The sequence was generated by a statistician who was not involved in the rest of the study. Assignments were enclosed in sequentially numbered, identical sealed envelopes. The randomization code was provided to the vaccine administrator, who was aware of study group assignments. Blinding was maintained as all subjects and investigators who participated in the assessment of safety or immunogenicity were unaware of the assignments. All vaccines had a similar appearance as they were clear solutions of equal volume. Assignment of subjects to vaccination groups was performed by means of stratified blocked randomization using http://randomization.com.
The study conditions were in compliance with the Declaration of Helsinki 13, and the recommendations and guidelines as published in the International Conference on Harmonization Harmonized Tripartite Guideline for Good Clinical Practice 14. The study protocol, the patient information sheet, the informed consent form and all other appropriate study‐related documents were reviewed by the Ethics Committee for Clinical Pharmacology of the Medical Research Council and approved by the National Institute of Pharmacy of Hungary, and is available upon request. All volunteers signed the patient information sheet and the informed consent form. The study was registered with the EU drug regulatory authorities and the European Medicines Agency (EMA) (https://eudract.ema.europa.eu/) under clinical trial registration number 2011–003166‐32.
Laboratory tests
Serum antibody titres against the vaccine virus strain were measured in duplicate by HI, using chicken red blood cells and following standard procedures 15. All serological tests were performed in duplicate; where there was a discrepancy, a third test was performed. The coefficient of variation for the tests was 4.5%. All serological tests were performed at a central laboratory (Department of Virology, National Centre for Epidemiology, Budapest, Hungary).
Interventions
Baseline evaluations on day 0 included obtaining demographic data and medical history, and performing a complete physical examination. Blood samples were drawn for baseline HI for all three vaccine virus strains. All groups received 0.5 ml of the vaccine assigned to them by injection into the deltoid muscle. On day 21, a medical history and the list of medications used during the days since the last visit were obtained, a physical examination was performed and blood samples were drawn for HI. Safety variables were collected at the follow‐up visits by taking a history and performing a physical examination, and by telephone interviews on days 1, 2, 3 and 7. In addition, subjects were asked to take their temperature on days 1, 2 and 3, with thermometers supplied by the study centre. The definition of fever was a temperature >38°C (100.4°F) orally. Diary cards were provided, and patients were requested to call if any side effects arose. All vaccinations took place in the period 5–17 October 2011. No interim analysis of data from the trial was planned or performed.
Statistics
All data analyses were carried out according to a pre‐established analysis plan. Safety and immunogenicity were prospectively identified as co‐primary objectives. A sample size of 32 per age group per vaccine group gives a power of 70% to detect a twofold difference in the ratio of geometric mean titres (GMT) between Groups 1, 2 and 3 separately vs. Group 4, with a one‐sided alpha value of 10%, assuming a geometric standard deviation of 3.17 [exp(mean)] of the standard deviations of the 21‐day postvaccination log(titres) of the three different strains, measured during the official yearly licensing study containing the same virus strains as the current study vaccines for the seasonal influenza vaccine used in Hungary, performed according to the CPMP/BWP/214/96 guidelines 9. The calculation was performed using the power calculation method specified by Dunnett et al. 16
We gave HI titres below the limit of detection (1:10) an arbitrary intermediate value of one in five. The geometric mean of duplicate results for each specified time was used for the calculation. The immunogenicity study outcomes were assessed according to EU Committee for Human Medicinal Products (CHMP) requirements concerning seasonal influenza vaccines as follows: (i) postvaccination seropositivity rate (percentage of subjects with titres of ≥40, the titre required by CHMP); (ii) the post‐ to prevaccination GMT ratio; and (iii) the proportion of subjects seroconverting – that is, displaying a ≥4‐fold titre increase postvaccination – and postvaccination titres of at least 1:40 9. The HI titre distributions were described with reverse cumulative distribution curves and were tested using the nonparametric Kruskal–Wallis test as appropriate to test for differences between groups. A P‐value of <0.05 was considered significant. A step‐down Dunnett test procedure was performed on 21 post vaccination day 21 GMTs, comparing Group 4 (15 μg HA) to Groups 1, 2 and 3 (3.5 μg HA, 6 μg HA and 9 μg HA, respectively).
Simultaneous two‐sided 95% confidence intervals (CIs) were calculated for the differences between percentages of subjects achieving seroconversion or a significant increase in antibody titre between Group 4 and Groups 1, 2 and 3 (3.5 μg HA, 6 μg HA and 9 μg HA, respectively) with Dunnett adjustment, and a single‐step multiple comparison was performed 17, where seroconversion is defined as the percentage of subjects with either a prevaccination HI titre <1:10 and a postvaccination HI titre >1:40, or a prevaccination HI titre >1:10 and a minimum fourfold rise in postvaccination HI antibody titre 9, 12 The calculation was performed using the R package binMto (asymptotic simultaneous confidence intervals for many‐to‐one comparisons of proportions), using function ‘Confidence intervals for many‐to‐one comparisons of proportions’. The statistical null hypothesis regarding the dose–effect relationships was that for all three vaccine virus strains, seroconversion % (3.5 μg vaccine) – seroconversion % (15 μg vaccine) ≥ 0; seroconversion % (6 μg vaccine) – seroconversion % (15 μg vaccine) ≥ 0; and seroconversion % (9 μg vaccine) – seroconversion % (15 μg vaccine) ≥ 0. Similarly, for post‐/prevaccination GMT titre ratios, log (GMT ratio 9 μg vaccine) – log(GMT R 15 μg vaccine)≥ 0; log(GMT R 6 μg vaccine) – log(GMT R 15 μg vaccine) ≥ 0 and log(GMT R 3.5 μg vaccine) – log(GMT R 15 μg vaccine) ≥ 0.
The drug nomenclature used conforms to the Br J Pharmacol's Concise Guide to Pharmacology 2015/16 18.
Results
All 256 participants entering the study were vaccinated as planned, and provided safety information intention to treat (IT) (Figure 1). Their data were used for the safety evaluation. A total of 254 subjects attended the last control visit on Days 21–28. Their data were used for immunogenicity evaluation per protocol (PP) (Table 1). There were no significant differences between the groups in terms of age, ethnicity, previous influenza vaccination status or baseline HI titres to any of the vaccine virus strains.
Figure 1.
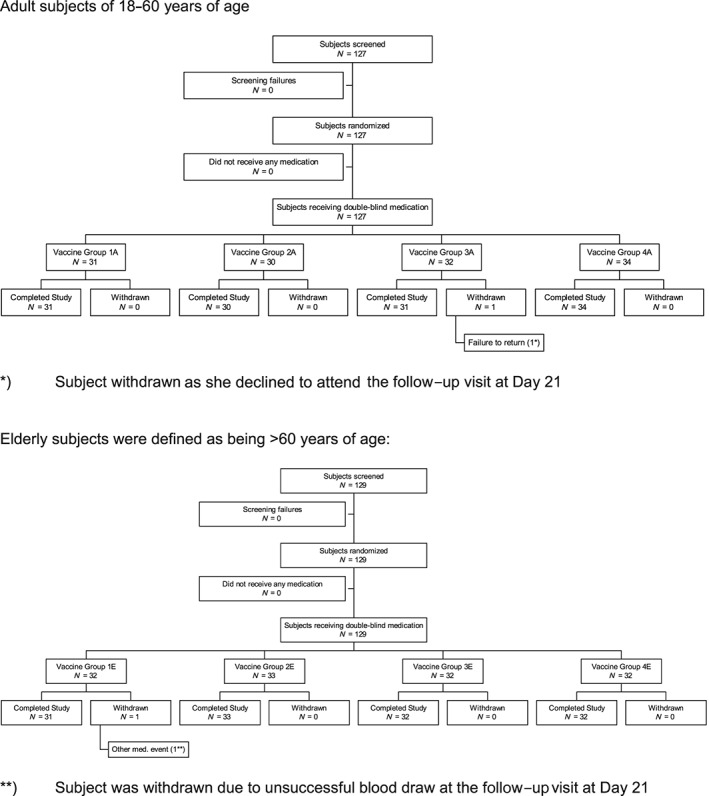
Study flowchart
Table 1.
Demographic characteristics of the study population
| Population | n | Mean age ± SD (years) | Median (years) | Min. (years) | Max. (years) |
|---|---|---|---|---|---|
| Vaccine group 1 (3.5 μg) | |||||
| Total | 62 | 57.9 ± 17.6 | 60 | 18 | 90 |
| 18–60 years | 31 | 43.7 ± 12.5 | 49 | 18 | 59 |
| 60+ years | 31 | 72.0 ± 7.9 | 70 | 61 | 90 |
| Males | 24 | 56.2 ± 16.4 | 59 | 25 | 86 |
| Females | 38 | 58.9 ± 18.5 | 62 | 18 | 90 |
| Vaccine group 2 (6 μg) | |||||
| Total | 63 | 59.1 ± 14.0 | 60 | 27 | 85 |
| 18–60 years | 30 | 47.0 ± 8.8 | 47 | 27 | 59 |
| 60+ years | 33 | 70.1 ± 6.8 | 70 | 60 | 85 |
| Males | 28 | 58.5 ± 14.4 | 62 | 27 | 77 |
| Females | 35 | 59.6 ± 13.8 | 58 | 36 | 85 |
| Vaccine group 3 (9 μg) | |||||
| Total | 63 | 59.5 ± 15.4 | 60 | 25 | 85 |
| 18–60 years | 31 | 46.8 ± 11.0 | 48 | 25 | 59 |
| 60+ years | 32 | 71.8 ± 6.5 | 72 | 60 | 85 |
| Males | 24 | 57.6 ± 15.8 | 59 | 25 | 85 |
| Females | 39 | 60.6 ± 15.3 | 62 | 25 | 85 |
| Vaccine group 4 (15 μg) | |||||
| Total | 66 | 55.6 ± 17.9 | 58 | 18 | 86 |
| 18–60 years | 34 | 41.0 ± 11.3 | 40 | 18 | 59 |
| 60+ years | 32 | 71.0 ± 7.6 | 70 | 60 | 86 |
| Males | 20 | 51.0 ± 16.8 | 55 | 21 | 79 |
| Females | 46 | 57.6 ± 18.2 | 62 | 18 | 86 |
| All subjects | |||||
| Total | 254 | 58.0 ± 16.3 | 60 | 18 | 90 |
| 18–60 years | 126 | 44.5 ± 11.2 | 46 | 18 | 59 |
| 60+ years | 128 | 71.2 ± 7.2 | 71 | 60 | 90 |
| Males | 96 | 56.1 ± 15.8 | 59 | 21 | 86 |
| Females | 158 | 59.1 ± 16.6 | 61 | 18 | 90 |
Max, maximum; Min, minimum; SD, standard deviation
Immunogenicity
There was no statistical null hypothesis regarding the immunogenicity objective ‘A’, which was analysed descriptively. Our descriptive statistical analysis showed that all four vaccines with 3.5, 6, 9 and 15 μg HA per strain fulfilled all three CHMP immunogenicity criteria for the evaluation of seasonal influenza vaccines, as determined in the CPMP/BWP/214/96 guideline, in terms of all three vaccine virus strains and both age groups, 21 days after vaccination (Table 2) 9.
Table 2.
Immunogenicity findings for each vaccine dose and age groups in face of EU Committee for Human Medicinal Products licensing criteria, with 95% confidence interval, where applicable 9
| Age group (years) | Strain | CPMP criterion | Values measured on Day 21 | |||
|---|---|---|---|---|---|---|
| 3.5 μg | 6 μg | 9 μg | 15 μg | |||
| 18–60 | H1N1 | i) >40% | 67.7% | 60.0% | 58.1% | 64.7% |
| ii) >2.5 | 4.045 (3.070; 5.328) | 3.482 (2.627, 4.614) | 5.115 (3.125, 8.371) | 4.429 (3.127, 6.271) | ||
| iii) >70% | 93.6% | 100.0% | 96.8% | 97.1% | ||
| H3N2 | i) >40% | 45.2% | 56.7% | 64.5% | 73.5% | |
| ii) >2.5 | 3.616 (2.348, 5.571) | 4.046 (2.708, 6.044) | 4.471 (3.128, 6.391) | 4.474 (3.226, 6.206) | ||
| iii) >70% | 90.3% | 100.0% | 100.0% | 100.0% | ||
| B | i) >40% | 54.8% | 60.0% | 45.2% | 61.8% | |
| ii) >2.5 | 4.679 (3.057, 7.160) | 3.442 (2.551, 4.644) | 4.138 (2.764, 6.196) | 4.252 (2.940, 6.148) | ||
| iii) >70% | 93.6% | 96.7% | 87.1% | 100.0% | ||
| 60+ | H1N1 | i) >30% | 54.8% | 60.6% | 53.1% | 62.5% |
| ii) >2.0 | 3.344 (2.294, 4.876) | 4.489 (2.940, 6.854) | 4.756 (3.050, 7.418) | 5.021 (3.206, 7.862) | ||
| iii) >60% | 93.6% | 93.9% | 90.6% | 100.0% | ||
| H3N2 | i) >30% | 48.4% | 63.6% | 59.4% | 62.5% | |
| ii) >2.0 | 3.093 (2.080, 4.598) | 4.731 (3.034, 7.378) | 4.705 (3.048, 7.262) | 5.076 (3.310, 7.784) | ||
| iii) >60% | 96.8% | 97.0% | 100.0% | 96.9% | ||
| B | i) >30% | 45.2% | 48.5% | 46.9% | 53.1% | |
| ii) >2.0 | 2.674 (1.942, 3.683) | 3.678 (2.271, 5.956) | 3.629 (2.239, 5.881) | 3.589 (2.628, 4.902) | ||
| iii) >60% | 80.7% | 93.9% | 87.5% | 84.4% | ||
Each value met the licensing criteria, as follows. For adult subjects: (i) the number of seroconversions or significant (≥4‐fold) increases in HI antibody titre should be >40%; (ii) the postvaccination/prevaccination geometric mean titre ratio (increase) should be >2.5‐fold; and (iii) the proportion of subjects achieving an HI titre of ≥40 should be >70%. For elderly subjects: (i) the number of seroconversions or significant (i.e. ≥4‐fold) increases in HI antibody titre should be >30%; (ii) the postvaccination/prevaccination geometric mean titre ratio (increase) should be >2.0‐fold; and (iii) the proportion of subjects achieving an HI titre of ≥40 should be >60% 9. CPMP, European Committee for Proprietary Medicinal Products
For the experimental vaccines with 6 μg and 9 μg HA per strain, there were no significant differences in terms of any of the CHMP licensing immunogenicity criteria, including postvaccination titres, seroconversion or a significant increase in antibody titres, and post‐/prevaccination GMT ratios measured 21 days after vaccination, in any of the vaccine virus strains, compared with the licensed vaccine with 15 μg HA per strain, in either the adult or elderly patient groups.
When comparing the experimental vaccine with 3.5 μg HA per strain with the licensed vaccine with 15 μg HA per strain, there were no significant differences between any of the immunogenicity licensing criteria regarding any of the vaccine strains, with the exception of one aspect. A significant difference was found between the proportion of subjects showing seroconversion or a significant increase in antibody titres 21 days after vaccination for the vaccine strain H3N2 in the age group 18–60 years as follows: the proportion of subjects seroconverted with 3.5 μg – the proportion of subjects seroconverted with 15 μg = −0.28 (95% CI –0.51, −0.03). Interestingly, in the age group >60 years and for other outcomes, no significant differences were found between the 3.5 μg and 15 μg dose groups (Table 3).
Table 3.
The number of adverse reactions observed during the study in each vaccine dose group. Data are shown as n (%)
| Reaction n (%) | Group 3.5 μg (n = 63) | Group: 6 μg (n = 63) | Group: 9 μg (n = 64) | Group: 15 μg, licensed vaccine (n = 66) |
|---|---|---|---|---|
| Chills | 1 (1.6%) | 3 (4.8%) | 2 (3.1%) | 0 |
| Headache | 4 (6.3%) | 1 (1.6%) | 4 (6.3%) | 2 (3%) |
| Malaise | 5 (7.9%) | 3 (4.8%) | 3 (4.7%) | 3 (4.5%) |
| Myalgia | 4 (6.3%) | 1 (1.6%) | 5 (7.8%) | 2 (3%) |
| Pain in extremity | 0 | 0 | 0 | 1 (1.5) |
| Pyrexia | 2 (3.2%) | 2 (3.2%) | 4 (6.3%) | 0 |
| Vaccination site discolouration | 0 | 1 (1.6%) | 0 | 0 |
| Vaccination site erythema | 5 (7.9%) | 7 (11.1%) | 2 (3.1% | 4 (6.1%) |
| Vaccination site haematoma | 1 (1.6%) | 0 | 0 | 0 |
| Vaccination site pain | 15 (23.8%) | 16 (25.4%) | 17 (26.6%) | 12 (18.2%) |
| Vaccination site rigidity | 5 (7.9%) | 7 (11.1%) | 7 (10.9%) | 5 (7.6%) |
| Vaccination site swelling | 4 (6.3%) | 8 (12.7%) | 11 (17.2%) | 5 (7.6%) |
Safety
Administration of all investigational vaccines was well tolerated by the study participants. No clinically significant changes were observed in the physical condition or vital signs of the volunteers. All possibly or probably related adverse events were mild or, in some cases, moderate, and resolved completely within a few days without requiring medical intervention. No severe or serious adverse reactions were observed. Using the Fisher exact test, no statistically significant difference was found in the frequency of adverse reactions occurring in Groups 1, 2 and 3 compared with Group 4, which received the licensed vaccine with a well‐documented safety profile (Table 4) 8.
Table 4.
Dunnett‐adjusted simultaneous confidence intervals for (Seroconversiondose – Seroconversion15 μg), where dose can be 3.5, 6 or 9 μg
| Age group (years) and virus strain | Null hypothesis (μg) | Estimate | 95% CI (lower) | 95% CI (upper) |
|---|---|---|---|---|
| 18–60, H1N1 | H0,3.5 | 0.03 | –0.21 | 0.27 |
| H0,6 | –0.05 | –0.29 | 0.20 | |
| H0,9 | –0.07 | –0.31 | 0.18 | |
| 18–60, H3N2 | H0,3.5 | –0.28 | –0.51 | –0.03* |
| H0,6 | –0.17 | –0.40 | 0.08 | |
| H0,9 | –0.09 | –0.32 | 0.15 | |
| 18–60, B | H0,3.5 | –0.07 | –0.31 | 0.18 |
| H0,6 | –0.02 | –0.26 | 0.23 | |
| H0,9 | –0.17 | –0.40 | 0.09 | |
| 60+, H1N1 | H0,3.5 | –0.08 | –0.32 | 0.18 |
| H0,6 | –0.02 | –0.26 | 0.23 | |
| H0,9 | –0.09 | –0.33 | 0.16 | |
| 60+, H3N2 | H0,3.5 | –0.14 | –0.38 | 0.12 |
| H0,6 | 0.01 | –0.23 | 0.25 | |
| H0,9 | –0.03 | –0.27 | 0.22 | |
| 60+, B | H0,3.5 | –0.08 | –0.33 | 0.18 |
| H0,6 | –0.05 | –0.29 | 0.20 | |
| H0,9 | –0.06 | –0.31 | 0.19 |
H0,dose was rejected when the upper limit of the 95% confidence interval (CI) was less than 0. No significant differences were found, except for 3.5 μg for influenza H3N2 in the age group 18–60 years.
p < 0.05
Discussion
In the present study, all vaccine doses fulfilled all immunogenicity licensing criteria for the EMA and the US FDA for seasonal influenza vaccines in both the adult and elderly subgroups, and were safe and tolerable 9, 12. However, we found that the 3.5 μg dose was significantly less immunogenic than the currently licensed 15 μg vaccine in the case of the adult group, for the H3N2 strain. This was a somewhat unexpected finding, and it can possibly be explained by higher exposure rates to H3N2 during the 1968 pandemic in the elderly subject group 19. We therefore concluded that there is no clear dose–response relationship for immunogenicity between 6 μg and 15 μg HA per strain for the seasonal trivalent influenza vaccine studied in the present trial in adult and elderly patients.
Although most of the current influenza vaccines are split or subunit vaccines, a renewed interest in whole‐virion vaccines has developed, at least in the case of monovalent prepandemic vaccines, when the traditional unadjuvanted split or subunit vaccines has not delivered the desired immunogenicity and doses up to 90 μg have been required 20. In unprimed individuals, inactivated whole‐virion vaccines are more immunogenic and induce protective antibody responses at a lower antigen dose than other formulations such as split‐virus or subunit vaccines, possibly by Toll‐like receptor signalling 21. We and others have shown that using whole‐virion vaccines can overcome the insufficient immunogenicity seen with split and subunit vaccines in the case of influenza A H5N1 21, 22, 23, 24.
Decreased immunogenicity has also been observed in the case of elderly patients, who require doses up to four times higher for the traditional, unadjuvanted split‐virus seasonal influenza vaccine 2. Using higher doses not only decreases production capacity, but also results in more vaccine reactions 2. Thus, it was a logical step to examine the immunogenicity of whole‐virion vaccines in elderly patients, such as in the present study.
Similarly, an interest in using adjuvants has recently developed in the case of monovalent prepandemic influenza vaccines, where unadjuvanted split or subvirion vaccines do not provide sufficient immunogenicity. We and others have consistently shown that using adjuvants can significantly boost immunogenicity to overcome the challenge presented, even by new, mutated influenza virus strains 25, 26, 27, 28. As we did not test the whole‐virion vaccine without adjuvants, we are unable to determine whether the immunogenicity results seen in the present trial at reduced doses were due to using a whole‐virion vaccine, using an adjuvant or both.
A potential weakness of our study was the low statistical power for detecting differences in immunogenicity or adverse events. However, we made careful power calculations and sample‐size determinations, as described above. Another possible limitation was that we did not test the effects of dose reduction in paediatric patients of younger than 18 years of age. However, in an earlier study we had found that a whole‐virion, adjuvanted monovalent prepandemic influenza vaccine containing 6 μg HA was safe and effective in children 29. All of our subjects were Caucasian, so our results may not be generalizable to other groups.
Our data strongly suggest that a significant dose reduction in seasonal trivalent influenza vaccines is possible in adult and elderly patients when using whole‐virion vaccines and/or adjuvants. No additional benefits are expected when using doses exceeding 6 μg HA per strain in vaccines prepared by the technology described in this and our earlier study 6. Thus, licensing studies conducted by our group for a seasonal trivalent influenza vaccine with 6 μg HA per strain are currently under way in Hungary, involving adult and elderly patients. This could have significant economic and public health implications.
Competing Interests
There are no competing interests to declare. The study was funded by Omninvest Ltd, Budapest, Hungary. The funding source had no role in the conduct of the study or the preparation of the manuscript.
This study was presented in part as a poster at the 9th Vaccine and ISV conference in Seoul, South Korea, October 2015.
Vajo, Z. , Balaton, G. , Vajo, P. , Kalabay, L. , Erdman, A. , and Torzsa, P. (2017) Dose sparing and the lack of a dose–response relationship with an influenza vaccine in adult and elderly patients – a randomized, double‐blind clinical trial. Br J Clin Pharmacol, 83: 1912–1920. doi: 10.1111/bcp.13289.
The trial was registered with the European Union Drug Regulatory Authorities, European Medicines Agency, European Clinical Trials Database. (http://eudract.emea.europa.eu/) under clinical trial registration number 2011‐003166‐32.
References
- 1. World Health Organization . Technical report series, No. 927, Annex 3. Recommendations for the production and control of influenza vaccine, 2005. [online]. Available at http://www.who.int/biologicals/publications/trs/areas/vaccines/influenza/ANNEX 3 InfluenzaP99‐134.pdf (last accessed 12 May 2015).
- 2. Couch RB, Winokur P, Brady R, Belshe R, Chen WH, Cate TR, et al. Safety and immunogenicity of a high dosage trivalent influenza vaccine among elderly subjects. Vaccine 2007; 25: 7656–7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chaloupka I, Schuler A, Marschall M, Meier‐Ewert H. Comparative analysis of six European influenza vaccines. Eur J Clin Microbiol Infect Dis 1996; 15: 121–127. [DOI] [PubMed] [Google Scholar]
- 4. Kempe A, Daley MF, Stokley S, Crane LA, Beaty BL, Barrow J, et al. Impact of a severe influenza vaccine shortage on primary care practice. Am J Prev Med 2007; 33: 486–491. [DOI] [PubMed] [Google Scholar]
- 5. Vajo Z, Tamas F, Sinka L, Jankovics I. Safety and immunogenicity of a 2009 pandemic influenza a H1N1 vaccine when administered alone or simultaneously with the seasonal influenza vaccine for the 2009–10 influenza season: a multicentre, randomised controlled trial. Lancet 2010a; 375: 49–55. [DOI] [PubMed] [Google Scholar]
- 6. Vajo Z, Tamas F, Jankovics I. A reduced‐dose seasonal trivalent influenza vaccine is safe and immunogenic in adult and elderly patients in a randomised controlled trial. Clin Vaccine Immunol 2012; 19: 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Committee for Medicinal Products for Human use . BWP ad‐hoc influenza working group. EU recommendations for the seasonal influenza vaccine composition for the season 2011/2012 [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Other/2011/03/WC500103708.pdf (last accessed 12 May 2015).
- 8. Vajo Z, Kosa L, Szilvasy I, Pauliny Z, Bartha K, Visontay I, et al. Yearly licensing studies from 1997 to 2007 of the inactivated whole virus seasonal influenza vaccine fluval – a useful approach to pandemic vaccine development even in less well developed countries? Influenza Other Respir Viruses 2008; 2: 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. European Committee for Proprietary Medicinal Products . Note for guidance on harmonisation of requirements for influenza vaccines, 1997, (CPMP/BWP/214/96) [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003945.pdf (last accessed 12 March 1997).
- 10. Wood JM, Schild GC, Newman RW, Seagroatt V. An improved single‐radial‐immunodiffusion technique for the assay of influenza haemagglutinin antigen: application for potency determinations of inactivated whole virus and subunit vaccines. J Biol Stand 1977; 5: 237–247. [DOI] [PubMed] [Google Scholar]
- 11. European Directorate for the Quality of Medicines and Health Care . European Pharmacopia, 5.8 CD, 7/2007, 2007; 3406–7. Strasbourg, France: European Directorate for the Quality of Medicines and Health Care.
- 12. Food and Drug Administration, Center for Biologics Evaluation and Research . Guidance for industry. Clinical data needed to support the licensure of seasonal inactivated influenza vaccines. US Department of Health and Human Services, 2007. [online]. Available at http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Vaccines/ucm091990.pdf (last accessed 12 May 2015)
- 13. World Medical Association, Declaration of Helsinki. Ethical principles for medical research involving human subjects [online] . Available at http://www.wma.net/en/30publications/10policies/b3/17c.pdf (last accessed 12 May 2015).
- 14. European Union . Good clinical practice [online]. Available at http://ec.europa.eu/health/files/eudralex/vol‐10/3cc1aen_en.pdf (last accessed 12 May 2015).
- 15. Klimov A, Cox N. Serologic diagnosis of influenza virus infections by hemagglutination inhibition, 2003; 7.1–7.5. Influenza Laboratory Course. Centres for Disease Control and Prevention, Atlanta, GA.
- 16. Dunnett CW, Horn M, Vollandt R. Sample size determination in step‐down and step‐up multiple tests for comparing treatments with a control. J Stat Plan Infer 2001; 97: 367–384. [Google Scholar]
- 17. Horn M, Vollandt R, Dunnett CW. Sample size determination for testing whether an identified treatment is best. Biometrics 2000; 56: 879–881. [DOI] [PubMed] [Google Scholar]
- 18. Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 2015; 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taubenberger JK, Morens DM. Influenza: the once and future pandemic. Public Health Rep 2010; 125: 16–26. [PMC free article] [PubMed] [Google Scholar]
- 20. Treanor JJ, Campbell JD, Zangwill KM, Rowe T, Wolff M. Safety and immunogenicity of an inactivated subvirion influenza a (H5N1) vaccine. N Engl J Med 2006; 354: 1343–1351. [DOI] [PubMed] [Google Scholar]
- 21. Geeraedts F, Goutagny N, Hornung V, Severa M, de Haan A, Pool J, et al. Superior immunogenicity of inactivated whole virus H5N1 influenza vaccine is primarily controlled by toll‐like receptor signalling. PLoS Pathog 2008; 4: e1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Budimir N, Huckriede A, Meijerhof T, Boon L, Gostick E, Price DA, et al. Induction of heterosubtypic cross‐protection against influenza by a whole inactivated virus vaccine: the role of viral membrane fusion activity. PLoS One 2012; 7: e30898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Furuya Y. Return of inactivated whole‐virus vaccine for superior efficacy. Immunol Cell Biol 2012; 90: 571–578. [DOI] [PubMed] [Google Scholar]
- 24. Vajo Z, Wood J, Kosa L, Szilvasy I, Paragh G, Pauliny Z, et al. A single‐dose influenza a (H5N1) vaccine safe and immunogenic in adult and elderly patients: an approach to pandemic vaccine development. J Virol 2010; 84: 1237–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen WH, Jackson LA, Edwards KM, Keitel WA, Hill H, Noah DL, et al. Safety, reactogenicity, and immunogenicity of inactivated monovalent influenza a(H5N1) virus vaccine administered with or without AS03 adjuvant. Open Forum Infect Dis 2014; 1: ofu091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Belshe RB, Frey SE, Graham IL, Anderson EL, Jackson LA, Spearman P, et al. Immunogenicity of avian influenza a/Anhui/01/2005(H5N1) vaccine with MF59 adjuvant: a randomised clinical trial. JAMA 2014; 312: 1420–1428. [DOI] [PubMed] [Google Scholar]
- 27. Fazekas G, Martosne‐Mendi R, Jankovics I, Szilvasy I, Vajo Z. Cross‐reactive immunity to clade 2 strains of influenza virus a subtype H5N1 induced in adults and elderly patients by Fluval, a prototype pandemic influenza virus vaccine derived by reverse genetics, formulated with a phosphate adjuvant, and directed to clade 1 strains. Clin Vaccine Immunol 2009; 16: 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vajo Z, Kosa L, Visontay I, Jankovics M, Jankovics I. Inactivated whole virus influenza a (H5N1) vaccine. Emerg Infect Dis 2007; 13: 807–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vajo Z, Kosa L, Szilvasy I, Pauliny Z, Bartha K, Visontay I, et al. Safety and immunogenicity of a prepandemic influenza a (H5N1) vaccine in children. Pediatr Infect Dis J 2008b; 27: 1052–1056. [DOI] [PubMed] [Google Scholar]