

Abstract
Key points
The highly variable phenotypes observed in patients with classic Bartter's syndrome (BS) remain unsatisfactorily explained. The wide spectrum of functional severity of CLCNKB mutations may contribute to the phenotypic variability, and the genotype–phenotype association has not been established.
Low‐level expression of the human ClC‐Kb channel in mammalian cells impedes the functional study of CLCNKB mutations, and the underlying cause is still unclear.
The human ClC‐Kb channel is highly degraded by proteasome in human embryonic kidney cells. The C‐terminal in‐frame green fluorescent protein fusion may slow down the proteasome‐mediated proteolysis. Barttin co‐expression necessarily improves the stability, membrane trafficking and gating of ClC‐Kb.
CLCNKB mutations in barttin‐binding sites, dimer interface or selectivity filter often have severe functional consequences.
The remaining chloride conductance of the ClC‐Kb mutant channel significantly correlates with the phenotypes, such as age at diagnosis, plasma chloride concentration, and the degree of calciuria in patients with classic BS.
Abstract
Mutations in the CLCNKB gene encoding the human voltage‐gated chloride ClC‐Kb (hClC‐Kb) channel cause classic Bartter's syndrome (BS). In contrast to antenatal BS, classic BS manifests with highly variable phenotypes. The functional severity of the mutant channel has been proposed to explain this phenomenon. Due to difficulties in the expression of hClC‐Kb in heterologous expression systems, the functional consequences of mutant channels have not been thoroughly examined, and the genotype–phenotype association has not been established. In this study, we found that hClC‐Kb, when expressed in human embryonic kidney (HEK) cells, was unstable due to degradation by proteasome. In‐frame fusion of green fluorescent protein (GFP) to the C‐terminus of the channel may ameliorate proteasome degradation. Co‐expression of barttin increased protein abundance and membrane trafficking of hClC‐Kb and markedly increased functional chloride current. We then functionally characterized 18 missense mutations identified in our classic BS cohort and others using HEK cells expressing hClC‐Kb‐GFP. Most CLCNKB mutations resulted in marked reduction in protein abundance and chloride current, especially those residing at barttin binding sites, dimer interface and selectivity filter. We enrolled classic BS patients carrying homozygous missense mutations with well‐described functional consequences and clinical presentations for genotype–phenotype analysis. We found significant correlations of mutant chloride current with the age at diagnosis, plasma chloride concentration and urine calcium excretion rate. In conclusion, hClC‐Kb expression in HEK cells is susceptible to proteasome degradation, and fusion of GFP to the C‐terminus of hClC‐Kb improves protein expression. The functional severity of the CLCNKB mutation is an important determinant of the phenotype in classic BS.
Keywords: classic Bartter''s syndrome, CLCNKB gene, ClC‐Kb chloride channel
Key points
The highly variable phenotypes observed in patients with classic Bartter's syndrome (BS) remain unsatisfactorily explained. The wide spectrum of functional severity of CLCNKB mutations may contribute to the phenotypic variability, and the genotype–phenotype association has not been established.
Low‐level expression of the human ClC‐Kb channel in mammalian cells impedes the functional study of CLCNKB mutations, and the underlying cause is still unclear.
The human ClC‐Kb channel is highly degraded by proteasome in human embryonic kidney cells. The C‐terminal in‐frame green fluorescent protein fusion may slow down the proteasome‐mediated proteolysis. Barttin co‐expression necessarily improves the stability, membrane trafficking and gating of ClC‐Kb.
CLCNKB mutations in barttin‐binding sites, dimer interface or selectivity filter often have severe functional consequences.
The remaining chloride conductance of the ClC‐Kb mutant channel significantly correlates with the phenotypes, such as age at diagnosis, plasma chloride concentration, and the degree of calciuria in patients with classic BS.
Abbreviations
- BS
Bartter's syndrome
- DCT
distal convoluted tubule
- GFP
green fluorescent protein
- GS
Gitelman's syndrome
- HEK
human embryonic kidney
- hClC‐Kb
human voltage‐gated chloride ClC‐Kb channel
Introduction
Bartter's syndrome (BS), a genetically heterogeneous renal tubular disorder, shares a defective salt reabsorption in the loop of Henle characterized by renal salt wasting, hypokalaemia, metabolic alkalosis and secondary hyperaldosteronism. Patients with BS can be divided into antenatal BS with SLC12A1, KCNJ1, BSND or the recently identified MAGED2 mutation or classic BS with CLCNKB mutation (Hebert, 2003; Laghmani et al. 2016). Virtually all patients with antenatal BS exhibit severe symptoms, such as maternal polyhydramnios, preterm birth and growth retardation. In contrast, patients with classic BS show highly variable courses of disease, ranging from an early‐onset and severe antenatal BS to a late‐onset and mild Gitelman's syndrome (GS), another inherited renal tubulopathy with defective sodium chloride cotransporter in distal convoluted tubule (DCT). The mechanism underlying the phenotypic heterogeneity of classic BS remains elusive.
The human CLCNKB gene encodes a voltage‐gated chloride channel ClC‐Kb, which belongs to the ClC chloride channel family (Jentsch et al. 2002). ClC‐Kb and its closest homologue, ClC‐Ka, earn their names due to the abundant expression in the kidneys, and they are responsible for the transepithelial chloride transport in renal tubules. Both ClC‐Kb and ClC‐Ka require an essential β‐subunit barttin encoded by the BSND gene to function properly. Human mutations in the BSND gene lead to antenatal BS accompanied by sensorineural deafness (Birkenhäger et al. 2001). Despite the 85% amino acid homology, ClC‐Kb and ClC‐Ka show different gating properties and a differential distribution in renal tubules with ClC‐Ka mostly expressed in the medulla and ClC‐Kb mainly found in the cortex, and they are suggested to be overlapped in the ascending limb of Henle's loop (Uchida & Sasaki, 2005). The compensation from the overlapped ClC‐Ka has been proposed as the underlying mechanism of the milder phenotype in classic BS. In Clcnk2 (the mouse orthologue of the human CLCNKB gene) knockout mice, Clcnk1 (the mouse orthologue of the human CLCNKA gene) was barely expressed in renal tubules other than the thin ascending limb and did not compensate for the genetic ablation of Clcnk2 (Hennings et al. 2017). In addition, the preserved function of the mutant human ClC‐Kb (hClC‐Kb) channel may alleviate the severity of this phenotype. A previous study reported that CLCNKB mutations with milder functional outcome were linked to older age at diagnosis of classic BS (Keck et al. 2013). The genotype–phenotype association has not been established in classic BS.
To date, more than 50 CLCNKB point mutations have been reported (Simon et al. 1997; Andrini et al. 2015). Only 20 of these mutations have been functionally validated and showed variable degrees (27–100%) of reduction in chloride current (Waldegger & Jentsch, 2000; Estevez et al. 2001; Yu et al. 2009; Keck et al. 2013; Andrini et al. 2014). Of note, these results were difficult to compare because of different expression systems and experimental conditions. We need a large‐scale functional characterization of CLCNKB mutations in the same mammalian experimental conditions. However, the major obstacle to using mammalian cells for study of hClC‐Kb is the poor hClC‐Kb protein expression. In this study, we optimized hClC‐Kb expression in human embryonic kidney (HEK)‐293 cells and then examined the functional consequences of 18 uncharacterized CLCNKB mutations. Phenotypes collected from previous patients carrying homozygous CLCNKB point mutations were used to correlate with the remaining chloride current of the corresponding mutation. The results indicate that in‐frame C‐terminal green fluorescent protein (GFP) fusion may ameliorate proteasome degradation and, together with barttin co‐expression, enabled the functional study of hClC‐Kb in mammalian cells. The remaining chloride current of the hClC‐Kb mutant was significantly correlated with the age at diagnosis, plasma chloride concentration and the degree of calciuria in classic BS.
Methods
Plasmid DNA constructs and transient expression of hClC‐Kb channel in cultured mammalian cells
To test the efficiency of hClC‐Kb protein expression in HEK‐293 cells, the human CLCNKB variant 1 gene (RefSeq: NM_000085.1) purchased from Origene (RG219199, Origene, Rockville, MD, USA) was cloned into different mammalian expression vectors, including pCMV6‐AC‐GFP with in‐frame C‐terminal GFP (PS100010, Origene), pCMV6‐AC‐entry with C‐terminal Myc‐DDK tag (PS100001, Origene), pEGFP‐C2 with in‐frame N‐terminal GFP (6083‐1, Clontech, Mountain View, CA, USA), and bicistronic pIRES‐hrGFP with coexisting GFP expression (240031, Agilent, Santa Clara, CA, USA). The human CLCNKB point mutations were generated by site‐directed mutagenesis (PfuUltra II Fusion HotStart, Agilent Technologies) and confirmed by sequencing.
The HEK‐293 cells were cultured as described previously (Lazrak et al. 2006). Cells were transfected with 0.5 μg wild‐type or mutant cDNA of hClC‐Kb and 0.5 μg barttin (RC213125, Origene) using 3 μl X‐tremeGENE HP DNA transfection reagents (Roche, Indianapolis, IN, USA). The total amount of DNA was minimized (maximal cDNA used for transfection ≤1.0 μg) to avoid saturation of protein expression in this study. The total amount of plasmid DNA in each experimental group was balanced using pCMV6‐AC‐entry empty vector.
Immunoblotting and surface biotinylation assay
Transfected, cultured HEK‐293 cells were incubated with RIPA buffer (150 mm NaCl, 50 mm Tris‐HCl, 5 mm EDTA, 1% Triton X‐100, 0.5% deoxycholic acid and 0.1% SDS) containing protease inhibitor cocktail (Complete Mini, Roche). The cell extracts were centrifuged after shaking for 30 min on the rotator at 4°C. Protein concentrations of the supernatant were measured by the Bradford assay using BSA as a standard. Equal amounts of lysates were mixed with Laemmli sample buffer and then separated by SDS‐PAGE under reducing conditions. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes, blocked with 5% non‐fat milk, and incubated with the anti‐CLCNKB (AV34916, Sigma, St Louis, MO, USA; 1:2000 dilution) or the anti‐turboGFP (TA150041, Origene, 1:4000 dilution) to detect protein expression of hClC‐Kb. Anti‐β‐actin antibody was used to detect endogenous β‐actin as a loading control. Bound antibodies were detected using ECL detection reagent (Pierce, Rockford, IL, USA). Densitometry was performed using ImageJ.
For biotinylation of cell surface hClC‐Kb, transfected HEK cells (per 35 mm well) were washed with 1 ml ice‐cold PBS three times and incubated with 1 ml PBS containing 1.5 mg ml−1 EZ‐link‐NHS‐SS‐biotin (Thermo Scientific, Waltham, MA, USA) for 2 h at 4°C. After quenching with glycine‐containing PBS for 20 min, cells were lysed in a RIPA buffer containing protease inhibitor cocktail for 30 min. Biotinylated proteins were precipitated by streptavidin‐agarose beads (Thermo Scientific) for 2 h at 4°C. Beads were subsequently washed three times with Tris‐buffered saline containing 1% Triton X‐100. Biotin‐labelled proteins were eluted in sample buffer, separated by SDS‐PAGE, and transferred to PVDF membranes for Western blotting using anti‐tGFP antibody. Biotinylation experiments were performed three times with similar results.
To inhibit proteasome activity, HEK cells transfected with designated CLCNKB constructs for 32–36 h were incubated with 20 μm MG‐132 for 0, 2, 4, 6 or 8 h. The protein lysates of these cells were blotted by anti‐CLCNKB antibody.
Electrophysiological recording of hClC‐Kb channel in HEK cells
About 24–36 h after transfection, cells were trypsinized and plated on poly‐l‐lysine‐coated coverslips. The whole cell hClC‐Kb/Barttin currents were recorded by using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA) as previously described (Lazrak et al. 2006). The pipette resistance was around 3–5 MΩ. Transfected cells were identified by green fluorescence using epifluorescence microscopy. In whole‐cell current recordings, the pipette solution contained (in mm) 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes (pH 7.4); the bath solution contained 115 NaCl, 2 MgCl2, 5 EGTA, 5 Hepes (pH 7.4). The cell membrane capacitance and series resistance were monitored and compensated for (>75%) electronically. The voltage protocol consisted of 0 mV holding potential for 50 ms and 200 ms steps from −145 to 135 mV in 20 mV increments. Data were sampled at 5 kHz with a 2 kHz low‐pass filter. ClampX 10.2 software (Axon Instruments) was used for data acquisition. Results are shown as means ± SEM (n = 6–8). Each experiment was repeated two to four times.
Genotype–phenotype analysis in patients with classic BS
Clinical data for patients with classic BS were collected from previous studies, including our patients (Simon et al. 1997; Bettineli et al. 2007; Lin et al. 2009; Vargas‐Poussou et al. 2011; García Castaño et al. 2013; Keck et al. 2013; Al‐Shibli et al. 2014; Andrini et al. 2014, 2015). Those patients with whole CLCNKB gene deletion, compound heterozygous mutation or mutation with unclear functional consequence (W610X) or obvious intra‐familial heterogeneity in phenotypes, such as P124L and A204T, were excluded. The remaining chloride conductance (% of wild type) of the mutant channel recorded by whole‐cell patch clamp was defined as the genotype of each CLCNKB mutation. The phenotypes, including age at diagnosis, plasma potassium, chloride, magnesium, bicarbonate, renin and aldosterone, and urine calcium excretion, were independently correlated with the genotype of the corresponding CLCNKB mutation.
Statistical analysis
All results are expressed as mean ± SEM. The difference between groups was assessed using Student's t test. Associations of the remaining chloride conductance of mutant hClC‐Kb channels and continuous phenotype variables were assessed by both Pearson and Spearman analysis. A P value less than 0.05 (*) was considered to be statistically significant.
Results
In‐frame C‐terminal GFP insertion and barttin co‐expression stabilized the hClC‐Kb protein in HEK‐293 cells
We first examined the protein expression of hClC‐Kb in HEK‐293 cells. The anti‐CLCNKB antibody detected two major immunoreactive bands at ∼100 and ∼200 kDa in HEK cells with hClC‐Kb‐GFP expression, which represented the monomer and multimer of hClC‐Kb‐GFP, respectively (Fig. 1 A, lane 2). The bands slightly bigger than the monomer (∼130 kDa) were probably due to the post‐translational modification, as previously reported (Janssen et al. 2009). The band at ∼55 kDa also found in HEK cells with mock transfection was non‐specific (lane 1). Comparing the protein expressions of hClC‐Kb‐myc and hClC‐Kb‐GFP (lanes 3 and 4), the in‐frame GFP fusion in the C‐terminus remarkably enhanced the protein expression of hClC‐Kb. The HEK cells transfected with pIRES‐hrGFP/hClC‐Kb (lane 5) expressed very few hClC‐Kb proteins despite a high green fluorescence under microscopy, indicating that a separately expressed GFP did not reproduce the effect of in‐frame C‐terminal GFP. Introducing a Q513X nonsense mutation into hClC‐Kb‐GFP abolished the expression of hClC‐Kb and resulted in a low molecular‐weight smear (∼40 kDa), probably due to proteolysis of unstable protein (lane 6). In contrast, the truncated amino acid 1–512 of hClC‐Kb followed by an in‐frame GFP in the C‐terminus was robustly expressed (lane 7). Taken together, the hClC‐Kb protein was unstable in HEK‐293 cells, and the in‐frame C‐terminal GFP fusion stabilized the hClC‐Kb protein. Of note, the effect of the GFP in‐frame fusion in the N‐terminus of hClC‐Kb (GFP‐hClC‐Kb) was less efficient (lane 8).
Figure 1. Functional studies of human ClC‐Kb (hClC‐Kb) in a mammalian expression system.
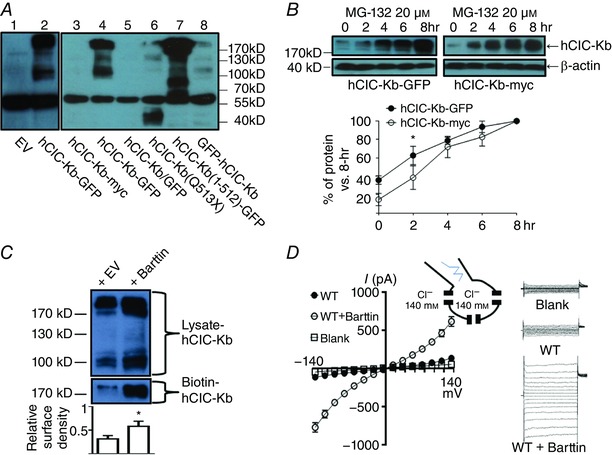
A, protein expressions of human ClC‐Kb (hClC‐Kb) in HEK‐293 cells transfected with different CLCNKB constructs were detected by Western blot using anti‐CLCNKB antibody. EV: empty vector, hClC‐Kb‐GFP: wild‐type CLCNKB in pCMV6‐AC‐GFP, hClC‐Kb‐myc: wild‐type CLCNKB in pCMV6‐AC‐entry, hClC‐Kb/GFP: wild‐type CLCNKB in pIRES‐hrGFP, hClC‐Kb(Q513X): Q513X mutant CLCNKB in pCMV6‐AC‐GFP, hClC‐Kb(1–512)‐GFP: the truncated amino acid 1–512 of CLCNKB in pCMV6‐AC‐GFP, GFP‐hClC‐Kb: wild‐type CLCNKB in pEGFP‐C2. The estimated molecular weights of hClC‐Kb‐myc and hClC‐Kb/GFP were ∼75 kDa, hClC‐Kb‐GFP and GFP‐hClC‐Kb were ∼102 kDa, hClC‐Kb (Q513X) was ∼56 kDa and hClC‐Kb (1–512)‐GFP was ∼83 kDa. B, protein levels of hClC‐Kb‐GFP (left panel) or hClC‐Kb‐myc (right panel) in HEK‐293 cells incubated with 20 μm MG‐132 for different time periods (0∼8 h) were detected by anti‐CLCNKB antibody. The protein level in different time points was standardized to the level at 8 h. The rate of hClC‐Kb protein accumulation induced by MG‐132 was compared between hClC‐Kb‐GFP and hClC‐Kb‐myc. C, comparison of the protein expression and membrane insertion of the hClC‐Kb channel in the absence (+ EV) or presence (+ barttin) of barttin. The surface density of hClC‐Kb was estimated by the ratio of biotin‐labelled hClC‐Kb (biotin‐ClC‐Kb, only multimer form present) and hClC‐Kb in total cell lysate (lysate‐ClC‐Kb, both monomer and multimer forms present). D, I–V relationship curves of wild‐type (WT) hClC‐Kb channels with or without co‐expression of barttin. The current measured in cells transfected with empty vector was designated as blank. The configuration of ruptured symmetrical whole‐cell recording of chloride current and representative currents from hClC‐Kb, hClC‐Kb + barttin and mock‐transfected cells are shown. Gels shown are representatives of three experiments with similar results. The abundance of each band in the gel was measured by densitometry by Image J. * P < 0.05 by unpaired two‐tailed Student's t test. [Color figure can be viewed at wileyonlinelibrary.com]
To study whether proteasome degraded hClC‐Kb, hClC‐Kb‐GFP or hClC‐Kb‐myc transfected HEK‐293 cells were incubated with a proteasome inhibitor, MG‐132. Two hours of incubation of MG‐132 significantly increased the protein amounts of hClC‐Kb, hClC‐Kb‐GFP or hClC‐Kb‐myc, which reached the plateau at approximately 8 h (Fig. 1 B). Of note, hClC‐Kb‐myc protein, which was barely detected in the basal state (Fig. 1 A, lane 3, and 0 h of Fig. 1 B), became abundantly expressed in the presence of MG‐132. The rate of hClC‐Kb‐myc protein accumulation after MG‐132 treatment was higher than hClC‐Kb‐GFP, supporting the notion that C‐terminal GFP fusion may mitigate proteasome‐mediated hClC‐Kb degradation.
Barttin has been known to exert multiple effects on ClC‐K channels. Here, we showed that co‐expression of barttin caused a ∼1.5‐fold increase in protein abundance of hClC‐Kb‐GFP (Fig. 1 C, upper blot), consistent with previous reports (Hayama et al. 2003; Janssen et al. 2009). As expected from published literature indicating that barttin stimulates the membrane trafficking of hClC‐Kb, the ratio of cell surface to total abundance of hClC‐Kb increased 2‐fold when barttin was co‐expressed (Fig. 1 C, lower blot, and bar chart). Importantly, HEK cells with expression of both barttin and hClC‐Kb displayed a robust, characteristic bi‐directionally rectifying hClC‐Kb current while cells with hClC‐Kb alone yielded a small current, not significantly different from the background leak (Fig. 1 D). In summary, barttin is indispensable for proper functioning of hClC‐Kb.
Mutations in barttin interacting motifs led to a significant functional impairment
The α‐helix B and J lining the outer lateral surface of the hClC‐K channel has been proposed as the barttin‐binding sites. We studied the functional consequences of A61D and A77T mutations occurring in α‐helix B and G296D, S297R and Q303P in α‐helix J (Fig. 2 A). Protein expressions of A61D and A77T were 40% and 10% of the wild type, respectively, and co‐expression of barttin had only a subtle effect on these two mutants (Fig. 2 B, lane 2 and 3 vs. lane 1). G296D, S297R and Q303P mutations caused a milder decrease in the protein expression of hClC‐Kb (87, 94 and 60% vs. the wild type, respectively; lanes 4–6 vs. lane 1) and a similarly reduced response to barttin co‐expression. Using a biotinylation assay, we estimated that A61D, G296 and Q303P mutations significantly disrupted the membrane insertion of hClC‐Kb by 41, 40 and 57%, respectively (Fig. 6). In patch clamp recordings, A61D, A77T, G296D, S297R and Q303P mutant currents were 16, 3, 27, 61 and 27% of the wild‐type current, respectively (Fig. 2 C).
Figure 2. CLCNKB mutations in the barttin‐interacting zone.
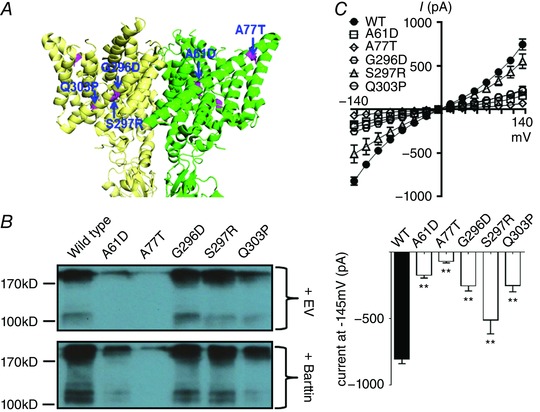
A, locations of the mutations (highlighted in magenta) in α‐helix B in the green subunit and α‐helix J in the yellow subunit. The homology model of hClC‐Kb based on a eukaryotic CLC transporter (PDB ID: 3ORG) shows the lateral view of two subunits. B, protein expression of wild‐type versus mutant hClC‐Kb channel [upper blot, total transfected plasmids were balanced by empty vector (EV)], and the responses to barttin co‐expression (lower blot). Gel shown is representative of three experiments with similar results. C, I–V relationship curves of wild‐type (WT) and mutant hClC‐Kb channels with barttin co‐expression (upper graph). The bar graph represents the wild‐type and mutant hClC‐Kb current (pA) at holding potential −145 mV (mean ± SEM, n ≥ 6 for each). ** P < 0.01 between wild‐type and each mutation by unpaired two‐tailed Student's t test.
Figure 6. Membrane abundance of mutant hClC‐Kb channels.
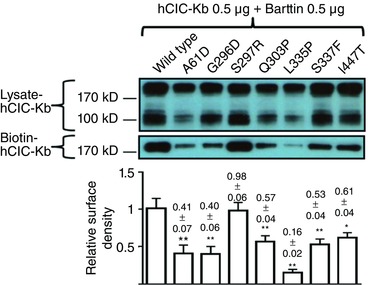
All hClC‐Kb constructs were co‐transfected with barttin. Lysate‐ and Biotin‐hClC‐Kb represent hClC‐Kb channels in HEK cell lysates and eluted from a mixture of HEK cell lysates and streptavidin‐agarose beads, respectively. The bar graph at the bottom is the relative surface density of the hClC‐Kb channel (normalized to wild‐type). The mean ± SEM from three separate experiments is shown on top of each bar. * P < 0.05 and ** P < 0.01 for each group versus wild‐type. The gel shown is representative of three experiments with similar results. The abundance of each band in the gel was measured by densitometry by the Image J program. [Color figure can be viewed at wileyonlinelibrary.com]
Mutations in the dimer interface had severe functional consequences
Amino acids P216 in the G–H linker and A242 in α‐helix I were near the dimer interface of two hClC‐Kb subunits (Fig. 3 A). Of note, the Western blot of P216L hClC‐Kb displayed an inverse dimer‐to‐monomer ratio with the monomer being the predominant form (Fig. 3 B, lane 2 vs. lane 1), suggesting that this mutation disrupted the normal dimerization of hClC‐Kb. The A242E mutant had a reduced protein expression (9% of the wild type) and no response to barttin co‐expression (lane 3 vs. lane 1). Both mutations correspondingly led to a massive reduction of hClC‐Kb current (8 and 3% of the wild‐type current for P216L and A242E mutants, respectively) (Fig. 3 C).
Figure 3. CLCNKB mutations in the dimer interface.
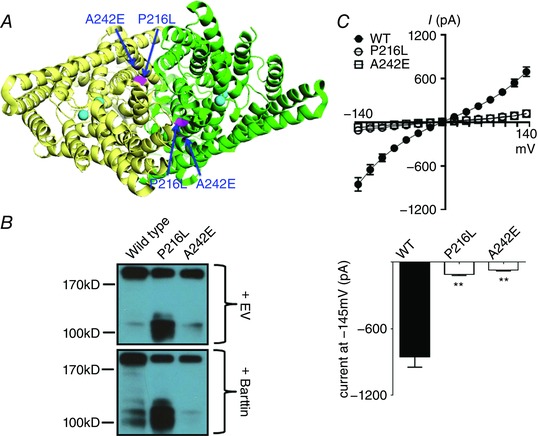
A, locations of the mutations (highlighted in magenta) in both subunits in external view from the top of the hClC‐Kb model (PDB ID: 3ORG). Four chloride ions in the permeation pore of two subunits are coloured cyan. B, Western blot of wild‐type versus mutant hClC‐Kb channel in the absence (+EV) or presence (+ Barttin) of barttin co‐expression. C, I–V relationship curves (upper graph) and current at holding potential of −145 mV (lower graph) of wild‐type (WT) and mutant hClC‐Kb channels with barttin co‐expression (mean ± SEM, n ≥ 6 for each). ** P < 0.01 between wild‐type and each mutation by unpaired two‐tailed Student's t test.
Most mutations around the selectivity filter were severe hypo‐functional
Eleven of 50 known point mutations were all around the selectivity filter at the outer surface of the hClC‐Kb channel. Here we studied six of these mutations, G424R, G437C, E442G, I447T, G470E and G470R, in mammalian cells (Fig. 4 A). These mutations, except I447T, resulted in marked reductions in protein level and in response to barttin co‐expression (G424R: 44%, G437C: 35%, E442G: 29%, I447T: 95%, G470E: 34%, G470R: 41% of wild‐type protein level) (Fig. 4 B). All of these mutants had very low whole‐cell chloride current (G424R: 6.3%, G437C: 6.6%, E442E: 7.8%, G470E: 6.6%, G470R: 7.5%), except I447T (55% of wild‐type current) (Fig. 4 C). The I447T mutation significantly impaired the surface insertion of the channel (61% of wild type, Fig. 6).
Figure 4. CLCNKB mutations around the selectivity filter.
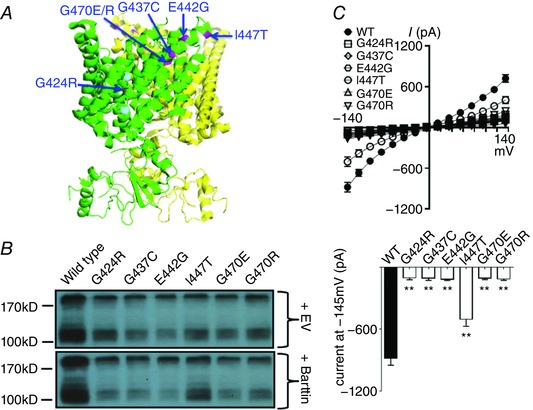
A, locations of the mutations (highlighted in magenta) in or around the selectivity filter in oblique view with green subunit in front (PDB ID: 3ORG). B, Western blot of wild‐type versus mutant hClC‐Kb channel in the absence (+ EV) or presence (+ Barttin) of barttin co‐expression. C, I–V relationship curves (upper graph) and current at holding potential of −145 mV (lower graph) of wild‐type (WT) and mutant hClC‐Kb channels with barttin co‐expression (mean ± SEM, n ≥ 6 for each). ** P < 0.01 between wild‐type and each mutation by unpaired two‐tailed Student's t test.
Other mutations had milder functional consequences
We also studied the G120V mutation in the C–D linker, V149E in α‐helix E, G164C in E–F linker, and L335P and S337F in α‐helix K (Fig. 5 A). Compared to wild‐type protein, the amounts of mutant proteins, except S337F, were lower (G120V: 57%, V149E: 51%, G164C: 36%, L335P: 57%, S337F: 95% of wild‐type level, respectively) (Fig. 5 B). The whole‐cell currents of mutant channels were 28% (G120V), 48% (V149E), 31% (G164C), 46% (L335P) and 69% (S337F) of the wild‐type current (Fig. 5 C). L335P and S337F mutations significantly reduced the membrane trafficking of the channel (16 and 53% of wild‐type, respectively, Fig. 6). Taken together, these mutations led to milder functional losses.
Figure 5. Other CLCNKB mutations.
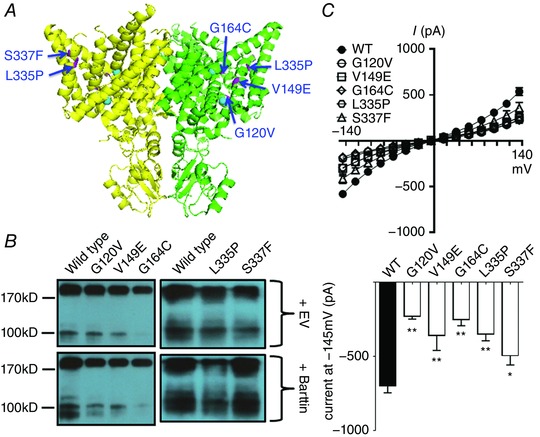
A, locations of the mutations (highlighted in magenta) in lateral view of the hClC‐Kb model (PDB ID: 3ORG). B, Western blot of wild‐type versus mutant hClC‐Kb channel in the absence (+EV) or presence (+Barttin) of barttin co‐expression. C, I–V relationship curves (upper graph) and current at holding potential of −145 mV (lower graph) of wild‐type (WT) and mutant hClC‐Kb channels with barttin co‐expression (mean ± SEM, n ≥ 6 for each). * P < 0.05 and ** P < 0.01 between wild‐type and each mutation by unpaired two‐tailed Student's t test.
Functional severity of the CLCNKB mutation correlated with the phenotypic severity of classic BS
To avoid ambiguity, we enrolled only patients with homozygous CLCNKB point mutations whose functional relevance has been clearly studied and quantified (Table 1) (Simon et al. 1997; Bettineli et al. 2007; Lin et al, 2009; Vargas‐Poussou et al. 2011; García Castaño et al. 2013; Keck et al. 2013; Al‐Shibli et al. 2014; Andrini et al. 2014, 2015) and excluded those with compound heterozygous mutations or mutations with unknown or inconsistent functional outcome. Early functional studies in oocytes were also excluded due to the non‐mammalian experimental conditions. Our results showed that the remaining chloride conductance of the mutant hClC‐Kb channel had significantly positive correlations with age at diagnosis and plasma chloride concentration (Fig. 7 A and C), and a significantly negative correlation with urinary calcium excretion rate (urine calcium to creatinine molar ratio) (Fig. 7 B). Otherwise, there was no correlation between the functional severity of mutant hClC‐Kb and plasma magnesium, potassium, bicarbonate (Fig. 7 D–F), renin or aldosterone levels (data not shown).
Table 1.
Phenotypes of enrolled classic BS patients carrying homozygous CLCNKB point mutations
| Mutation | Remaining Cl− current (% of WT) | Age at diagnosis (years) | Serum Cl− (mm) | Serum K+ (mm) | Serum Mg2+ (mm) | Serum HCO3 − (mm) | Renin (*) | Aldo (*) | PGE (mm) | Urine Ca2+/Cr (mmol mmol–1) | Urine Cl− (mmol day–1) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L81P | 35 | 0.4 | NA | 1.9 | 1.2 | 36 | 10 | 0 | NA | 1.29 | NA | Keck et al. (2013) |
| R92W | 67 | 20 | NA | 2.0 | 0.67 | 30 | NA | NA | NA | NA | 427.2 | Vargas‐Poussou et al. (2011) |
| G164C | 31 | 14 | 89 | 2.4 | 0.61 | 33 | NA | NA | 140 | 0.42 | NA | Al‐Shibli et al. (2014) |
| G164C | 31 | 8 | 96 | 2.3 | 0.92 | 31 | NA | NA | 70 | 0.55 | NA | Al‐Shibli et al. (2014) |
| G164C | 31 | 11 | 96 | 2.7 | 0.71 | 32 | NA | NA | 74 | 0.06 | NA | Al‐Shibli et al. (2014) |
| G164C | 31 | 8 | 89 | 2.3 | 0.85 | 32 | NA | NA | 44 | NA | NA | Al‐Shibli et al. (2014) |
| V170M | 60 | 43 | 99 | 2.74 | 1.03 | 34 | 0 | 0 | NA | 0.15 | NA | Andrini et al. (2014) |
| V170M | 60 | 24 | 97 | 2.8 | 0.77 | 32 | 6.5 | 1.2 | NA | 0.31 | NA | Andrini et al. (2014) |
| V170M | 60 | 60 | NA | 2.9 | 0.78 | NA | 4.3 | 2.5 | NA | NA | NA | Andrini et al. (2014) |
| V170M | 60 | 37 | 95 | 2.9 | 0.78 | 36 | 2 | 4 | NA | 0.49 | NA | Andrini et al. (2014) |
| A242E | 3 | 0.2 | 90 | 2.8 | 0.8 | 30 | NA | NA | NA | 1.13 | NA | Bettineli et al. (2007) |
| A242E | 3 | 0.7 | 94 | 2.5 | 0.91 | 32 | NA | NA | NA | 0.93 | NA | Bettineli et al. (2007) |
| G246R | 10 | 0.5 | 76 | 3.0 | 0.9 | 33 | NA | NA | NA | 0.8 | NA | Keck et al. (2013) |
| G296D | 27 | 0.1 | NA | 2.8 | NA | NA | NA | NA | NA | NA | NA | Vargas‐Poussou et al. (2011) |
| S297R | 61 | 0.6 | NA | NA | low | NA | NA | NA | NA | high | NA | Konrad et al. (2000) |
| A349D | 10 | 0.1 | NA | 2.4 | NA | 32 | NA | NA | NA | 1.4 | NA | Simon et al. (1997) |
| G424E | 6.3 | 13 | 91 | 2.8 | 0.78 | 32 | 2.3 | NA | NA | NA | NA | Keck et al. (2013) |
| I447T | 55 | 46 | 98 | 3.0 | 0.64 | 31 | NA | NA | NA | 0.07 | 157 | Vargas‐Poussou et al. (2011) |
| G470E | 6.6 | 0.75 | 93 | 2.5 | NA | 27 | NA | NA | NA | NA | NA | Lin et al. (2009) |
*Times above the upper limit for age. Aldo, aldosterone; NA, not available; PGE, urinary prostaglandin E2; WT, wild‐type.
Figure 7. The analysis of genotype–phenotype associations in patients with classic BS.
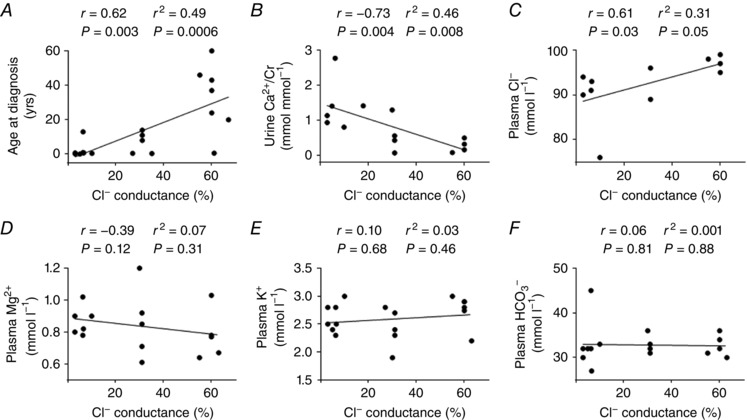
The remaining chloride conductance of mutant channels was plotted against phenotypic variables, including: A, age at diagnosis; B, urine calcium to creatinine ratio (Ca2+/Cr); C, plasma chloride (Cl−); D, plasma magnesium (Mg2+); E, plasma potassium (K+); and F, plasma bicarbonate (HCO3 −) concentrations. The r and P values of Spearman's test and the r 2 and P values of Pearson's test are shown. The CLCNKB mutations included for analysis were L81P, R92W, G164C, V170M, A242E, G246R, G296D, S297R, A349D, G424E, I447T and G470E. The phenotypes of the corresponding patients are listed in Table 1.
Discussion
In this study, we noted that hClC‐Kb protein expression was inefficient in HEK‐293 cells, probably due to active proteasome‐mediated degradation. By adding the in‐frame GFP to the C‐terminus of hClC‐Kb and co‐transfecting barttin, the protein expression and macroscopic chloride current of hClC‐Kb‐GFP were significantly enhanced in HEK cells. Functional validation of 18 different missense CLCNKB mutations identified in classic BS demonstrated that the protein abundance and chloride current were markedly reduced in the mutants residing at barttin‐binding sites, dimer interface and selectivity filter. We also found that the remaining basolateral hClC‐Kb conductance correlated with the age at onset, plasma chloride concentration and urine calcium excretion rate, suggesting that the functional severity of CLCNKB genotypes could affect the disease phenotype in classic BS.
Since cloning of the CLCNKB gene (Kieferle et al. 1994), studies of hClC‐Kb have been limited by the lack of a reliable heterologous expression system. Chimeric hClC‐Kb/ratClC‐K1 expressed in oocytes was first used despite the mixed properties with ratClC‐K1 (Waldegger & Jentsch, 2000). The discovery of barttin enhanced the hClC‐Kb/barttin current in oocytes and allowed functional studies of CLCNKB mutations (Estevez et al. 2001). However, hClC‐Kb currents measured in oocytes were somehow different from those in mammalian cells, probably due to some unknown regulatory factors (Fahlke & Fischer, 2010; Imbrici et al. 2014). In mammalian cells, the hClC‐Kb with N‐terminal yellow fluorescent protein fusion was efficiently expressed (Scholl et al. 2006; Janssen et al. 2009) while the hClC‐Kb‐V5‐His protein was barely detected by immunohistochemical staining, nor by Western blotting (Cho et al. 2013). We here demonstrate that hClC‐Kb was unstable in HEK‐293 cells, and C‐terminal GFP fusion may cause hClC‐Kb protein to be more resistant to proteasome‐mediated proteolysis. Recent studies have reported that epithelial ClC channels, ClC‐1 and ClC‐2, were bound and regulated by cereblon, a novel Cullin 4A E3 ligase substrate receptor (Hohberger & Enz, 2009; Chen et al. 2015). Also, ClC‐Ka was shown to be activated by serum‐ and glucocorticoid‐dependent kinase‐mediated Nedd4‐2 phosphorylation, which in turn avoids the ubiquitination and proteolysis of ClC‐Ka (Embark et al. 2004). Further study is needed to confirm the proteasome‐mediated proteolysis for hClC‐Kb.
Barttin has been known to increase both total and surface amounts of hClC‐Kb. We showed that barttin increased the extra bands of hClC‐Kb‐GFP around the size of 100–130 kDa, which probably represented the core‐glycosylated and complex‐glycosylated hClC‐Kb, respectively (Maulet et al. 1999; Janssen et al. 2009). Glycosylation of hClC‐Kb may stabilize protein structure and resist proteolysis. Moreover, co‐expression of barttin dramatically boosted the bi‐directionally rectifying hClC‐Kb/barttin current. Neither increased protein expression nor membrane trafficking of hClC‐Kb could fully explain this large enhancement of the chloride current, which was probably related to changes in the gating properties of the hClC‐Kb/barttin channel (Waldegger et al. 2002; Janssen et al. 2009). Our study supports the notion that barttin is essential for proper functioning of hClC‐Kb through physical stabilization, efficient membrane transport and channel gating.
Based on the functional results of mutant hClC‐Kb channels, nine mutations (A61D, A77T, P216L, A242E, G424R, G437C, E442G, G470E and G470R) caused massive reductions of the chloride current. Of note, these nine mutations were clustered into three functionally important domains, including the barttin‐interacting α‐helix B, the proposed dimer interface and the selectivity filter. It is still unclear how barttin interacts with hClC‐Kb, but the hydrophobic residues in helices B and J may form hydrophobic interactions with the hydrophobic residues of barttin transmembrane domain 1 (Tajima et al. 2007; Wojciechowski et al. 2015). Both A61D and A77T replaced the hydrophobic alanine with charged or hydroxyl amino acid. These mutations may disrupt the interaction between barttin and hClC‐Kb and thus reduce the barttin‐induced protein modifications, trafficking and gating of hClC‐Kb. The functional hClC‐Kb channel must form a stable dimer (Waldegger & Jentsch, 2000; Keck et al. 2013). Several hydrophobic residues on helices H, I, P and Q facing the inner side of the hClC‐Kb subunit may form strong hydrophobic interactions with the counterpart subunit (Fig. 8) (Andrini et al. 2015). The P216L mutation showed a larger band of monomers, suggesting that this mutation impairs the dimerization of hClC‐Kb. Other mutations around the dimerization site (A242E, G246R) all caused a huge reduction in protein level and chloride current, probably due to the instability and rapid degradation of the hClC‐Kb monomer. The α‐helix N forms the critical part of the hClC‐Kb selectivity filter and is loaded with pathogenic mutations. These mutations nearly always ensure a severe functional impairment, probably due to the enhanced proteolysis and abnormal gating of hClC‐Kb (Keck et al. 2013).
Figure 8. Hydrophobic interactions between the proposed dimer interface of the hClC‐Kb channel.
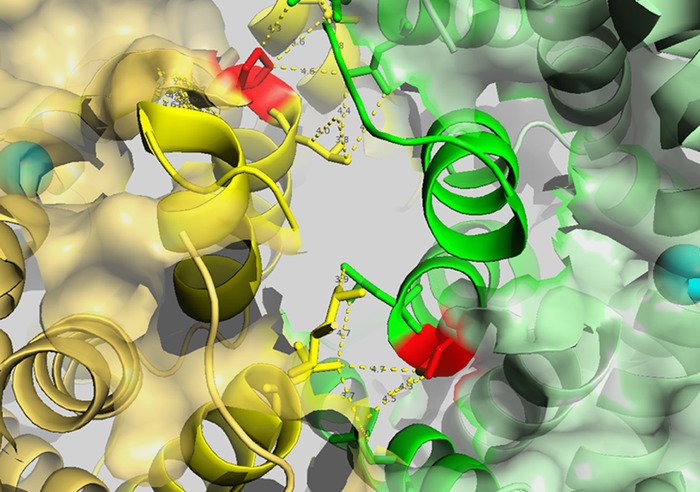
The homology model of hClC‐Kb based on a eukaryotic CLC transporter (PDB ID: 3ORG) showing the top view of two subunits, coloured green and yellow. The critical amino acid P216 is marked in red. The hydrophobic residues in helices H and I of one subunit could potentially interact with the hydrophobic residues in helices P and Q of the other subunit (highlighted by yellow dashed lines).
Previous studies found a poor correlation between the type of CLCNKB mutations and the phenotype, probably due to small patient numbers, lack of functional results or detailed laboratory records, or enrolling most patients with the same founder mutation (Konrad et al. 2000; Zelikovic et al. 2003; Brochard et al. 2009; Lee et al. 2012; García Castaño et al. 2017). Families sharing the same founder mutation, such as A204T in Spanish, R438H in Bedouin and W610X in Asians, or large CLCNKB gene deletion displayed variable phenotypes, suggesting inter‐ and intra‐familial heterogeneities (Zelikovic et al. 2003; Rodriguez‐Soriano et al. 2005; Lee et al. 2012; García Castaño et al. 2017). These phenotypic heterogeneities could be due to various degrees of compensation through alternative basolateral chloride efflux. In addition, other regulatory mechanisms of ClC‐K, such as blood pH, plasma calcium concentration and the probable proteasome‐mediated proteolysis, may alter the abundance and activity of mutant hClC‐Kb channels and cause phenotypic heterogeneity (Waldegger & Jentsch, 2000; Nissant et al. 2006; Pinelli et al. 2016).
In this study, we found that patients with severe mutations were diagnosed early in their infancy (Table 1). In contrast, patients with mild mutations (R92W, V170M and I447T, but not S297R) remained undiagnosed until early adulthood. Defective tubular chloride absorption due to an impaired hClC‐Kb channel can lead to urinary chloride wasting and more obvious hypochloraemia, a hallmark of classic BS (Jeck et al. 2005). Indeed, we revealed a significant correlation between the functional severity of CLCNKB mutations and plasma chloride concentration. Hypercalciuria, the typical feature of antenatal BS, was noted in patients with severe mutations, including A242E, A349D, G424E, R438H and L81P. Interestingly, hypocalciuria, the phenotypic character of GS, could be observed in patients with mild CLCNKB mutations, such as G164C, V170M and I447T. Accordingly, the age at diagnosis, plasma chloride concentration and urine calcium excretion rate, all of which have often been used to differentiate BS from GS, are highly correlated with the functional severity of CLCNKB mutations. In line with this conclusion, patients with homozygous gross deletion of CLCNKB often had a more severe phenotype and hypercalciuria (Simon et al. 1997; Konrad et al. 2000, Schurman et al. 2001; García Castaño et al. 2017). In type IV BS, mutations with preserved barttin function (e.g. G47R) were correlated with adult‐onset and less pronounced renal symptoms (Janssen et al. 2009; Riazuddin et al. 2009), also supporting the genotype–phenotype association in classic BS since barttin and ClC‐Kb function together. It is hypothesized that classic BS can mimic either antenatal BS or GS since hClC‐Kb is expressed in both thick ascending limb of Henle's loop and DCT. With the current finding that patients carrying mild CLCNKB mutations manifested GS‐like older age at diagnosis, hypocalciuria and less hypochloraemia, we speculate that DCT may be more vulnerable to the loss of basolateral ClC‐Kb than Henle's loop. Other clinical features of classic BS, such as plasma potassium, magnesium, bicarbonate, renin, aldosterone and even neuromuscular symptoms, were not correlated with the functional severity of mutations, probably related to dietary and environmental influences and different volume status (Bettineli et al. 2009).
There are some limitations of this study. Our functional study could be limited by the potential effect of tandem GFP on hClC‐Kb. We were unable to compare the properties of hClC‐Kb‐myc and hClC‐Kb‐GFP since hClC‐Kb‐myc was poorly expressed. According to previous studies, the fused fluorescent protein did not affect the current–voltage curve of the hClC‐Kb channel (Scholl et al. 2006). In addition, the hClC‐Kb current in our study was bi‐directionally rectifying, consistent with previous findings in mammalian cells (Janssen et al. 2009; Keck et al. 2013; Stölting et al. 2015). In contrast, the ClC‐Kb currents measured in oocytes were more outwardly rectifying than inwardly rectifying (Waldegger & Jentsch, 2000; Estevez et al. 2001). In genotype–phenotype analysis, our results could be limited by excluding patients carrying compound heterozygous mutations, mutations with inconsistent or unknown functional outcome, and founder mutations.
Conclusions
Our hClC‐Kb mammalian expression system was efficient for the functional validation of hClC‐Kb mutant channels. Taking advantage of the largest number of functional results of CLCNKB mutations, we reveal the functionally important domains and severe mutational spots of the hClC‐Kb channel and establish the genotype–phenotype association in classic BS. Future studies are warranted to provide more regulatory mechanisms on the hClC‐Kb channel and clarify the roles of each chloride channel or transporter in renal tubules.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All experiments were performed in the laboratory of CJC at National Defense Medical Center, Taipei, Taiwan. Study design and the experimental approach were discussed among all authors. CJC conceived the idea for the project, conducted the patch clamp experiments and wrote the paper. YFL conducted the MG‐132 experiments. JCC constructed wild‐type and mutant CLCNKB plasmids for expression and performed Western blot and biotinylation assays. CLH revised the manuscript for important intellectual content and advised on the project throughout. SHL diagnosed patients, screened gene mutation, collected clinical information, discussed the results and edited the manuscript. All authors listed approved the final version of the manuscript submitted for publication, support the integrity and accuracy of the results, and qualify for authorship.
Funding
This work was supported in part by grants from MOST of Taiwan (MOST 103‐2628‐B‐016‐001‐MY3 to CJC), and from Tri‐Service General Hospital (TSGH‐C103‐119, TSGH‐C105‐111, TSGH‐C106‐094 to CJC; TSGH‐C105‐110, TSGH‐C106‐091 to SHL). CLH is supported by grants from the NIH, USA (DK109887).
Acknowledgements
The authors thank Michel Baum for editing the manuscript.
Linked articles This article is highlighted by a Perspective by Imbrici et al. To read this Perspective, visit https://doi.org/10.1113/JP274645.
This is an Editor's Choice article from the 15 August 2017 issue.
References
- Al‐Shibli A, Yusuf M, Abounajab I & WIllems PJ (2014). Mixed Bartter‐Gitelman syndrome: an inbred family with a heterogeneous phenotype expression of a novel variant in the CLCNKB gene. Springerplus 3, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrini O, Keck M, L'Hoste S, Briones R, Mansour‐Hendili L, Grand T, Sepúlveda FV, Blanchard A, Lourdel S, Vargas‐Poussou R & Teulon J (2014). CLCNKB mutations causing mild Bartter syndrome profoundly alter the pH and Ca2+ dependence of ClC‐Kb channels. Pflugers Arch 466, 1713–1723. [DOI] [PubMed] [Google Scholar]
- Andrini O, Keck M, Briones R, Lourdel S, Vargas‐Poussou R & Teulon J (2015). ClC‐K chloride channels: emerging pathophysiology of Bartter syndrome type 3. Am J Physiol Renal Physiol 308, F1324–F1334. [DOI] [PubMed] [Google Scholar]
- Bettineli A, Borsa N, Bellantuono R, Syrèn ML, Calabrese R, Edefonti A, Komninos J, Santostefano M, Beccaria L, Pela I, Bianchetti MG & Tedeschi S (2007). Patients with biallelic mutations in the chloride channel gene CLCNKB: long‐term management and outcome. Am J Kidney Dis 49, 91–98. [DOI] [PubMed] [Google Scholar]
- Birkenhäger R, Otto E, Schürmann MJ, Vollmer M, Ruf EM, Maier‐Lutz I, Beekmann F, Fekete A, Omran H, Feldmann D, Milford DV, Jeck N, Konrad M, Landau D, Knoers NV, Antignac C, Sudbrak R, Kispert A & Hildebrandt F (2001). Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet 29, 310–314. [DOI] [PubMed] [Google Scholar]
- Brochard K, Boyer O, Blanchard A, Loirat C, Niaudet P, Macher MA, Deschenes G, Bensman A, Decramer S, Cochat P, Morin D, Broux F, Caillez M, Guyot C, Novo R, Jeunemaître X & Vargas‐Poussou R (2009). Phenotype–genotype correlation in antenatal and neonatal variants of Bartter syndrome. Nephrol Dial Transplant 24, 1455–1464. [DOI] [PubMed] [Google Scholar]
- Chen YA, Peng YJ, Hu MC, Huang JJ, Chien YC, Wu JT, Chen TY & Tang CY (2015). The cullin 4A/B‐DDB1‐cereblon E3 ubiquitin ligase complex mediates the degradation of CLC‐1 chloride channels. Sci Rep 5, 10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Lee BH & Cheong HI (2013). Translational read‐through of a nonsense mutation causing Bartter syndrome. J Korean Med Sci 28, 821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embark HM, Böhmer C, Palmada M, Rajamanickam J, Wyatt AW, Wallisch S, Capasso G, Waldegger P, Seyberth HW, Waldegger S & Lang F (2004). Regulation of CLC‐Ka/barttin by the ubiquitin ligase Nedd4‐2 and the serum‐ and glucocorticoid‐dependent kinases. Kidney Int 66, 1918–1925. [DOI] [PubMed] [Google Scholar]
- Estevez R, Boettger T, Stein V, Birkenhäger R, Otto E, Hildebrandt F & Jentsch TJ (2001). Barttin is a Cl− channel β‐subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 414, 558–561. [DOI] [PubMed] [Google Scholar]
- Fahlke C & Fischer M (2010). Physiology and pathophysiology of ClC‐K/barttin channels. Front Physiol 1, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García Castaño A, Pérez de Nanclares G, Madariaga L, Aguirre M, Madrid A, Nadal I, Navarro M, Lucas E, Fijo J, Espino M, Espitaletta Z, Castaño L, Ariceta G & Renal Tube Group (2013). Genetics of type III Bartter syndrome in Spain, proposed diagnostic algorithm. PLoS One 8, e74673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García Castaño A, Pérez de Nanclares G, Madariaga L, Aguirre M, Madrid Á, Chocrón S, Nadal I, Navarro M, Lucas E, Fijo J, Espino M, Espitaletta Z, García Nieto V, Barajas de Frutos D, Loza R, Pintos G, Castaño L; RenalTube Group & Ariceta G (2017). Poor phenotype‐genotype association in a large series of patients with Type III Bartter syndrome. PLoS One 12, e0173581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayama A, Rai T, Sasaki S & Uchida S (2003). Molecular mechanisms of Bartter syndrome caused by mutations in the BSND gene. Histochem Cell Biol 119, 485–493. [DOI] [PubMed] [Google Scholar]
- Hebert SC (2003). Bartter syndrome. Curr Opin Nephrol Hypertens 12, 527–532. [DOI] [PubMed] [Google Scholar]
- Hennings JC, Andrini O, Picard N, Paulais M, Huebner AK, Cayuqueo IK, Bignon Y, Keck M, Cornière N, Böhm D, Jentsch TJ, Chambrey R, Teulon J, Hübner CA & Eladari D (2017). The ClC‐K2 chloride channel is critical for salt handling in the distal nephron. J Am Soc Nephrol 28, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohberger B & Enz R (2009). Cereblon is expressed in the retina and binds to voltage‐gated chloride channels. FEBS Lett 583, 633–637. [DOI] [PubMed] [Google Scholar]
- Imbrici P, Liantonio A, Gradogna A, Pusch M & Camerino DC (2014). Targeting kidney CLC‐K channels: pharmacological profile in a human cell line versus Xenopus oocytes. Biochim Biophys Acta 1838, 2484–2491. [DOI] [PubMed] [Google Scholar]
- Janssen AG, Scholl U, Domeyer C, Nothmann D, Leinenweber A & Fahlke C (2009). Disease‐causing dysfunctions of barttin in Bartter syndrome type IV. J Am Soc Nephrol 20, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck N, Schlingmann KP, Reinalter SC, Kömhoff M, Peters M, Waldegger S & Seyberth HW (2005). Salt handling in the distal nephron: lessons learned from inherited human disorders. Am J Physiol Regul Integr Comp Physiol 288, R782–R795. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Stein V, Weinreich F & Zdebik AA (2002). Molecular structure and physiological function of chloride channels. Physiol Rev 82, 503–568. [DOI] [PubMed] [Google Scholar]
- Keck M, Andrini O, Lahuna O, Burgos J, Cid LP, Sepúlveda FV, L'hoste S, Blanchard A, Vargas‐Poussou R, Lourdel S & Teulon J (2013). Novel CLCNKB mutations causing Bartter syndrome affect channel surface expression. Hum Mutat 34, 1269–1278. [DOI] [PubMed] [Google Scholar]
- Kieferle S, Fong P, Bens M, Vandewalle A & Jentsch TJ (1994). Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc Natl Acad Sci USA 91, 6943–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad M, Vollmer M, Lemmink HH, van den Heuvel LP, Jeck N, Vargas‐Poussou R, Lakings A, Ruf R, Deschênes G, Antignac C, Guay‐Woodford L, Knoers NV, Seyberth HW, Feldmann D & Hildebrandt F (2000). Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol 11, 1449–1459. [DOI] [PubMed] [Google Scholar]
- Laghmani K, Beck BB, Yang SS, Seaayfan E, Wenzel A, Reusch B, Vitzthum H, Priem D, Demaretz S, Bergmann K, Duin LK, Göbel H, Mache C, Thiele H, Bartram MP, Dombret C, Altmüller J, Nürnberg P, Benzing T, Levtchenko E, Seyberth HW, Klaus G, Yigit G, Lin SH, Timmer A, de Koning TJ, Scherjon SA, Schlingmann KP, Bertrand MJ, Rinschen MM, de Backer O, Konrad M & Kömhoff M (2016). Polyhydramnios, transient antenatal Bartter's syndrome, and MAGED2 mutations. N Engl J Med 374, 1853–1863. [DOI] [PubMed] [Google Scholar]
- Lazrak A, Liu Z & Huang CL (2006). Antagonistic regulation of ROMK by long and kidney‐specific WNK1 isoforms. Proc Natl Acad Sci USA 103, 1615–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CM, Tsai JD, Lo YF, Yan MT, Yang SS & Lin SH (2009). Chronic renal failure in a boy with classic Bartter's syndrome due to a novel mutation in CLCNKB coding for the chloride channel. Eur J Pediatr 168, 1129–1133. [DOI] [PubMed] [Google Scholar]
- Lee BH, Cho HY, Lee H, Han KH, Kang HG, Ha IS, Lee JH, Park YS, Shin JI, Lee DY, Kim SY, Choi Y & Cheong HI (2012). Genetic basis of Bartter syndrome in Korea. Nephrol Dial Transplant 27, 1516–1521. [DOI] [PubMed] [Google Scholar]
- Maulet Y, Lambert RC, Mykita S, Mouton J, Partisani M, Bailly Y, Bombarde G & Feltz A (1999). Expression and targeting to the plasma membrane of xClC‐K, a chloride channel specifically expressed in distinct tubule segments of Xenopus laevis kidney. Biochem J 340, 737–743. [PMC free article] [PubMed] [Google Scholar]
- Nissant A, Paulais M, Lachheb S, Lourdel S & Teulon J (2006). Similar chloride channel in the connecting tubule and cortical collecting duct of the mouse kidney. Am J Physiol Renal Physiol 290, F1421–F1429. [DOI] [PubMed] [Google Scholar]
- Pinelli L, Nissant A, Edwards A, Lourdel S, Teulon J & Paulais M (2016). Dual regulation of the native ClC‐K2 chloride channel in the distal nephron by voltage and pH. J Gen Physiol 148, 213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin S, Anwar S, Fischer M, Ahmed ZM, Khan SY, Janssen AG, Zafar AU, Scholl U, Husnain T, Belyantseva IA, Friedman PL, Riazuddin S, Friedman TB & Fahlke C. (2009). Molecular basis of DFNB73: mutations of BSND can cause nonsyndromic deafness or Bartter syndrome. Am J Hum Genet 85, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Soriano J, Vallo A, Perez de Nanclares G, Bilbao JR & Castaño L (2005). A founder mutation in the CLCNKB gene causes Bartter syndrome type III in Spain. Pediatr Nephrol 20, 891–896. [DOI] [PubMed] [Google Scholar]
- Scholl U, Hebeisen S, Janssen AG, Müller‐Newen G, Alekov A & Fahlke C (2006). Barttin modulates trafficking and function of ClC‐K channels. Proc Natl Acad Sci USA 103, 11411–11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurman SJ, Perlman SA, Sutphen R, Campos A, Garin EH, Cruz DN & Shoemaker LR (2001). Genotype/phenotype observations in African Americans with Bartter syndrome. J Pediatr 139, 105–110. [DOI] [PubMed] [Google Scholar]
- Simon DB, Bindra RS, Mansfield TA, Nelson‐Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez‐Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E & Lifton RP (1997). Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat Genet 17, 171–178. [DOI] [PubMed] [Google Scholar]
- Stölting G, Bungert‐Plümke S, Franzen A & Fahlke C (2015). Carboxyl‐terminal truncations of ClC‐Kb abolish channel activation by Barttin via modified common gating and trafficking. J Biol Chem 290, 30406–30416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima M, Hayama A, Rai T, Sasaki S & Uchida S (2007). Barttin binds to the outer lateral surface of the ClC‐K2 chloride channel. Biochem Biophys Res Commun 362, 858–864. [DOI] [PubMed] [Google Scholar]
- Uchida S & Sasaki S (2005). Function of chloride channels in the kidney. Annu Rev Physiol 67, 759–778. [DOI] [PubMed] [Google Scholar]
- Vargas‐Poussou R, Dahan K, Kahila D, Venisse A, Riveira‐Munoz E, Debaix H, Grisart B, Bridoux F, Unwin R, Moulin B, Haymann JP, Vantyghem MC, Rigothier C, Dussol B, Godin M, Nivet H, Dubourg L, Tack I, Gimenez‐Roqueplo AP, Houillier P, Blanchard A, Devuyst O & Jeunemaitre X (2011). Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol 22, 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldegger S & Jentsch TJ (2000). Functional and structural analysis of ClC‐K chloride channels involved in renal disease. J Biol Chem 275, 24527–24533. [DOI] [PubMed] [Google Scholar]
- Waldegger S, Jeck N, Barth P, Peters M, Vitzthum H, Wolf K, Kurtz A, Konrad M & Seyberth HW (2002). Barttin increases surface expression and changes current properties of ClC‐K channels. Pflugers Arch 444, 411–418. [DOI] [PubMed] [Google Scholar]
- Wojciechowski D, Fischer M & Fahlke C (2015). Tryptophan scanning mutagenesis identifies the molecular determinants of distinct barttin functions. J Biol Chem 290, 18732–18743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Xu C, Pan X, Ren H, Wang W, Meng X, Huang F & Chen N (2009). Identification and functional analysis of novel mutations of the CLCNKB gene in Chinese patients with classic Bartter syndrome. Clin Genet 77, 155–162. [DOI] [PubMed] [Google Scholar]
- Zelikovic I, Szargel R, Hawash A, Labay V, Hatib I, Cohen N & Nakhoul F (2003). A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int 63, 24–32. [DOI] [PubMed] [Google Scholar]