

Abstract
Key points
Mechanotransduction in endothelial cells is a central mechanism in the regulation of vascular tone and vascular remodelling
Mechanotransduction and vascular function may be affected by high sugar levels in plasma because of a resulting increase in oxidative stress and increased levels of advanced glycation end‐products (AGE).
In healthy young subjects, 2 weeks of daily supplementation with 3 × 75 g of sucrose was found to reduce blood flow in response to passive lower leg movement and in response to 12 W of knee extensor exercise.
This vascular impairment was paralleled by up‐regulation of platelet endothelial cell adhesion molecule (PECAM)‐1, endothelial nitric oxide synthase, NADPH oxidase and Rho family GTPase Rac1 protein expression, an increased basal phosphorylation status of vascular endothelial growth factor receptor 2 and a reduced phosphorylation status of PECAM‐1. There were no measurable changes in AGE levels.
The findings of the present study demonstrate that daily high sucrose intake markedly affects mechanotransduction proteins and has a detrimental effect on vascular function.
Abstract
Endothelial mechanotransduction is important for vascular function but alterations and activation of vascular mechanosensory proteins have not been investigated in humans. In endothelial cell culture, simple sugars effectively impair mechanosensor proteins. To study mechanosensor‐ and vascular function in humans, 12 young healthy male subjects supplemented their diet with 3 × 75 g sucrose day−1 for 14 days in a randomized cross‐over design. Before and after the intervention period, the hyperaemic response to passive lower leg movement and active knee extensor exercise was determined by ultrasound doppler. A muscle biopsy was obtained from the thigh muscle before and after acute passive leg movement to allow assessment of protein amounts and the phosphorylation status of mechanosensory proteins and NADPH oxidase. The sucrose intervention led to a reduced flow response to passive movement (by 17 ± 2%) and to 12 W of active exercise (by 9 ± 1%), indicating impaired vascular function. A reduced flow response to passive and active exercise was paralleled by a significant up‐regulation of platelet endothelial cell adhesion molecule (PECAM‐1), endothelial nitric oxide synthase, NADPH oxidase and the Rho family GTPase Rac1 protein expression in the muscle tissue, as well as an increased basal phosphorylation status of vascular endothelial growth factor receptor 2 and a reduced phosphorylation status of PECAM‐1. The phosphorylation status was not acutely altered with passive leg movement. These findings indicate that a regular intake of high levels of sucrose can impair vascular mechanotransduction and increase the oxidative stress potential, and suggest that dietary excessive sugar intake may contribute to the development of vascular disease.
Keywords: vascular function, passive leg movement, mechanosensor
Key points
Mechanotransduction in endothelial cells is a central mechanism in the regulation of vascular tone and vascular remodelling
Mechanotransduction and vascular function may be affected by high sugar levels in plasma because of a resulting increase in oxidative stress and increased levels of advanced glycation end‐products (AGE).
In healthy young subjects, 2 weeks of daily supplementation with 3 × 75 g of sucrose was found to reduce blood flow in response to passive lower leg movement and in response to 12 W of knee extensor exercise.
This vascular impairment was paralleled by up‐regulation of platelet endothelial cell adhesion molecule (PECAM)‐1, endothelial nitric oxide synthase, NADPH oxidase and Rho family GTPase Rac1 protein expression, an increased basal phosphorylation status of vascular endothelial growth factor receptor 2 and a reduced phosphorylation status of PECAM‐1. There were no measurable changes in AGE levels.
The findings of the present study demonstrate that daily high sucrose intake markedly affects mechanotransduction proteins and has a detrimental effect on vascular function.
Abbreviations
- AGE
advanced glycation end‐products
- eNOS
endothelial nitric oxide synthase
- NO
nitric oxide
- NOX
NADPH oxidase
- NOx
nitrite and nitrate
- PECAM‐1
platelet endothelial cell adhesion molecule
- PGI2
prostacyclin
- Rac1
Rho family GTPase Rac1
- ROS
reactive oxygen species
- sRAGE
soluble receptor of advanced glycation end products
- VEGF‐R2
vascular endothelial growth factor receptor 2
- VE‐cadherin
vascular endothelial cadheri
Introduction
The vascular system is precisely controlled, allowing for a tight coupling between oxygen delivery and oxygen demand in most organs throughout the body (Andersen & Saltin, 1985; Joyner & Casey, 2015). In conditions with a mismatch between oxygen delivery and oxygen demand, an impaired regulation of vascular tone (i.e. vascular dysfunction) is often the underlying cause. Vascular dysfunction commonly develops with a sedentary lifestyle, obesity and ageing (McVeigh et al. 1992; Taddei et al. 2001; Seals et al. 2011) and is good predictor of cardiovascular disease (Widlansky et al. 2003; Deanfield et al. 2007; Green et al. 2011).
The regulation of vascular tone involves a complex integration of stimuli from sympathetic nerve activity, locally produced or circulating compounds, as well as mechanical forces, including shear stress (Lamontagne et al. 1992). Shear stress is the frictional force of flowing blood applied to the endothelial layer lining the blood vessels. Studies on endothelial cells in culture have demonstrated that shear stress is sensed by a complex of mechanosensors consisting of vascular endothelial growth factor receptor 2 (VEGF‐R2), vascular endothelial cadherin (VE‐cadherin) and platelet endothelial cell adhesion molecule 1 (PECAM‐1) (Osawa et al. 2002; Shay‐Salit et al. 2002; Tzima et al. 2005). Mechanical deformation of the mechanosensor complex leads to phosphorylation of all three proteins (Tzima et al. 2005) and subsequent downstream activation of intracellular signal cascades (Fleming & Busse, 1999). Ultimately, activation of the cascade leads to increased formation of nitric oxide (NO) via phosphorylation and activation of endothelial nitric oxide synthase (eNOS), in particular serine residue 1177 (Jin et al. 2003; Fleming et al. 2005; Tzima et al. 2005). NO in turn causes a cGMP‐dependent decrease in calcium levels and sensitivity in adjacent smooth muscle cells. This leads to shear stress induced vasodilatation.
It is well established that impaired vascular function in hypertension, diabetes and ageing is related to a dysfunctional NO system (Taddei et al. 2006). This dysfunction does not appear to be related to a reduced expression of eNOS (Donato et al. 2009; Nyberg et al. 2012) but rather to a reduced mechanical or chemical activation of eNOS or to an excessive removal of NO via reaction with reactive oxygen species (ROS) (Gryglewski et al. 1986; Guzik et al. 2002; Fleming & Busse, 2003). Shear stress induced activation of eNOS in the microcirculation and how it may be altered in disease has been poorly explored in vivo and the available data in humans are limited.
Hyperglycaemia acutely impairs shear stress induced vascular regulation in healthy humans (Kawano et al. 1999; Zhu et al. 2007) and incubation of bovine endothelial cells with high glucose suppresses both ACh‐ and shear stress induced activation of eNOS and NO formation (Connell et al. 2007). Moreover, diabetic subjects with chronic hyperglycaemia present an impaired macrovascular shear stress response (Naylor et al. 2011). One of the mechanisms underlying this effect in diabetic patients could be glycation of the mechanosensor proteins. Long‐term high blood glucose levels have been shown to induce elevated levels of advanced glycation end‐products (AGE) (Fiorentino et al. 2013). Glycation of the mechanosensitive proteins caused by the presence of AGE leads to decreased mechanosensor sensitivity (Otero et al. 2001; Liu et al. 2012; Naser et al. 2013). This link has been established in hypertension, diabetes and ageing (Goh & Cooper, 2008; Seals et al. 2011; Liu et al. 2016) and may in turn lead to impaired eNOS activation and NO production (Soro‐Paavonen et al. 2010; Naser et al. 2013). In addition, high glucose levels are associated with an increased level of ROS in the vascular system, which also may affect the bioavailability of NO (Cosentino et al. 1997; Inoguchi et al. 2000; Cosentino et al. 2003). A period of experimental manipulation of blood glucose via intake of simple sugars would therefore be a relevant approach for manipulating endothelial mechanotransduction and vasodilatation in humans to allow study of the underlying mechanisms of vascular function.
Shear stress induced vasodilatation can be evaluated in humans by the flow response to passive movement of the lower leg (Hellsten et al. 2008; Mortensen et al. 2012). Passive movement of the lower leg causes a rapid biphasic increase in femoral arterial blood flow and a passive stretch of the muscle in the absence of muscle activity or metabolic disturbances (Hellsten et al. 2008; Mortensen et al. 2012). Both shear stress (Pohl et al. 1986) and stretch probably contribute to the flow response, although the stretch effect is probably short lasting (Venturelli et al. 2017). The response to passive movement is highly NO‐dependent, as indicated by an almost completely abolished flow response in the presence of an NO synthase blocker (Mortensen et al. 2012). Thus, passive leg movement induced hyperaemia is a valid method for studying the impact of mechanotransduction in the vascular system in vivo.
The two hypotheses of the present study are that, in healthy young men, the increase in blood flow during passive leg movement would be associated with phosphorylation of endothelial mechanotransductor proteins and eNOS and that 14 days of high sucrose intake would impair mechanosensing during passive leg movement with a consequent reduction in mechanosensor phosphorylation and blood flow.
Methods
Ethical approval
The present study was approved by the Ethics Committee of Copenhagen and Frederiksberg communities (H‐6‐2014‐085) and was conducted in accordance with the latest guidelines of the Declaration of Helsinki. Written informed consent was obtained from all subjects before enrollment in the study.
Subjects
Twelve young healthy men (aged 20–25 years) were recruited. All subjects were of normal weight (body mass index < 28 kg m–2), non‐smokers and without known chronic diseases.
Intervention
The study was of a randomized, cross‐over design. Each subject completed two 14 day periods of either a control or a high sucrose diet, with a 30 day washout period between the end of the first and the start of the second period. Before the first experimental day, subjects were randomly allocated to start with either the control or the sucrose diet in a balanced order. During the control treatment, subjects maintained their normal diet and during the sucrose treatment subjects supplemented their normal diet with three daily doses of 75 g of sucrose dissolved in 200 ml of water. The sucrose diet introduced a daily increase in energy intake of 886 kcal. With normal diet and energy expenditure maintained during the 2 week intervention, this corresponds to 1.58 kg of body fat. Each daily dose of sucrose was ingested after a meal, morning, midday and afternoon. Subjects registered the time of sucrose ingestion and any discomfort experienced. A 7 day diet diary was completed before the start of the intervention and normal diets of the subjects were maintained from enrollment to end of the intervention. All analyses were blinded to the investigators.
Pre‐testing
Before the first experimental day, subjects visited the laboratory where pulmonary maximal oxygen uptake () was determined (Oxycon Pro; Viasys Healhtcare, Hoechberg, Germany) with an incremental exercise test on a mechanically braked cycle ergometer (Monark Ergomedic 839E; Monark, Vansbro, Sweden). Subjects completed a 5 min warm‐up at 125 W, after which the workload was increased by 25 W min−1 until exhaustion. was calculated as the average of the three highest consecutive 15 s values. For recognition of true , three of the following five criteria had to be met: individual perception of exhaustion, respiratory exchange rate > 1.15, curve plateau, heart rate approaching age‐predicted maximum, and inability to maintain pedalling frequency above 80 rpm. Verbal encouragement was given throughout the test. To confirm normal glucose metabolism and liver function, blood samples were drawn from the antecubital vein and analysed for glucose, HbA1c, insulin and hepatic enzymes at the clinical biochemical unit at the main hospital in Copenhagen (Rigshospitalet) (Table 1).
Table 1.
Subject characteristics and blood values
| Age (years) | 22.8 ± 0.9 | – | |
| Height (cm) | 186.1 ± 1.6 | – | |
| (ml O2 min−1) | 4130 ± 141 | – | |
| (ml O2 min−1 kg−1) | 50.4 ± 1.9 | – | |
| Control | After sucrose | P | |
| Body weight (kg) | 82.2 ± 3.3 | 83.5 ± 3.5 | 0.21 |
| Systolic blood pressure (mmHg) | 116.4 ± 2.8 | 118.3 ± 2.3 | 0.43 |
| Diastolic blood pressure (mmHg) | 66.6 ± 2.0 | 69.9 ± 1.6 | 0.09 |
| Rest heart rate (beats min–1) | 55.0 ± 1.9 | 56.6 ± 2.6 | 0.32 |
| Erythrocytes (1012 l−1) | 4.7 ± 0.1 | 4.8 ± 0.1 | 0.52 |
| Hemoglobin (mmol l−1) | 8.5 ± 0.1 | 8.7 ± 0.1 | 0.38 |
| Plasma endothelin‐1 (pg ml−1) | 2.6 ± 0.1 | 2.3 ± 0.1 | 0.21 |
| Fasting blood glucose (mmol l−1) | 5.1 ± 0.1 | 4.9 ± 0.1 | 0.07 |
| HbA1c (mmol l−1) | 5.4 ± 0.1 | 5.3 ± 0.1 | 0.09 |
| Insulin (pmol l−1) | 38.0 ± 4.4 | 45.5 ± 5.5 | 0.18 |
| QUICKI | 0.44 ± 0.01 | 0.43 ± 0.01 | 0.34 |
| Total cholesterol (mmol l−1) | 3.7 ± 0.2 | 3.8 ± 0.2 | 0.25 |
| HDL cholesterol (mmol l−1) | 1.3 ± 0.1 | 1.3 ± 0.1 | 0.72 |
| LDL cholesterol (mmol l−1) | 2.2 ± 0.2 | 2.4 ± 0.2 | 0.06 |
| Triglycerides (mmol l−1) | 0.7 ± 0.1 | 0.7 ± 0.1 | 0.64 |
, maximal oxygen optake; QUICKI, quantitative insulin sensitivity check index (Katz et al. 2000). HDL, high‐density lipoprotein; LDL, low‐density lipoprotein. Data are presented as the mean ± SD.
Experimental days
On experimental days, subjects arrived at the laboratory after an overnight fast. Subjects rested in the supine position and a catheter (DB Venflon Pro Safety, 18 GA; Becton Dickinson Infusion Therapy AB, Stockholm, Sweden) was inserted into the antecubital vein for blood sampling. Blood was drawn at rest, after 20 min of passive leg movement (Mortensen et al. 2012), and before and after active knee extensor exercise. Two microdialysis probes (CMA63 with 30 mm membrane length and 20 kDa cut‐off; M Dialysis AB, Stockholm, Sweden) were inserted into the musculus vastus lateralis under local anaesthesia (xylocaine, ∼3 ml, 20 mg ml−1; AstraZeneca, Gothenburg, Sweden). After insertion of the probes, subjects rested for 120 min to allow the muscle to attain a resting state and to recover from insertion trauma. The probes were perfused with Ringer acetate buffer (Fresenius Kabi AB, Oslo, Norway) at a rate of 5 ml min−1 and, to determine the relative exchange over the membrane, a small amount (2.7 nm) of [2‐3H] labelled adenosine was added to the perfusate to allow for calculation of probe recovery. After the 120 min of initial rest, blood pressure was measured three consecutive times with an automatic sphygmomanometer (M7; OMRON, Vernon Hills, IL, USA) on the left and right upper arm and microdialysate was collected for 20 min of subsequent rest. Microdialysate was then collected during 20 min of passive leg movement, excluding the first 2 min to account for delay in the probe perfusate. Immediately after collection, samples were weighed and triplicates of 5 ml of dialysate were allocated into 3 ml of Ultimate Gold scintillation liquid (Perkin Elmer, Waltham, MA, USA). The remaining dialysate was frozen at −80°C. Probe recovery (PR) was calculated as [PR = (dpminfusate–dpmdialysate/dpminfusate)], where dpm denotes disintegrations per minute (Scheller & Kolb, 1991; Jansson et al. 1994). The probe recovery was used to obtain an estimate of probe recovery at rest and during passive movement. The main purpose for this estimation was to take into account changes in recovery in going from rest to movement. Accordingly, the resulting values are presented as estimated interstitial concentrations. The [2‐3H] adenosine activity of the dialysate was measured on a liquid scintillation counter (Tri‐Carb 2910 TR; Perkin Elmer).
Femoral arterial blood flow was measured at rest, during passive leg movement and during active knee extensor exercise (at 12, 18 and 24 W) with ultrasound Doppler (GE Vivid E9; GE Healthcare, Pittsburgh, PA, USA) equipped with a linear probe (L5) operating at an imaging frequency of 9 MHz and a Doppler frequency of 4.2 MHz. The site of blood velocity measurements in the common femoral artery was distal to the inguinal ligament but above the bifurcation into the superficial and profound femoral branch to avoid turbulence from the bifurcation. All recordings were obtained at the lowest possible insonation angle and always below 60°. The sample volume was maximized by choosing the widest section of the vessel and the measurements were made without interference of the vessel walls. A low‐velocity filter (velocities < 1.8 m s–1) rejected noises caused by turbulence at the vascular wall. Doppler tracings and B‐mode images were recorded continuously and Doppler tracings were averaged over 45 s. Resting femoral arterial blood flow was measured before passive leg movement and before active knee extensor exercise. During the 20 min passive leg movement, femoral arterial blood flow was measured at 15 s intervals during the initial 3 min, after 4 and 5 min, and at 2.5 min intervals from 7.5 to 20 min. After 30 min of rest, active knee extensor exercise was performed for 3 min at each load, allowing for steady‐state blood flow. Femoral arterial blood flow was measured during the final minute of each exercise bout.
Immediately before and immediately after the 20 min passive leg movement bout, a muscle biopsy was obtained from the musculus vastus lateralis under local anaesthesia (xylocaine, ∼5 ml, 20 mg ml−1; AstraZeneca) using the percutaneous needle biopsy technique with suction (Bergström, 1975). The biopsies were immediately frozen in liquid nitrogen. Frozen samples were stored at 80°C until further analysis.
Quantification of protein expression
Biopsies were freeze‐dried and dissected free from fat, blood and connective tissue. Approximately 5 mg dry weight of muscle tissue was homogenized in a fresh batch buffer (10% glycerol, 20 mm sodium‐pyrophosphate, 150 mm NaCl, 50 mm Hepes, 1% Nonidet P‐40, 20 mm β‐glycerophosphate and proteolytic inhibitors) two times for 30 s (Qiagen Tissuelyser II; Retsch, Haan, Germany). After rotation end‐over‐end for 1 h, the samples were centrifuged for 30 min at 17 500 g at 4°C and the lysates were collected as the supernatant. Protein concentrations were determined in the lysates using BSA standards (Pierce Reagents, Rockford, Il, USA). The lysates were diluted to appropriate protein concentrations in a concentrated sample buffer (0.5 m Tris‐base, dithiothreitol, SDS, glycerol and bromphenol blue) and equal amounts of total protein were loaded for each sample in different wells on precasted Tris‐HCL gels (Bio‐Rad, Hercules, CA, USA). For comparisons, samples from the same subject were always loaded on the same gel. After gel electrophoresis, the proteins were transferred (semidry) to a polyvinylidene difluoride membrane (Immobilon Transfer Membrane; Millipore, Billerica, MA, USA), which was incubated with ∼10 ml of primary antibody overnight and then washed three times for 5 min in Tris‐buffered saline‐Tween before incubation with secondary antibody for 1 h. The membranes were incubated with the primary antibodies: VEGF‐R2 (sc‐19530; Santa Cruz Biotechnology, Santa Cruz, CA, USA); VEGF‐R2 Tyr1175 (2478, Cell Signaling Technology, Danvers, MA, USA); eNOS (BD 610287, BD Biosciences, San Jose, CA, USA‐9; eNOS Ser1177 (Calbiochem 482737; Merck Milipore, Darmstadt, Germany); PECAM‐1 (AF806; R&D Systems Inc., Minneapolis, MN, USA); PECAM‐1 Tyr 713 (BS4666; Bioworld Technology, St Louis Park, MN, USA); VE‐cadherin (ab33168; Abcam, Cambridge, UK); VE‐cadherin Tyr731 (NBP1‐51393; Novus Biologicals, Littleton, CO, USA); NADPH oxidase (NOX) p67 (610912; BD Biosciences, San Jose, CA, USA); S‐nitrosocysteine (ab94930; Abcam); and Rho family GTPase Rac1 (Rac1) (610651; BD Biosciences). Secondary antibodies used were goat anti‐rabbit or rabbit anti‐goat HRP‐conjugated antibodies (P‐0448 and P‐0449; DakoCytomation, Glostrup, Denmark; dilution 1:5000). Following detection and quantification (ChemiDoc MP system; Bio‐Rad), the protein content was expressed in arbitrary units. To control for loading differences, the blots were also analysed for glyceraldehyde 3‐phosphate dehydrogenase (ab9484; Abcam), actin (A2066; Sigma‐Aldrich, St Louis, MO, USA) or α‐tubulin (ab4076, Abcam).
To confirm that the mechanosensor proteins PECAM‐1, VE‐cadherin and VEGF‐R2 were not present in skeletal muscle cells, pure human primary skeletal muscle cell cultures (human skeletal muscle cells; PromoCell, Heidelberg, Germany) were run on a gel together with whole human muscle homogenate (both samples loaded with the same protein concentration). Blots are shown in Fig. 3 C.
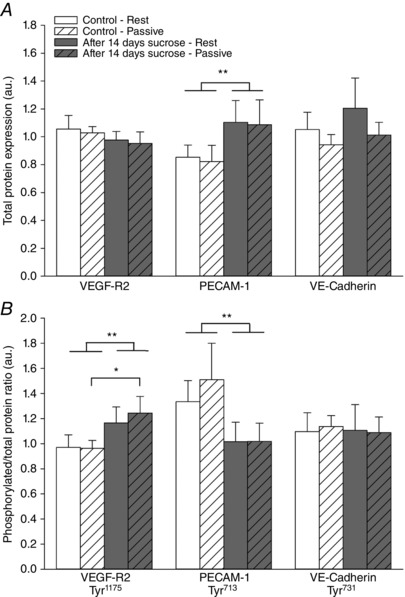
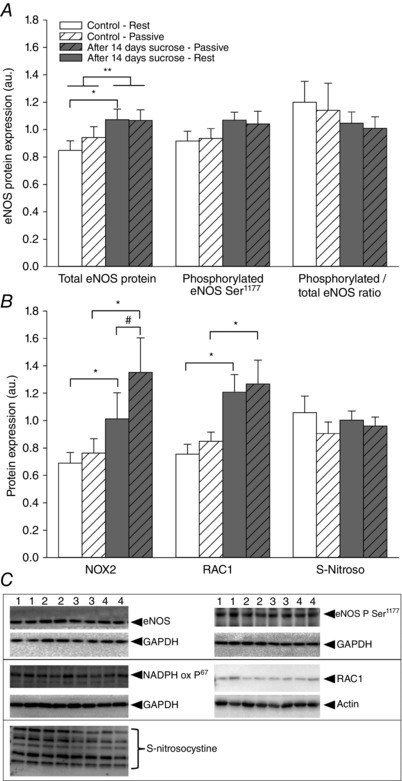
Figure 3. Protein expression and phosphorylated/total protein ratio of mechanosensor complex before and after 14 days of sucrose intervention.
A, protein expression of VEGF‐R2, PECAM‐1 and VE‐cadherin. B, phosphorylated VEGF‐R2 (tyr1175), PECAM‐1 (tyr713) and VE‐cadherin (tyr731) to total protein ratio in muscle homogenates from musculus vastus lateralis at rest and after 20 min of passive leg movement. C, expression of VE‐CAD, VEGF‐R2 and PECAM‐1 in isolated primary skeletal muscle cells (SkMu Cell) and skeletal muscle tissue homogenate (SkMu Tissue). D, representative blots are shown from one subject in duplicates from rest (1) and after passive leg movement (2) under the control situation and from rest (3) and after passive leg movement (4) after the sucrose intervention. Data are presented as the mean ± SD (n = 12). *Significantly different compared to before the sucrose intervention during the condition of passive leg movement. **Overall significant effect of the sucrose intervention.
Analysis of skeletal muscle mRNA content: RNA isolation, reverse transcription and PCR
Total RNA was isolated from the muscle biopsies using TRIzol reagent in accordance with the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). First‐strand cDNA was synthesized from 1 μg of total RNA by SuperScript™ II Reverse Transcriptase (Invitrogen) as described previously (Pilegaard et al. 2000). The mRNA content of VEGF‐R2, eNOS and PECAM‐1, was determined by real‐time RT‐PCR (ABI PRISM 7900 Sequence Detection System; Applied Biosystems, Foster City, CA, USA). The cDNAs were amplified using TaqMan Gene expression assays from Applied Biosystems. For each sample, the amount of target gene mRNA was normalized to β‐actin mRNA content. The effect of the experimental condition on the level of β‐actin mRNA was determined statistically and no significant effect was found with acute exercise.
Analysis of NOx, ET‐1, AGE and RAGE in plasma
The stable metabolites of NO in plasma, nitrite and nitrate (NOx), were measured using a fluorometric EIA kit (Cayman Chemical Co., Ann Harbor, MI, USA). ET‐1 and soluble receptor of advanced glycation end products (sRAGE) in plasma were measured with a Quantikine ELISA kit (R&D Systems). AGE in plasma was measured with ELISA kits (Oxyselect Advanced Glycation End Products Competitive ELISA kit; Cell Biolabs, San Diego, CA, USA).
Analysis of NOx and prostacyclin (PGI2) in microdialysate
NOx and the stable metabolites of of PGI2, 6‐keto prostaglandin F1α, in microdialysate were measured using fluorometric kits (Cayman Chemical Co.). Interstitial concentrations were estimated based on the relative recovery of [2‐3H] adenosine in the microdialysis probe. Based on previous findings, it was assumed that the relative recovery of [2‐3H] adenosine is similar to the recovery of other compounds (Höffner et al. 2003).
Statistical analysis
Subject number is based on power calculations of selected primary outcome measures. These include average differences and variability in femoral arterial blood flow in response to passive leg movement and protein expression and phosphorylation of eNOS in muscle homogenates from musculus vastus lateralis (Altman, 1980). The area under the curve for the analysis of total blood flow to the leg during the passive leg movement was calculated using the trapezoidal rule. Significance level for all tests was set at an α‐level of P < 0.05 at a power level of 0.8. Data are reported as the mean ± SD.
A linear mixed‐model approach (RStudio, version 0.99.903; RStudio, Boston, MA, USA) was used to investigate the effects of (i) the sucrose intervention and (ii) passive leg movement and active exercise. Fixed factors were ‘intervention’ (control or sucrose) and ‘time’ (rest, passive leg movement and active exercise). Subjects were specified as a repeated factor and identifier of random variation. The homogeneity of variance and normal distribution was confirmed through residual and Q–Q plots. Pairwise differences were identified using the Tukey's honestly significant difference post hoc procedure.
Results
Compliance with the intervention
Based on self‐reports, the subjects exhibited very high compliance (100%) with the intervention, resulting in no deviations from the sucrose ingestion protocol or their normal diet during the intervention period.
Body composition, blood values and blood pressure
Under control conditions, subjects had a body mass index of 23.7 ± 0.7 kg m−2, in addition to normal blood pressure, blood glucose, insulin and lipid profiles (Table 1). The sucrose intervention did not affect body weight, body composition, blood pressure, plasma endothelin‐1, fasting blood glucose, insulin, calculated QUICKI index or lipid profile (Table 1). Hepatic enzymes were not affected by the sucrose intervention (data not shown).
Blood flow response to passive leg movement
Femoral arterial blood flow was increased at 15 s of passive leg movement and remained elevated throughout the 20 min period (P = 0.029) (Fig. 1). After the sucrose intervention, the blood flow response was significantly lower compared to the control condition (P = 0.033). The area under the curve comprising total blood flow during the 20 min period was 17% lower under the sucrose condition compared to the control condition (10.4 ± 1 vs. 12.5 ± 1 l; P = 0.018).
Figure 1. Femoral arterial blood flow during rest and passive leg movement.
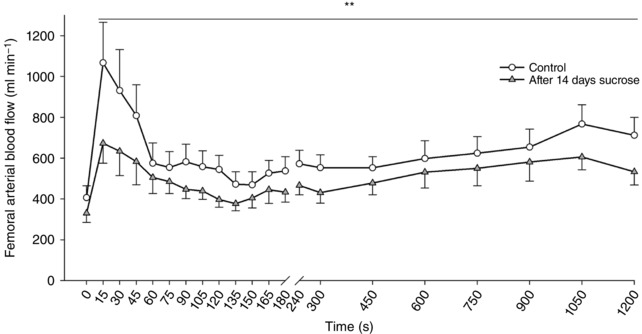
Blood flow response to 20 min of passive leg movement under control conditions (open circles) and after 2 weeks of high sucrose intake (grey triangles). Data are presented as the mean ± SD (n = 12). **Overall significant effect of the sucrose intervention compared to the control condition.
Blood flow response to active exercise
Femoral arterial blood flow at rest was no different between the two conditions (Fig. 2). During active exercise, blood flow increased at 12, 24 and 36 W under both conditions (P < 0.0001). Flow was lower at 12 W after the sucrose intervention compared to the control condition (P = 0.033) (Fig. 2).
Figure 2. Femoral arterial blood flow during active one leg knee extensor exercise.
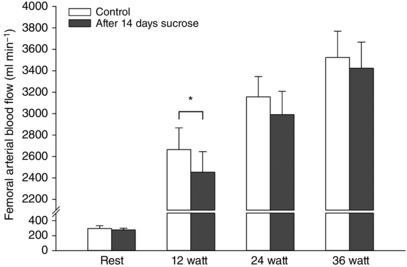
Blood flow response to 12, 24 and 36 W exercise under control conditions (open bars) and after 2 weeks of high sucrose intake (grey bars). Data are presented as the mean ± SD (n = 12). *Significantly different compared to the control condition.
Protein expression and phosphorylation of mechanosensors
Baseline protein levels of VEGF‐R2 and VE‐cadherin were no different between the two conditions, whereas baseline protein PECAM‐1 levels were higher overall after the sucrose intervention (P = 0.022) (Fig. 3 A). There was no effect of acute passive leg movement on content of any of the proteins. The ratio of phosphorylation at tyrosine residue 1175 to total protein of VEGF‐R2 was increased after the sucrose intervention overall and was higher after passive leg movement subsequent to the sucrose intervention compared to the control (P = 0.014) (Fig. 3 B). The ratio of phosphorylated PECAM‐1 at tyrosine residue 713 to total protein of PECAM‐1 was reduced after the sucrose intervention (P = 0.037) (Fig. 3 B). The phosphorylation status of VE‐cadherin at tyrosine residue 731 was no different between the sucrose intervention and the control condition. Passive leg movement did not change the phosphorylation status of any of the mechanosensor proteins in the control or after the sucrose intervention (Fig. 3 B).
Comparison of PECAM1, VE‐cadherin and VEGF‐R2 protein expression in whole muscle homogenates compared to purified primary skeletal muscle cells confirmed a lack of expression of these proteins in the skeletal muscle cells (Fig. 3 C)
Protein expression and phosphorylation eNOS and NOX
eNOS protein content was higher after the sucrose intervention (P = 0.015) (Fig. 4 A). As expected, the protein expression of eNOS was not affected by passive leg movement under either condition. Total phosphorylation of eNOS at serine residue 1177 and the ratio to eNOS protein were no different after the sucrose intervention and were not affected by passive leg movement (Fig. 4 A).
Figure 4. Protein expression and phosphorylation of eNOS, NOX, Rac1 and S‐nitrosocystine before and after 14 days of sucrose intervention.
A, protein expression of eNOS, phosphorylated eNOS (ser1177) and phosphorylated to total eNOS ratio. B, NOX subunit p‐67, Rac1 and S‐nitrosocystine in muscle homogenates from musculus vastus lateralis at rest and after 20 min of passive leg movement. C, representative blots are shown from one subject in duplicates from rest (1) and after passive leg movement (2) under the control situation and from rest (3) and after passive leg movement (4) after the sucrose intervention. Data are presented as the mean ± SD (n = 12). *Significantly different compared to before the sucrose intervention under the same condition. **Overall significantly different compared to before the sucrose intervention.
NOX subunit p67 levels was markedly increased after the sucrose intervention (P < 0.001) (Fig. 4 B) but unaffected by passive leg movement under both conditions.
Protein expression of Rac1 and S‐nitrosocysteine
The protein expression of Rac1 levels was higher after the sucrose intervention (P < 0.001) (Fig. 4 B) and was not changed from rest to acute passive leg movement for either of the conditions. The protein expression of S‐nitrosocysteine was not changed by the sucrose intervention or from rest to acute passive leg movement under any condition (Fig. 4 B).
Skeletal muscle homogenate mRNA levels
eNOS, PECAM‐1 and VEGF‐R2 mRNA levels were higher after the sucrose intervention (P < 0.001) (Fig. 5) and were not changed from rest to acute passive leg movement under any condition.
Figure 5. Skeletal muscle mRNA levels of eNOS, PECAM‐1 and VEGR‐R2 before and after 14 days of sucrose intervention.
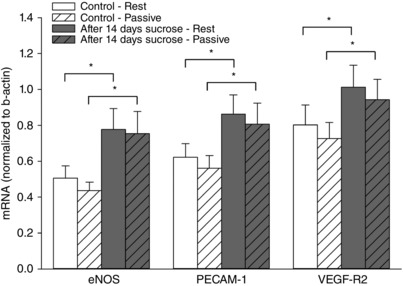
mRNA levels of eNOS, PECAM‐1 and VEGF‐R2 in muscle homogenates from musculus vastus lateralis at rest and after 20 min of passive leg movement. Data are presented as the mean ± SD (n = 12). *Significantly different compared to before the sucrose intervention under the same condition. There was no significant effect of passive leg movement.
Plasma AGE and sRAGE
Plasma concentrations of AGE and sRAGE were no different between the control condition and the sucrose intervention (Fig. 6).
Figure 6. Plasma concentrations of AGE and sRAGE before and after 14 days of sucrose intervention.
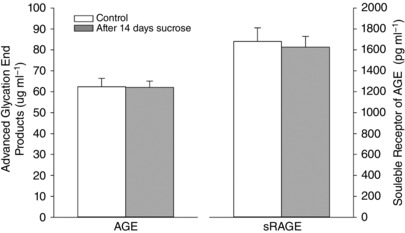
Plasma concentration of AGE and sRAGE collected at rest. Data are presented as the mean ± SD (n = 12). There was no significant effect of the sucrose intervention on AGE and sRAGE levels.
Skeletal muscle interstitial and plasma NOx and PGI2 levels
The muscle interstitial concentration of NOx was higher overall for all conditions (P = 0.024) and during passive movement (P = 0.017) after the sucrose intervention compared to the control condition (Fig. 7). Interstitial NOx was not altered acutely by passive leg movement or by active exercise compared to at rest. Plasma levels of NOx were not affected by the sucrose intervention (82.2 ± 11 and 86.6 ± 8 μm, control and sucrose, respectively).
Figure 7. Skeletal muscle interstitial nitrite and nitrate before and after 14 days of sucrose intervention.
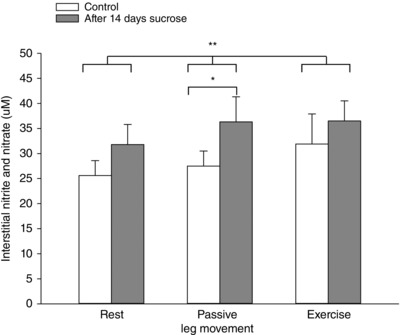
Muscle interstial levels of the stable metabolites of nitric oxide, nitrite and nitrate (NOx) at rest, during 20 min of passive leg movement and active one leg knee extensor exercise. Data are presented as the mean ± SD (n = 12). *Significantly different compared to before the sucrose intervention during passive leg movement. **Overall significantly different compared to before the sucrose intervention.
Interstitial PGI2 in musculus vastus lateralis was not changed with the sucrose intervention, but levels were increased during passive leg movement (P = 0.032) and active exercise (P = 0.027) compared to resting conditions (Fig. 8).
Figure 8. Skeletal muscle interstitial 6‐keto prostaglandin F1α before and after 14 days of sucrose intervention.
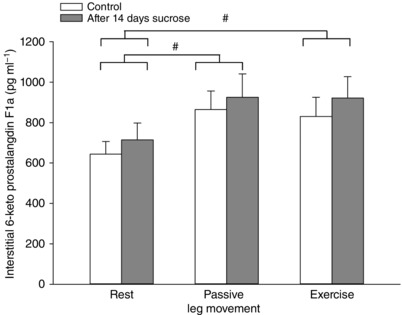
Muscle interstitial levels of the stable metabolites of PGI2, 6‐keto prostaglandin F1α, at rest, during 20 min of passive leg movement and active one leg knee extensor exercise. Data are presented as the mean ± SD (n = 12). #Overall significantly different compared to resting conditions. There was no significant effect of the sucrose intervention.
Discussion
The principal findings of the present study were that 14 days of supplementation of the diet with sucrose attenuated vascular function in young healthy individuals, as indicated by a lower hyperaemic response to both passive leg movement and active exercise. This impairment in vascular function was paralleled by an up‐regulation of PECAM‐1, eNOS, NOX and Rac1 protein expression in whole muscle tissue, as well as by an increased phosphorylation status of VEGF‐R2 and a reduced phosphorylation status of PECAM‐1. We propose that the impaired hyperaemic response after the sucrose intervention was the result of a reduced activation of eNOS caused by impairments in the mechanosensory complex and enhanced removal of NO because of increased superoxide formation by NOX. These findings indicate that regular intake of large amounts of sucrose, as commonly observed in unhealthy diets, is detrimental to vascular health and function and may be one of the underlying causes of cardiovascular disease.
Another novel finding was that there was no detectable increase in phosphorylation status of the mechanosensory complex in response to increased shear stress induced by acute passive leg movement. This finding is in contrast to cell culture findings showing increased phosphorylation with added shear stress and may also reflect the continuous shear stress influence on endothelial cells in vivo, rendering them less sensitive to experimental changes in flow.
Two weeks of high sucrose intake impairs vascular function
The supplementation of a normal diet with 3 × 75 g of sucrose results in a repeated transient ∼50% and 700% increase in blood glucose and insulin, respectively (Jameel et al. 2014). The response curve to sucrose is similar to that of a standard oral glucose tolerance test and blood glucose returns to initial levels after 2 h in the morning and after 3 h at midday and in the evening (Jarrett et al. 1972; Carroll & Nestel, 1973). The present intervention thus led to high blood glucose levels during at least 8 h every day and, as hypothesized, the diet had adverse effects on vascular function in young healthy subjects. This was indicated by a marked reduction in the blood flow response to passive leg movement and a reduction in blood flow during low intensity exercise. The hyperaemia in response to passive leg movement has previously been shown to be almost completely dependent on the NO system (Mortensen et al. 2012). Thus, the present finding of a reduced blood flow response to passive leg movement after high sucrose intake indicates that a high sucrose intake interferes with the function of the NO system, either by affecting NO formation, NO availability or NO responsiveness.
The detrimental effect of the sucrose intervention on vascular function was further confirmed by our finding that blood flow during low intensity active exercise also was decreased after the sucrose trial compared to the control condition. Interventions that reduce exercise hyperaemia are unusual because the oxygen requirement during exercise provides a strong and multifactorial impact on several interacting vasodilator systems, allowing for redundancy, including between the NO and the prostacyclin systems (Boushel et al. 2002; Hillig et al. 2003; Mortensen et al. 2007). However, a compensatory formation of prostacyclin during exercise was not evident after the sucrose intervention, as indicated by similar interstitial prostacyclin levels in in the control and sucrose conditions.
Chronic high blood glucose levels have been linked to increased endothelin‐1 vasoconstrictor tone in humans (Diehl et al. 2013) and NO has been shown to inhibit the expression and release of endothelin‐1; thus, reduced NO availability may increase endothelin‐1 levels (Bourque et al. 2011). However, endothelin‐1 did not appear to have influenced the impaired vascular reponse in the present study because levels were similar in the sucrose and control intervention.
It should also be noted that, considering the limited muscle mass involved in knee extensions, the observed reductions in blood flow after the sugar intervention were probably not a result of alterations in cardac function, as also confirmed by similar heart rates in the sucrose and control conditions.
Vascular mechanosensors are widely affected by high sucrose ingestion
Our functional data suggest an impairment in the NO system, which was not related to the amount of eNOS. By contrast, the enzyme was up‐regulated both at the mRNA and protein level. This finding fits well with previous observations of up‐regulation of eNOS in endothelial cells collected from the brachial artery of aged subjects (Seals et al. 2011) and may reflect a compensatory mechanism for the impaired function of the NO system. Therefore, the reduced response to passive leg movement after the sucrose intervention may have been related to limitations in the signal transduction leading to activation of eNOS (Fleming & Busse, 1999). Tyrosine phosphorylation of PECAM‐1 is central to the translation of shear stress‐induced activation of eNOS (Fleming et al. 2005). In the present study, we show that the impaired flow response to passive leg movement is paralleled by a marked reduction in basal phosphorylation status of PECAM‐1 at tyrosine residue 713, suggesting that this mechanosensor was negatively affected by the sucrose intervention.
An additional explanation for the lower shear stress response after the sucrose intervention is the parallel increase in basal phosphorylation of VEGF‐R2 serine residue 1175. The enhanced phosphorylation of VEGF‐R2 is in agreement with observations in endothelial cell cultures, showing that the glucose induced phosphorylation occurs via the Src family kinases and is dependent on oxidative stress (Warren et al. 2014). Importantly, phosphorylation was shown to result in impaired trafficking of VEGF‐R2 to the cell membrane with a consequent reduction in available receptors at the cell surface (Warren et al. 2014). Such an effect could reduce shear stress sensing and the increased VEGF‐R2 phosphorylation after the sucrose intervention may partly explain the observed lower flow response during passive movement.
Regulation and activation of eNOS
The period of high sucrose intake led to an increase in eNOS expression both at the mRNA and protein level. This observation is in agreement with observations in endothelial cell cultures showing that incubation of cells with high glucose enhances eNOS expression (Cosentino et al. 1997). The mechanisms behind the up‐regulation of eNOS could, in the present set‐up, be related to oxidized low‐density lipoprotein and/or hydrogen peroxide because these have been shown to increase eNOS gene transcription and prolong the half‐life of eNOS mRNA, respectively (Cosentino et al. 1997; Ramasamy et al. 1998; Drummond et al. 2000).
We found no changes in phosphorylation of eNOS serine residue 1177, which is known to be an important site of activation (Dimmeler et al. 1999; Fisslthaler et al. 2000), although one or several of the other mechanisms of eNOS activation could have been impaired, in particular those influenced by oxidative stress, as described below.
High sucrose intake leads to high NOX activity
NO reacts rapidly with superoxide radicals, resulting in the formation of peroxynitrite, and NO is no longer available for vasodilatation. A high concentration of superoxide can also lead to uncoupling of eNOS, which leads to reduced NO formation and increased superoxide formation by the enzyme (Fleming & Busse, 2003). The most important source of superoxide radicals in the vasculature is NOX in endothelial cells. High glucose levels are known to be associated with increased levels of oxidative stress in endothelial cell cultures in part via up‐regulation (Cosentino et al. 1997, 2003) and increased activity (Inoguchi et al. 2000) of NOX. In the present study, the sucrose intervention resulted in a two‐fold increase of the regulatory subunit p67 on NOX compared to the control condition. This was also paralleled by an increased protein expression of the NOX assembler Rac1 suggesting increased activity of the enzyme. NOX is known to be present both in endothelial cells and in skeletal muscle (Cocks et al. 2012, 2016) and the observed increases could have occurred in either or both tissues.
Thus, the level of superoxide production was probably enhanced and the bioavailability of NO was correspondingly reduced. We could, however, not detect an increase in S‐nitrosocysteine levels in the muscle samples. An increased level of S‐nitrosocysteine is suggested to reflect an increased formation of peroxynitrite (Hlaing & Clément, 2014; Hsieh et al. 2014). The data should, however, be interpreted with caution because the sensitivity of the assay is limited and sensitivity may also be low considering that the measurements were made on whole muscle homogenates of which endothelial cells make up only a small fraction.
AGE are not associated with reduced vascular function
High levels of AGE have been linked to reduced mechanotransduction (Otero et al. 2001; Soro‐Paavonen et al. 2010; Liu et al. 2012; Naser et al. 2013) and, based on previous findings, a diet high in sucrose was hypothesized to increase the circulating levels of AGE (Schalkwijk et al. 2004). Plasma AGE levels were, however found to be unaltered after the sucrose intervention. One explanation could be that the diet intervention was too short and that a limited AGE formation plasma was removed by RAGE. The RAGE levels of the young healthy subjects in this project were relatively high compared to the general male population (Prakash et al. 2015). Thus, the changes observed in vascular function were not related to changes in AGE plasma concentration.
Phosphorylation of the mechanosensor complex
The present study attempted to assess the acute effect of passive movement and thereby shear stress on the activation status of the mechanosensor complex comprising PECAM‐1, VEGF‐R2 and VE‐cadherin. Interestingly, activation of the mechanosensor complex was not detectable after 20 min of passive leg movement. This finding contrasts with findings from cell culture studies in which endothelial cells (of varying origin) have been cultured and stimulated with laminar fluid shear stress (Osawa et al. 1997; Orsenigo et al. 2012; dela Paz et al. 2013). The lack of effect of passive movement induced hyperaemia on phosphorylation of the mechanosensor proteins could be related to the endothelial cells in vivo continuously experiencing large variations in shear stress according to heart cycle, muscle activation and changes in flow, and thus the increased flow during passive leg movement provides a relatively small stimulus. By contrast, cells in culture are habituated to static no‐flow conditions and, subsequently, they are exposed to shear stress that probably provides a greater stimulus than passive movement in the in vivo setting.
Conclusions
In the present study, we demonstrate that a diet high in sucrose impairs vascular function in young healthy subjects. The impaired vascular function was paralleled by alterations of the endothelial mechanosensory protein complex, suggesting that vascular function was compromised at the site of mechanosensing in addition to potential disruption of the NO system. Interference of eNOS was also indicated by a compensatory up‐regulation of this protein. A probable mechanism behind a reduced NO availability was inactivation of eNOS and/or removal of NO due to increased superoxide formation from the up‐regulation of NOX.
Another important finding was that acute passive leg movement did not lead to measureable activation of the mechanosensor complex or changes in NO formation despite leading to increased blood flow. Prostacyclin formation, on the other hand, is increased during passive leg movement to the same extent as during low‐intensity active exercise.
Perspective
A single bottle of sweetened soft drink alone can contain ∼50 g of simple sugars and, in western societies, soft drinks are often a companion throughout the day. The findings of the present study indicate that the regular intake of large amounts of sucrose impairs vascular mechanosensory proteins, increases the oxidative stress potential and disrupts the regulation of vascular tone. Impaired vascular regulation is the first step on the way to vascular disease and the findings of the present study indicate that a sucrose‐rich diet can be a factor contributing to the development of vascular disease.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All authors were resonsible for the conception and design of the study; the collection of data; and the drafting of the article or revising it critically for important intellectual content. LG, NM, ML, LS, EAR and YH were responsible for the analysis and interpretation of data. All authors approved the final version and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The study was supported by The Danish Council for Independent Research – Medical Sciences (#4004‐00363) and the Danish Toyota Foundation (#KJ/BG 8876F). Lasse Gliemann and Lykke Sylow was suppoted by a postdoctoral fellowship from the The Danish Council for Independent Research – Medical Sciences (##6110‐00632 and #4004‐00233). The support from these foundations is gratefully acknowledged.
Translational perspectives
The present study shows that dietary sugar impairs vascular function in healthy humans. The translation of the mechanical stress that flowing blood applies to the blood vessel wall into an intracellular signal, termed ‘mechanotransduction’, is important for vascular function. Sugar could impair this mechanosensing; therefore, we aimed to evaluate the impact of a diet high in simple sugars on vascular function in healthy young men. We hypothesized that endothelial mechanotransductor proteins are important for vascular regulation and that a high sucrose diet impairs vascular function in part by decreasing mechanotransduction in vascular endothelial cells. Young healthy men were supplemented with three daily 75 g sucrose drinks for 2 weeks. The sucrose intervention greatly affected vascular function, which is a novel finding, emphasizing the impact of sugar intake on the risk of vascular diseases. Moreover, our molecular findings indicate that the regular intake of high amounts of sucrose disrupts the normal regulation of blood vessels by impairing vascular mechanosensor proteins. The findings of the present study suggest that a sucrose‐rich diet can contribute to the development of vascular disease. Future studies are encouraged to investigate whether sugar‐induced vascular dysfunction is reversible after several years of ‘sugar abuse’ and more work is needed to fully understand the mechanisms involved in dietary‐induced vascular diseases.
Acknowledgements
We thank Gemma Kroos and Karina Olsen for their skilled technical assistance.
This is an Editor's Choice article from the 15 August 2017 issue.
References
- Altman DG (1980). Statistics and ethics in medical research: III how large a sample? Br Med J 281, 1336–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P & Saltin B (1985). Maximal perfusion of skeletal‐muscle in man. J Physiol 366, 233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergström J (1975). Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest 35, 609–616. [PubMed] [Google Scholar]
- Bourque SL, Davidge ST & Adams MA (2011). The interaction between endothelin‐1 and nitric oxide in the vasculature: new perspectives. Am J Physiol Regul Integr Comp Physiol 300, R1288–R1295. [DOI] [PubMed] [Google Scholar]
- Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M & Kjaer M (2002). Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol 543, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KF & Nestel PJ (1973). Diurnal variation in glucose tolerance and in insulin secretion in man. Diabetes 22, 333–348. [DOI] [PubMed] [Google Scholar]
- Cocks M, Shaw CS, Shepherd SO, Fisher JP, Ranasinghe A, Barker TA & Wagenmagers AJM (2016). Sprint interval and moderate‐intensity continuous training have equal benefits on aerobic capacity, insulin sensitivity, muscle capillarisation and endothelial eNOS/NAD(P)Hoxidase protein ratio in obese men. J Physiol 594, 2307–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocks M, Shepherd SO, Shaw CS, Achten J, Costa ML & Wagenmagers AJM (2012). Immunofluorescence microscopy to assess enzymes controlling nitric oxide availability and microvascular blood flow in muscle. Microcirculation 19, 642–651. [DOI] [PubMed] [Google Scholar]
- Connell P, Walshe T, Ferguson G, Gao W, O'Brien C & Cahill PA (2007). Elevated glucose attenuates agonist‐ and flow‐stimulated endothelial nitric oxide synthase activity in microvascular retinal endothelial cells. Endothelium 14, 17–24. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Eto M, De Paolis P, van der Loo B, Bachschmid M, Ullrich V, Kouroedov A, Delli Gatti C, Joch H, Volpe M & Lüscher TF (2003). High glucose causes upregulation of cyclooxygenase‐2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation 107, 1017–1023. [DOI] [PubMed] [Google Scholar]
- Cosentino F, Hishikawa K, Katusic ZS & Lüscher TF (1997). High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation 96, 25–28. [DOI] [PubMed] [Google Scholar]
- Deanfield JE, Halcox JP & Rabelink TJ (2007). Endothelial function and dysfunction: testing and clinical relevance. Circulation 115, 1285–1295. [DOI] [PubMed] [Google Scholar]
- Diehl KJ, Templeton DL, Ma J, Weil BR, Greiner JJ, Stauffer BL & DeSouza CA (2013). Impaired fasting blood glucose is associated with increased endothelin‐1 vasoconstrictor tone. Atherosclerosis 229, 130–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R & Zeiher AM (1999). Activation of nitric oxide synthase in endothelial cells by Akt‐dependent phosphorylation. Nature 399, 601–605. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Gano LB, Eskurza I, Silver AE, Gates PE, Jablonski K & Seals DR (2009). Vascular endothelial dysfunction with aging: endothelin‐1 and endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol 297, H425–H432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GR, Cai H, Davis ME, Ramasamy S & Harrison DG (2000). Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res 86, 347–354. [DOI] [PubMed] [Google Scholar]
- Fiorentino TV, Prioletta A, Zuo P & Folli F (2013). Hyperglycemia‐induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr Pharm Des 19, 5695–5703. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Dimmeler S, Hermann C, Busse R & Fleming I (2000). Phosphorylation and activation of the endothelial nitric oxide synthase by fluid shear stress. Acta Physiol Scand 168, 81–88. [DOI] [PubMed] [Google Scholar]
- Fleming I & Busse R (1999). Signal transduction of eNOS activation. Cardiovasc Res 43, 532–541. [DOI] [PubMed] [Google Scholar]
- Fleming I & Busse R (2003). Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol 284, R1–R12. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Dixit M & Busse R (2005). Role of PECAM‐1 in the shear‐stress‐induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci 118, 4103–4111. [DOI] [PubMed] [Google Scholar]
- Goh S‐Y & Cooper ME (2008). Clinical review: the role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab 93, 1143–1152. [DOI] [PubMed] [Google Scholar]
- Green DJ, Jones H, Thijssen D, Cable NT & Atkinson G (2011). Flow‐mediated dilation and cardiovascular event prediction: does nitric oxide matter? Hypertension 57, 363–369. [DOI] [PubMed] [Google Scholar]
- Gryglewski RJ, Palmer RM & Moncada S (1986). Superoxide anion is involved in the breakdown of endothelium‐derived vascular relaxing factor. Nature 320, 454–456. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, West NEJ, Pillai R, Taggart DP & Channon KM (2002). Nitric oxide modulates superoxide release and peroxynitrite formation in human blood vessels. Hypertension 39, 1088–1094. [DOI] [PubMed] [Google Scholar]
- Hellsten Y, Rufener N, Nielsen JJ, Høier B, Krustrup P & Bangsbo J (2008). Passive leg movement enhances interstitial VEGF protein, endothelial cell proliferation, and eNOS mRNA content in human skeletal muscle. ‐ PubMed ‐ NCBI. Am J Physiol Regul Integr Comp Physiol 294, R975–R982. [DOI] [PubMed] [Google Scholar]
- Hillig T, Krustrup P, Fleming I, Osada T, Saltin B & Hellsten Y (2003). Cytochrome P450 2C9 plays an important role in the regulation of exercise‐induced skeletal muscle blood flow and oxygen uptake in humans. J Physiol 546, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlaing KH & Clément M‐V (2014). Formation of protein S‐nitrosylation by reactive oxygen species. Free Radic Res 48, 996–1010. [DOI] [PubMed] [Google Scholar]
- Höffner L, Nielsen JJ, Langberg H & Hellsten Y (2003). Exercise but not prostanoids enhance levels of vascular endothelial growth factor and other proliferative agents in human skeletal muscle interstitium. J Physiol 550, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H‐J, Liu C‐A, Huang B, Tseng AH & Wang DL (2014). Shear‐induced endothelial mechanotransduction: the interplay between reactive oxygen species (ROS) and nitric oxide (NO) and the pathophysiological implications. J Biomed Sci 21, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H & Nawata H (2000). High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C‐dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49, 1939–1945. [DOI] [PubMed] [Google Scholar]
- Jameel F, Phang M, Wood LG & Garg ML (2014). Acute effects of feeding fructose, glucose and sucrose on blood lipid levels and systemic inflammation. Lipids Health Dis 13, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson PA, Veneman T, Nurjhan N & Gerich J (1994). An improved method to calculate adipose tissue interstitial substrate recovery for microdialysis studies. Life Sciences 54, 1621–1624. [DOI] [PubMed] [Google Scholar]
- Jarrett RJ, Baker IA, Keen H & Oakley NW (1972). Diurnal variation in oral glucose tolerance: blood sugar and plasma insulin levels morning, afternoon, and evening. Br Med J 1, 199–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z‐G, Ueba H, Tanimoto T, Lungu AO, Frame MD & Berk BC (2003). Ligand‐independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res 93, 354–363. [DOI] [PubMed] [Google Scholar]
- Joyner MJ & Casey DP (2015). Regulation of increased blood flow (hyperemia) to muscles during exercise: a hierarchy of competing physiological needs. Physiol Rev 95, 549–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G & Quon MJ (2000). Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab 85, 2402–2410. [DOI] [PubMed] [Google Scholar]
- Kawano H, Motoyama T, Hirashima O, Hirai N, Miyao Y, Sakamoto T, Kugiyama K, Ogawa H & Yasue H (1999). Hyperglycemia rapidly suppresses flow‐mediated endothelium‐dependent vasodilation of brachial artery. J Am Coll Cardiol 34, 146–154. [DOI] [PubMed] [Google Scholar]
- Lamontagne D, Pohl U & Busse R (1992). Mechanical deformation of vessel wall and shear stress determine the basal release of endothelium‐derived relaxing factor in the intact rabbit coronary vascular bed. Circ Res 70, 123–130. [DOI] [PubMed] [Google Scholar]
- Liu H, Yu S, Zhang H & Xu J (2012). Angiogenesis impairment in diabetes: role of methylglyoxal‐induced receptor for advanced glycation endproducts, autophagy and vascular endothelial growth factor receptor 2. ed. Ushio‐Fukai M. PLoS ONE 7, e46720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yu M, Zhang L, Cao Q, Song Y, Liu Y & Gong J (2016). Soluble receptor for advanced glycation end products mitigates vascular dysfunction in spontaneously hypertensive rats. Mol Cell Biochem 419, 165–176. [DOI] [PubMed] [Google Scholar]
- McVeigh GE, Brennan GM, Johnston GD, McDermott BJ, McGrath LT, Henry WR, Andrews JW & Hayes JR (1992). Impaired endothelium‐dependent and independent vasodilation in patients with type 2 (non‐insulin‐dependent) diabetes mellitus. Diabetologia 35, 771–776. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Askew CD, Walker M, Nyberg M & Hellsten Y (2012). The hyperaemic response to passive leg movement is dependent on nitric oxide: a new tool to evaluate endothelial nitric oxide function. J Physiol 590, 4391–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez‐Alonso J, Damsgaard R, Saltin B & Hellsten Y (2007). Inhibition of nitric oxide and prostaglandins, but not endothelial‐derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol 581, 853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naser N, Januszewski AS, Brown BE, Jenkins AJ, Hill MA & Murphy TV (2013). Advanced glycation end products acutely impair ca(2+) signaling in bovine aortic endothelial cells. Front Physiol 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor LH, Green DJ, Jones TW, Kalic RJ, Suriano KL, Shah M, Hopkins N & Davis EA (2011). Endothelial function and carotid intima‐medial thickness in adolescents with type 2 diabetes mellitus. J Pediatr 159, 971–974. [DOI] [PubMed] [Google Scholar]
- Nyberg M, Gliemann L, Thaning P, Hellsten Y & Mortensen SP (2012). Role of nitric oxide and prostanoids in the regulation of leg blood flow and blood pressure in humans with essential hypertension: effect of high‐intensity aerobic training. J Physiol 590, 1481–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY, Franco D, Kurtcuoglu V, Poulikakos D, Baluk P, McDonald D, Grazia Lampugnani M & Dejana E (2012). Phosphorylation of VE‐cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 3, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M, Masuda M, Harada N, Lopes RB & Fujiwara K (1997). Tyrosine phosphorylation of platelet endothelial cell adhesion molecule‐1 (PECAM‐1, CD31) in mechanically stimulated vascular endothelial cells. Eur J Cell Biol 72, 229–237. [PubMed] [Google Scholar]
- Osawa M, Masuda M, Kusano K‐I & Fujiwara K (2002). Evidence for a role of platelet endothelial cell adhesion molecule‐1 in endothelial cell mechanosignal transduction: is it a mechanoresponsive molecule? J Cell Biol 158, 773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero K, Martínez F, Beltrán A, González D, Herrera B, Quintero G, Delgado R & Rojas A (2001). Albumin‐derived advanced glycation end‐products trigger the disruption of the vascular endothelial cadherin complex in cultured human and murine endothelial cells. Biochem J 359, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dela Paz NG, Melchior B & Frangos JA (2013). Early VEGFR2 activation in response to flow is VEGF‐dependent and mediated by MMP activity. Biochem Biophys Res Commun 434, 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilegaard H, Ordway GA, Saltin B & Neufer PD (2000). Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. AJP: Endocrinol Metab 279, E806–E814. [DOI] [PubMed] [Google Scholar]
- Pohl U, Holtz J, Busse R & Bassenge E (1986). Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension 8, 37–44. [DOI] [PubMed] [Google Scholar]
- Prakash J, Pichchadze G, Trofimov S & Livshits G (2015). Age and genetic determinants of variation of circulating levels of the receptor for advanced glycation end products (RAGE) in the general human population. Mech Ageing Dev 145, 18–25. [DOI] [PubMed] [Google Scholar]
- Ramasamy S, Parthasarathy S & Harrison DG (1998). Regulation of endothelial nitric oxide synthase gene expression by oxidized linoleic acid. J Lipid Res 39, 268–276. [PubMed] [Google Scholar]
- Schalkwijk CG, Stehouwer CDA & van Hinsbergh VWM (2004). Fructose‐mediated non‐enzymatic glycation: sweet coupling or bad modification. Diabetes Metab Res Rev 20, 369–382. [DOI] [PubMed] [Google Scholar]
- Scheller D & Kolb J (1991). The internal reference technique in microdialysis: a practical approach to monitoring dialysis efficiency and to calculating tissue concentration from dialysate samples. J Neurosci Methods 40, 31–38. [DOI] [PubMed] [Google Scholar]
- Seals DR, Jablonski KL & Donato AJ (2011). Aging and vascular endothelial function in humans. Clin Sci 120, 357–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay‐Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E & Resnick N (2002). VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci USA 99, 9462–9467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soro‐Paavonen A, Zhang W‐Z, Venardos K, Coughlan MT, Harris E, Tong DCK, Brasacchio D, Paavonen K, Chin‐Dusting J, Cooper ME, Kaye D, Thomas MC & Forbes JM (2010). Advanced glycation end‐products induce vascular dysfunction via resistance to nitric oxide and suppression of endothelial nitric oxide synthase. J Hypertens 28, 780–788. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Sudano I & Salvetti A (2001). Endothelial dysfunction in hypertension. J Cardiovasc Pharmacol 38, S11. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Versari D & Salvetti A (2006). Endothelium, aging, and hypertension. Curr Hypertens Rep 8, 84–89. [DOI] [PubMed] [Google Scholar]
- Tzima E, Irani‐Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H & Schwartz MA (2005). A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431. [DOI] [PubMed] [Google Scholar]
- Venturelli M, Cè E, Limonta E, Bisconti AV, Devoto M, Rampichini S & Esposito F (2017). Central and peripheral responses to static and dynamic stretch of skeletal muscle: mechano‐ and metaboreflex implications. J Appl Physiol 122, 112–120. [DOI] [PubMed] [Google Scholar]
- Warren CM, Ziyad S, Briot A, Der A & Iruela‐Arispe ML (2014). A ligand‐independent VEGFR2 signaling pathway limits angiogenic responses in diabetes. Sci Signal 7, ra1–ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlansky ME, Gokce N, Keaney JF & Vita JA (2003). The clinical implications of endothelial dysfunction. J Am Coll Cardiol 42, 1149–1160. [DOI] [PubMed] [Google Scholar]
- Zhu W, Zhong C, Yu Y & Li K (2007). Acute effects of hyperglycaemia with and without exercise on endothelial function in healthy young men. Eur J Appl Physiol 99, 585–591. [DOI] [PubMed] [Google Scholar]