

Abstract
Key points
Comprehensive imaging analyses of glucose transporter 4 (GLUT4) behaviour in mouse skeletal muscle was conducted.
Quantum dot‐based single molecule nanometry revealed that GLUT4 molecules in skeletal myofibres are governed by regulatory systems involving ‘static retention’ and ‘stimulus‐dependent liberation’.
Vital imaging analyses and super‐resolution microscopy‐based morphometry demonstrated that insulin liberates the GLUT4 molecule from its static state by triggering acute heterotypic endomembrane fusion arising from the very small GLUT4‐containing vesicles in skeletal myofibres.
Prior exposure to exercise‐mimetic stimuli potentiated this insulin‐responsive endomembrane fusion event involving GLUT4‐containing vesicles, suggesting that this endomembranous regulation process is a potential site related to the effects of exercise.
Abstract
Skeletal muscle is the major systemic glucose disposal site. Both insulin and exercise facilitate translocation of the glucose transporter glucose transporter 4 (GLUT4) via distinct signalling pathways and exercise also enhances insulin sensitivity. However, the trafficking mechanisms controlling GLUT4 mobilization in skeletal muscle remain poorly understood as a resuly of technical limitations. In the present study, which employs various imaging techniques on isolated skeletal myofibres, we show that one of the initial triggers of insulin‐induced GLUT4 translocation is heterotypic endomembrane fusion arising from very small static GLUT4‐containing vesicles with a subset of transferrin receptor‐containing endosomes. Importantly, pretreatment with exercise‐mimetic stimuli potentiated the susceptibility to insulin responsiveness, as indicated by these acute endomembranous activities. We also found that AS160 exhibited stripe‐like localization close to sarcomeric α‐actinin and that insulin induced a reduction of the stripe‐like localization accompanying changes in its detergent solubility. The results of the present study thus provide a conceptual framework indicating that GLUT4 protein trafficking via heterotypic fusion is a critical feature of GLUT4 translocation in skeletal muscles and also suggest that the efficacy of the endomembranous fusion process in response to insulin is involved in the benefits of exercise.
Keywords: exercise, GLUT4, heterotypic endomembrane fusion, insulin, skeletal myofibers
Key points
Comprehensive imaging analyses of glucose transporter 4 (GLUT4) behaviour in mouse skeletal muscle was conducted.
Quantum dot‐based single molecule nanometry revealed that GLUT4 molecules in skeletal myofibres are governed by regulatory systems involving ‘static retention’ and ‘stimulus‐dependent liberation’.
Vital imaging analyses and super‐resolution microscopy‐based morphometry demonstrated that insulin liberates the GLUT4 molecule from its static state by triggering acute heterotypic endomembrane fusion arising from the very small GLUT4‐containing vesicles in skeletal myofibres.
Prior exposure to exercise‐mimetic stimuli potentiated this insulin‐responsive endomembrane fusion event involving GLUT4‐containing vesicles, suggesting that this endomembranous regulation process is a potential site related to the effects of exercise.
Abbreviations
- AS160
Akt substrate of 160 kDa
- DMEM
Dulbecco's modified Eagle's medium
- EDL
extensor digitorum longus
- FWHM
full‐width‐at‐half‐maximum
- GLUT4
glucose transporter 4
- MSD
mean‐square displacement
- QD
quantum dot
- RabGAP
Rab GTPase‐activating protein
- RT
room temperature
- SIM
structured illumination microscopy
- STED
stimulated‐emission depletion
- Tbc1d
Tre‐2/BUB2/CDC16 domain
- Tf
transferrin
- TfR
transferrin receptor
- TGN
trans‐Golgi network
- T‐tubule
transverse tubule
Introduction
Insulin plays a pivotal role in glucose homeostasis (Saltiel & Kahn, 2001). In target tissues such as skeletal muscle and adipose, insulin induces the redistribution of the glucose transporter glucose transporter 4 (GLUT4) from intracellular storage compartments to the cell surface via GLUT4 translocation (Kanzaki, 2006). Skeletal muscle is the major systemic glucose disposal site (DeFronzo et al. 1981) and not only insulin, but also exercise induces GLUT4 translocation via distinct proximal signalling cascades (Goodyear & Kahn, 1998). Importantly, exercise‐induced GLUT4 translocation is often normal, even in the presence of insulin resistance (Wojtaszewski et al. 1999). Moreover, exercise plays a beneficial role in muscle insulin potency, particularly augmented GLUT4 translocation (Richter et al. 1982; Cartee et al. 1989; Hansen et al. 1998). Unveiling the details of GLUT4 behaviour in the skeletal muscle would thus be critical for understanding systemic glucose homeostasis, type 2 diabetes and the mechanistic details of the benefits of exercise.
Despite progress in unraveling signalling cascades crucial for GLUT4 translocation, a major challenge inherent to GLUT4 research in skeletal muscle remains, partly as a result of the characteristic properties of myofibres. Notably, myofibres have a highly‐ordered multinucleate subcellular architecture with two plasma membrane domain types, sarcolemma and transverse (T)‐tubules, with insulin and exercise/muscle contraction both inducing the redistribution of endomembrane‐localized GLUT4 molecules to the membrane domains (Ploug et al. 1998; Lauritzen et al. 2006).
Recently, we developed a novel GLUT4 nanometry method for quantifying intracellular GLUT4 behaviour based on single molecule imaging with quantum dot (QD) fluorescent nanocrystals. With this method, we can dissect intracellular GLUT4 trafficking processes into discrete and experimentally traceable steps (Fujita et al. 2010). Taking advantage of these novel capabilities, we quantitatively determined the behavioural nature of GLUT4 molecules in 3T3L1 adipocytes and identified at least three sites of insulin action. Among these, insulin‐dependent GLUT4 liberation from its static states is the most important element of insulin action in the entire GLUT4 trafficking itinerary because this initial process is a prerequisite allowing GLUT4 to reach the plasma membrane. In addition, we revealed the roles of two key Tre‐2/BUB2/CDC16 domain (Tbc1d) family Rab GTPase‐activating proteins (RabGAPs), the Akt substrate of 160 kDa (AS160)/Tbc1d4 (hereafter AS160) and Tbc1d1 (Miinea et al. 2005; Roach et al. 2007), both of which are critical for insulin‐stimulated glucose uptake into skeletal muscle (Chadt et al. 2015) in the process of the stimulus‐responsive liberation of static GLUT4 (Hatakeyama & Kanzaki, 2011,2013b). All of these experiments employed cultured cell lines, such that a fundamental question remains. Are GLUT4 molecules in skeletal muscle fibres indeed governed by regulatory mechanisms similar to those revealed in cultured cell lines, such as ‘static retention’ and ‘stimulus‐dependent liberation’, and, if so, what are the underlying mechanisms?
In the present study, we aimed to clarify the initial effects of insulin on GLUT4 behaviours in isolated murine skeletal myofibres employing various cutting‐edge imaging techniques. Specifically, our QD‐based single molecule nanometry in isolated skeletal myofibres demonstrated that they are indeed endowed with essentially the same GLUT4 regulatory systems as those in cultured cell lines. Super‐resolution microscopy‐based morphometry unexpectedly revealed that insulin promptly increases the sizes of intracellular GLUT4‐containing structures, such that endomembrane fusion events between very small static GLUT4‐containing vesicles and transferrin receptor (TfR)‐containing endosomes are facilitated in at least a portion of isolated skeletal myofibres obtained from soleus and extensor digitorum longus (EDL) muscles of mice. We propose that acute endosomal fusion serves as one of the initial triggers for releasing GLUT4 molecules from the static state in response to insulin in skeletal myofibres.
Methods
Ethical approval
All of the animal experiments were performed in accordance with the regulations of Tohoku University (2014BeLMO‐003). Animals were given access to food and water ad libitum.
Mice and skeletal myofibre isolation
Myc‐GLUT4‐EGFP transgenic mice were generated by carrying a myc‐GLUT4‐EGFP cDNA under the control of the human skeletal α‐actin promoter (Brennan & Hardeman, 1993). Male and female transgenic or wild‐type mice were housed at 22–24°C under a 12:12 h light/dark cycle with standard chow and water provided ad libitum. The mice were killed at 8–20 weeks of age by cervical dislocation, and single or partially‐digested myofibres were isolated from soleus and EDL muscles by collagenase digestion. For live‐imaging, isolated myofibres were plated onto glass‐bottom dishes (No. 1S, thickness, 0.16–0.19 mm; Matsunami Glass, Kishiwada, Japan) coated with laminin, and cultured overnight in a humidified atmosphere of 5% CO2/95% air at 37°C in Dulbecco's modified Eagle's medium (DMEM) containing glucose (1 mg ml−1) supplemented with 20% fetal bovine serum, 2 mm GlutaMAX (Thermo Fisher Scientific, Waltham, MA, USA) and 1 mm sodium pyruvate. Prior to stimulation, the myofibres were starved with serum‐free DMEM containing glucose (1 mg ml−1) supplemented with 2 mm GlutaMAX and 1 mm sodium pyruvate, which had been bubbled with 95% O2/5% CO2. For imaging experiments, the myofibres were immersed in phenol red‐free DMEM containing glucose (1 mg ml−1) supplemented with 2 mm GlutaMAX and 1 mm sodium pyruvate, which had been bubbled with 95% O2/5% CO2.
Antibodies and western blotting
Anti‐myc monoclonal antibody (9E10) was purified from hybridoma culture supernatants. Antibody against GLUT4 was provided by Dr Hiroshi Shibata (Gunma University, Maebashi, Japan). Anti‐Tbc1d1 antibodies, for experimental use, were generated by immunizing rabbits with KLH‐conjugated peptides (H‐CSKKEPRTKQ‐OH), followed by immunoaffinity purification using SulfoLink Coupling Gel (Pierce, Rockford, IL, USA) or purchased from Protein Tech (Chicago, IL, USA) (22124‐1‐AP). Anti‐phospho‐Tbc1d1 (Ser237) antibody was generated by immunizing rabbits with KLH‐conjugated peptides (H‐CRPMRKSF‐pS‐QPGLRS‐OH) and purified as described above. Anti‐Akt (#9272), anti‐phospho‐Akt (Ser473) (#9271), anti‐AMPKα (#2603), anti‐phospho‐AMPKα (Thr172) (#2535) and anti‐phospho‐AS160 (Thr642) (#4288) antibodies were obtained from Cell Signaling (Beverly, MA, USA). Anti‐AS160 antibodies for western blotting and immunofluorescence were purchased from Abcam (Cambridge, MA, USA) (ab91242), Millipore (Billerica, MA, USA) (ABS54), and Santa Cruz Biotechnology (Santa Cruz, CA, USA) (sc‐47470 and sc‐47467). Other antibodies were: anti‐BIN1 (Santa Cruz Biotechnology) (sc‐8534), anti‐TfR (Leinco Tech, Fenton, MO, USA) (C275), anti‐sarcomeric α‐actinin (Sigma) (A7811), HRP‐conjugated secondary antibodies (Pierce) and Alexa Fluor‐conjugated secondary antibodies (Thermo Fisher Scientific). For western blotting, muscle tissues were homogenized employing a Bead Homogenizer (MS‐100R; Tomy Digital Biology, Tokyo, Japan) at 4°C for 15 s × 2 with three zirconium beads (2 mm) at a speed setting of 4500 rpm in lysis buffer (50 mm Tris‐HCl, pH 7.4, 150 mm NaCl, 20 mm sodium pyrophosphate, 10 mm NaF, 2 mm sodium orthovanadate, 1 mm EDTA, 1% Triton X‐100, 1 μg ml−1 pepstatin, 5 μg ml−1 leupeptin, 1 mm phenylmethylsulfonyl fluoride, 6500 IU ml–1 aprotinin, phosphatase inhibitor mixture‐1; Sigma). After end‐over‐end rotation of the homogenates for 30 min, lysate supernatants were collected by centrifugation (21,880 × g) for 20 min at 4°C, and protein concentrations were measured using the bicinchonic acid method with BSA as the standard (Pierce). Total protein and phosphorylation levels of relevant proteins were determined by conventional immunoblotting techniques with loading of equal amounts of proteins. PVDF membranes (Immobilon‐P; Millipore) were blocked for 2 h at room temperature (RT) with 5% BSA in TBS containing 0.1% Tween 20. After sequential incubations with first and second antibodies, chemiluminescence (SuperSignal West Femto; Thermo Fisher Scientific) was detected with an ImageQuant LAS4000 mini (GE Healthcare Life Sciences, Little Chalfont, UK) imaging system.
QD labelling
For preparation of QD‐conjugated anti‐myc antibody, first, the Fab' fragment of anti‐myc antibody was prepared with a Mouse IgG1 Fab and F(ab')2 Preparation Kit (Pierce), followed by mild reduction with 2‐mercaptoethanolamine. Maleimide‐activated QD655 was also prepared by incubating Qdot 655 ITK Amino (PEG) Quantum dots (Thermo Fisher Scientific) with sulfo‐SMCC (Pierce) at RT for 1 h. The resultant Fab' fragment of anti‐myc antibodies and maleimide‐activated QD655 was mixed, and the final concentrations of the QD655‐conjugated antibodies were determined by measuring optical density at 638 nm and then using the formula A = εcL, where A is the absorbance, ε is the molar extinction coefficient (800 000 M–1 cm–1), c is the molar concentration and L is the path length. For cellular labelling, the cells were serum‐starved and incubated in the presence of 1.5–5 nm QD655‐conjugated antibodies for 1 h. The cells were then extensively washed for at least 3 h to remove unbound QD‐labelled antibodies. Labelling of TfR with QD was performed using QD‐conjugated transferrin (Tf), which had been prepared with Tf‐biotin and streptavidin‐QD (Thermo Fisher Scientific) in the presence of excess (100×) biotin. For cellular labelling, cells were treated with the Tf‐QD conjugates for 15 min, and images were acquired for up to 30 min.
Single molecule imaging and particle tracking
Single molecule imaging of QD was performed with an inverted microscope (IX81; Olympus, Tokyo, Japan) equipped with an electron‐multiplying charge‐coupled device camera (iXon Ultra; Andor Technology, Belfast, UK), a Nipkow disk confocal unit (CSU‐X1; Yokogawa, Tokyo, Japan), a motorized xy stage (Sigma Koki, Tokyo, Japan), a z‐drift compensation module (Olympus) and an oil‐immersion objective lens (UPLSAPO100xO, NA 1.4; Olympus) at 20 frames s–1. Imaging was performed at ∼30°C using a stage heater and a lens heater (Tokai Hit, Fujinomiya, Japan). QD655 fluorescence was excited at 488 or 532 nm with a solid‐state laser and detected via a 655/12 bandpass filter (Semrock, Rochester, NY, USA). Single particle tracking was performed using G‐Count (G‐Angstrom, Sendai, Japan) with a two‐dimensional Gaussian fitting mode. We tracked each particle successfully fitted within a 13 × 13 pixel region‐of‐interest for at least 30 frames. When the signal in a frame was lost because of blinking, no fitting was performed until reappearance of the bright spot. When a bright spot did not reappear within 10 frames, tracking was aborted. We typically tracked 50–150 particles per cell and obtained the images in at least three independent experiments. We evaluated movements with mean velocities and mean‐square displacements (MSD) (Fujita et al. 2010). The velocities for individual particle movements were calculated by linear fit of the displacement during four frames. Mean velocities were first calculated in a cell, and then averaged among cells under the same treatment conditions. We found that QD in fixed cells showed non‐negligible velocities (∼0.19 μm s−1), which was attributed to unavoidable instrumental noise. Thus, when representing mean velocities, we used corrected values instead of raw data by subtracting the noise. Because the velocities were calculated based on the movements within short periods of time and single molecule fluorescence with a limited signal‐to‐noise ratio, such relatively large noise was inevitable. Although our approach does allow clear observation of stimulation‐dependent changes in speeds under such conditions, we also calculated MSD showing relatively long time‐scale behaviours, aiming to clearly determine the actions triggered by insulin stimulation. MSD values of individual particles were calculated with:
where N is the total number of positions measured, Δt is the time interval of successive images, τ is the time lag that can calculated by nΔt where n is integer from 1 to N – 1, and p is the position vector of the molecule, respectively.
Imaging of endomembrane fusion
Imaging of endosomal fusion of myc‐GLUT4‐containing vesicles with TfR‐containing vesicles was performed using a fluorometric avidin‐biotin binding assay (Emans et al. 1995). To prepare streptavidin‐BODIPY‐conjugated anti‐myc antibodies, first, recombinant streptavidin (ProSpec, Rehovot, Israel) was mixed with BODIPY FL‐NHS (Thermo Fisher Scientific) at a 1:2 molar ratio in 0.1 m sodium bicarbonate buffer (pH 8.2) and incubated at RT for 1 h. Unreacted BODIPY FL‐NHS was then removed by dialysis in PBS. The degree of labelling was ∼1.7 mol BODIPY mol–1 streptavidin. Next, streptavidin‐BODIPY was maleimide‐activated in the presence of a 10‐fold excess of sulfo‐SMCC (Pierce) at RT for 30 min, and desalted with PBS containing 5 mm EDTA. The maleimide‐activated streptavidin‐BODIPY was mixed with sulfhydryl‐anti‐myc antibodies, which had been prepared separately by reacting anti‐myc antibodies with 2‐mercaptoethanolamine, for 30 min at RT. For labelling, cells were treated with 4 μg ml−1 streptavidin‐BODIPY FL‐conjugated anti‐myc antibodies for 1 h. Then, the cells were extensively washed, followed by an additional incubation for at least 1 h. After incubation, the cells were treated with 10 μg ml−1 Tf‐biotin for 15 min and then extensively washed. Imaging experiments were performed with an SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany) equipped with an inverted microscope, a white‐light pulsed laser, highly sensitive hybrid detectors and an oil‐immersion objective lens (HC PL APO CS2 63×, NA 1.40). Excitation was at 488 nm and fluorescence was acquired at 495–600 nm. Images were acquired as a t series at intervals of 10 s. Note that we obtained merged fluorescence of BODIPY and EGFP as a result of the similar fluorescence spectra of BODIPY and EGFP.
Immunofluorescence, super‐resolution imaging and image analysis
Myofibres were fixed with 2% paraformaldehyde in PBS for 15 min, and permeabilized with P‐buffer consisting of 0.5% saponin and 1% BSA in PBS for 10 min. The myofibres were then blocked with P‐buffer supplemented with 5% calf serum. Primary and Alexa Fluor‐conjugated secondary antibodies were diluted in P‐buffer and incubated overnight at 4°C and for 1 h at RT, respectively. The samples were mounted in ProLong Gold antifade reagent (Thermo Fisher Scientific). Confocal and stimulated emission depletion (STED) images were taken using a gated STED microscope system (SP8 gSTED; Leica) equipped with an inverted microscope, a white‐light pulsed laser (for excitation), a continuous wave laser (for STED), highly sensitive hybrid detectors and an oil‐immersion objective lens (HC PL APO 100× Oil STED, NA 1.40). Excitation and STED were at 488 and 592 nm, respectively, and fluorescence was acquired at 495–585 nm with time‐gated detection (1.5–6.5 ns). Structured illumination microscopy (SIM) images were acquired with the N‐SIM system (Nikon, Tokyo, Japan) equipped with an inverted microscope, an electron‐multiplying charge‐coupled device camera (iXon3; Andor Technology) and an oil‐immersed objective lens (CFI Apo TIRF 100×, NA 1.49). Excitation was at 488 and 561 nm with solid‐state lasers (Sapphire; Coherent Inc., Santa Clara, CA, USA). Images were acquired by 3D SIM mode as a z‐series at intervals of 0.1 μm and SIM reconstructions were performed using NIS elements AR (Nikon). For measuring particle area, fluorescence images were binarized by Otsu's method and particle sizes were measured using the Analyse Particles plug‐in of Fiji (Schindelin et al. 2012). For measuring particle diameters, intensity profiles of particles were obtained along a defined line, followed by fitting with Gaussian (confocal images) or Lorentzian (STED images) functions using the Levenberg–Marquardt algorithm in ORIGIN (OriginLab Corp., Northampton, MA, USA), and full‐width‐at‐half‐maximum (FWHM) diameters were obtained by the fitted functions. For co‐localization analyses, background fluorescence of the acquired images was measured within the cell‐free area adjacent to the cell of interest and its mean + 3 SD value was subtracted from the entire image. Then, quantitative co‐localization analysis was performed with Coloc2 plug‐in of Fiji. Amplitudes of stripe‐like structures were determined with custom‐written Matlab scripts (MathWorks Inc., Natick, MA, USA). First, power spectra of the immunofluorescence images were obtained by 2D‐FFT and their radial profiles were calculated. Then, the maximum peak amplitude between spatial frequencies of 0.4–0.6 μm–1 (which corresponds to intervals of 1.7–2.5 μm) was obtained.
Statistical analysis
Data with error bars are the mean ± SEM unless otherwise indicated. The Mann–Whitney U test and Dunnett's multiple comparison test were used for comparisons between two groups and among three or more groups of mean values, respectively. P < 0.05 was considered statistically significant. Statistical tests were performed with SPSS, version 22 (IBM Corp., Armonk, NY, USA) or ORIGIN (OriginLab Corp.).
Results
GLUT4 nanometry in single myofibres
In these experiments, we endeavoured to apply GLUT4 nanometry (which has been successfully employed in cultured cell lines) to isolated mouse skeletal myofibres. First, we generated myc‐GLUT4‐EGFP transgenic mice using α‐actin promotors to express the proteins in skeletal muscle and then isolated single myofibres from soleus (slow‐twitch) and EDL (fast‐twitch) muscles by collagenase digestion (Fig. 1 A). We labelled intracellular myc‐GLUT4‐EGFP with QD‐conjugated anti‐myc antibodies (Fig. 1 B) and tracked the intracellular movements of individual QD signals in myofibres employing a method already established for cultured cell lines (Fujita et al. 2010). We observed GLUT4 behaviour just below the sarcolemma, at a depth of 1–4 μm above the glass surface.
Figure 1. Single molecule nanometry in isolated mouse skeletal myofibres.
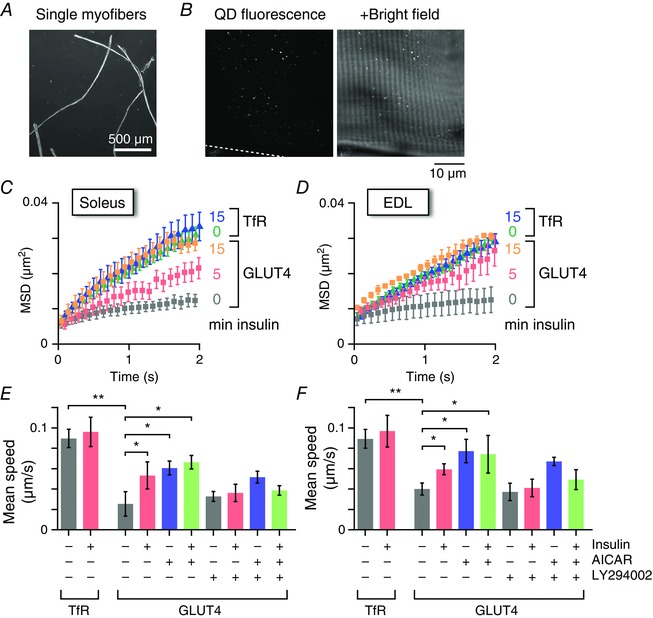
A, micrograph of isolated EDL myofibres. B, fluorescence (left) and overlaid bright field (right) images of an EDL myofibre isolated from a myc‐GLUT4‐EGFP transgenic mouse and then labelled with QD655‐conjugated anti‐myc antibodies. C and D, mean MSD curves of intracellular movements of GLUT4 or TfR before and after insulin stimulation (100 nm; 5 or 15 min) in soleus (C) and EDL (D) myofibres. E and F, mean speed of intracellular movement of GLUT4 or TfR under the indicated conditions. Some myofibres were pretreated with LY294002 (50 μm) for 30 min before stimulation with insulin (100 nm) or AICAR (1 mm) for 5 min. * P < 0.05, * * P < 0.01 (n = 4–6).
The intracellular movement of GLUT4 was significantly restricted compared to that of TfR, a well‐established recycling pathway marker, in both soleus and EDL muscles (Fig. 1 C–F), indicating the existence of a stationary compartment, especially for GLUT4 in skeletal myofibres, similar to that in 3T3‐L1 adipocytes. We next analysed the actions of insulin mediating GLUT4 behaviour and found significant increases in GLUT4 movement with as little as 5 min of insulin stimulation in both soleus and EDL myofibres and, in addition, the insulin‐induced responses were abolished in the presence of the phosphoinositide 3‐kinase inhibitor LY294002 (Fig. 1 C–F), suggesting that insulin promptly stimulates the release/liberation of static GLUT4. By contrast, TfR intracellular activities were unaffected by insulin stimulation. An AMP kinase activator, AICAR, also induced significant liberation of static GLUT4, although we observed no significant additive effects of insulin and AICAR on GLUT4 liberation (Fig. 1 E and F).
Phosphorylation and localization of AS160 and Tbc1d1 in myofibres
To confirm intramyofibre signalling status with 5 min exposure to stimuli, western blot and immunofluorescence analyses of signalling intermediates related to GLUT4 translocation including AS160 and Tbc1d1 were performed. Phosphorylation of these signalling intermediates was induced by each stimulus, although AICAR‐dependent responses such as phorphorylations of AMPK and Tbc1d1 were relatively weak (Fig. 2 A). The insulin‐responsive phosphorylation of Akt and AS160 was abolished in the presence of LY294002 (see below). We found no obvious additive effects on AS160 or Thr642 phosphorylation in response to treatment with insulin and AICAR (Fig. 2 A), which is consistent with a previous observation (Kramer et al. 2006), although that previous study used prolonged stimulation. Interestingly, we observed marked increases in total AS160 solubilized into supernatants from muscle homogenates after insulin stimulation, suggesting that insulin induced changes in AS160 states lead to Triton X‐100 soluble fractions. Such subcellular localization changes of AS160 in skeletal muscles were observed by employing a fractionation method (Zheng & Cartee, 2016).
Figure 2. Proximal signalling and localization of AS160 and Tbc1d1.
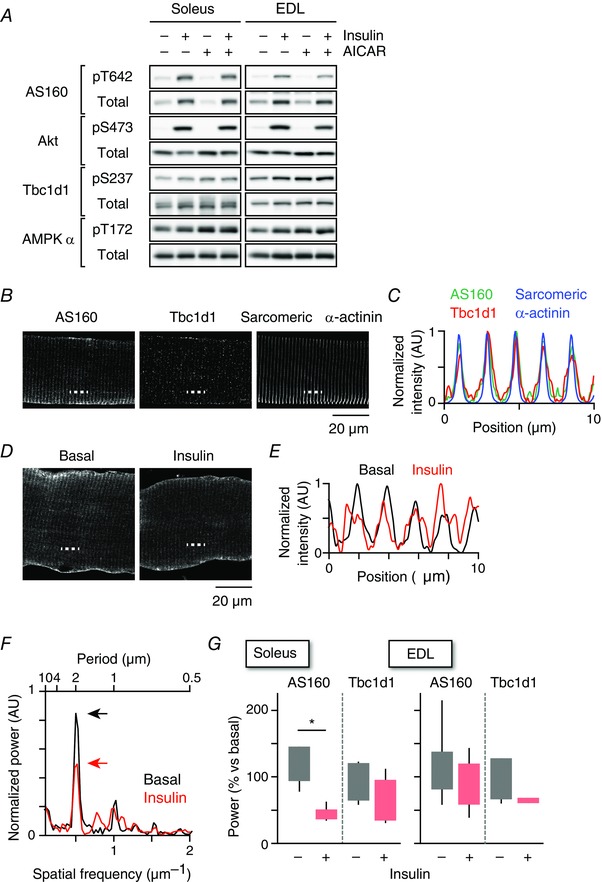
A, western blotting analyses of proximal signalling related to GLUT4 translocation. B, immunofluorescence images of AS160, Tbc1d1 and sarcomeric α‐actinin in EDL myofibres. C, intensity profiles of AS160 (green), Tbc1d1 (red) and sarcomeric α‐actinin (blue) along the dashed line shown in (B). D, immunofluorescence images of AS160 before and after insulin stimulation (100 nm, 15 min) in soleus myofibre. E, intensity profiles of AS160 before (black) and after (red) insulin stimulation along the dashed line in (D). F, power spectra calculated from the images shown in (D) by 2D‐FFT before (black) and after (red) insulin stimulation. G, quantification of changes in stripe‐like structures of AS160 and Tbc1d1 after insulin stimulation in soleus (left) and EDL (right) myofibres. * P < 0.05 (n = 6).
Immunofluorescence analyses revealed that both AS160 and Tbc1d1 exhibited stripe‐like structures and co‐localized with a z‐band marker sarcomeric α‐actinin (Fig. 2 B and C). Similar staining patterns of AS160 and Tbc1d1 were observed using antibodies generated from different epitopes. Insulin stimulation resulted in a reduction of these stripe‐like localizations of AS160 (Fig. 2 D and E). By quantifying the amplitudes of the stripe‐like structures of AS160 and Tbc1d1 (Fig. 2 F), we found significantly reduced AS160 (but not Tbc1d1) localization in the stripe‐like structures in soleus muscle after insulin stimulation. Localization of AS160 in the stripe‐like structures in EDL muscle also tended to diminish with insulin stimulation (Fig. 2 G), although the decreases were not statistically significant.
Insulin increases sizes of GLUT4‐containing structures and their co‐localization with TfR
To further assess initial kinetic actions of insulin in GLUT4 trafficking in skeletal muscle, we next analysed morphological and functional signatures of GLUT4‐containing elements after brief (5 min) insulin stimulation. First, we analysed the structures of endogenous GLUT4‐containing elements in myofibres of wild‐type mice using STED super‐resolution microscopy because most GLUT4‐containing structures are assumed to be smaller than the diffraction limit of conventional fluorescence microscopy (∼200 nm) (Rodnick et al. 1992). With STED microscopy, we detected a number of very small GLUT4‐containing structures (Fig. 3 A), some of which could even be resolved into two (or more) GLUT4‐containing structures in proximity (Fig. 3 B and C). Intriguingly, insulin increased the sizes of GLUT4‐containing structures after 5 min of stimulation (Fig. 4 A). These GLUT4‐containing structures showed significant enlargement in both soleus and EDL muscles, although the former appeared to be more sensitive to this effect (Fig. 4 B and C). As expected, insulin‐induced enlargement of the GLUT4‐containing structures was blunted in the presence of LY294002. The frequency distribution of the areas was best‐fitted by the sum of two exponential functions, characterized by smaller [(0.441 ± 0.013) × 10–2 μm2] and larger [(5.43 ± 0.16) × 10–2 μm2] components (Fig. 4 D) and insulin significantly increased the larger component in 12% of soleus and 5% of EDL muscles (Fig. 4 E). Assuming spherical particles, the estimated maximum diameters of the two components are 75 ± 6 nm and 263 ± 22 nm and ∼10–40 of the smaller structures are required to generate the larger one. We also measured FWHM diameters of the observed structures and confirmed the acute effects of insulin (Fig. 4 F). The median diameter of GLUT4‐containing structures, even after insulin stimulation (∼138 nm), remains below the diffraction limit. Therefore, the present study is the first to obtain such findings employing morphometric evaluations with super‐resolution microscopy. AICAR also enlarged the structures examined (Fig. 4 B–F), although we detected no significant additive effects of insulin and AICAR.
Figure 3. Comparison of confocal and STED images of GLUT4‐containing structures.
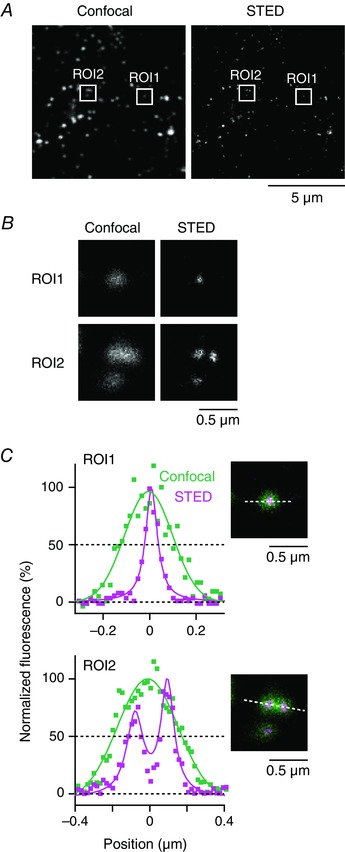
A, confocal (left) and STED (right) images of endogenous GLUT4 in EDL muscle. These images were obtained from the same region with the same acquisition setting, except for the excitation/STED laser power. B, magnified images of boxed region‐of‐interest (ROI) in (A). C, intensity profiles of confocal (green) and STED (magenta) images along the dashed line of each ROI (inset). Continuous lines represent Gaussian (confocal) or Lorentzian (STED) fitting of the profiles. In ROI1, FWHM diameters are 243 nm (confocal) and 62 nm (STED), respectively. In ROI2, the intensity profile obtained from the confocal image was best‐fitted with a single Gaussian function for which the FWHM diameter is 384 nm, whereas that from the STED image was best‐fitted with a sum of two Lorentzian functions for which the FWHM diameters are 108 and 85 nm with an interpeak spacing of 173 nm.
Figure 4. Brief insulin stimulation enlarges GLUT4‐containing structures in myofibres.
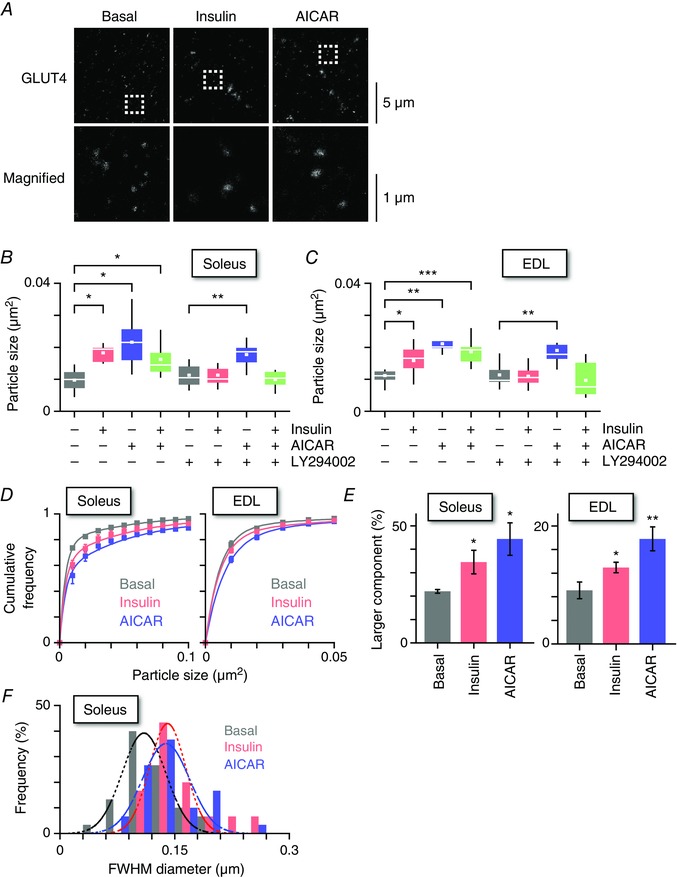
A, STED images of endogenous GLUT4 in soleus myofibres of wild‐type mice (upper) and magnified images of boxed regions (lower) in the basal state (left) and after 5 min of insulin (middle, 100 nm) or AICAR (right, 1 mm) stimulation. Box plots of the particle sizes are shown under the indicated conditions in soleus (B) and EDL (C) myofibres. Some myofibres were pretreated with LY294002 (50 μm) for 30 min before stimulation with insulin (100 nm) or AICAR (1 mm) for 5 min. The white line and the square represent median and mean values, respectively. * P < 0.05, * * P < 0.01, * * * P < 0.01 (n = 7–11). D, distributions of GLUT4‐containing structures in the basal state (grey) and after 5 min of insulin (red, 100 nm) or AICAR (blue, 1 mm) stimulations in soleus (left) and EDL (right) muscles. Smooth lines are double‐exponential curves that are characterized by smaller (0.441 × 10–2 μm2) and larger (5.43 × 10–2 μm2) components. E, fractions of the larger component of GLUT4‐containing structures in the basal state (grey) and after 5 min of insulin (red, 100 nm) or AICAR (blue, 1 mm) stimulations in soleus (left) and EDL (right) muscles. * P < 0.05, * * P < 0.01 (n = 4–9). F, FWHM diameters of GLUT4‐containing vesicles in soleus myofibres in the basal state (black) and after 5 min of insulin (red, 100 nm) or AICAR (blue, 1 mm) stimulation. Dashed lines represent Gaussian fitting of the histograms for each treatment condition. Median diameters in the basal, insulin and AICAR‐stimulated conditions were 0.115, 0.138 and 0.133 μm, respectively.
Importantly, double‐immunofluorescence staining analysis using structured illumination super‐resolution microscopy revealed that 5 min of insulin stimulation resulted in statistically significant increases in co‐localization of GLUT4 with TfR (Fig. 5 A and B) in both soleus and EDL muscles (Fig. 5 C). Of note, no significant increase in co‐localization of GLUT4 with a T‐tubule protein, BIN1, was detected after 5 min of insulin stimulation, whereas prolonged insulin stimulation (30 min) did increase their co‐localization (Fig. 6 A and B).
Figure 5. Brief insulin stimulation increases co‐localization of GLUT4 with TfR in myofibres.
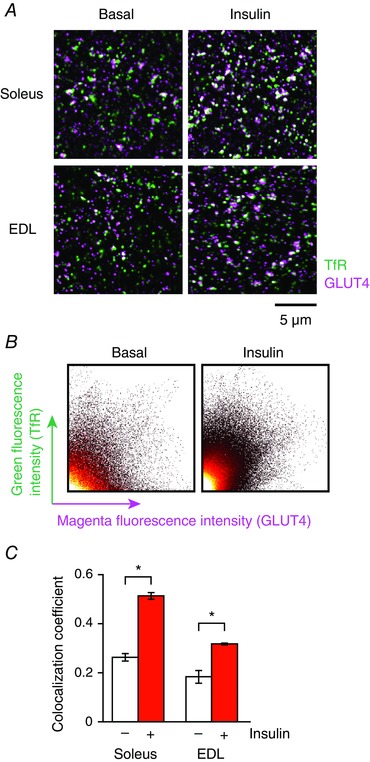
A, immunofluorescence images of TfR (green) and GLUT4 (magenta) before (left) and after 5 min of insulin stimulation (right, 100 nm) in isolated soleus (upper) and EDL (lower) myofibres. B, scatter plot analysis of GLUT4‐TfR co‐localization in soleus muscle shown in (A). C, changes in co‐localization coefficient of GLUT4 with TfR after 5 min of insulin stimulation (100 nm). * P < 0.05 (n = 5).
Figure 6. Immunofluorescence of endogenous GLUT4 and T‐tubule protein BIN1.
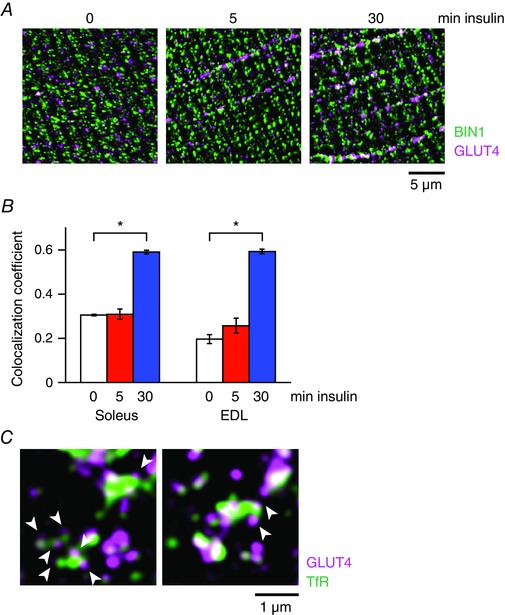
A, immunofluorescence images of a T‐tubule protein BIN1 (green) and GLUT4 (magenta) in soleus myofibres of wild‐type mice before (left) and after 5 min (middle) and 30 min (right) of insulin stimulation (100 nm). B, changes in the colocalization coefficient between BIN1 and GLUT4 after insulin stimulation (100 nm) in soleus and EDL muscles. * P < 0.001 (n = 5). C, immunofluorescence images of TfR (green) and GLUT4 (magenta) in soleus myofibres after 5 min of insulin stimulation (100 nm). Arrowheads indicate small GLUT4‐containing vesicles surrounding a large TfR‐containing endosome.
Insulin stimulates heterotypic fusion of GLUT4‐containing vesicles and the TfR‐containing compartment
GLUT4 behaviour observations in skeletal myofibres showed that insulin promptly induces (i) increases in the speed of GLUT4 movement; (ii) enlargement of GLUT4‐containing structures; and (iii) increases in co‐localization with TfR. These findings suggest that, initially, insulin stimulates endomembrane fusion of several GLUT4‐containing vesicles with other types of endosomes, including those of the TfR‐containing compartment. As noted, ∼10–40 of the small vesicles are required for enlargement of GLUT4‐containing structures to occur solely via homotypic fusion. More probably, a few small GLUT4‐containing vesicles heterotypically fuse with a large TfR‐containing endosome. Immunofluorescence analysis clearly demonstrated that TfR‐containing endosomes were, in some cases, surrounded by a few small GLUT4‐containing structures (Fig. 6 C). To test this hypothesis directly, we attempted to visualize such fusion events using a fluorometric assay, which is based on enhancement of the fluorescence of BODIPY‐conjugated streptavidin (BODIPY/streptavidin) upon biotin binding (Fig. 7 A) (Emans et al. 1995). As expected, BODIPY or BODIPY/streptavidin fluorescence was enhanced in the presence of biotin or biotinylated Tf, respectively (Fig. 7 B and C). Therefore, if such fusion events occur, enhancement of BODIPY fluorescence will be observed (Fig. 7 D).
Figure 7. Brief insulin stimulation induces fusion of GLUT4‐containing vesicles with TfR‐containing vesicles in myofibres.
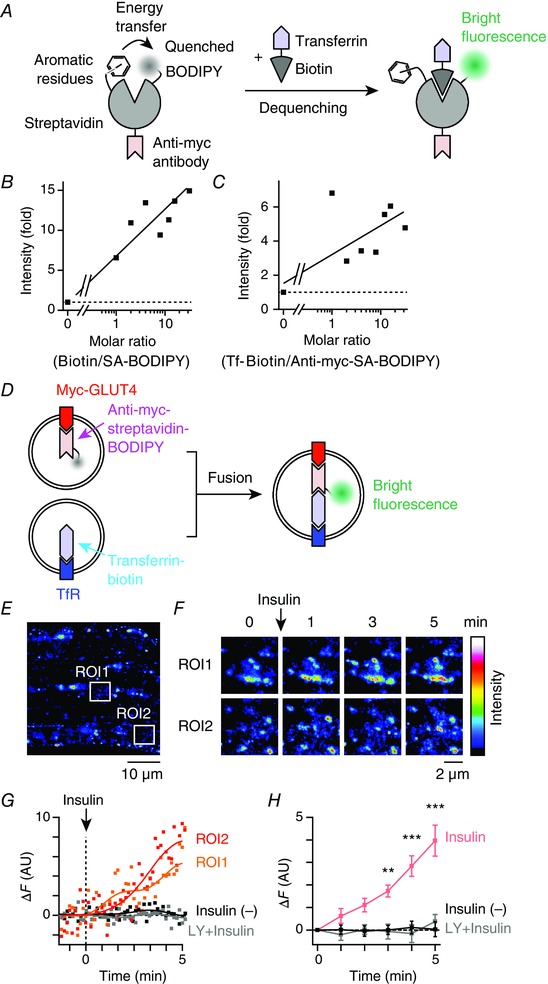
A, schema for the proposed mechanism of BODIPY fluorescence dequenching by biotin binding to streptavidin. In the absence of biotin binding, BODIPY fluorescence is quenched as a result of aromatic residues of streptavidin. B, enhancement of streptavidin‐BODIPY fluorescence in the presence of biotin in vitro. Continuous line represents linear regression of the data. C, enhancement of anti‐myc‐streptavidin‐BODIPY fluorescence in the presence of transferrin‐biotin in vitro. Continuous line represents linear regression of the data. D, schema for the fusion experiments. Prior to stimulation, myc‐GLUT4‐EGFP and TfR were labelled with anti‐myc‐streptavidin/BODIPY and transferrin‐biotin, respectively. After myc‐GLUT4‐EGFP‐containing vesicles fuse with TfR‐containing vesicles, biotin‐streptavidin binding dequenches BODIPY fluorescence, such that increases in green fluorescence can be observed. E, fluorescence image of myc‐GLUT4‐EGFP + anti‐myc‐streptavidin/BODIPY in EDL myofibres of myc‐GLUT4‐EGFP transgenic mouse. F, changes in green fluorescence of boxed regions in (E). Images are the maximum intensity projections of five successive frames. G, time courses of changes in green fluorescence (ΔF) of boxed regions in (E). Myofibres were stimulated with insulin (100 nm) at time 0. For comparison, ΔF is shown in myofibres pretreated with LY294002 (50 μm) for 30 min (grey), as well as in non‐stimulated myofibres (black). Continuous lines represent fast Fourier transform filter smoothing curves. H, mean changes in green fluorescence in the insulin‐stimulated myofibres (red), those pretreated with LY294002 (50 μm) for 30 min (grey) and non‐stimulated myofibres (black). * * P < 0.01, * * * P < 0.001 vs. before stimulation (t = 0, n = 4).
The myc‐GLUT4 molecules in isolated EDL myofibres obtained from the transgenic mice were labelled in advance with BODIPY/streptavidin‐conjugated anti‐myc antibodies. The labelled myc‐GLUT4 was then allowed to recycle back to its stationary compartments. Next, endogenous TfR was labelled with biotinylated Tf, and insulin‐induced changes in fluorescence intensity reflecting their fusion activities were monitored (Fig. 7 E–G). The fluorescence spectrum of BODIPY is similar to that of EGFP. We thus observed merged fluorescence of these two compounds in this set of experiments. Insulin stimulation elicited a gradual increase in fluorescence intensity within several myofibre regions, presumably resulting from facilitated endosomal fusion between BODIPY/streptavidin‐labelled GLUT4‐containing vesicles and a biotinylated‐Tf‐conjugated TfR‐containing compartment. This enhancement was statistically significant after 3 min of insulin stimulation (Fig. 7 H). Importantly, such enhancement was completely abolished by treatment with the phosphoinositide 3‐kinase inhibitor LY294002 and was not observed in the control cells without insulin stimulation (Fig. 7 G and H). These results provide compelling evidence that insulin can rapidly trigger heterotypic endosomal fusion, at least between GLUT4‐containing vesicles and TfR‐containing endosomes in skeletal myofibres. Of note, this approach can specifically detect initial fusion of these two distinct vesicle types. However, analysing post‐fusion events is not possible because the complex formed between myc‐GLUT4 and TfR undergoes strong biotin‐streptavidin binding and thus cannot be dissociated.
Exercise‐mimetic stimuli enhance insulin sensitivity in skeletal muscle
Exercise is well‐known to enhance insulin‐responsive GLUT4 translocation and glucose transport (Richter et al. 1982; Cartee et al. 1989). To determine whether exercise‐mimetic stimuli delivered to skeletal myofibres can modulate insulin susceptibility, we performed STED microscopy‐based morphometric analysis, which is an accurate quantitative insulin dose‐responsive analytical method designed especially for broad areas of myofibres. Myofibres were first stimulated with or without AICAR for 30 min, washed, allowed to recover for 1 h, and then stimulated with various insulin concentrations for 5 min (Fig. 8 A). After being washed for 1 h, the sizes of GLUT4‐containing structures showed recovery to those in the basal control state (Fig. 8 B and C), although slight phosphorylation of AMPK and its substrate (Ser237 of Tbc1d1) remained (Fig. 8 D and E). We found that, with AICAR pretreatment, insulin‐induced enlargement of GLUT4‐containing structures was more prominent even at lower concentrations of insulin in both soleus and EDL muscles, and that these actions were abolished in the presence of LY294002 (Fig. 8 B and C). Insulin sensitivity was observed to be more enhanced in soleus than in EDL muscles at lower concentrations of insulin. Although it cannot be completely ruled out that remaining phosphorylation of AMP kinase‐related signals might be involved in the susceptibility to insulin, these observations demonstrated that AICAR‐pretreatment was capable of inducing enhanced insulin‐responsiveness as assessed by enlargement of GLUT4‐containing structures. It is noteworthy that proximal insulin‐related signalling events such as phosphorylation of Akt and its substrate (Thr642 of AS160) did not differ significantly between the presence and absence of AICAR pretreatment (Fig. 8 D).
Figure 8. Sequential stimulations with insulin after AICAR treatment enhanced insulin sensitivity in myofibres.
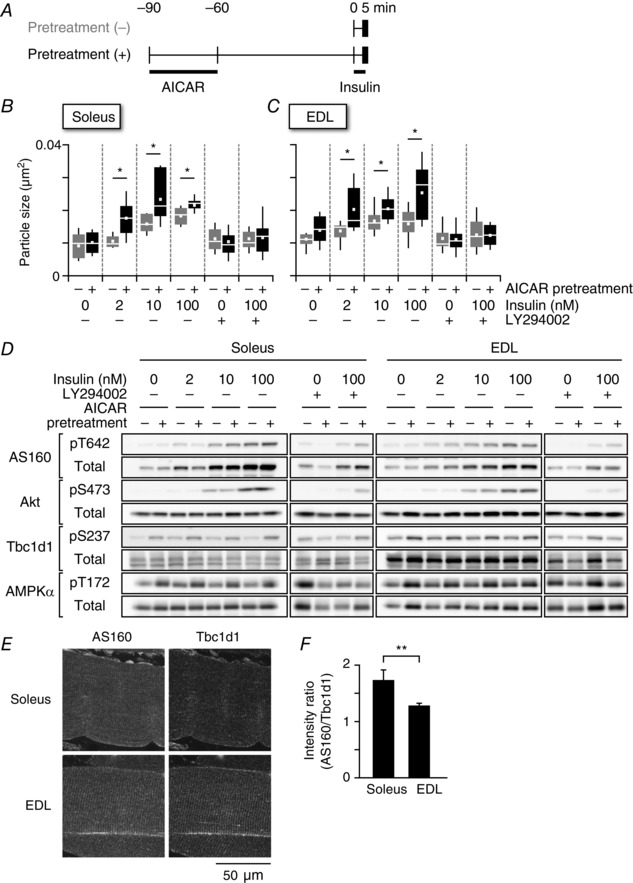
A, treatment protocols for analysing the effects of AICAR pretreatment (1 mm) on the sizes of GLUT4‐containing structures. Box plots of the particle sizes of endogenous GLUT4‐containing vesicles are shown under the indicated conditions in soleus (B) or EDL (C) myofibres. Some myofibres were pretreated with LY294002 (50 μm) prior to the 30 min of insulin stimulation (0–100 nm) for 5 min. The white line and square represent median and mean values, respectively. * P < 0.05 (n = 7–11). D, western blotting analyses of protein phosphorylation and expression under the indicated conditions. E, immunofluorescence images of AS160 (left, green on the right) and Tbc1d1 (middle, magenta on the right) in soleus (upper) and EDL (lower) muscles. F, fluorescence intensity ratios of AS160 to Tbc1d1 in soleus and EDL myofibres. * P < 0.05 (n = 5).
We previously found that Tbc1d1 can mediate beneficial effects of exercise on muscle insulin potency (Hatakeyama & Kanzaki, 2013b). Furthermore, we recently revealed that the co‐operative actions of two RabGAPs (AS160 and Tbc1d1) modulate the insulin sensitivity of GLUT4 trafficking activity (Hatakeyama H & Kanzaki M, manuscript in preparation) based on detailed analyses using a cell‐based reconstitution model (Hatakeyama & Kanzaki, 2011). Consistent with previous studies employing western blotting (Taylor et al. 2008), the relative ratios of AS160 to Tbc1d1 were higher in soleus than in EDL muscles, based on the fluorescence intensities of AS160 and Tbc1d1 (Fig. 8 E and F), suggesting that insulin susceptibility is modulated by the expression ratio of these RabGAPs.
Discussion
The findings of the present study demonstrate that GLUT4 molecules segregated within very small vesicles residing in the specialized stationary compartment and were efficiently liberated upon (and/or just beforehand) stimulus‐facilitated endosomal fusion with a subset of travelling endosomes containing TfR in skeletal myofibres (Fig. 9). These initial activities involving endomembranous regulation are potentiated by combinatorial stimuli such as insulin plus exercise‐related signals in myofibres. Thus, our comprehensive imaging analyses employing skeletal myofibres, and focusing on insulin‐elicited initial GLUT4 behaviour, strongly supports the novel concept that GLUT4 ‘protein trafficking’ via heterotypic fusion is a feature of the stimuli‐dependent initial trigger for GLUT4 translocation, at least in skeletal muscles. Whether or not adipocytes possess a similar capacity to initiate GLUT4 translocation remains unknown. However, the findings of the present study provide novel insights into GLUT4 translocation, which has been intensively investigated under the canonical concept based on ‘GLUT4‐vesicle trafficking’, in that most GLUT4‐vesicles are destined to directly reach their final destinations (sarcolemma and T‐tubules). Although our data do not allow the relative contributions of ‘protein’ and ‘vesicle’ trafficking to the initial round of GLUT4 translocation as a whole to be determined, morphometry‐based estimations suggest that 12% of very small GLUT4‐containing vesicles can immediately (∼5 min) undergo heterotypic fusion in soleus myofibres (Fig. 4 E). Although this fraction appears to be small, it is important to note that this value was derived from a ‘snapshot’ of super‐resolution images at specific time points and that much greater enhancements of GLUT4 kinetics, as well as of co‐localization/fusion indices with TfR, were actually observed. Because the remaining small GLUT4 vesicles may still have the ability to fuse with larger compartments and GLUT4 molecules are highly condensed into these specialized small vesicles, much larger numbers of total GLUT4 molecules might be translocated via heterotypic endosomal fusion events during the initial round of stimulation in living myofibres. Of note, the insulin‐responsive actions exerted on small GLUT4‐containing vesicles are enhanced after exercise‐mimetic stimuli (see below).
Figure 9. Schematic depiction of heterotypic endosomal fusion induced by insulin in skeletal myofibres.
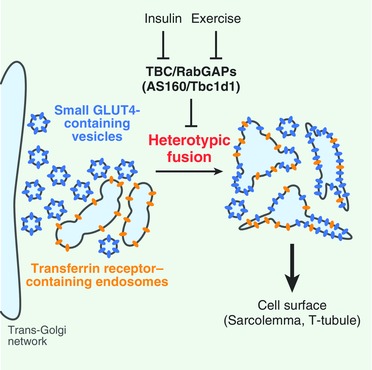
Comprehensive imaging analyses of GLUT4 behaviour in skeletal muscle revealed that insulin triggers acute heterotypic endomembrane fusion of GLUT4‐containing vesicles with a subset of TfR‐containing endosomes. The insulin‐responsive actions exerted on small GLUT4‐containing vesicles are enhanced after exercise‐mimetic stimuli, possibly regulated by functional co‐operation between AS160 and Tbc1d1. The findings provide a conceptual framework indicating that GLUT4 ‘protein’ trafficking via heterotypic fusion is a critical feature of GLUT4 translocation in skeletal muscles and also suggest that the insulin‐responsive efficacy of the endomembranous fusion process is involved in the benefits of exercise.
Although the molecular mechanisms underlying heterotypic fusion are unknown, an array of fusion and tethering machineries functioning at various stages in regulating the endomembrane system, probably governed by AS160/Tbc1d1‐directed regulation of Rab GTPases, must be involved in these processes. Generally, the specificity and efficiency of the endomembrane system are determined by co‐ordinated actions of SNARE proteins, tethering factors and Rab GTPases (Cai et al. 2007). Several SNARE proteins, including VAMP3 (Riggs et al. 2012); VAMP4, syntaxin 6, syntaxin 13 and Vti1a (Brandhorst et al. 2006); and VAMP8 (Behrendorff et al. 2011) reportedly function in endosomal fusion processes and some have also been implicated in the intracellular GLUT4 trafficking (Williams & Pessin, 2008), as well as GLUT4 translocation (Randhawa et al. 2004; Zhao et al. 2009). Regarding tethering factors, one of the coiled‐coil type golgin tethering factors localized in the trans‐Golgi network (TGN), golgin‐97, is required for sortilin‐mediated static GLUT4 behaviour in concert with SNARE proteins such as syntaxin 6 and Vti1a (Hatakeyama & Kanzaki, 2011). Other recently identified tethering complexes, such as GARP (Bonifacino & Hierro, 2011), EARP (Schindler et al. 2015) and TUG protein (Bogan et al. 2003), may be involved in the initial insulin action. In any case, AS160 and Tbc1d1 exhibit potent RabGAP effects on multiple Rab GTPases, including Rab 8, 10 and 14 (Miinea et al. 2005; Roach et al. 2007), which are all crucially involved in several endomembrane trafficking events, including tethering, fusion and sorting, particularly among the TGN, endosomes and plasma membrane. Thus, once these Rab GTPases are activated in co‐ordination, upon catalytic release from RabGAP‐mediated suppression, a variety of their downstream effectors co‐operatively control not only the canonical fusion event at the plasma membrane (Chen et al. 2012), but also (as revealed in the present study) the initial heterotypic endosomal fusion process.
GLUT4 dynamics in skeletal myofibres
Early immunoelectron and immunofluorescence microscopy studies provided spatial information regarding GLUT4 localization. GLUT4 molecules are present in several membranous compartments, including the TGN, endosomes and T‐tubules, and insulin and contraction both induced the redistribution of the subcellular localization of GLUT4 (Rodnick et al. 1992; Ploug et al. 1998). Ploug et al. (1998) found ∼77% of GLUT4 to be present in small tubulovesicular structures, which can be further divided into TfR‐positive (∼52%) and TfR‐negative elements, whereas the remainder was found in larger structures that were partially co‐localized with a TGN protein, TGN38. It was proposed that the TfR‐negative elements are somewhat smaller than the TfR‐positive ones and the smaller TfR‐negative elements were not associated with the Golgi complex based on co‐localization with TGN38. However, skeletal muscle has a non‐conventional Golgi complex, comprised of a network of smaller elements (Ralston et al. 1999), and the TGN has multiple biochemically and functionally distinct subdomains (Gleeson et al. 2004). Therefore, we can reasonably speculate that the small TfR‐negative elements at least partially comprise subdomains derived from the TGN. Specific TGN subdomains such as golgin‐97‐localizing regions (Hatakeyama & Kanzaki, 2011) or those enriched in syntaxin 6 and syntaxin 16 but not TGN38 (Shewan et al. 2003) are critical intermediate transits for GLUT4 trafficking. Co‐localization analysis of these proteins might thus be an optimal approach with respect to analysing their structures.
Consistent with our observations, Ploug et al. (1998) also demonstrated that insulin stimulation induced decreases in the TfR‐negative smaller elements and concomitant increases in the TfR‐positive larger structures, although their observations were made after prolonged insulin stimulation (20 min). Extensive efforts have been made to determine how GLUT4 cycles between intracellular compartments and the plasma membrane, although most such studies employed 3T3L1 adipocytes because these cells are easily handled experimentally. Although the densely‐packed GLUT4 in perinuclear regions of mononucleate adipocytes can, on occasion, hinder efforts to analyse GLUT4 trafficking in detail, an early electron microscopic study indicated that insulin exerts marked effects on endosome morphology in adipose tissues (Slot et al. 1991). Furthermore, a biochemical fractionation study also demonstrated that insulin stimulation (15 min) altered the buoyant density (from high to low) of GLUT4‐containing vesicles of adipocytes, as well as those of skeletal muscles obtained from rats (Kandror et al. 1995). Thus, further analyses based on the emerging concept that insulin‐evoked acute endomembrane fusion activities are a key feature for triggering GLUT4 translocation would clearly be worthwhile.
Recent studies have employed intravital imaging to make major contributions to revealing the temporal regulation of GLUT4 trafficking in skeletal (quadriceps) muscles expressing GLUT4‐EGFP (Lauritzen et al. 2006), demonstrating that both large (> 1 μm) and small (< 1 μm) GLUT4‐containing structures tend to be stationary in the basal state. However, it is important to note that the GLUT4‐containing structures observed comprised numerous GLUT4 molecules, with some even smaller than 1 μm in diameter, such that whether individual GLUT4 molecules were stationary in the basal state could be not determined. By contrast, our single molecule nanometry can directly address the movements of individual GLUT4 molecules with high positional precision. We thereby succeeded in demonstrating individual GLUT4 behaviours and consequently their total dynamics in detail. Our current instrument cannot simultaneously visualize individual GLUT4 with QD together with the structures of GLUT4‐containing elements, such that improvements enabling dual‐colour imaging (Hatakeyama & Kanzaki, 2013a) are necessary for further clarification of movement–structure relationships. Furthermore, although our observations were consistently made 1–4 μm above the glass surface where the majority of GLUT4‐containing vesicles localize (Ploug et al. 1998), imaging or analytical approaches allowing evaluation of three‐dimensional movements (Ram et al. 2008; Gardini et al. 2015) are required for future studies.
In the present study, the stimulatory effects of insulin and AICAR on the movement and sizes of GLUT4‐containing structures were similarly observed in both soleus and EDL muscles (Figs 1 and 3), whereas AICAR effects on glucose uptake are reported to be less pronounced in soleus than in EDL muscles (Lai et al. 2014; Jensen et al. 2015). One possible reason for this discrepancy might reflect the processes observed and the time scale analysed (i.e. initial events occurring within several minutes vs. overall glucose uptake after several tens of minutes, or more, of stimulation). Multiple composite mechanisms would presumably be involved in the overall glucose uptake, especially after prolonged stimulation, and the actions of insulin and AICAR could differ among such processes between soleus and EDL muscles. Similarly, such a mechanism could also explain why no significant additive effects of insulin and AICAR were observed, as obviously are seen in glucose uptake (Hayashi et al. 1998).
A potential site of action involved in the benefits of exercise: stimulus‐evoked endomembranous activities arising from small GLUT4‐containing vesicles
One important aspect of skeletal muscle glucose metabolism is the effects of exercise (i.e. a single exercise session can lead to enhanced insulin‐responsive glucose transport) (Richter et al. 1982; Cartee et al. 1989). Although these processes are reportedly mediated by increased GLUT4 translocation without enhanced proximal insulin signalling (e.g. Akt activity) (Hansen et al. 1998; Wojtaszewski et al. 1999; Maarbjerg et al. 2011), several studies have suggested that AS160 phosphorylation mediates in vivo effects of exercise (i.e. exercise‐induced phosphorylation of AS160 remained elevated several hours after the exercise concomitant with insulin‐stimulated glucose uptake, whereas such sustained phosphorylation was not detected in Tbc1d1) (Funai et al. 2009; Treebak et al. 2009; Pehmoller et al. 2012).
Although ex vivo contraction also enhances insulin‐stimulated glucose uptake, especially in the presence of serum (Cartee and Holloszy, 1990) via mechanisms apparently distinct from sustained phosphorylation (Funai et al. 2010), we have demonstrated the enhancement of insulin sensitivity after exercise‐mimetic stimuli in the absence of serum. One reason for this discrepancy might involve differences in the observed processes between the studies, in that we focused specifically on the initial processes, such as GLUT4 movement or the sizes of GLUT4‐containing structures, whereas other analyses employed overall glucose uptake assays. Thus, we can reasonably speculate that the main actions of humoral factors in serum are mediated by processes other than those that we observed. Another possibility is the concentration of insulin, in that we stimulated myofibres or cells with super‐physiological insulin (> 1 nm) concentrations, whereas post‐exercise enhancement of insulin‐stimulated glucose uptake is observable at lower concentrations (Richter et al. 1982). This might also explain the discrepancy between our observations and recent results obtained with isolated muscle indicating that the enhancing effects of previous AICAR stimulation on low‐concentration insulin‐dependent glucose uptake in the presence of serum were more pronounced in EDL than in soleus muscles (Kjobsted et al. 2015).
We also found that soleus muscle demonstrated more insulin‐sensitive responses and had a higher ratio of AS160 to Tbc1d1 than EDL muscle, which is consistent with previous observations of differences in insulin‐induced glucose uptake between these two muscles (James et al. 1985; Henriksen et al. 1990), as well as the western blotting results for AS160 and Tbc1d1 expression (Taylor et al. 2008). Given that obesity‐related mutants of Tbc1d1 within the phosphotyrosine binding domain (R125W) showed decreased insulin‐dependent glucose uptake without significant phosphorylation changes in skeletal muscle (An et al. 2010), additional mechanisms that presumably depend on the functional phosphotyrosine binding domain must be involved in these processes. Indeed, our QD‐based single molecule nanometry demonstrated that Tbc1d1 has characteristic intrinsic properties such as the capacity to acquire temporal insulin responsiveness after exercise‐mimetic stimuli (Hatakeyama & Kanzaki, 2013b). Thus, functional co‐operation between these two RabGAPs modulating the initial GLUT4 behaviour is apparently a key factor in the benefits of exercise. We identified localization and solubility changes of AS160 in response to insulin, especially in the soleus muscle (Fig. 2), and it is possible that such a shift in localization might modulate functional co‐operation between these two RabGAPs. Future studies focusing on these issues are needed to reveal the molecular mechanism underlying the benefits of exercise.
In summary, we have clearly demonstrated the initial effects of insulin on GLUT4 behaviour in skeletal myofibres. Stimulus‐dependent redistribution of membrane proteins is an essential aspect of cellular functions, and GLUT4 is one of the best studied proteins among the various molecules undergoing such processes. To date, we have provided clear evidence of the critical steps in GLUT4 trafficking systems, including their stationary behavioural nature, as well as stimulus‐responsive liberation and fusion with endomembrane systems presumably mediated by the unlocking of RabGAP‐catalysed inhibitions of certain trafficking activities. Similar regulatory mechanisms may play roles in other stimulus‐dependent trafficking in various cell types. Further clarification of these processes is anticipated with respect to providing insights for our understanding of not only GLUT4 behaviour and type 2 diabetes, but also exercise physiology and intracellular trafficking in general.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
MK and HH conceived and designed the experiments. HH performed the imaging experiments and analysed the data. MK and HH wrote the manuscript and approved the final version of the manuscript.
Funding
This work was supported in part by grants from the Japan Society for the Promotion of Science (nos. 26670099 to MK and 25713010 to HH), as well as the Program for Fostering Researchers for the Next Generation in the Project for Establishing a Consortium for the Development of Human Resources in Science and Technology (to HH), the Banyu Life Science Foundation International (to HH) and the Takeda Science Foundation (to MK).
Acknowledgements
We thank Fumie Wagatsuma and Natsumi Emoto for technical assistance. We thank Drs Edna Hardeman (University of New South Wales) and Hiroshi Shibata (Gunma University) for providing the human α‐actin promoter plasmid and the anti‐GLUT4 antibody, respectively.
References
- An D, Toyoda T, Taylor EB, Yu H, Fujii N, Hirshman MF & Goodyear LJ (2010). TBC1D1 regulates insulin‐ and contraction‐induced glucose transport in mouse skeletal muscle. Diabetes 59, 1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrendorff N, Dolai S, Hong W, Gaisano HY & Thorn P (2011). Vesicle‐associated membrane protein 8 (VAMP8) is a SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) selectively required for sequential granule‐to‐granule fusion. J Biol Chem 286, 29627–29634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan JS, Hendon N, McKee AE, Tsao TS & Lodish HF (2003). Functional cloning of TUG as a regulator of GLUT4 glucose transporter trafficking. Nature 425, 727–733. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS & Hierro A (2011). Transport according to GARP: receiving retrograde cargo at the trans‐Golgi network. Trends Cell Biol 21, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandhorst D, Zwilling D, Rizzoli SO, Lippert U, Lang T & Jahn R (2006). Homotypic fusion of early endosomes: SNAREs do not determine fusion specificity. Proc Natl Acad Sci U S A 103, 2701–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan KJ & Hardeman EC (1993). Quantitative analysis of the human alpha‐skeletal actin gene in transgenic mice. J Biol Chem 268, 719–725. [PubMed] [Google Scholar]
- Cai H, Reinisch K & Ferro‐Novick S (2007). Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell 12, 671–682. [DOI] [PubMed] [Google Scholar]
- Cartee GD, Young DA, Sleeper MD, Zierath J, Wallberg‐Henriksson H & Holloszy JO (1989). Prolonged increase in insulin‐stimulated glucose transport in muscle after exercise. Am J Physiol Endocrinol Metab 256, E494–E499. [DOI] [PubMed] [Google Scholar]
- Chadt A, Immisch A, de Wendt C, Springer C, Zhou Z, Stermann T, Holman GD, Loffing‐Cueni D, Loffing J, Joost HG & Al‐Hasani H (2015). Deletion of both Rab‐GTPase‐activating proteins TBC1D1 and TBC1D4 in mice eliminates insulin‐ and AICAR‐stimulated glucose transport [corrected]. Diabetes 64, 746–759. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang Y, Zhang J, Deng Y, Jiang L, Song E, Wu XS, Hammer JA, Xu T & Lippincott‐Schwartz J (2012). Rab10 and myosin‐Va mediate insulin‐stimulated GLUT4 storage vesicle translocation in adipocytes. J Cell Biol 198, 545–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J & Felber JP (1981). The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30, 1000–1007. [DOI] [PubMed] [Google Scholar]
- Emans N, Biwersi J & Verkman AS (1995). Imaging of endosome fusion in BHK fibroblasts based on a novel fluorimetric avidin‐biotin binding assay. Biophys J 69, 716–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Hatakeyama H, Watanabe TM, Sato M, Higuchi H & Kanzaki M (2010). Identification of three distinct functional sites of insulin‐mediated GLUT4 trafficking in adipocytes using quantitative single molecule imaging. Mol Biol Cell 21, 2721–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funai K, Schweitzer GG, Castorena CM, Kanzaki M & Cartee GD (2010). In vivo exercise followed by in vitro contraction additively elevates subsequent insulin‐stimulated glucose transport by rat skeletal muscle. Am J Physiol Endocrinol Metab 298, E999–E1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funai K, Schweitzer GG, Sharma N, Kanzaki M & Cartee GD (2009). Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased postexercise insulin sensitivity in rat skeletal muscle. Am J Physiol Endocrinol Metab 297, E242–E251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini L, Capitanio M & Pavone FS (2015). 3D tracking of single nanoparticles and quantum dots in living cells by out‐of‐focus imaging with diffraction pattern recognition. Sci Rep 5, 16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson PA, Lock JG, Luke MR & Stow JL (2004). Domains of the TGN: coats, tethers and G proteins. Traffic 5, 315–326. [DOI] [PubMed] [Google Scholar]
- Goodyear LJ & Kahn BB (1998). Exercise, glucose transport, and insulin sensitivity. Annu Rev Med 49, 235–261. [DOI] [PubMed] [Google Scholar]
- Hansen PA, Nolte LA, Chen MM & Holloszy JO (1998). Increased GLUT‐4 translocation mediates enhanced insulin sensitivity of muscle glucose transport after exercise. J Appl Physiol (1985) 85, 1218–1222. [DOI] [PubMed] [Google Scholar]
- Hatakeyama H & Kanzaki M (2011). Molecular basis of insulin‐responsive GLUT4 trafficking systems revealed by single molecule imaging. Traffic 12, 1805–1820. [DOI] [PubMed] [Google Scholar]
- Hatakeyama H & Kanzaki M (2013a). Development of dual‐color simultaneous single molecule imaging system for analyzing multiple intracellular trafficking activities. Conf Proc IEEE Eng Med Biol Soc 2013, 1418–1421. [DOI] [PubMed] [Google Scholar]
- Hatakeyama H & Kanzaki M (2013b). Regulatory mode shift of Tbc1d1 is required for acquisition of insulin‐responsive GLUT4‐trafficking activity. Mol Biol Cell 24, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Kurth EJ, Winder WW & Goodyear LJ (1998). Evidence for 5′ AMP‐activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 47, 1369–1373. [DOI] [PubMed] [Google Scholar]
- Henriksen EJ, Bourey RE, Rodnick KJ, Koranyi L, Permutt MA & Holloszy JO (1990). Glucose transporter protein content and glucose transport capacity in rat skeletal muscles. Am J Physiol Endocrinol Metab 259, E593–E598. [DOI] [PubMed] [Google Scholar]
- James DE, Kraegen EW & Chisholm DJ (1985). Muscle glucose metabolism in exercising rats: comparison with insulin stimulation. Am J Physiol Endocrinol Metab 248, E575–E580. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Ross FA, Kleinert M, Sylow L, Knudsen JR, Gowans GJ, Hardie DG & Richter EA (2015). PT‐1 selectively activates AMPK‐gamma1 complexes in mouse skeletal muscle, but activates all three gamma subunit complexes in cultured human cells by inhibiting the respiratory chain. Biochem J 467, 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandror KV, Coderre L, Pushkin AV & Pilch PF (1995). Comparison of glucose‐transporter‐containing vesicles from rat fat and muscle tissues: evidence for a unique endosomal compartment. Biochem J 307, 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki M (2006). Insulin receptor signals regulating GLUT4 translocation and actin dynamics. Endocr J 53, 267–293. [DOI] [PubMed] [Google Scholar]
- Kjobsted R, Treebak JT, Fentz J, Lantier L, Viollet B, Birk JB, Schjerling P, Bjornholm M, Zierath JR & Wojtaszewski JF (2015). Prior AICAR stimulation increases insulin sensitivity in mouse skeletal muscle in an AMPK‐dependent manner. Diabetes 64, 2042–2055. [DOI] [PubMed] [Google Scholar]
- Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K, Hirshman MF & Goodyear LJ (2006). Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55, 2067–2076. [DOI] [PubMed] [Google Scholar]
- Lai YC, Kviklyte S, Vertommen D, Lantier L, Foretz M, Viollet B, Hallen S & Rider MH (2014). A small‐molecule benzimidazole derivative that potently activates AMPK to increase glucose transport in skeletal muscle: comparison with effects of contraction and other AMPK activators. Biochem J 460, 363–375. [DOI] [PubMed] [Google Scholar]
- Lauritzen HP, Ploug T, Prats C, Tavare JM & Galbo H (2006). Imaging of insulin signaling in skeletal muscle of living mice shows major role of T‐tubules. Diabetes 55, 1300–1306. [DOI] [PubMed] [Google Scholar]
- Maarbjerg SJ, Sylow L & Richter EA (2011). Current understanding of increased insulin sensitivity after exercise – emerging candidates. Acta Physiol (Oxf) 202, 323–335. [DOI] [PubMed] [Google Scholar]
- Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS & Lienhard GE (2005). AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase‐activating protein domain. Biochem J 391, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehmoller C, Brandt N, Birk JB, Hoeg LD, Sjoberg KA, Goodyear LJ, Kiens B, Richter EA & Wojtaszewski JF (2012). Exercise alleviates lipid‐induced insulin resistance in human skeletal muscle‐signaling interaction at the level of TBC1 domain family member 4. Diabetes 61, 2743–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploug T, van Deurs B, Ai H, Cushman SW & Ralston E (1998). Analysis of GLUT4 distribution in whole skeletal muscle fibers: identification of distinct storage compartments that are recruited by insulin and muscle contractions. J Cell Biol 142, 1429–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston E, Lu Z & Ploug T (1999). The organization of the Golgi complex and microtubules in skeletal muscle is fiber type‐dependent. J Neurosci 19, 10694–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram S, Prabhat P, Chao J, Ward ES & Ober RJ (2008). High accuracy 3D quantum dot tracking with multifocal plane microscopy for the study of fast intracellular dynamics in live cells. Biophys J 95, 6025–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa VK, Thong FS, Lim DY, Li D, Garg RR, Rudge R, Galli T, Rudich A & Klip A (2004). Insulin and hypertonicity recruit GLUT4 to the plasma membrane of muscle cells by using N‐ethylmaleimide‐sensitive factor‐dependent SNARE mechanisms but different v‐SNAREs: role of TI‐VAMP. Mol Biol Cell 15, 5565–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Garetto LP, Goodman MN & Ruderman NB (1982). Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. J Clin Invest 69, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs KA, Hasan N, Humphrey D, Raleigh C, Nevitt C, Corbin D & Hu C (2012). Regulation of integrin endocytic recycling and chemotactic cell migration by syntaxin 6 and VAMP3 interaction. J Cell Sci 125, 3827–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach WG, Chavez JA, Miinea CP & Lienhard GE (2007). Substrate specificity and effect on GLUT4 translocation of the Rab GTPase‐activating protein Tbc1d1. Biochem J 403, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnick KJ, Slot JW, Studelska DR, Hanpeter DE, Robinson LJ, Geuze HJ & James DE (1992). Immunocytochemical and biochemical studies of GLUT4 in rat skeletal muscle. J Biol Chem 267, 6278–6285. [PubMed] [Google Scholar]
- Saltiel AR & Kahn CR (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806. [DOI] [PubMed] [Google Scholar]
- Schindler C, Chen Y, Pu J, Guo X & Bonifacino JS (2015). EARP is a multisubunit tethering complex involved in endocytic recycling. Nat Cell Biol 17, 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Rueden CT, Hiner MC & Eliceiri KW (2012). Fiji: an open‐source platform for biological‐image analysis. Nature methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewan AM, van Dam EM, Martin S, Luen TB, Hong W, Bryant NJ & James DE (2003). GLUT4 recycles via a trans‐Golgi network (TGN) subdomain enriched in Syntaxins 6 and 16 but not TGN38: involvement of an acidic targeting motif. Mol Biol Cell 14, 973–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ, Gigengack S, Lienhard GE & James DE (1991). Immuno‐localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol 113, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EB, An D, Kramer HF, Yu H, Fujii NL, Roeckl KS, Bowles N, Hirshman MF, Xie J, Feener EP & Goodyear LJ (2008). Discovery of TBC1D1 as an insulin‐, AICAR‐, and contraction‐stimulated signaling nexus in mouse skeletal muscle. J Biol Chem 283, 9787–9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treebak JT, Frosig C, Pehmoller C, Chen S, Maarbjerg SJ, Brandt N, MacKintosh C, Zierath JR, Hardie DG, Kiens B, Richter EA, Pilegaard H & Wojtaszewski JF (2009). Potential role of TBC1D4 in enhanced post‐exercise insulin action in human skeletal muscle. Diabetologia 52, 891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D & Pessin JE (2008). Mapping of R‐SNARE function at distinct intracellular GLUT4 trafficking steps in adipocytes. J Cell Biol 180, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JF, Higaki Y, Hirshman MF, Michael MD, Dufresne SD, Kahn CR & Goodyear LJ (1999). Exercise modulates postreceptor insulin signaling and glucose transport in muscle‐specific insulin receptor knockout mice. J Clin Invest 104, 1257–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Yang L, Lopez JA, Fan J, Burchfield JG, Bai L, Hong W, Xu T & James DE (2009). Variations in the requirement for v‐SNAREs in GLUT4 trafficking in adipocytes. J Cell Sci 122, 3472–3480. [DOI] [PubMed] [Google Scholar]
- Zheng X & Cartee GD (2016). Insulin‐induced Effects on the subcellular localization of AKT1, AKT2 and AS160 in rat skeletal muscle. Sci Rep 6, 39230. [DOI] [PMC free article] [PubMed] [Google Scholar]