

Abstract
During health, animal sleep is regulated by an internal clock and by the duration of prior wakefulness. During sickness, sleep is regulated by cytokines released from either peripheral cells or from cells within the nervous system. These cytokines regulate central nervous system neurons to induce sleep. Recent research in the invertebrates Caenorhabditis elegans and Drosophila melanogaster has led to new insights into the mechanism of sleep during sickness. Sickness is triggered by exposure to environments such as infection, heat, or ultraviolet light irradiation, all of which cause cellular stress. Epidermal growth factor is released from stressed cells and signals to activate central neuroendocrine cell(s). These neuron(s) release neuropeptides including those containing an amidated arginine(R)‐phenylalanine(F) motif at their C‐termini (RFamide peptides). Importantly, mechanisms regulating sickness sleep are partially distinct from those regulating healthy sleep. We will here review key findings that have elucidated the central neuroendocrine mechanism of sleep during sickness. Adaptive mechanisms employed in the control of sickness sleep may play a role in correcting cellular homeostasis after various insults. We speculate that these mechanisms may play a maladaptive role in human pathological conditions such as in the fatigue and anorexia associated with autoimmune diseases, with major depression, and with unexplained chronic fatigue.
Keywords: epidermal growth factor, C. elegans, cellular stress, cytokines, D. melanogaster, sickness, sleep
Introduction
The modern era of sleep research began in earnest in the 1950s with the discovery of rapid eye movement (REM) sleep. Much of the focus of this research has been on understanding natural or healthy sleep. Natural sleep is the sleep that occurs with an approximately 24 h (circadian) periodicity and is regulated by the time spent awake prior to sleep. The dominant model to explain natural sleep is called the two process model, whereby the interaction of circadian and homeostatic mechanisms regulate the timing and depth of sleep (Borbely, 1982).
But in addition to the sleep that occurs on a daily basis, we have all experienced the sensation of being sleepy and fatigued during an acute infection or other illness. Sleepiness is one of the cardinal manifestations of sickness behaviour, which also includes anorexia (lack of eating despite ample food), social withdrawal, malaise, and, in homeotherms, fever. Mechanisms of sickness sleep remain poorly understood. Fundamental questions include: (1) What are the mechanisms regulating sleep during sickness and are they the same mechanisms that regulate sleep during health? (2) What is the function of sickness sleep? (3) Do mechanisms involved in sickness sleep play a role in conditions of human pathological sleepiness or fatigue?
We here review mechanistic studies of sickness sleep in mammals, and then describe in detail recent findings from studies of the invertebrates Caenorhabditis elegans and Drosophila melanogaster, which shed light on the first two questions. We end by speculating on the possible role of inappropriate activation of sickness sleep pathways in human pathological conditions.
Cytokines are released in response to cell injury and can promote sleep
Cytokines are broadly defined as small proteins that are released from one cell and affect the physiology of other cells in a paracrine or endocrine fashion. The field of cytokine research started with studies of the immune system and as a result, much of this research has focused on the immune modulating properties of these signalling proteins. Many of these proteins are called ‘interleukins’ because they signal between white blood cells. Over 35 distinct interleukins have been described. Other proteins traditionally referred to as cytokines include the anti‐viral interferons and tumour necrosis factor α (TNFα). Based on the broad definition of cytokines as small proteins involved in cell–cell communication, we include in this category also signalling proteins initially identified in research outside the immune system. Examples of such signalling proteins are epidermal growth factor (EGF) family members, transforming growth factor β (TGFβ) family members, and other hormones and growth factors.
Though undoubtedly a key function of cytokines is in immune system modulation, a role outside the immune system was suggested when some of the classical cytokines as well as their receptors were found in central nervous system cells (Eriksson et al. 2000). Although several cytokines have been demonstrated to affect the brain, most of the focus in this regard has been on two cytokines, interleukin 1β (IL1β) and TNFα.
IL1β and TNFα are strongly induced in response to cellular injury, which can be caused by infection, surgical trauma, ischaemia, head injury and other insults. This occurs both in the periphery (Cohen, 1977) and in the central nervous system (Merrill, 1987; Stoll et al. 2002). Following peripheral injection of the Gram‐negative bacterial cell wall component lipopolysaccharide (also known as endotoxin) into the peritoneal cavity of rats, IL1β is strongly induced in several areas of the brain (Eriksson et al. 2000). Among these areas are the circumventricular organs that, because of a breakdown in the blood–brain barrier, can be involved in the mechanism by which central neurons detect changes in the chemistry of blood. Injection of lipopolysaccharide, IL1β, or TNFα into the peritoneal cavity of mammals all result in the induction of a sickness syndrome, which includes increased sleep, fever and anorexia. The sleep response of peripheral IL1β injection is attenuated when the vagus nerve is severed (Hansen & Krueger, 1997), suggesting that detection of these chemicals and cytokines by the brain occurs partially via peripheral neural pathways. The effect of IL1β on sleep is independent of its effect on fever, since its anatomical site of action for fever induction is distinct from that for sleep enhancement (Walter et al. 1989) and because the IL1β pyrogenic effects can be pharmacologically blocked without affecting its somnogenic effects (Krueger et al. 1984). Moreover, minute injections of endotoxin cause an increase in non‐REM sleep without causing a fever (Mullington et al. 2000).
In addition to IL1β and TNFα, there are several other cytokines implicated in the regulation of sleep, primarily based on their somnogenic effects noted when injected into the cerebrospinal fluid of mammals. We focus here on one class of such cytokines, the epidermal growth factors, since this is relevant to the invertebrate research we review below.
Epidermal growth factor
Members of the epidermal growth factor (EGF) family of proteins include 11 small proteins with a similar amino acid repeat sequence that includes six cysteine residues that form three disulfide bonds (Dreux et al. 2006). While much of the early research has focused on developmental roles for EGFs, several of these proteins are expressed in adult brains and have roles in the function of the nervous system. Neurally expressed EGFs include EGF, heparin‐binding EGF (HB‐EGF), transforming growth factor α (TGFα), and the four neuregulins (NRG1–4). EGF family members signal via one of four closely related receptor tyrosine kinases, which belong to the ErbB family of receptors. The receptors include EGFR (ErbB‐1), HER2 (ErB‐2), HER3 (ErbB‐3) and HER4 (ErbB‐4).
Much like the classical cytokines such as IL1β and TNFα discussed above, EGF family members are also released in response to cellular injury and stress. HB‐EGF is released following hypoxic‐ischaemic injury (Jin et al. 2002), or following osmotic and oxidative injury (Fischer et al. 2004), and TGFα is released by osmotic stress (Kuper et al. 2007, 2009).
The effect of EGF family members on mammalian sleep has been studied in rabbits and rodents. In rabbits, the intracerebroventricular (ICV) administration of EGF results in an increase in non‐REM sleep (Kushikata et al. 1998). In rodents, injection of TGFα, EGF, or neuregulin‐1 into the cerebral ventricles results in the inhibition of both movement and feeding (Kramer et al. 2001; Snodgrass‐Belt et al. 2005). This inhibition of locomotion by TGFα is absent in mutant animals with defective EGF receptor (ErbB‐1) signalling (Kramer et al. 2001), indicating that the locomotion‐inhibiting effect of these EGFs is mediated by the EGF receptor (EGFR). A high concentration of EGFR is found in the subparaventricular zone (SPZ), a region in which a lesion causes disrupted circadian regulation of locomotion (Lu et al. 2001). The SPZ is also a major synaptic target of the suprachiasmatic nucleus (SCN) of the hypothalamus (Kramer et al. 2001).
The research on growth factors and cytokines that affect sleep during sickness provides compelling evidence for the involvement of these factors in this type of sleep. However, the target neurons affected by this signalling and the mechanism by which such neurons mediate the global animal sleep response remain unknown in mammals. The relevant neurons may reside in the hypothalamus, which is the area of the brain regulating all homeostatic behaviours, including sleep (Saper et al. 2005), but the neurons and their transmitters functioning in this regard remain unknown. Research in invertebrate animal models has elucidated the central neuroendocrine mechanisms of sickness sleep. This research is described in the following section.
Neural mechanisms of sickness sleep elucidated in invertebrates
Invertebrate sleep is defined behaviourally
The standard electrophysiological tool used to measure sleep in mammals, the electroencephalogram, cannot be used in fruit flies and round worms because of their small size and because their neural anatomy is significantly different from that of mammals. Instead, sleep in invertebrate species is defined behaviourally (Hendricks et al. 2000). Sleep is defined as a rapidly reversible quiescent behavioural state associated with reduced responsiveness.
Sleep in C. elegans during health and sickness
As in mammals, C. elegans sleep is observed under conditions of health and of sickness (Trojanowski & Raizen, 2016). There is a third quiescent behavioural state, which occurs in the setting of satiety (You et al. 2008), but detailed studies to test whether this is also a sleep state have not yet been reported.
Developing larvae enter a sleep state prior to each of the four moults, during a period known as lethargus (Singh & Sulston, 1978; Raizen et al. 2008). Sleep during lethargus, which we refer to as developmentally timed sleep (DTS), is analogous to the sleep that occurs on a 24 h periodicity in diurnal animals such as fruit flies or humans. The timing of DTS in nematodes is regulated by a homologue of the core circadian regulator PERIOD, which controls the timing of sleep in both mammals and fruit flies (Dunlap, 1999). Key regulators of DTS include proteins such as pigment dispersing factor (Choi et al. 2013) and neurotransmitters such as dopamine and GABA (Singh et al. 2014), which regulate sleep in similar fashions in fruit flies and mammals, indicating that DTS is homologous to circadian sleep in these other species.
The second type of sleep observed in C. elegans is called stress‐induced sleep (SIS) and is observed following exposures of the animals to environments that result in cellular injury and stress. Documented injurious exposures that induce sleep include bacterial pore‐forming toxins, heat shock (33–40°C), ethanol shock (5% ethanol by volume), cold shock (−15°C), and osmotic shock (500 mm NaCl) (Hill et al. 2014). In addition, we have found that exposures to short wave ultraviolet radiation also result in a sleep response (H. Debardeleben and D. Raizen, unpublished observations). Because of its relationship to cell injury and stress, we consider SIS to be homologous to sleep observed during sickness in mammals (Trojanowski & Raizen, 2016).
Figure 1 shows a schematic diagram of the mechanism of stress‐induced sleep in C. elegans. Stress‐induced sleep in C. elegans is characterized by a lack of feeding, lack of locomotion, reduced responsiveness to weak sensory stimuli, and movement in response to strong mechanical stimulation. Among stressors that induce SIS, heat shock is thus far the best studied. The relevant cytokine required for this behaviour is epidermal growth factor (Hill et al. 2014). Unlike mammals, which have 11 EGF family members and 4 EGFR family members, the C. elegans genome encodes only one EGF called LIN‐3 and only one EGFR called LET‐23 (Gupta et al. 2012).
Figure 1. Pathways for regulating sickness sleep.
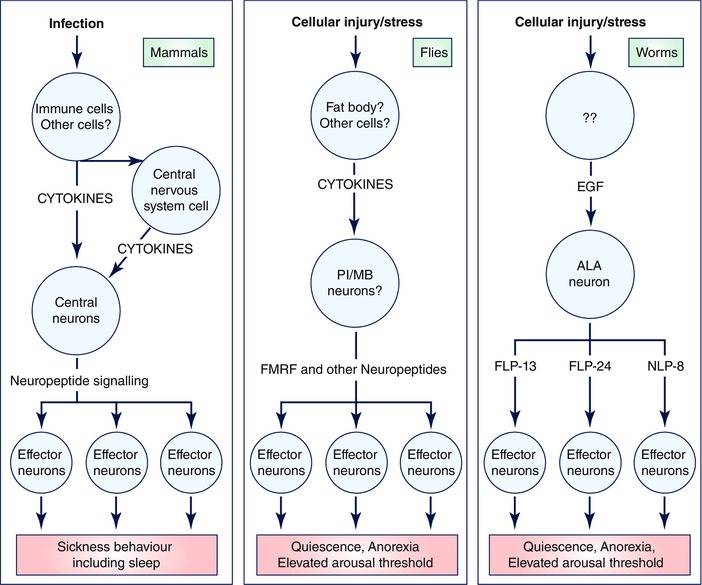
In mammals, infection is detected by immune cells, which secrete cytokines to activate, both directly and indirectly, central nervous system neurons that regulate sleep. In Drosphila melanogaster (flies), infection, tissue injury and possibly heat stress are detected by the extra‐neural fat bodies, which then signal via cytokines to the nervous system to regulate sleep. In Caenorhabditis elegans (worms), environmental exposures that cause cellular injury/stress including high heat, ultraviolet light and toxins result in activation of unidentified cells that release the cytokine epidermal growth factor (EGF). EGF activates the single ALA neuron, which then signals via the collective action of several neuropeptides to induce sub‐programmes of sleep including anorexia, movement quiescence and elevated arousal threshold.
Following heat shock, EGF/LIN‐3 is released from stressed cells by mechanisms that are yet to be defined (Hill et al. 2014). EGF binds to the EGFR/LET‐23 on a single interneuron called ALA. EGFR signals in ALA via phospholipase C‐γ and diacyl glycerol (Van Buskirk & Sternberg, 2007) and causes membrane depolarization and the release of a complex cocktail of neuropeptides (Nath et al. 2016). Among these neuropeptides are several characterized by a C‐terminus consisting of an amidated arginine (R)‐phenylalanine (F) motif (RFamide motif). The best characterized of these peptides is encoded by the gene flp‐13 (Nelson et al. 2014; Nath et al. 2016). The FLP‐13 protein is processed into seven distinct FMRF‐like peptides (Li & Kim, 2008). FLP‐13 neuropeptides released from ALA signal humorally to activate a G‐protein‐coupled receptor called DMSR‐1 (Iannacone et al. 2017). DMSR‐1 activation, probably by signalling via a Gi/o heterotrimeric G‐protein (Trojanowski et al. 2015), causes the inhibition of wake‐promoting neurons (Iannacone et al. 2017).
Other sleep‐modulating peptides released from ALA, which are encoded by the genes flp‐24 and nlp‐8 (Nath et al. 2016), may also act on DMSR‐1, or alternatively may act on other receptors. Overexpression of each of these neuropeptide‐encoding genes affects a particular subset of behaviours observed during sleep: flp‐13 primarily affects locomotion, feeding and defecation quiescence, nlp‐8 primarily affects defecation quiescence, and flp‐24 primarily affects sensory responsiveness and body movement quiescence (Nelson et al. 2014; Nath et al. 2016). Therefore, the sleep state triggered by the single ALA neuron stems from the collective action of several neuropeptides (Nath et al. 2016). This idea of the collective action of neuropeptides controlling sickness behaviour is relevant to our thinking about sickness sleep in mammals. If a similar distinct sub‐behaviours mechanism controls sickness behaviour in mammals, then one might observe fragments of sickness behaviour, such as only sleepiness, only anorexia, only malaise, or only social isolation, without other elements of the behaviour. Fragments of sickness behaviour may masquerade as other diseases such as depression or chronic fatigue (Fig. 2, see more below).
Figure 2. Inappropriate activation of the sickness sleep pathways can result in symptoms of sickness behaviour including sleepiness, social withdrawal and/or malaise.
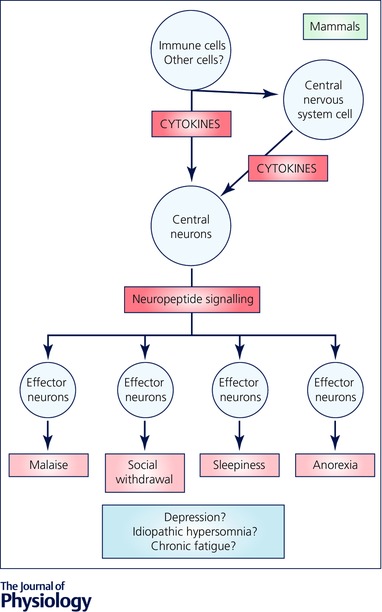
These symptoms may lead to the diagnosis of depression, idiopathic hypersomnia, or chronic fatigue. Sickness behaviour may occur due to inappropriate release of cytokines from peripheral or central cells, by inappropriate release of neuropeptides from central neuroendocrine cells, or by inappropriate activation of neurons mediating sub‐programmes of sickness behaviour. Possible activated pathways in these pathological states are highlighted in red.
Sleep in Drosophila melanogaster during health and sickness
As in mammals and C. elegans, two types of sleep have been described in the fruit fly Drosophila melanogaster. Sleep in Drosophila consists of behavioural quiescence associated with reduced responsiveness to stimuli. As in mammals, the amount and duration of sleep periods in Drosophila decline with age (Koh et al. 2006), and drugs such as caffeine and modafinil, which modify sleep in mammals, modify sleep in similar fashions in fruit flies (Hendricks et al. 2003; Wu et al. 2009). Potassium channels are critical for sleep both in Drosophila (Cirelli et al. 2005) and in mammals (Douglas et al. 2007). The neurochemistry of sleep regulation in fruit flies includes dopamine and GABA (Sehgal & Mignot, 2011), which regulate sleep in similar fashions in mammals and C. elegans. Thus, Drosophila sleep is fundamentally similar to mammalian sleep and lessons learned from studying sleep in Drosophila are relevant to our understanding of sleep in general.
In addition to the sleep observed in healthy fruit flies, which occurs with a diurnal rhythm locked to the day–night cycle and is regulated by circadian proteins such as PERIOD, flies also sleep in response to injury. The injurious stimuli reported include bacterial infection (Kuo et al. 2010), tissue trauma (Kuo et al. 2010) and high heat (Lenz et al. 2015). Following these injuries, fruit flies show increased sleep.
SIS in Drosophila is regulated by non‐neural as well as neural signalling. The transcription factor nuclear factor κ B (NFκB) is required in the fat body and not in neurons for SIS following tissue trauma or a bacterial infection (Kuo et al. 2010). The fat body, which is similar to the mammalian liver, then signals to the nervous system by means that have yet to be defined, but probably involve cytokines (Fig. 1).
Based on the C. elegans results described above, an attractive candidate for such cytokine signalling is EGF. Flies have one EGF receptor and four ligands for this receptor. Three of these four EGF ligands are processed by the protease RHOMBOID, which cleaves membrane‐bound ligands into their soluble, active form (Shilo, 2003). Overexpression of RHOMBOID, which elevates EGFR signalling due to ectopic secretion of EGF ligands, or overexpression of the EGF ligand SPITZ, each leads to increased fly sleep (Foltenyi et al. 2007). By contrast, reduction of EGF signalling causes reduced sleep (Foltenyi et al. 2007). The cells in which EGF receptor activation causes increased sleep remain unknown. One attractive candidate region is pars intercerebralis (PI), which is a region of neuropil rich with peptidergic neurons and in some aspects is similar to the mammalian hypothalamus (Foltenyi et al. 2007). Consistent with a possible role of the PI in regulating sleep is the observation that a reduction of EGF signalling in the PI (as well as probably other regions) causes reduced sleep (Foltenyi et al. 2007). An alternative site of activity of EGF signalling is the mushroom bodies, which have been shown to be key regulators of fly sleep (Joiner et al. 2006; Pitman et al. 2006).
Which neurotransmitters are required for SIS in Drosophila? The experiments in this regard were guided by observations in C. elegans of involvement of peptides that are similar to amidated phenylalanine‐methionine‐arginine‐phenylalanine (FMRFamide) neuropeptides. Mutations in the Drosophila gene encoding FMRFamide or in the gene encoding an FMRFamide receptor result in an impairment in SIS following heat shock or bacterial infection (Lenz et al. 2015). Future identification of the source of FMRFamide and of the location of the FMRFamide responsive cells in the fly brain will help define the neural circuitry that mediates sickness sleep in fruit flies.
Distinct and overlapping mechanisms of healthy sleep and sickness sleep
The behaviours observed during C. elegans DTS and SIS and during D. melanogaster healthy circadian sleep and sickness sleep appear identical. In all cases, there is quiescence of locomotion and of feeding and there is a reduced responsiveness to weak sensory stimuli yet rapid reversibility to strong stimulation. Despite the similar behavioural appearance of these states, some of the underlying mechanisms regulating these two types of sleep differ (Trojanowski et al. 2015).
In C. elegans, ALA is the key neuron required for SIS (Hill et al. 2014), but a different neuron called RIS is the key neuron required for DTS (Turek et al. 2013). FLP‐13 peptides, which are required for SIS, are not required for DTS (Nelson et al. 2014). Finally, anorexia during DTS results from a loss of pharyngeal muscle excitability, whereas anorexia during SIS results from a loss of excitability in the nervous system (Trojanowski et al. 2015). Whereas the mechanisms of behavioural quiescence differ between DTS and SIS, the mechanisms of elevated sensory arousal threshold appear to be similar (Cho & Sternberg, 2014). In both types of sleep, the elevated sensory arousal threshold is explained by changes in the physiology of both sensory neurons and the downstream interneurons. In sleeping nematodes, sensory neurons show reduced sensitivity (Schwarz et al. 2011; Cho & Sternberg, 2014) and interneurons show asynchronous activity (Cho & Sternberg, 2014).
In Drosophila, the mechanisms of healthy sleep and sickness sleep are also partially distinct. A mutation in the fr gene, which encodes a receptor for the neuropeptide FMRFamide, is associated with a normal circadian healthy sleep, but with an impaired sleep response after infectious or heat stress (Lenz et al. 2015).
While sleep under unperturbed conditions may be regulated partially distinctly from stress‐induced sleep, the situation may be different when sleep follows sleep deprivation. Sleep deprivation is associated with markers of cellular stress in multiple species including D. melanogaster (Shaw et al. 2000) and C. elegans (Driver et al. 2013). Therefore, sleep deprivation may induce a sickness sleep response. In Drosophila, while sleep deprivation causes cellular stress, infectious stress conversely leads to disruption of the normal circadian rhythmicity of sleep (Shirasu‐Hiza et al. 2007), suggesting a bidirectional relationship between sleep and cellular stress.
Is sickness sleep beneficial to the animal?
Does sickness sleep serve a function that benefits the animal? If this type of sleep is beneficial, then there should be (1) a correlation between the duration of sleep post infection/injury and recovery success, and (2) an impairment in recovery from the illness when sickness sleep is prevented.
To test the first prediction, Toth, Krueger, and colleagues followed up on their observation of enhanced survival from a bacterial infection in rabbits that showed strong enhanced sleep responses to the infectious challenge (Toth & Krueger, 1988). They performed a retrospective analysis of data derived from 100 rabbits infected with Escherichia coli, Staphylococcus aureus, or Candida albicans (Toth et al. 1993). This analysis confirmed that enhanced sleep following infectious exposure was associated with a more favourable morbidity and mortality outcome. In support of the second prediction that impaired sleep affects survival is the observation that total sleep deprivation in rats results in sepsis and death (Rechtschaffen et al. 1983). The deeper and more consolidated sleep that follows sleep deprivation leads to an enhanced ability to fight an infectious challenge in both mice (Renegar et al. 2000) and fruit flies (Kuo & Williams, 2014 a). The interpretation of the effects of sleep in these latter two experiments is potentially confounded by the enhancement of sleep by sleep deprivation, which is a variable that might contribute directly to cellular stress and hence sickness sleep, as discussed above.
In addition to these animal studies, which used complete sleep deprivation, studies on the effect of short term sleep deprivation on immune function parameters have been performed in humans. While the outcomes of these studies were limited to proxies for immune function such as antibody production, the data as a whole suggest that natural healthy sleep serves a benefit to the immune system (Marshall & Born, 2002). Whether sleep during sickness is also beneficial in recovery from the injury/stressor remains an open question in humans.
The question of whether sleep following the induction of cellular stress is beneficial has been addressed in both C. elegans and Drosophila. Three days after exposure to a high heat pulse of 40°C (the animals’ preferred cultivation temperature is <25°C), about 20% of C. elegans animals die. However, in C. elegans animals with impaired stress‐induced sleep (due to removal of the ALA, the key neuron required for SIS, Fig. 1) about 60% of the animals die. This enhanced lethal effect of heat shock is reversed by an independent genetic manipulation that makes the animals lacking ALA neuron function sleepy again (Hill et al. 2014). A similar experiment was carried out in animals with defective ALA function due to a mutation in the guanine nucleotide‐exchange factor VAV‐1, which is expressed in and functions in ALA. vav‐1 mutant animals, like ALA‐less animals, show reduced survival after heat shock exposure (Fry et al. 2016). Therefore, in C. elegans, sleep after cellular stress is beneficial to survival.
In Drosophila, the benefit of SIS to survival has been assessed in two fashions. Animals with defective SIS due to mutations that impair the function of the neuropeptide FMRFa or its receptor RF, show reduced survival after either heat exposure or infectious challenges (Lenz et al. 2015). To assess the benefit of sleep to survival following a bacterial challenge, Kuo and colleagues genetically manipulated flies to have more sleep post‐infection by either silencing wake‐promoting neurons via transgenic expression of a potassium channel or by sleep‐depriving the animals prior to infection. Flies induced to sleep more after infection survive bacterial infections better than flies that are not induced to sleep more (Kuo & Williams, 2014 a,b). Given the possible relationship between sleep deprivation and cellular stress discussed above, it will be of future interest to test whether Drosophila mutants that fail to increase their sleep after sleep deprivation also have impaired sleep in response to infectious stress and a reduced ability to survive infection.
While these experiments provide phenomenological evidence that sickness sleep is beneficial, they do not provide a mechanism for this benefit. One attractive hypothesis is that sickness sleep facilitates organismal recovery by diverting resources used for wake‐associated neural processing towards processes for fighting off the infection and repairing cells. Reallocation of metabolic resources during sleep was recently proposed also as a function for healthy sleep (Schmidt, 2014). There are potential experimental approaches to test the notion that sickness sleep serves the purpose of reallocation of metabolic resources. A prediction built on this hypothesis is that cellular metabolic resources will be used differently during sleep than during wakefulness. Specifically, during sickness sleep, these resources would be used to repair cellular damage or to combat the infectious challenge. Moreover, the trigger for induction of sickness sleep may involve a cellular mechanism sensing metabolic challenges. One potential sensor is the target of rapamycin (TOR) pathway, which has been demonstrated to affect resistance to infection in Drosophila (Allen et al. 2016).
Perspective: how do lessons learned from invertebrate research inform our studies of sickness sleep in mammals?
There are several lessons learned from the above invertebrate research relevant to our understanding of sickness behaviour in mammals.
First, the observation in both worms and flies that mechanisms regulating quiescence behaviour during sickness sleep are at least partially distinct from the mechanism regulating this behaviour during health, suggests that differences also exist in the regulation of these two types of mammalian sleep. The neural circuits and neuropeptides regulating circadian healthy sleep may be different from those regulating sickness sleep.
Second, the identification in C. elegans and Drosophila of a specific neuroendocrine mechanism for sickness sleep raises the question whether a similar mechanism exists in mammals. How do we identify such a neuroendocrine mechanism in mammals? Based on the C. elegans research, a central mammalian neuron regulating sickness behaviour should have the following properties. (1) It should express an epidermal growth factor receptor and be activated by an epidermal growth factor member. (2) It should signal by neuroendocrine mechanisms (i.e. not by synaptic mechanisms). (3) It should contain several neuropeptides including RFamide neuropeptides. Since there are only five RFamide peptides in mammals (Walker et al. 2009), testing each of these for regulation of sickness sleep is a feasible endeavour.
Finally, the observation that the C. elegans ALA neuron controls sickness behaviour via the collective action of several distinct neuropeptides (Nath et al. 2016) demonstrates that sub‐programmes of sickness behaviour can be activated separately. In mammals, sickness behaviour includes a range of behaviours in addition to sleep; these include anorexia, malaise and social withdrawal. Social withdrawal and malaise are cardinal symptoms of major depression, which in some patients also presents with excessive daytime sleepiness. In addition, there are patients with isolated excessive daytime sleepiness that is not explained by sleep curtailment or by any other known sleep disorder. These patients are often diagnosed with ‘idiopathic hypersomnia’, or ‘primary hypersomnia’. We suggest that the sleepiness of such patients may in fact be explained by the inappropriate activation of a particular neuropeptide signalling system that typically functions during sickness behaviour (Fig. 2). Support for our hypothesis would include the finding of elevated levels of such a somnogenic peptide in the cerebrospinal fluid (CSF) of these patients. Interestingly, Rye and colleagues identified a protein in the CSF of primary hypersomnia patients with a size consistent with that of neuropeptides (Rye et al. 2012). The nature of this neuropeptide(s), which the authors showed modulates GABAA currents (Rye et al. 2012), has not yet been reported.
It will be of future interest to translate specific mechanistic details elucidated in fruit flies and round worms into a mammalian system and the clinic. Specific questions to ask include: (1) Do injurious stimuli aside from infection, such as exposure to high heat or ultraviolet radiation induce sickness behaviour, including sleepiness? (2) Is there an increase in circulating EGF family members after exposure to infectious or other stressors? (3) Is there an increase in RFamide or other neuropeptides in the CSF of animals displaying sickness sleep behaviour and are such peptides required for sickness sleep behaviour?
In addition to their relevance to disorders of hypersomnolence and to major depression, answers to these questions are relevant to conditions associated with prominent cytokine activation in which malaise and fatigue are disabling symptoms. These include autoimmune disorders such as rheumatoid arthritis (Hewlett et al. 2005) and multiple sclerosis (Flachenecker et al. 2004; Giovannoni, 2006) as well as radiotherapy side effects (Hickok et al. 2005; Bower et al. 2009). Finally, most of the research on sleep induced by cellular injury in animal models has focused on the effects of acute infection or other stressors. Chronic or recurrent stress may result in allostatic changes in sleep that are distinct from those caused by a single acute stressor. Such allostatic stress responses may be relevant to the fragmented sleep observed in patients with neurodegenerative diseases (Ju et al. 2014), persistent parasitic infection (Lejon et al. 2013) and autoimmune disease (Abad et al. 2008).
Additional information
Competing interests
None declared.
Funding
D.M.R. was supported by National Institutes of Health (NIH) grants R01NS088432 and R21NS091500 from the National Institute of Neurological Diseases and Stroke (NINDS). K.C.D. was supported by T32‐ES019851 (PI: Trevor Penning) from the National Institute of Environmental Health Sciences (NIEHS; NIH). This publication was made possible in part by grant 1P30ES013508 from the NIEHS. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS or NINDS, NIH.
Biographies
Kristen Davis is a postdoctoral fellow in the department of Neurology at the University of Pennsylvania Perelman School of Medicine in Philadelphia, Pennsylvania, USA. She received her PhD degree from Virginia Commonwealth University in Richmond, Virginia, USA.
David Raizen is an Associate Professor of Neurology, Medicine, and Genetics at the University of Pennsylvania Perelman School of Medicine. He received his MD and PhD degrees from the University of Texas Southwestern Medical School in Dallas, Texas, USA.

This review was presented at the symposium “Animal models of sleep: from 100 billion to 302 neurons”, which took place at Physiology 2016, Dublin, Ireland, 29–31 July 2016.
References
- Abad VC, Sarinas PS & Guilleminault C (2008). Sleep and rheumatologic disorders. Sleep Med Rev 12, 211–228. [DOI] [PubMed] [Google Scholar]
- Allen VW, O'Connor RM, Ulgherait M, Zhou CG, Stone EF, Hill VM, Murphy KR, Canman JC, Ja WW & Shirasu‐Hiza MM (2016). period‐regulated feeding behaviour and TOR signaling modulate survival of infection. Curr Biol 26, 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbely AA (1982). A two process model of sleep regulation. Hum Neurobiol 1, 195–204. [PubMed] [Google Scholar]
- Bower JE, Ganz PA, Tao ML, Hu W, Belin TR, Sepah S, Cole S & Aziz N (2009). Inflammatory biomarkers and fatigue during radiation therapy for breast and prostate cancer. Clin Cancer Res 15, 5534–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JY & Sternberg PW (2014). Multilevel modulation of a sensory motor circuit during C. elegans sleep and arousal. Cell 156, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Chatzigeorgiou M, Taylor KP, Schafer WR & Kaplan JM (2013). Analysis of NPR‐1 reveals a circuit mechanism for behavioral quiescence in C. elegans . Neuron 78, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B & Tononi G (2005). Reduced sleep in Drosophila Shaker mutants. Nature 434, 1087–1092. [DOI] [PubMed] [Google Scholar]
- Cohen S (1977). The role of cell‐mediated immunity in the induction of inflammatory responses. Parke‐Davis Award Lecture, 1977. Am J Pathol 88, 502–528. [PMC free article] [PubMed] [Google Scholar]
- Douglas CL, Vyazovskiy V, Southard T, Chiu SY, Messing A, Tononi G & Cirelli C (2007). Sleep in Kcna2 knockout mice. BMC Biol 5, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreux AC, Lamb DJ, Modjtahedi H & Ferns GA (2006). The epidermal growth factor receptors and their family of ligands: their putative role in atherogenesis. Atherosclerosis 186, 38–53. [DOI] [PubMed] [Google Scholar]
- Driver RJ, Lamb AL, Wyner AJ & Raizen DM (2013). DAF‐16/FOXO regulates homeostasis of essential sleep‐like behaviour during larval transitions in C. elegans . Curr Biol 23, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap JC (1999). Molecular bases for circadian clocks. Cell 96, 271–290. [DOI] [PubMed] [Google Scholar]
- Eriksson C, Nobel S, Winblad B & Schultzberg M (2000). Expression of interleukin 1α and β, and interleukin 1 receptor antagonist mRNA in the rat central nervous system after peripheral administration of lipopolysaccharides. Cytokine 12, 423–431. [DOI] [PubMed] [Google Scholar]
- Fischer OM, Hart S, Gschwind A, Prenzel N & Ullrich A (2004). Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin‐binding epidermal growth factor. Mol Cell Biol 24, 5172–5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachenecker P, Bihler I, Weber F, Gottschalk M, Toyka KV & Rieckmann P (2004). Cytokine mRNA expression in patients with multiple sclerosis and fatigue. Mult Scler 10, 165–169. [DOI] [PubMed] [Google Scholar]
- Foltenyi K, Greenspan RJ & Newport JW (2007). Activation of EGFR and ERK by rhomboid signaling regulates the consolidation and maintenance of sleep in Drosophila . Nat Neurosci 10, 1160–1167. [DOI] [PubMed] [Google Scholar]
- Fry AL, Laboy JT, Huang H, Hart AC & Norman KR (2016). A conserved GEF for rho‐family GTPases acts in an EGF signaling pathway to promote sleep‐like quiescence in Caenorhabditis elegans . Genetics 202, 1153–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni G (2006). Multiple sclerosis related fatigue. J Neurol Neurosurg Psychiatry 77, 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta BP, Hanna‐Rose W & Sternberg PW (2012). Morphogenesis of the vulva and the vulval‐uterine connection. WormBook, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MK & Krueger JM (1997). Subdiaphragmatic vagotomy blocks the sleep‐ and fever‐promoting effects of interleukin‐1β. Am J Physiol 273, R1246–R1253. [DOI] [PubMed] [Google Scholar]
- Hendricks JC, Kirk D, Panckeri K, Miller MS & Pack AI (2003). Modafinil maintains waking in the fruit fly Drosophila melanogaster . Sleep 26, 139–146. [DOI] [PubMed] [Google Scholar]
- Hendricks JC, Sehgal A & Pack AI (2000). The need for a simple animal model to understand sleep. Prog Neurobiol 61, 339–351. [DOI] [PubMed] [Google Scholar]
- Hewlett S, Cockshott Z, Byron M, Kitchen K, Tipler S, Pope D & Hehir M (2005). Patients’ perceptions of fatigue in rheumatoid arthritis: overwhelming, uncontrollable, ignored. Arthritis Rheum 53, 697–702. [DOI] [PubMed] [Google Scholar]
- Hickok JT, Roscoe JA, Morrow GR, Mustian K, Okunieff P & Bole CW (2005). Frequency, severity, clinical course, and correlates of fatigue in 372 patients during 5 weeks of radiotherapy for cancer. Cancer 104, 1772–1778. [DOI] [PubMed] [Google Scholar]
- Hill AJ, Mansfield R, Lopez JM, Raizen DM & Van Buskirk C (2014). Cellular stress induces a protective sleep‐like state in C. elegans . Curr Biol 24, 2399–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannacone MJ, Beets I, Lopes LE, Churgin MA, Fang‐Yen C, Nelson MD, Schoofs L & Raizen DM (2017). The RFamide receptor DMSR‐1 regulates stress‐induced sleep in C. elegans. eLife 6, e19837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Mao XO, Sun Y, Xie L, Jin L, Nishi E, Klagsbrun M & Greenberg DA (2002). Heparin‐binding epidermal growth factor‐like growth factor: hypoxia‐inducible expression in vitro and stimulation of neurogenesis in vitro and in vivo . J Neurosci 22, 5365–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner WJ, Crocker A, White BH & Sehgal A (2006). Sleep in Drosophila is regulated by adult mushroom bodies. Nature 441, 757–760. [DOI] [PubMed] [Google Scholar]
- Ju YE, Lucey BP & Holtzman DM (2014). Sleep and Alzheimer disease pathology – a bidirectional relationship. Nat Rev Neurol 10, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K, Evans JM, Hendricks JC & Sehgal A (2006). A Drosophila model for age‐associated changes in sleep:wake cycles. Proc Natl Acad Sci USA 103, 13843–13847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Yang FC, Snodgrass P, Li X, Scammell TE, Davis FC & Weitz CJ (2001). Regulation of daily locomotor activity and sleep by hypothalamic EGF receptor signaling. Science 294, 2511–2515. [DOI] [PubMed] [Google Scholar]
- Krueger JM, Walter J, Dinarello CA, Wolff SM & Chedid L (1984). Sleep‐promoting effects of endogenous pyrogen (interleukin‐1). Am J Physiol 246, R994–R999. [DOI] [PubMed] [Google Scholar]
- Kuo TH, Pike DH, Beizaeipour Z & Williams JA (2010). Sleep triggered by an immune response in Drosophila is regulated by the circadian clock and requires the NFκB Relish. BMC Neurosci 11, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo TH & Williams JA (2014. a). Acute sleep deprivation enhances post‐infection sleep and promotes survival during bacterial infection in Drosophila . Sleep 37, 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo TH & Williams JA (2014. b). Increased sleep promotes survival during a bacterial infection in Drosophila . Sleep 37, 1077–1086, 1086A–1086D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuper C, Bartels H, Fraek ML, Beck FX & Neuhofer W (2007). Ectodomain shedding of pro‐TGF‐α is required for COX‐2 induction and cell survival in renal medullary cells exposed to osmotic stress. Am J Physiol Cell Physiol 293, C1971–C1982. [DOI] [PubMed] [Google Scholar]
- Kuper C, Steinert D, Fraek ML, Beck FX & Neuhofer W (2009). EGF receptor signaling is involved in expression of osmoprotective TonEBP target gene aldose reductase under hypertonic conditions. Am J Physiol Renal Physiol 296, F1100–F1108. [DOI] [PubMed] [Google Scholar]
- Kushikata T, Fang J, Chen Z, Wang Y & Krueger JM (1998). Epidermal growth factor enhances spontaneous sleep in rabbits. Am J Physiol 275, R509–R514. [DOI] [PubMed] [Google Scholar]
- Lejon V, Bentivoglio M & Franco JR (2013). Human African trypanosomiasis. Handb Clin Neurol 114, 169–181. [DOI] [PubMed] [Google Scholar]
- Lenz O, Xiong J, Nelson MD, Raizen DM & Williams JA (2015). FMRFamide signaling promotes stress‐induced sleep in Drosophila . Brain Behav Immun 47, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C & Kim K (2008). Neuropeptides. WormBook, 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Zhang YH, Chou TC, Gaus SE, Elmquist JK, Shiromani P & Saper CB (2001). Contrasting effects of ibotenate lesions of the paraventricular nucleus and subparaventricular zone on sleep‐wake cycle and temperature regulation. J Neurosci 21, 4864–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall L & Born J (2002). Brain‐immune interactions in sleep. Int Rev Neurobiol 52, 93–131. [DOI] [PubMed] [Google Scholar]
- Merrill JE (1987). Macroglia: neural cells responsive to lymphokines and growth factors. Immunol Today 8, 146–150. [DOI] [PubMed] [Google Scholar]
- Mullington J, Korth C, Hermann DM, Orth A, Galanos C, Holsboer F & Pollmacher T (2000). Dose‐dependent effects of endotoxin on human sleep. Am J Physiol Regul Integr Comp Physiol 278, R947–R955. [DOI] [PubMed] [Google Scholar]
- Nath RD, Chow ES, Wang H, Schwarz EM & Sternberg PW (2016). C. elegans stress‐induced sleep emerges from the collective action of multiple neuropeptides. Curr Biol 26, 2446–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MD, Lee KH, Churgin MA, Hill AJ, Van Buskirk C, Fang‐Yen C & Raizen DM (2014). FMRFamide‐like FLP‐13 neuropeptides promote quiescence following heat stress in Caenorhabditis elegans . Curr Biol 24, 2406–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman JL, McGill JJ, Keegan KP & Allada R (2006). A dynamic role for the mushroom bodies in promoting sleep in Drosophila . Nature 441, 753–756. [DOI] [PubMed] [Google Scholar]
- Raizen DM, Zimmerman JE, Maycock MH, Ta UD, You YJ, Sundaram MV & Pack AI (2008). Lethargus is a Caenorhabditis elegans sleep‐like state. Nature 451, 569–572. [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A, Gilliland MA, Bergmann BM & Winter JB (1983). Physiological correlates of prolonged sleep deprivation in rats. Science 221, 182–184. [DOI] [PubMed] [Google Scholar]
- Renegar KB, Crouse D, Floyd RA & Krueger J (2000). Progression of influenza viral infection through the murine respiratory tract: the protective role of sleep deprivation. Sleep 23, 859–863. [PubMed] [Google Scholar]
- Rye DB, Bliwise DL, Parker K, Trotti LM, Saini P, Fairley J, Freeman A, Garcia PS, Owens MJ, Ritchie JC & Jenkins A (2012). Modulation of vigilance in the primary hypersomnias by endogenous enhancement of GABAA receptors. Sci Transl Med 4, 161ra151. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE & Lu J (2005). Hypothalamic regulation of sleep and circadian rhythms. Nature 437, 1257–1263. [DOI] [PubMed] [Google Scholar]
- Schmidt MH (2014). The energy allocation function of sleep: a unifying theory of sleep, torpor, and continuous wakefulness. Neurosci Biobehav Rev 47, 122–153. [DOI] [PubMed] [Google Scholar]
- Schwarz J, Lewandrowski I & Bringmann H (2011). Reduced activity of a sensory neuron during a sleep‐like state in Caenorhabditis elegans . Curr Biol 21, R983–984. [DOI] [PubMed] [Google Scholar]
- Sehgal A & Mignot E (2011). Genetics of sleep and sleep disorders. Cell 146, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Cirelli C, Greenspan RJ & Tononi G (2000). Correlates of sleep and waking in Drosophila melanogaster . Science 287, 1834–1837. [DOI] [PubMed] [Google Scholar]
- Shilo BZ (2003). Signaling by the Drosophila epidermal growth factor receptor pathway during development. Exp Cell Res 284, 140–149. [DOI] [PubMed] [Google Scholar]
- Shirasu‐Hiza MM, Dionne MS, Pham LN, Ayres JS & Schneider DS (2007). Interactions between circadian rhythm and immunity in Drosophila melanogaster . Curr Biol 17, R353–355. [DOI] [PubMed] [Google Scholar]
- Singh K, Ju JY, Walsh MB, DiIorio MA & Hart AC (2014). Deep conservation of genes required for both Drosphila melanogaster and Caenorhabditis elegans sleep includes a role for dopaminergic signaling. Sleep 37, 1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RN & Sulston JE (1978). Some observations on moulting in Caenorhabditis elegans . Nematologica 24, 63–71. [Google Scholar]
- Snodgrass‐Belt P, Gilbert JL & Davis FC (2005). Central administration of transforming growth factor‐alpha and neuregulin‐1 suppress active behaviors and cause weight loss in hamsters. Brain Res 1038, 171–182. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S & Schroeter M (2002). Detrimental and beneficial effects of injury‐induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol 513, 87–113. [DOI] [PubMed] [Google Scholar]
- Toth LA & Krueger JM (1988). Alteration of sleep in rabbits by Staphylococcus aureus infection. Infect Immun 56, 1785–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth LA, Tolley EA & Krueger JM (1993). Sleep as a prognostic indicator during infectious disease in rabbits. Proc Soc Exp Biol Med 203, 179–192. [DOI] [PubMed] [Google Scholar]
- Trojanowski NF, Nelson MD, Flavell SW, Fang‐Yen C & Raizen DM (2015). Distinct mechanisms underlie quiescence during two Caenorhabditis elegans sleep‐like states. J Neurosci 35, 14571–14584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski NF & Raizen DM (2016). Call it Worm Sleep. Trends Neurosci 39, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turek M, Lewandrowski I & Bringmann H (2013). An AP2 transcription factor is required for a sleep‐active neuron to induce sleep‐like quiescence in C. elegans . Curr Biol 23, 2215–2223. [DOI] [PubMed] [Google Scholar]
- Van Buskirk C & Sternberg PW (2007). Epidermal growth factor signaling induces behavioral quiescence in Caenorhabditis elegans . Nat Neurosci 10, 1300–1307. [DOI] [PubMed] [Google Scholar]
- Walker RJ, Papaioannou S & Holden‐Dye L (2009). A review of FMRFamide‐ and RFamide‐like peptides in metazoa. Invert Neurosci 9, 111–153. [DOI] [PubMed] [Google Scholar]
- Walter JS, Meyers P & Krueger JM (1989). Microinjection of interleukin‐1 into brain: separation of sleep and fever responses. Physiol Behav 45, 169–176. [DOI] [PubMed] [Google Scholar]
- Wu MN, Ho K, Crocker A, Yue Z, Koh K & Sehgal A (2009). The effects of caffeine on sleep in Drosophila require PKA activity, but not the adenosine receptor. J Neurosci 29, 11029–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You YJ, Kim J, Raizen DM & Avery L (2008). Insulin, cGMP, and TGF‐β signals regulate food intake and quiescence in C. elegans: a model for satiety. Cell Metab 7, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]