

Abstract
Proteins of the conserved HORMA domain family, including the spindle assembly checkpoint protein MAD2 and the meiotic HORMADs, assemble into signaling complexes by binding short peptides termed “closure motifs”. The AAA+ ATPase TRIP13 regulates both MAD2 and meiotic HORMADs by disassembling these HORMA domain–closure motif complexes, but its mechanisms of substrate recognition and remodeling are unknown. Here, we combine X‐ray crystallography and crosslinking mass spectrometry to outline how TRIP13 recognizes MAD2 with the help of the adapter protein p31comet. We show that p31comet binding to the TRIP13 N‐terminal domain positions the disordered MAD2 N‐terminus for engagement by the TRIP13 “pore loops”, which then unfold MAD2 in the presence of ATP. N‐terminal truncation of MAD2 renders it refractory to TRIP13 action in vitro, and in cells causes spindle assembly checkpoint defects consistent with loss of TRIP13 function. Similar truncation of HORMAD1 in mouse spermatocytes compromises its TRIP13‐mediated removal from meiotic chromosomes, highlighting a conserved mechanism for recognition and disassembly of HORMA domain–closure motif complexes by TRIP13.
Keywords: AAA+ ATPase, HORMA domain, meiotic chromosome structure, spindle assembly checkpoint
Subject Categories: Cell Cycle, Structural Biology
Introduction
The HORMA domain is a structurally unique protein–protein interaction module with key functional roles in a number of eukaryotic signaling pathways. The HORMA name derives from the three protein families first shown to share this domain: Hop1, a meiotic chromosome‐associated protein; Rev7, a structural subunit of the trans‐lesion DNA polymerase ζ; and Mad2, a key player in the spindle assembly checkpoint (Aravind & Koonin, 1998; Rosenberg & Corbett, 2015). HORMA domains have since been identified in a second spindle assembly checkpoint protein, p31comet (Yang et al, 2007), and two autophagy‐signaling proteins, Atg13 (Jao et al, 2013) and Atg101 (Qi et al, 2015). The best‐understood HORMA domain protein, MAD2, can adopt two differently folded conformations. In its “closed” conformation, MAD2 wraps its C‐terminal “safety belt” region entirely around a bound peptide, called a MAD2‐interacting motif or closure motif; while in the “open” state, the MAD2 safety belt is folded back on the closure motif binding site, preventing partner binding (Fig 1A) (Luo et al, 2000, 2002; Sironi et al, 2002; Mapelli & Musacchio, 2007; Rosenberg & Corbett, 2015). Related HORMA domain proteins, including Hop1 and its orthologs (collectively termed meiotic HORMADs) and Rev7, have only been observed in the closed, closure motif‐bound conformation (Hara et al, 2010; Kikuchi et al, 2012; Kim et al, 2014). Nonetheless, the topology of these proteins’ complexes with closure motifs mean that they must at least transiently adopt an open or open‐like state in order to bind or dissociate from these motifs (Rosenberg & Corbett, 2015).
Figure 1. Structure of human TRIP13E253Q .
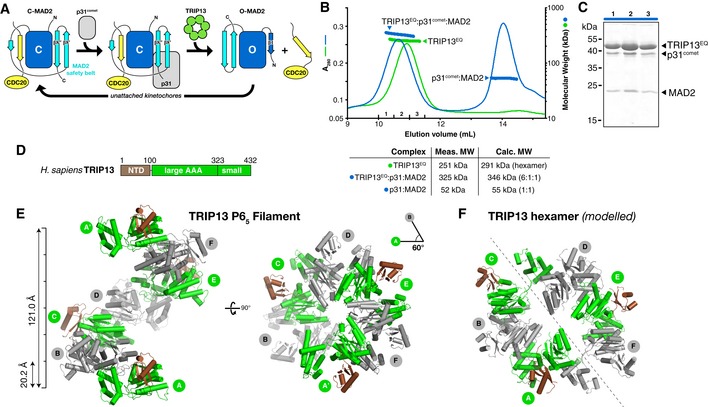
- Schematic of conformational changes in MAD2. The HORMA domain core is shown in dark blue with safety belt in cyan, and bound CDC20 closure motif in yellow. Closed MAD2 (C‐MAD2; unbound or CDC20‐bound) is recognized by p31comet, and this complex is recognized by TRIP13. TRIP13 then converts C‐MAD2 to the open state (O‐MAD2). The N‐terminal β1 strand stabilizes O‐MAD2 and becomes disordered in C‐MAD2.
- Size‐exclusion chromatography‐coupled multi‐angle light scattering (SEC‐MALS) analysis of TRIP13E253Q (green) and its complex with p31comet:MAD2 (blue), with measured and calculated molecular weights of each complex. Excess p31comet:MAD2 complex elutes at ˜14 ml.
- SDS–PAGE analysis of fractions from TRIP13E253Q: p31comet:MAD2 (fractions 1–3 noted in panel B).
- Domain structure of Homo sapiens TRIP13.
- Top and side views of the P65 filament formed in the crystals of TRIP13E253Q. See Fig EV1 for nucleotide binding and comparison with the Caenorhabditis elegans PCH‐2 structure.
MAD2 is a key player in the spindle assembly checkpoint (SAC), which monitors the status of chromosome–microtubule attachments in mitosis and meiosis, allowing chromosome segregation and anaphase onset only after all chromosomes are properly attached to spindle microtubules (reviewed in Primorac & Musacchio, 2013; Musacchio & Salmon, 2007; Musacchio, 2015; Lara‐Gonzalez et al, 2012). In the SAC, unattached kinetochores catalyze the assembly of a MAD2‐containing “mitotic checkpoint complex” (MCC), which delays anaphase onset by inhibiting the activity of the anaphase‐promoting complex/cyclosome (APC/C) (Hardwick et al, 2000; Fraschini et al, 2001; Sudakin et al, 2001). The key step of MCC assembly is the recruitment of open MAD2 (O‐MAD2) to unattached kinetochores, where it binds a kinetochore‐localized MAD1:MAD2 complex and is converted to closed MAD2 (C‐MAD2) (Howell et al, 2004; Shah et al, 2004; de Antoni et al, 2005; Mapelli & Musacchio, 2007). Conformational conversion of MAD2 occurs concomitantly with binding to a closure motif in CDC20, and the resulting MAD2‐CDC20 complex then binds BUBR1 to complete MCC assembly (Kulukian et al, 2009; Simonetta et al, 2009; Faesen et al, 2017).
While SAC activation and kinetochore‐mediated MCC assembly are mostly well understood, a key question has been whether and how the MCC, and particularly the MAD2‐CDC20 complex, is disassembled upon kinetochore attachment and SAC silencing. While one major mechanism for SAC inactivation is the ubiquitination and degradation of CDC20 in the MCC (reviewed in Primorac & Musacchio, 2013; Musacchio, 2015; Lara‐Gonzalez et al, 2012; Musacchio & Salmon, 2007), a second mechanism has recently been identified in which the MCC is directly disassembled through the joint action of a AAA+ ATPase, TRIP13, and the well‐known but functionally mysterious SAC antagonist p31comet. p31comet was first identified as a MAD2‐binding protein that negatively regulates the SAC (Habu et al, 2002), binding specifically to MAD2 in the closed conformation (Xia et al, 2004; Mapelli et al, 2006). Structural studies showed that p31comet is a HORMA domain protein, and forms a heterodimer with C‐MAD2 resembling the MAD2 dimer formed at unattached kinetochores (Yang et al, 2007). Subsequent work showed that p31comet binds C‐MAD2 in both soluble and APC/C‐bound MCC (Teichner et al, 2011; Westhorpe et al, 2011), and acts as an adapter to enable TRIP13 to convert this C‐MAD2 to the open conformation (Tipton et al, 2012; Wang et al, 2014; Ye et al, 2015), thereby dissociating it from CDC20 (Eytan et al, 2014; Miniowitz‐Shemtov et al, 2015). Later work showed that p31comet and TRIP13 are also involved in SAC activation, likely by maintaining a pool of soluble O‐MAD2 that can be recruited to kinetochores for MCC assembly (Ma & Poon, 2016; Nelson et al, 2015).1
Prior to its identification as a key component of the SAC, TRIP13 was known to regulate the MAD2‐related HORMAD proteins in meiosis (San‐Segundo & Roeder, 1999; Borner et al, 2008; Joshi et al, 2009; Wojtasz et al, 2009; Roig et al, 2010). The meiotic HORMADs, including Saccharomyces cerevisiae Hop1 and mammalian HORMAD1 and HORMAD2, associate with the meiotic chromosome axis and control multiple aspects of homologous chromosome interactions and meiotic recombination (Hollingsworth et al, 1990; Woltering et al, 2000; Wojtasz et al, 2009; Fukuda et al, 2010). Meiotic HORMADs assemble on the meiotic chromosome axis through binding of their N‐terminal HORMA domains to closure motifs in both their own C‐termini and, most likely, other meiotic chromosome axis proteins (Kim et al, 2014). Late in meiotic prophase, TRIP13 (Pch2 in S. cerevisiae and plants) mediates the removal of HORMADs from the chromosome axis (Borner et al, 2008; Joshi et al, 2009; Wojtasz et al, 2009; Lambing et al, 2015; Vader, 2015). This TRIP13‐mediated axis remodeling likely forms the basis of a feedback pathway that regulates recombination and ensures the fidelity of homolog pairing during meiosis (Wojtasz et al, 2009, 2012; Daniel et al, 2011; Keeney et al, 2014; Thacker et al, 2014; Stanzione et al, 2016). Given the structural similarities between meiotic HORMADs and MAD2, it is likely that TRIP13 uses a common physical mechanism on both substrates and directly disassembles meiotic HORMAD complexes, but thus far this idea has not been directly tested.
Here, we determine the high‐resolution crystal structure of human TRIP13, and use crosslinking mass spectrometry (XLMS) to show how this protein recognizes a p31comet:MAD2 substrate complex. Substrate binding positions the disordered MAD2 N‐terminus near the TRIP13 pore loops, which directly engage and then unfold MAD2 from its N‐terminus. Truncation of the MAD2 N‐terminus renders the protein refractory to TRIP13‐mediated conformational conversion in vitro, and in cells causes progressive defects in both SAC activation and silencing. Strikingly, constructs of Mus musculus HORMAD1 missing its disordered N‐terminus cannot be removed from the meiotic chromosome axis, indicating that TRIP13 directly removes these proteins from meiotic chromosomes through a common N‐terminal unfolding mechanism. Finally, we identify a conserved interaction network in both MAD2 and the meiotic HORMADs that directly links these protein's N‐termini to their C‐terminal safety belt regions. Disruption of this network destabilizes the closed HORMA domain conformation, suggesting that TRIP13‐mediated conformational conversion could be accomplished by unfolding as few as 15–20 residues of these proteins’ N‐terminal regions.
Results
Reconstitution of a TRIP13‐substrate complex
To better understand the molecular basis for MAD2 conformational conversion by TRIP13, we first sought to reconstitute the TRIP13 hexamer and its complex with p31comet and MAD2. Purified human TRIP13 adopts a mixture of oligomeric states in solution including monomers, dimers, and trimers, forming hexamers only in the presence of ATP or a non‐hydrolyzable analog, ATP‐γS (Appendix Fig S1A). In keeping with its expected ability to bind but not hydrolyze ATP, a Walker B motif mutant (E253Q) forms stable hexamers in the presence of either ATP or ATP‐γS (Appendix Fig S1C). We mixed Homo sapiens TRIP13E253Q·ATP, p31comet, and MAD2, and purified a stable complex of the three proteins (Fig 1B and C). Analysis of the complex by size‐exclusion chromatography/multi‐angle light scattering (SEC‐MALS) reveals a 6:1:1 stoichiometry in the TRIP13:p31comet:MAD2 complex, as we observed previously with an equivalent complex of M. musculus proteins (Ye et al, 2015). Thus, a TRIP13 hexamer interacts strongly with a single p31comet:MAD2 substrate complex.
Structure of TRIP13
We crystallized full‐length H. sapiens TRIP13E253Q and determined its structure in the absence of nucleotides to a resolution of 3.02 Å and in the presence of ATP to a resolution of 2.54 Å (Fig 1D–F and Table EV1). Crystallization trials with the TRIP13E253Q:p31comet:MAD2 complex yielded only crystals of TRIP13E253Q alone, precluding high‐resolution analysis of this complex (see Materials and Methods). The TRIP13 monomer closely resembles Caenorhabditis elegans PCH‐2 (Ye et al, 2015), with a small N‐terminal domain (NTD; residues 1–100) putatively involved in substrate recognition, followed by a AAA+ ATPase region with two subdomains, large and small, that together form the ATP‐binding site (Fig EV1A–C). The large AAA domain contains two loops that extend into the hexamer pore, referred to as pore loop 1 (residues ~213–227) and pore loop 2 (residues ~260–272) (Fig EV1B). Pore loop 1 contains a highly conserved tryptophan, W221, which we previously showed is critical for MAD2 conformational conversion and proper coupling of ATP hydrolysis to substrate engagement (Ye et al, 2015). In the ATP‐bound structure of TRIP13E253Q, we observe electron density for bound nucleotide in a conserved cleft between the large and small AAA subdomains. In our structure, the γ‐phosphate of ATP is surrounded by residues K185/T186 of the Walker A motif, D252/Q253 (E253 in wild‐type TRIP13) of the Walker B motif, and the Sensor I residue N300 (Fig EV1D). The Sensor II arginine, R386, is positioned close to the ATP α‐phosphate, but not within hydrogen‐bonding distance. The arginine‐finger residue R312 of the neighboring subunit is oriented away from ATP, with its guanidinium group ~17 Å from the ATP γ‐phosphate. Despite the presence of Mg2+ in the crystallization solution, we do not observe a bound Mg2+ ion in the active site.
Figure EV1. Structure and nucleotide binding of human TRIP13.

- Domain schematic of TRIP13, with N‐terminal domain in blue and the large and small AAA ATPase subdomains in green and red, respectively.
- Structure of a single TRIP13E253Q monomer, with domains colored as in panel (A). The two pore loops PL1 (pink; residues 217–226) and PL2 (green; residues 262–271) are disordered in the structure. PL1 contains W221, needed for coupling ATP hydrolysis to MAD2 engagement (Ye et al, 2015) and lysines 220, 227, and 231, which crosslink to MAD2 in the ternary complex (Fig 2A).
- Structural comparison of the PCH‐2 (PDB ID 4XGU) (Ye et al, 2015) and TRIP13 N‐terminal domains (1.72 Å r.m.s.d. over 61 Cα atom pairs). Disordered regions of TRIP13 (residues 52–54 and 78–88) are shown as dotted lines.
- Close‐up of ATP binding by TRIP13E253Q.
In both the presence and absence of ATP, crystals of TRIP13E253Q form in space group P65 with one copy of TRIP13 per asymmetric unit. Adjacent TRIP13 protomers pack equivalently to C. elegans PCH‐2 and related ATPases (Fig EV2A and B), but form a helical filament instead of a closed hexamer (Fig 1E). Similar helical filaments with P65 crystallographic symmetry have been observed for several related AAA+ protein remodelers that nonetheless function as hexamers in solution, including Vps4 (Scott et al, 2005; Inoue et al, 2008; Monroe et al, 2014; Caillat et al, 2015), fidgetin/spastin (Roll‐Mecak & Vale, 2008; Taylor et al, 2012; Peng et al, 2013), and others. In our previous structure of a closed hexamer of C. elegans PCH‐2, four of the six subunits adopt the “closed” conformation seen in our TRIP13 structure, in which the large and small AAA domains are closely juxtaposed, while two subunits adopt an “open” conformation with the small AAA subdomain rotated ~70° away from the large subdomain (Ye et al, 2015). By overlaying two sets of adjacent TRIP13 protomers (assembled from crystallographic symmetry in the P65 filament) onto the two “half‐hexamers” of C. elegans PCH‐2, we could roughly model a closed TRIP13 hexamer (Figs 1F and EV2C).
Figure EV2. Modeling a closed TRIP13 hexamer.
- Top and side views of the Caenorhabditis elegans PCH‐2 hexamer (chains A/B from PDB ID 4XGU) (Ye et al, 2015), with ATPase domains alternately colored gray and green, and NTDs in brown. Dashed line indicates the “half‐hexamer” used as template for modeling a TRIP13 hexamer, splitting the “open” conformation C/F chains between the large and small AAA subdomains.
- Overlay of consecutive subunits in C. elegans PCH‐2 (gray) and human TRIP13 (green/red). View is equivalent to the side view in panel (A), with NTDs removed for clarity. The large AAA subdomain of chain B was aligned between the two structures, with the location of chain A determined by non‐crystallographic (PCH‐2) or crystallographic (TRIP13) symmetry.
- Top and side views of a modeled TRIP13 hexamer. To model the TRIP13 hexamer, four consecutive subunits of the TRIP13 filament were aligned with either chains A, B or D, E of the C. elegans PCH‐2 hexamer structure (Ye et al, 2015) to generate “half‐hexamer” models (split according to the dashed lines in the top views of panels A and C). For each half‐hexamer, residues 1–320 of the first chain and 321–429 of the fourth chain, plus the entire second and third chains, were kept for the final “full‐hexamer” model. The two resulting “open conformation” chains in the final model, in which residues 1–320 and 321–429 were modeled independently, show a ~17 Å distance between Cα atoms of residues 320 and 321 (dotted lines).
Architecture and dynamics of a TRIP13:p31comet:MAD2 complex
We next investigated the overall architecture and dynamics of the TRIP13E253Q:p31comet:MAD2 complex using XLMS and hydrogen–deuterium exchange mass spectrometry (HDX‐MS). XLMS uses a bifunctional crosslinker to identify pairs of lysine side chains that are within 15–30 Å in a native complex (Herzog et al, 2012), and the mapped crosslinks can be considered as a set of distance restraints for assembling a rough physical model of the complex. After incubation of the TRIP13E253Q:p31comet:MAD2 complex with the crosslinker BS3 (Appendix Fig S2A), we detected crosslinks between TRIP13 and both p31comet and MAD2 (Fig 2A, and Tables EV2 and EV3). In related AAA+ ATPases including NSF and p97/Cdc48, the NTD is responsible for substrate recognition, and consistent with this, we found that three lysine residues in the TRIP13 NTD (K34, K35, and K44) crosslinked to p31comet (Fig 2A and D). These residues formed crosslinks with two p31comet residues in a disordered loop (K100 and K110) and one, K229, whose mutation to alanine has been demonstrated to disrupt TRIP13 binding both in vitro (Ye et al, 2015) and in cells (Ma & Poon, 2016) (Fig 2A and C). Thus, our crosslinking data are consistent with prior findings and indicate that p31comet is recognized directly by the TRIP13 NTD.
Figure 2. XLMS and HDX‐MS of the TRIP13E253Q:p31comet:MAD2 complex.
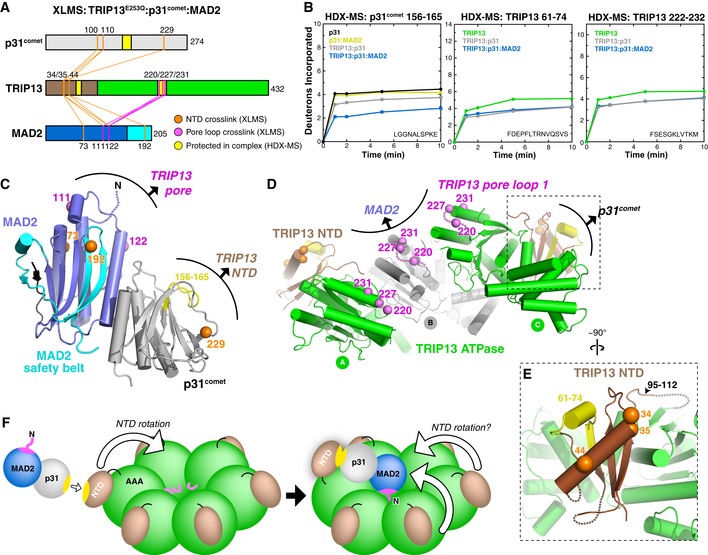
- Schematic of inter‐protein crosslinks detected by XLMS in the TRIP13E253Q:p31comet:MAD2 complex. Crosslinks to the TRIP13 NTD are shown in orange, and crosslinks to the TRIP13 pore loop are shown in pink. No crosslinks were detected between MAD2 and p31comet. See Tables EV2 and EV3 for complete lists of detected crosslinks.
- Deuterium uptake plots for the regions of p31comet (residues 156–165) and TRIP13 (residues 61–74 and 222–232) that are most protected upon complex formation, as measured by HDX‐MS (shown in yellow in panels A, C, and D).
- View of the MAD2:p31comet complex (PDB ID 2QYF) (Yang et al, 2007), with residues that crosslink to the TRIP13 NTD and pore loops shown as orange and pink spheres, respectively. Residues K100 and K110 of p31comet are in a disordered region and are not shown. Shown in yellow is the p31comet region most protected by TRIP13 binding (residues 156–165).
- Pore side view of three adjacent subunits in the TRIP13E253Q filament, with NTD and pore loop residues crosslinking to MAD2 and p31comet shown as orange and pink spheres, respectively. Pore loop residues 217–226 are disordered in the TRIP13E253Q structure, so the position of K220 is approximate. Residues 61–74, highly protected upon p31comet binding, are shown in yellow.
- Close‐up view of the TRIP13 NTD (brown), showing the juxtaposition of p31comet‐interacting regions identified by XLMS (K34, K35, K44; orange spheres) and HDX‐MS (61–74; yellow).
- Model of MAD2:p31comet binding to a TRIP13 hexamer, based on crystal structures and crosslinking data. Yellow surfaces of p31comet and TRIP13 correspond to the regions highlighted in yellow in panels (C and D). The pink surface on MAD2 indicates the TRIP13 pore loop‐interacting surface defined by K111 and K122.
While p31comet crosslinked exclusively to the TRIP13 NTD, MAD2 formed crosslinks with both the TRIP13 NTD and three residues in pore loop 1 (referred to hereafter simply as the “pore loop”): K220, K227, and K231 (Fig 2A, C, and D). The two MAD2 residues that crosslink to the TRIP13 pore loop, K111 and K122, are located on the same face of MAD2 (Fig 2C), suggesting that this surface is positioned close to the TRIP13 hexamer pore in the ternary complex. Given the known structure of the p31comet:MAD2 complex (Yang et al, 2007) and our model of the TRIP13 hexamer, we could use these data to roughly model a ternary TRIP13:p31comet:MAD2 complex (Fig 2F). In this model, initial recognition of p31comet by the TRIP13 NTD is followed by a rotation of the NTD to enable MAD2 to interact with the TRIP13 pore loops. While our crystal structure shows the TRIP13 NTDs docked against the outer face of the AAA+ domains, the NTDs of related ATPases like NSF are highly mobile and can swing “upward” upon substrate binding to enable engagement by the AAA+ domain pore loops (Zhao et al, 2015). Like NSF, it is possible that multiple TRIP13 NTDs swing upward to engage MAD2 once it is bound; this could explain the direct crosslinks we observe between the TRIP13 NTD and other surfaces of MAD2 (Fig 2A and D).
We next performed HDX‐MS, which provides information on solvent accessibility and dynamics by measuring exchange of main‐chain amide hydrogen atoms with solvent deuterium atoms (Hoofnagle et al, 2003; Skinner et al, 2012). We compared the TRIP13E253Q:p31comet:MAD2 complex to each individual protein, as well as binary TRIP13E253Q:p31comet and p31comet:MAD2 complexes (Figs 2B–E and EV3, and Appendix Fig S2). While we were unable to examine changes in MAD2 between different states due to poor sequence coverage (data not shown), we detected changes in both TRIP13E253Q and p31comet upon complex formation that provide information about how these two proteins interact. We previously showed that p31comet residues P228/K229 and the loop encompassing residues 157–162 constitute a conserved TRIP13‐binding site (Ye et al, 2015). Our HDX‐MS data show that p31comet residues 156–165 are significantly protected from hydrogen–deuterium exchange upon TRIP13 binding, and even more protected in the ternary TRIP13:p31comet:MAD2 complex (Fig 2B). Peptides encompassing the established MAD2‐binding site of p31comet were protected upon binding of MAD2, and were also further protected in the ternary TRIP13E253Q:p31comet:MAD2 complex (not shown).
Figure EV3. Bimodal peptides in TRIP13E253Q HDX‐MS.
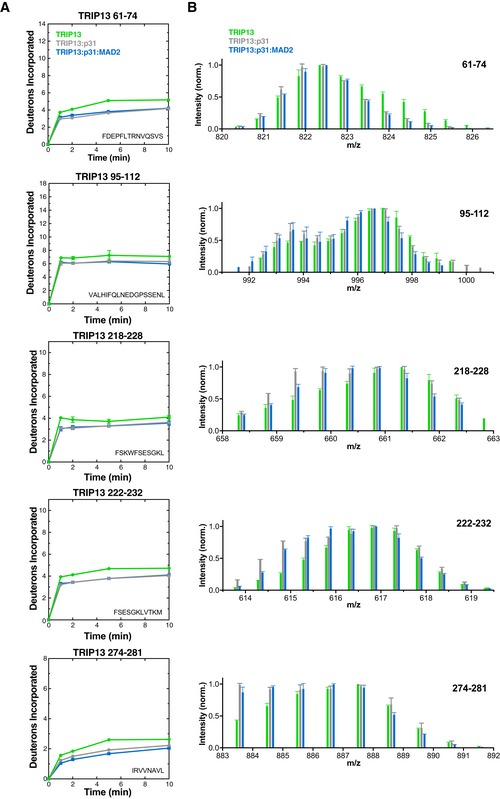
- Deuterium uptake plots for TRIP13 peptides showing bimodality in HDX‐MS analysis of one or more states (TRIP13E253Q alone green, TRIP13E253Q:p31comet gray, TRIP13E253Q:p31comet:MAD2 blue). Error bars represent standard deviation from triplicate measurements.
- Sticks (MS peak height versus mass/charge ratio, average and standard deviation of three independent samples) for TRIP13 peptides shown in panel (A). Each set of data shows peaks at the same m/z ratios; datasets are shifted on the graph for clarity.
In TRIP13E253Q, two regions were strongly protected upon binding either p31comet or the p31comet:MAD2 complex: residues 61–74 and 95–112 in the protein's NTD, and several peptides in the TRIP13 pore loops. In our structure of TRIP13, residues 61–74 reside in the NTD adjacent to residues K34, K35, and K44 (Fig 2E), supporting a role for this surface as the p31comet binding site. TRIP13 residues 95–112 constitute the NTD‐ATPase domain linker, and protection of this region upon p31comet binding (Fig EV3) suggests either decreased mobility or direct protection from solvent upon substrate binding. The TRIP13 95–112 peptide also shows significant bimodality in its HD exchange profile in all samples (Fig EV3 and Appendix Fig S2C). A bimodal HD exchange profile suggests that this region can adopt at least two differently solvent‐accessible conformations in solution, supporting the idea of TRIP13 NTD mobility.
The TRIP13 pore loops were also significantly protected from HD exchange upon association with either p31comet or the p31comet:MAD2 complex (Figs 2B and EV3). Two peptides from this loop (residues 218–228 and 222–232) showed both significant bimodality in their HD exchange profile and significant protection upon substrate binding, even in the presence of p31comet alone. These data suggest that the pore loops can adopt multiple conformations in solution that differ in their solvent accessibility and that p31comet, despite its inability to engage and activate TRIP13's ATPase activity (Ye et al, 2015), nonetheless affects the structure of TRIP13 upon binding. We propose that p31comet binding may “prime” TRIP13 for proper MAD2 engagement and remodeling, subtly affecting the overall TRIP13 hexamer structure, and imparting significant additional order onto the pore loops in particular.
TRIP13 pore loops directly engage the disordered MAD2 N‐terminus
Our XLMS data show that MAD2 residues K111 and K122 are positioned near the TRIP13 pore loops in the ternary complex. MAD2 possesses a short 10‐ to 15‐amino acid disordered region at its N‐terminus that extends from the same surface as K111 and K122 in the C‐MAD2 conformation (Fig 2C). We therefore tested whether the TRIP13 pore loops directly interact with the MAD2 N‐terminus using a cysteine crosslinking assay similar to one previously described for the AAA+ ATPase Vps4 (Fig 3A) (Yang et al, 2015). We engineered MAD2 and TRIP13 constructs lacking solvent‐exposed cysteines (MAD2ΔCys and TRIP13ΔCys), and re‐introduced single cysteines in the TRIP13 pore loop (W221C) and in the MAD2 N‐terminus (S6C), αA helix (A17C, S25C, or S40C), or at native internal cysteine sites (C79, C106) (Fig 3B). We mixed TRIP13W221C and TRIP13ΔCys at a 1:8 molar ratio in the presence of ATP or the non‐hydrolyzable analog ATP‐γS to generate TRIP13 hexamers with ≤ 1 pore loop cysteine and then reduced these cysteines with DTT. We then activated cysteines in MAD2 using DTNB (Ellman's reagent), incubated with p31comet and pre‐assembled TRIP13 hexamers, and then detected crosslinks by non‐reducing SDS–PAGE (Fig 3C). We found that MAD2S6C, with a cysteine in the disordered N‐terminus, robustly formed crosslinks with TRIP13 in the presence of either ATP or ATP‐γS, indicating that the MAD2 N‐terminus is directly engaged by the TRIP13 pore loops. MAD2 mutants with internal cysteines all showed crosslink formation in the presence of ATP, but not ATP‐γS, indicating that ATP hydrolysis is required to processively unfold MAD2 (Fig 3C). In all cases, MAD2‐TRIP13 crosslinks depended on cysteines in both proteins, required p31comet, and disappeared upon the addition of reducing agent (Fig 3D and Appendix Fig S3). These data show that TRIP13 interacts with the disordered MAD2 N‐terminus upon initial association with p31comet:MAD2, and can then unfold at least 106 residues of MAD2 (205 residues total) in the presence of ATP.
Figure 3. Cysteine crosslinking detects TRIP13 pore loop–MAD2 engagement.
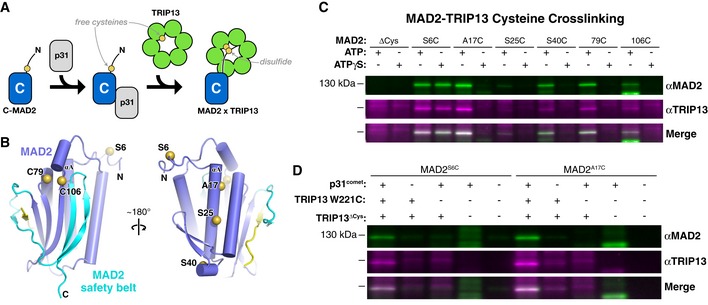
- Experimental schematic to detect crosslinking between single cysteines on MAD2 and the TRIP13 pore loop. See Appendix Fig S3A–C for biochemical verification that all MADs and TRIP13 cysteine mutants are functional.
- Two views of C‐MAD2 (from PDB ID 2V64) (Mapelli et al, 2007) with bound closure motif in yellow and safety belt region in cyan. Residues mutated to cysteine for crosslinking experiments (S6, A17, S25, and S40 on helix αA, plus native cysteines C79 and C106) are shown as yellow spheres.
- Western blots of non‐reducing SDS–PAGE analysis of TRIP13‐MAD2 crosslinking reactions (anti‐MAD2 green, anti‐TRIP13 magenta) in the presence of ATP or the non‐hydrolyzable ATP‐γS. See Appendix Fig S3D for full gels. The crosslinked band migrating at ˜120 kDa is detected by antibodies to both proteins, and disappears upon addition of DTT (Appendix Fig S3E).
- Validation of the requirement for TRIP13 W221C and p31comet for MAD2‐TRIP13 crosslinking.
TRIP13 requires the disordered MAD2 N‐terminus for conformational conversion
We have previously shown that TRIP13 and p31comet can together convert C‐MAD2 to O‐MAD2 (Ye et al, 2015), explaining their ability to disassemble MCC and inactivate the SAC (Teichner et al, 2011; Westhorpe et al, 2011; Eytan et al, 2014; Wang et al, 2014; Miniowitz‐Shemtov et al, 2015). To test whether TRIP13 engagement of the MAD2 N‐terminus is required for conformational conversion, we designed a series of N‐terminal truncations of MAD2 (Fig 4A). In structures of C‐MAD2, the first residue of helix αA is L13 (Luo et al, 2002; Sironi et al, 2002); in O‐MAD2, residues 11–15 are in the β1 strand, and αA begins at residue A17 (Luo et al, 2000; Mapelli et al, 2007). We designed truncations of M. musculus MAD2 starting at residues M1 (full‐length; FL), S6 (ΔN5), I11 (ΔN10), and S16 (ΔN15), and successfully purified all but ΔN10 (see Materials and Methods). All MAD2 constructs also contained the R133A mutation, which disrupts MAD2 homodimer formation and enables separation of O‐MAD2 and C‐MAD2 populations by anion‐exchange chromatography (see, e.g., Appendix Fig S7) (Sironi et al, 2001; Luo et al, 2004). When expressed in Escherichia coli, FL and ΔN5‐MAD2 adopt both O‐MAD2 and C‐MAD2 conformations, while ΔN15‐MAD2 adopts solely the C‐MAD2 conformation as previously reported (Mapelli et al, 2007), likely because this construct lacks the β1 strand needed to stabilize the O‐MAD2 conformation. Both ΔN5‐MAD2 and ΔN15‐MAD2 strongly bind a closure motif‐containing CDC20127‐147 peptide (Fig 4B) [again, as previously reported for ΔN15‐MAD2; (Mapelli et al, 2006)] and bind p31comet (Fig 4C), and equivalent H. sapiens MAD2 truncations behave identically (Appendix Fig S4B and C). Thus, removing up to 15 residues from the MAD2 N‐terminus does not affect the protein's ability to bind a closure motif peptide and adopt the closed conformation in vitro.
Figure 4. The MAD2 N‐terminus is required for TRIP13 recognition and conformational conversion.
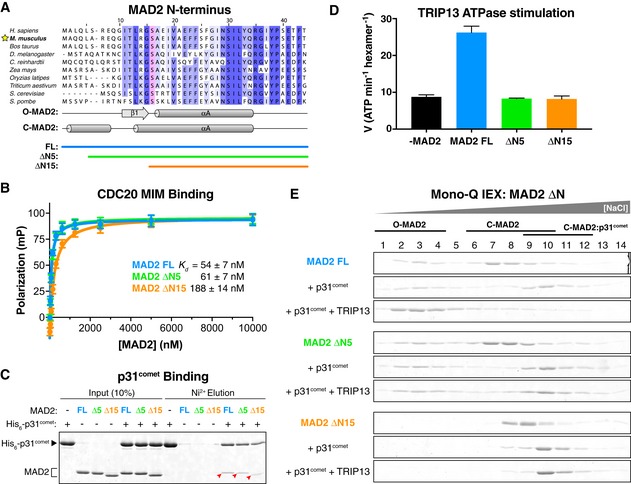
- Fluorescence polarization assay showing binding of MAD2 constructs to a closure motif‐containing peptide (CDC20127‐147). Error bars represent standard deviation from triplicate measurements.
- Pull‐down assay showing binding of MAD2 constructs to p31comet (red arrowheads).
- ATP hydrolysis by Mus musculus TRIP13 in the presence of p31comet and MAD2 FL, ΔN5, or ΔN15. No stimulation was observed in the absence of p31comet (not shown). See Appendix Fig S4 for similar characterization of Homo sapiens MAD2 FL, ΔN5, and ΔN15. Error bars represent standard deviation from triplicate measurements.
- Mono‐Q ion‐exchange chromatography measuring conformational conversion of MAD2 FL (blue), ΔN5 (green), and ΔN15 (orange). Each MAD2 construct was incubated either alone (top), with p31comet (middle), or with p31comet, TRIP13, and ATP (bottom) then separated by ion‐exchange to determine the amounts of O‐MAD2 and C‐MAD2. See Fig EV4 for sample ion‐exchange elution profiles and gels showing p31comet.
Figure EV4. Ion‐exchange separation of MAD2 conformers.
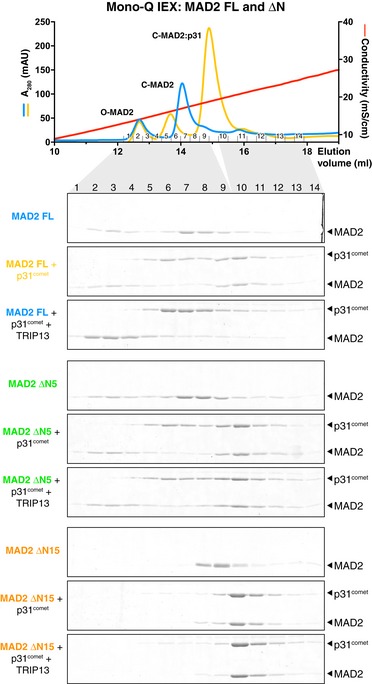
Top: UV absorbance (280 nm) and conductivity traces, and associated fractions from Mono‐Q ion‐exchange separation of O‐ and C‐MAD2. Samples shown are Mus musculus MAD2 FL (blue, same as Fig 4E, top panel) and MAD2 FL + p31comet (yellow, same as Fig 4E, second panel). Bottom: Mono‐Q ion‐exchange chromatography measuring conformational conversion of MAD2 FL (blue), ΔN5 (green), and ΔN15 (orange). Gels shown are the same as in Fig 4E, but are cropped to show both p31comet and MAD2.
We previously showed that p31comet and MAD2 together stimulate the ATPase activity of M. musculus TRIP13 and that mutation of the conserved pore loop tryptophan residue W221 to alanine uncouples ATPase stimulation from substrate binding (Ye et al, 2015). We next tested the ability of our MAD2 ΔN mutants to stimulate TRIP13 and found that, while FL‐MAD2 stimulated TRIP13's ATPase activity roughly threefold in the presence of p31comet, neither ΔN5‐MAD2 nor ΔN15‐MAD2 stimulated TRIP13 (Fig 4D). Equivalent assays with human proteins again showed identical results (Appendix Fig S4D). These data indicate that TRIP13's ATPase activity is tightly coupled to engagement of the MAD2 N‐terminal tail by the TRIP13 pore loops. Finally, we directly tested for TRIP13‐mediated conformational conversion of MAD2 using anion‐exchange chromatography as previously described (Ye et al, 2015). We incubated purified C‐MAD2 FL, ΔN5, and ΔN15 with p31comet, TRIP13, and ATP and then separated the reaction products on a Mono‐Q ion‐exchange column. While FL‐MAD2 was robustly converted to O‐MAD2, we detected no conversion of either ΔN5‐MAD2 or ΔN15‐MAD2 (Fig 4E). Thus, the disordered MAD2 N‐terminus is required to stimulate the ATPase activity of TRIP13, and for TRIP13‐mediated MAD2 conformational conversion.
The spindle assembly checkpoint is compromised in cells containing ΔN‐MAD2
RNAi knockdown of TRIP13 in cells, or depletion of TRIP13 from cell extracts, has been shown to cause defects in MCC disassembly, compromising inactivation of the SAC and delaying anaphase onset (Wang et al, 2014). Conversely, deleting TRIP13 entirely in cells compromises activation of the SAC, likely by eliminating the pool of O‐MAD2 necessary for recruitment to kinetochores and assembly into the MCC (Ma & Poon, 2016). As ΔN‐MAD2 cannot be properly engaged by TRIP13 in vitro, its effects may mimic that of TRIP13 knockdown or deletion. To explore this idea, we inhibited endogenous MAD2 expression in DLD1 cells by RNAi targeting the 3′ UTR and then rescued with either full‐length or ΔN‐MAD2 constructs (Fig 5A and B). While ΔN5‐ and ΔN10‐MAD2 expressed at near‐wild‐type levels in these cells (Appendix Fig S5), ΔN15‐MAD2 showed poor expression and was not examined further (not shown). We first examined the O‐MAD2/C‐MAD2 ratio in these cells. Similar to prior findings in HeLa cells (Luo et al, 2004), we found that FL‐MAD2 overwhelmingly adopted the open conformation in unsynchronized DLD1 cells (Fig 5C). In contrast, ΔN5‐ and ΔN10‐MAD2 were ~50% and nearly 100% closed, respectively (Fig 5C), in close agreement with the phenotype of TRIP13‐deleted cells (Ma & Poon, 2016). These data support the idea that truncating the MAD2 N‐terminus compromises TRIP13‐mediated conformational conversion, and suggest that SAC activation may be compromised in these cells as well.
Figure 5. MAD2 N‐terminal truncations are defective in SAC activation and silencing.

- Schematic of the MAD2 N‐terminus, with secondary structure and limits of full‐length, ΔN5, and ΔN10 mutants.
- Schematic of experiments testing function of MAD2 truncations in SAC activation and silencing. Nocodazole was added only for the experiment shown in (E). See Appendix Fig S5 for Western blot showing MAD2 expression in each strain.
- Mono‐Q ion‐exchange chromatography analysis of soluble MAD2 conformation in cells. Fractions are equivalent to Fig 4E.
- Analysis of unperturbed mitotic timing (nuclear envelope breakdown to anaphase) in MAD2‐RNAi and rescue cell lines. P‐values were calculated using an unpaired two‐tailed t‐test. For each sample, black lines indicate mean and SEM (standard error of the mean) from 50 cells.
- MAD2 ΔN5 and ΔN10 show partial defects in SAC activation, as measured by the percentage of mitotic cells remaining arrested in nocodazole (added 1 h prior to filming).
We next examined mitotic timing in cells expressing ΔN‐MAD2. RNAi knockdown of MAD2 causes a significant shortening of the time between nuclear envelope breakdown (NEBD) and anaphase onset, indicating that the SAC cannot be activated in the absence of MAD2 (Fig 5D). SAC activation is rescued by expression of FL‐MAD2, but rescue with ΔN5‐ and ΔN10‐MAD2 had a more variable effect. In agreement with our ion‐exchange chromatography results showing that nearly all ΔN10‐MAD2 is in the C‐MAD2 state, these cells showed a SAC activation defect nearly as strong as that observed in the MAD2 RNAi‐alone cells. ΔN5‐MAD2, in contrast, showed wide variability in mitotic timing, with many cells showing short‐to‐normal timing, but a small percentage showing extremely delayed anaphase onset (Fig 5D). This indicates that while many cells expressing ΔN5‐MAD2 cannot activate the SAC, some may retain enough O‐MAD2 to assemble MCC and activate the SAC. Because ΔN5‐MAD2 is refractory to TRIP13‐mediated disassembly, SAC inactivation could therefore be compromised in these cells. Finally, we examined whether ΔN‐MAD2 could support SAC activation in the presence of the microtubule poison nocodazole. Nocodazole was added 1 h prior to the start of time‐lapse filming, and the percentage of mitotic cells at time zero that remained arrested in mitosis was followed for several hours. While RNAi knockdown of MAD2 caused a complete loss of SAC activation, as indicated by a quick exit from mitosis by all cells, full‐length MAD2 fully rescued SAC activation and nocodazole‐mediated mitotic arrest (Fig 5E). As in our previous assay, ΔN5‐ and ΔN10‐MAD2 had an intermediate effect, with ~60% and ~40% of cells, respectively, showing robust SAC activation (Fig 5E).
N‐terminal truncation of HORMAD1 compromises its removal from the meiotic chromosome axis
Prior to its identification as a key component of the spindle assembly checkpoint, TRIP13 was implicated in meiotic chromosome axis remodeling, specifically the removal of MAD2‐related HORMAD1 and HORMAD2 from the chromosome axis upon synaptonemal complex assembly (Wojtasz et al, 2009). While a direct interaction between these proteins and TRIP13 has not yet been demonstrated, we sought to test whether truncating their disordered N‐termini would compromise their removal from meiotic chromosomes. We designed truncations of M. musculus HORMAD1 roughly equivalent to ΔN5‐MAD2 (ΔN5‐HORMAD1) and ΔN10‐MAD2 (ΔN20‐HORMAD1; Fig 6A) and then introduced vectors encoding these proteins into testes of HORMAD1 −/− mice. We examined chromosome localization of eGFP‐tagged HORMAD1 constructs in late zygotene/early pachytene, when HORMAD1/2 are being removed from regions of homolog pairs that have undergone synapsis. We found that full‐length HORMAD1‐eGFP localizes to chromosome axes early in meiotic prophase, and is then efficiently removed upon homolog synapsis, as judged by a lack of co‐localization with the synaptonemal complex protein SYCP1 (Fig 6B). Truncation of the HORMAD1 N‐terminus did not affect initial localization of the protein to chromosome axes, but caused a progressive defect in synapsis‐linked removal: While full‐length HORMAD1‐eGFP was never co‐localized with SYCP1, ΔN5‐HORMAD1‐eGFP showed at least partial co‐localization with SYCP1 in 32% of cells, and ΔN20‐HORMAD1 showed co‐localization with SYCP1 in 88% of cells (Fig 6C and D). We found that bulky N‐terminal tags on HORMAD1 also significantly disrupt its removal from the chromosome axis (Appendix Fig S6); these tags may disrupt recognition and engagement by TRIP13. Overall, these results strongly support the idea that TRIP13 mediates removal of HORMAD1 from meiotic chromosomes directly, using a HORMA domain‐remodeling mechanism similar to the one we outlined for MAD2.
Figure 6. HORMAD1 N‐terminal truncations are defective in TRIP13‐mediated removal from the meiotic chromosome axis.
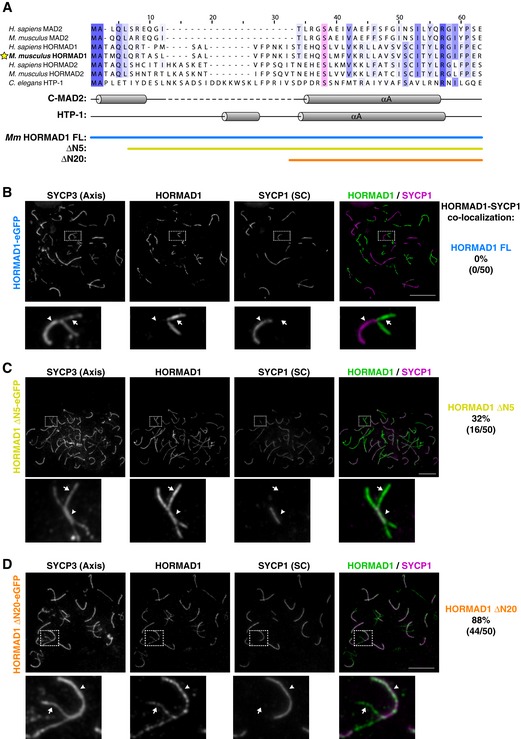
- Sequence alignment of the N‐termini of mammalian MAD2, HORMAD1, and HORMAD2 with Caenorhabditis elegans HTP‐1. The highly conserved αA serine (S38 in HTP‐1; see Fig 7) is highlighted in pink. Below, secondary structure of C‐MAD2 and HTP‐1 is shown, along with limits of full‐length (blue), ΔN5 (yellow), and ΔN20 (orange) constructs of Mus musculus HORMAD1.
- Chromosome localization of SYCP3 (chromosome axis), SYCP1 (synaptonemal complex/SC), and full‐length HORMAD1 in late zygotene/early pachytene mouse spermatocytes.
- Chromosome localization of ΔN5‐HORMAD1. Cells shown are examples where the protein is not efficiently removed upon synapsis.
- Chromosome localization of ΔN20‐HORMAD1. Cells shown are examples where the protein is not efficiently removed upon synapsis.
A conserved hydrogen‐bond network stabilizes the C‐MAD2 conformation
While our crosslinking data show that TRIP13 can unfold at least 106 residues of MAD2, our earlier measurements of ATP usage by TRIP13 suggested that on average, conformational conversion of MAD2 is accomplished using only 8–10 ATP molecules (Ye et al, 2015), likely not enough to mediate complete MAD2 unfolding. Examination of the structure of C‐MAD2 reveals that the C‐terminal safety belt β‐hairpin (strands β8′ and β8″) interacts directly with the N‐terminal αA helix (residues ~13–35) through a network of buried hydrogen bonds (Figs 7A and EV5A and B), suggesting that disruption of this network by limited N‐terminal unfolding could mediate MAD2 conformational conversion. Supporting this idea, prior work showed that mutation of MAD2 safety belt residues T188 or H191, which interact directly with S16 on the αA helix, causes MAD2 to preferentially adopt the open conformation (Yang et al, 2008). All three residues are highly conserved in MAD2 orthologs, and an equivalent buried hydrogen‐bond network is also conserved in the meiotic HORMAD proteins (Figs 7B and EV5C and D) (Kim et al, 2014).
Figure 7. A conserved hydrogen‐bond network stabilizes the closed HORMA domain conformation.
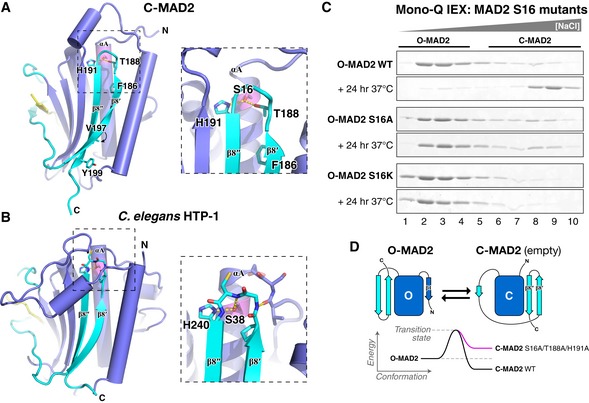
- Structure of Caenorhabditis elegans HTP‐1 (green, safety belt pink) bound to HIM‐3 closure motif (yellow) (PDB ID 4TZO) (Kim et al, 2014). HTP‐1 S38 forms a conserved hydrogen‐bond network with H240 and the main‐chain amide of C239. See Fig EV5 for sequence alignments showing conservation of these residues in both protein families.
- Mono‐Q ion‐exchange chromatography showing equilibrium O‐MAD2/C‐MAD2 ratios for MAD2 wild type, S16A, and S16K. See Appendix Fig S7 for initial ion‐exchange purification of MAD2 S16A and S16K.
- Proposed energy landscape of O‐MAD2/C‐MAD2 in wild‐type (black) or point mutants disrupting the αA‐safety belt hydrogen‐bond network (pink).
Figure EV5. Conservation of the αA‐β8′‐β8″ hydrogen‐bond network.

- Sequence alignment of MAD2 proteins, showing conservation of S16, T188, and H191.
- Structure of Caenorhabditis elegans HTP‐1 (green, safety belt pink) bound to HIM‐3 closure motif (yellow) (PDB ID 4TZO) (Kim et al, 2014). HTP‐1 S38 forms a conserved hydrogen‐bond network with H240 and the main‐chain amide of C239.
- Sequence alignment of meiotic HORMAD proteins, showing conservation of S38 and H240.
To test the idea that disruption of the buried hydrogen‐bond network linking MAD2's N‐terminus to the safety belt promotes the open conformation, we mutated MAD2 S16 to alanine or lysine and examined the equilibrium open/closed ratio of these mutants by ion‐exchange chromatography. We purified O‐MAD2 (which in wild‐type MAD2 is stable at 4°C) for each variant and then incubated each protein for 24 h at 37°C. With wild‐type MAD2, this treatment results in near‐complete spontaneous conversion to the C‐MAD2 form (Fig 7C). Replacement of S16 with a bulky lysine (S16K) probably sterically inhibits docking of the β8′‐β8″ hairpin, and this mutant adopted exclusively the open conformation, even after 24 h at 37°C (Fig 7C). The S16A mutant, by contrast, adopted both open and closed conformations when purified from E. coli (Appendix Fig S7), but purified O‐MAD2S16A showed no conversion to the closed conformation after 24 h at 37°C (Fig 7C). Thus, disrupting the conserved hydrogen‐bond network linking helix αA to the β8′‐β8″ hairpin favors the open conformation of MAD2, likely by destabilizing the closed conformation (Fig 7D). These data suggest that TRIP13 could act by unfolding a limited stretch of the MAD2 N‐terminus, perhaps as few as 15–20 residues, which would disrupt the αA‐β8′‐β8″ hydrogen‐bond network and mediate a closed‐to‐open conformational conversion without complete MAD2 unfolding.
Discussion
Here, we outline the molecular mechanism of HORMA domain conformational conversion and complex disassembly by the conserved AAA+ ATPase TRIP13. Our crosslinking data indicates that TRIP13 initially binds p31comet via its N‐terminal domain, which in related AAA+ remodelers like NSF is also responsible for substrate recognition (Zhao et al, 2015). Similar to these enzymes, the TRIP13 NTD may “flip up” upon substrate binding to allow the TRIP13 pore loops to directly engage the N‐terminus of MAD2 (Fig 2F). Our data show that TRIP13 is capable of unfolding at least 106 residues of MAD2, but we also show that a much more limited unfolding of the MAD2 N‐terminus could mediate conformational conversion by disrupting interactions that stabilize the C‐terminal safety belt in the C‐MAD2 conformation. In this mode, TRIP13 would unfold the MAD2 N‐terminal region to mediate safety belt release from the HORMA domain core and then release MAD2 to allow relaxation to the open conformation (Fig 8). An important question in this case is how TRIP13 would release MAD2 after limited unfolding. Our data show that the TRIP13 hexamer is relatively unstable (Appendix Fig S1), perhaps because this protein possesses a single AAA+ ATPase region as opposed to tandem AAA+ domains in related remodelers such as NSF and p97/Cdc48. This inherent instability, plus the fact that p31comet specifically recognizes the closed MAD2 conformation (Xia et al, 2004; Yang et al, 2007), could mean that the ternary TRIP13:p31comet:MAD2 complex is destabilized upon partial MAD2 unfolding, promoting disassembly of the complex and release of MAD2.
Figure 8. Model for HORMA domain remodeling by MAD2.
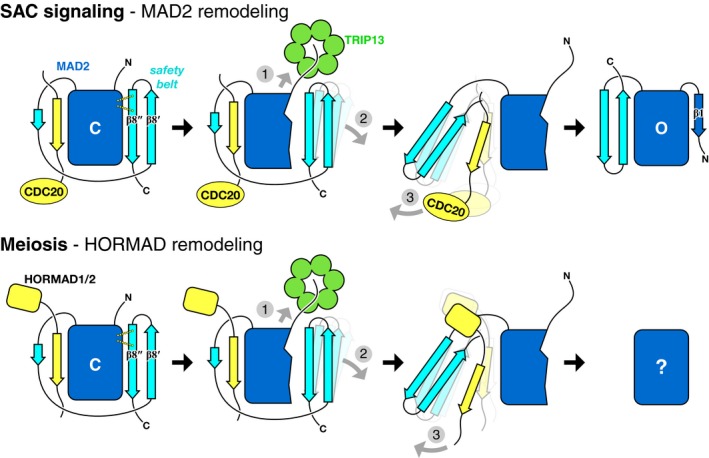
Proposed mechanism of TRIP13 in the spindle assembly checkpoint (top) and meiosis (bottom). In both cases, TRIP13 (green) recognizes HORMA domain proteins (MAD2 or meiotic HORMADs; blue) in the closed conformation, bound to closure motifs of binding partners (yellow; CDC20 to MAD2, HORMAD C‐termini for HORMADs). Limited unfolding of the HORMA domain N‐terminus disrupts a hydrogen‐bond network stabilizing the C‐terminal safety belt (1), mediating its dissociation from the HORMA domain core (2), followed by release of the bound closure motif (3). Following release of the MAD2 N‐terminus by TRIP13, this region forms strand β1 to stabilize the open conformation. The conformation of unbound meiotic HORMADs is currently unknown.
Our data show that N‐terminal truncation of M. musculus HORMAD1 disrupts its TRIP13‐mediated removal from the chromosome axis, suggesting a shared unfolding mechanism with MAD2 (Fig 8). Supporting this idea, the residues comprising the N‐terminus–safety belt hydrogen‐bond network are conserved in meiotic HORMADs from budding yeast to mammals (Fig EV5). A hypothetical mechanism in which TRIP13 unfolds these proteins’ N‐termini to destabilize the closed conformation and allow closure motif dissociation can explain how meiotic HORMADs are removed from the chromosome axis, but several questions remain. First, as mentioned earlier, meiotic HORMADs have not been shown to possess a stable open conformation equivalent to O‐MAD2. It may be that because of the unique requirements of the spindle assembly checkpoint, MAD2's open conformation has evolved to be more stable than its equivalent in the meiotic HORMADs. Nonetheless, as both assembly and disassembly of HORMAD–closure motif complexes would require conformational changes in these proteins’ safety belt region (Kim et al, 2014), we support the idea that these proteins can adopt an open‐like state, even if this state is transient.
A second question regarding the meiotic HORMADs involves the mechanism of initial recognition by Pch2/TRIP13. The key aspect of Pch2/TRIP13 function in meiosis is that it removes meiotic HORMADs specifically from synapsed chromosomes/regions, serving as a feedback mechanism controlling DNA breakage and crossover levels on a per‐chromosome basis (Borner et al, 2008; Joshi et al, 2009; Wojtasz et al, 2009; Thacker et al, 2014; Lambing et al, 2015; Vader, 2015). A recent finding that in rice, p31comet is localized to the synaptonemal complex suggests that this protein might also be involved in the recognition and removal of meiotic HORMADs, and in dictating the specificity for synapsed chromosome regions (Ji et al, 2016). We have so far been unable to demonstrate direct interactions between mammalian p31comet and meiotic HORMADs in vitro, however (data not shown), suggesting either that this mechanism is not highly conserved or involves additional factors. Moreover, S. cerevisiae Pch2 regulates the meiotic HORMAD protein Hop1 without p31comet, as this organism lacks a p31comet ortholog (Borner et al, 2008; Joshi et al, 2009). Based on our structural modeling of the TRIP13:p31comet:MAD2 complex, we suggest that TRIP13 may be able to recognize and remodel the meiotic HORMAD proteins directly, if they form homodimers similar in structure to the p31comet:C‐MAD2 dimer. There is currently no evidence for dimerization of meiotic HORMADs either in vitro or in vivo, but the fact that all other HORMA domain proteins are known to dimerize is suggestive of such a capability (Rosenberg & Corbett, 2015). It may be that meiotic HORMADs dimerize, or alternatively bind p31comet, only in response to structural changes at the meiotic chromosome axis that occur upon synaptonemal complex assembly; this could explain how Pch2/TRIP13 specifically removes meiotic HORMADs from synapsed regions of chromosomes.
Could TRIP13 regulate other HORMA domain proteins, including Rev7 and the autophagy proteins Atg13 and Atg101? While Atg101 forms a stable open‐like state (Qi et al, 2015) and is therefore unlikely to be a TRIP13 substrate, both Rev7 and Atg13 form the closed conformation and bind closure motifs: Rev7 binds Rev3 (Hara et al, 2010; Kikuchi et al, 2012) and Atg13 likely binds Atg9 (Suzuki et al, 2015). Close inspection of their structures, however, reveals that the hydrogen‐bond network between helix αA and the β8′‐β8″ hairpin does not exist in these proteins, having been replaced by hydrophobic interactions. TRIP13 was previously found to bind Rev7 in a high‐throughput yeast two‐hybrid screen (Venkatesan et al, 2009), but our attempts to reproduce this result have been unsuccessful (data not shown). Overall, the evidence so far suggests that TRIP13 does not regulate either Rev7 or the autophagy‐signaling HORMA domain proteins. Finally, TRIP13's S. cerevisiae ortholog Pch2 (which probably does not act on Mad2 due to a lack of p31comet in S. cerevisiae) has also been reported to interact with several non‐HORMA domain proteins including Orc1 and Xrs2 (Ho & Burgess, 2011; Vader et al, 2011), leaving open the possibility of a wider array of Pch2/TRIP13 substrates. Notably, the S. cerevisiae Pch2 NTD is significantly diverged from that of other TRIP13 orthologs (not shown), suggesting potential additional substrate‐recognition abilities.
Materials and Methods
For full methods, see the Appendix.
Cloning and protein purification
All proteins were cloned into pET‐based vectors with N‐terminal His6 tags cleavable by TEV protease or, in the case of MAD2 variants for biochemical assays, the SUMO isopeptidase Ulp1. Proteins were expressed in E. coli strain Rosetta2(DE3)pLysS (EMD Millipore) except for TRIP13, which was expressed in E. coli LOBSTR (Kerafast, Inc.) (Andersen et al, 2013). Proteins were purified by Ni2+ affinity (Ni‐NTA; Qiagen) and ion‐exchange (HiTrap SP HP or HiTrap Q HP; GE Healthcare) chromatography, His6 tags were cleaved by incubation with TEV protease (Kapust et al, 2001) at 4°C overnight, and His6‐SUMO tags were cleaved by incubation with S. cerevisiae Ulp1 at 4°C overnight. Proteins were concentrated and stored at 4°C for crystallization, or at −80°C for biochemical assays.
Crystallization and structure determination
For crystallization, TRIP13E253Q was concentrated to 15 mg/ml and exchanged into a buffer containing 25 mM Tris–HCl pH 7.5, 0.2 M NaCl, 5 mM MgCl2, and 1 mM TCEP. For crystallization without ATP, TRIP13E253Q was mixed 1:1 in hanging drop format with well solution containing 0.1 M Tris–HCl pH 8.5, 0.8 M LiCl, and 4% PEG 3350. Crystals (~90 × 90 × 50 μm) were cryoprotected by addition of 20% glycerol and flash‐frozen in liquid nitrogen. For ATP‐bound TRIP13E253Q, 1 mM ATP was added to the protein solution; then protein was mixed 1:1 in hanging drop format with well solution containing 0.1 M Tris–HCl pH 8.5, 0.2 M KCl, 10% pentaerythritol propoxylate. Crystals (~100 × 100 × 60 μm) were cryoprotected by addition of 10% glycerol and 12% pentaerythritol propoxylate (22% total) and flash‐frozen in liquid nitrogen.
X‐ray diffraction datasets for TRIP13E253Q apo and TRIP13E253Q·ATP were collected on NE‐CAT beamline 24ID‐E at the Advanced Photon Source (Argonne National Laboratory, Argonne IL; see Appendix for support statement). Phasing was performed by molecular replacement with PHASER (McCoy et al, 2007), using a structure of C. elegans PCH‐2 (PDB ID 4XGU) (Ye et al, 2015) with most side chains removed. Initial models were iteratively rebuilt in COOT (Emsley et al, 2010) and refined using phenix.refine (Adams et al, 2010) with a consistent free‐R set across multiple structures (Table EV1).
TRIP13E253Q:p31comet:MAD2 complex assembly and crosslinking mass spectrometry
For assembly of the TRIP13E253Q:p31comet:MAD2 complex, proteins were mixed in a final molar ratio of 2:1:1 (three copies of p31comet:MAD2 per TRIP13E253Q hexamer) in a buffer containing 20 mM HEPES pH 7.5, 0.3 M NaCl, 5 mM MgCl2, 5% glycerol, and 1 mM ATP and then incubated for 60 min at 4°C. Complexes were injected onto a Superdex 200 Increase 10/300 GL size‐exclusion column (GE Life Sciences) and fractions representing the TRIP13E253Q hexamer plus associated p31comet and MAD2 were pooled and concentrated to 1 mg/ml. Crosslinking was performed using isotopically coded D0/D12 BS3 (bis‐sulfosuccinimidylsuberate; Creative Molecules) for 30 min at 30°C, followed by quenching in 100 mM NH4HCO3. Crosslinked proteins were digested with Lys‐C and trypsin, purified by reversed‐phase chromatography, and analyzed by tandem mass spectrometry (Orbitrap Elite, Thermo Scientific) (Herzog et al, 2012). Fragment ion spectra were searched and crosslinks identified by the dedicated software program xQuest (Rinner et al, 2008; Walzthoeni et al, 2015).
Hydrogen–deuterium exchange mass spectrometry
HD exchange experiments were conducted with a Waters Synapt G2S system; 5 μl of samples containing 10 μM individual proteins or complexes in exchange buffer (300 mM NaCl, 20 mM Tris pH 7.5, 10% glycerol, 1 mM DTT) was mixed with 55 μl of the same buffer made with D2O for several deuteration times (0, 1, 2, 5, 10 min) at 15°C. The exchange was quenched for 2 min at 1°C with an equal volume of quench buffer (3 M guanidine HCl, 0.1% formic acid). Proteins were cleaved with pepsin and separated by reverse‐phase chromatography and then directed into a Waters SYNAPT G2s quadrupole time‐of‐flight (qTOF) mass spectrometer. Peptides were identified using PLGS version 2.5 (Waters, Inc.), and relative deuterium uptake was calculated using DynamX version 2.0 (Waters Corp.).
ATPase assays
ATPase reactions contained assay buffer (25 mM Tris–HCl at pH 7.5, 200 mM NaCl, 10 mM MgCl2, 1 mM DTT, 5% glycerol) plus 250 μM ATP, and were incubated 30 min at 37°C. Reactions were stopped and triplicate samples were assayed using the ADP‐Glo kinase assay kit (Promega) in 384‐well microplates using a TECAN (Mannedorf, Switzerland) Infinite M1000 spectrophotometer. Most assays were performed with 100 or 200 nM TRIP13 (monomer concentration); below this concentration ATPase activity was highly inconsistent, potentially due to dissociation of the TRIP13 hexamer. For most assays, p31comet and MAD2 were added in excess, at 2 and 5 μM, respectively. Data were converted to K cat using a standard curve for ADP, and fit using PRISM version 7 (GraphPad Software, La Jolla, CA, USA).
Protein interaction assays
For MAD2‐p31comet binding assays, equimolar amounts of proteins were incubated 60 min at 20°C, bound to Ni‐NTA resin (Qiagen), and then analyzed by SDS–PAGE. For fluorescence polarization assays, N‐terminal FITC‐labeled CDC20 closure motif peptide (residues 127–147; EAKILRLSGKPQNAPEGYQNR) was resuspended in binding buffer (25 mM Tris–HCl pH 7.5, 200 mM NaCl, 5% glycerol, 1 mM DTT, 0.1% NP‐40). Fifty microliter reactions containing 50 nM peptide plus 40 nM–40 μM MAD2 (FL or N‐terminal truncations) were incubated 30 min at room temperature, and then, fluorescence polarization was read in 384‐well plates using a TECAN Infinite M1000 PRO fluorescence plate reader. All binding curves were done in triplicate. Binding data were analyzed with GraphPad Prism v. 7 using a single‐site binding model.
Cysteine crosslinking
Cysteine crosslinking assays were performed essentially as described (Yang et al, 2015). Cysteines in MAD2 were activated by addition of 5,5′‐dithiobis‐(2‐nitrobenzoic acid) (DTNB) and then buffer‐exchanged into DTT‐ and DTNB‐free crosslinking buffer. TRIP13W221C and TRIP13ΔCys were mixed in a 1:8 molar ratio in the presence of 1 mM ATP and 5 mM DTT and then buffer‐exchanged to DTT‐free buffer. p31comet was prepared as TRIP13. TRIP13 (20 μM monomers), MAD2 (5 μM), and p31comet (5 μM) were mixed in the presence of 1 mM ATP or ATP‐γS for 1 min at 30°C, and then, reactions were quenched and analyzed by non‐reducing PAGE and Western blotting.
Ion‐exchange chromatography and MAD2 conversion assays
Examination of MAD2 conformation by ion‐exchange chromatography, and MAD2 conformational conversion assays, was carried out largely as described (Ye et al, 2015).
Time‐lapse imaging
Full‐length or N‐terminally truncated MAD2 was incorporated into parental Flp‐In TRex‐DLD‐1 parental cells that stably express mRFP‐tagged histone H2B (H2B–mRFP) (Holland et al, 2010; Han et al, 2013) using FRT/Flp‐mediated recombination (Tighe et al, 2004), and expression induced with 1 μg/ml doxycycline. siRNAs were directed against the 3′ untranslated region of Mad2, not present in Flp‐In constructs.
To determine unperturbed mitotic timing, cells were seeded onto CELLSTAR μClear 96‐well plate (Greiner bio‐one) with 50 nM of oligonucleotides using Lipofectamine RNAiMAX (Thermo Fisher); 24 h after transfection, doxycycline was added to express MAD2 for 24 h before analyzing by time‐lapse microscopy. Cells were maintained at 37°C and 5% CO2 and images were collected using a CQ1 confocal image cytometer (Yokogawa) with a 40× objective at 10‐min time intervals. Movies were assembled and analyzed using ImageJ (National Institutes of Health) software. To measure mitotic slippage, nocodazole was added 1 h before filming.
Mouse spermatocyte protein localization
Proteins were expressed in spermatocytes by injection and electroporation of CMV‐driven constructs into testes of live juvenile CD1 mice (13dpp) according to published protocols (Shoji et al, 2005; Shibuya et al, 2014). Spermatocytes were collected 24 h after electroporation, and nuclear surface spreads were carried out as described previously (Peters et al, 1997).
Data availability
The crystal structures reported here are available at the Protein Data Bank (http://www.rcsb.org) under the accession codes 5VQ9 (TRIP13E253Q Apo; https://doi.org/10.2210/pdb5vq9/pdb) and 5VQA (TRIP13E253Q·ATP; https://doi.org/10.2210/pdb5vqa/pdb). Diffraction datasets for both structures are available at the SBGrid Data Bank (https://data.sbgrid.org) under the accession codes 409 (TRIP13E253Q Apo; https://doi.org/10.15785/sbgrid/409) and 410 (TRIP13E253Q·ATP; https://doi.org/10.15785/sbgrid/410).
Author contributions
QY purified proteins, determined the 3D structure of TRIP13, performed TRIP13‐p31comet‐MAD2 crosslinking for XLMS, and performed all biochemical assays. SCR performed HDX‐MS. DHK performed time‐lapse imaging assays under the guidance of DWC. ID performed HORMAD1 localization assays under the guidance of AT. GH performed XLMS under the guidance of FH. KDC conceived of the study, cloned expression constructs, interpreted data, and wrote the manuscript with input from QY, SCR, DHK, and DWC.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File
Acknowledgements
The authors thank the staff of APS NE‐CAT beamline 24ID‐E for assistance with data collection, E. A. Komives for advice and assistance with HDX‐MS, H. Zhou for the gift of Ulp1 protease, J. H. Hurley for advice on cysteine crosslinking experiments, and A. Desai, A. Shiau, and members of the Corbett laboratory for helpful discussions and critical reading of the manuscript. D.W.C. acknowledges funding from the Ludwig Institute for Cancer Research and the National Institutes of Health (R01 GM29513). K.D.C. acknowledges funding from the Ludwig Institute for Cancer Research, the National Institutes of Health (R01 GM104141), and the March of Dimes Foundation (General Research Award 1‐FY14‐251). K.D.C., F.H., and A.T. acknowledge further collaborative funding from the Human Frontiers Science Program (RGP0008/2015).
The EMBO Journal (2017) 36: 2419–2434
Note
Correction added on 15 August 2017 after first online publication: The last sentence in this paragraph (which includes a new reference Nelson et al, 2015) was added.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen KR, Leksa NC, Schwartz TU (2013) Optimized E. coli expression strain LOBSTR eliminates common contaminants from His‐tag purification. Proteins 81: 1857–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon ED, Musacchio A (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol 15: 214–225 [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV (1998) The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem Sci 23: 284–286 [DOI] [PubMed] [Google Scholar]
- Borner GV, Barot A, Kleckner N (2008) Yeast Pch2 promotes domainal axis organization, timely recombination progression, and arrest of defective recombinosomes during meiosis. Proc Natl Acad Sci USA 105: 3327–3332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillat C, Macheboeuf P, Wu Y, McCarthy AA, Boeri‐Erba E, Effantin G, Göttlinger HG, Weissenhorn W, Renesto P (2015) Asymmetric ring structure of Vps4 required for ESCRT‐III disassembly. Nat Commun 6: 8781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel K, Lange J, Hached K, Fu J, Anastassiadis K, Roig I, Cooke HJ, Stewart AF, Wassmann K, Jasin M, Keeney S, Toth A (2011) Meiotic homologue alignment and its quality surveillance are controlled by mouse HORMAD1. Nat Cell Biol 13: 599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eytan E, Wang K, Miniowitz‐Shemtov S, Sitry‐Shevah D, Kaisari S, Yen TJ, Liu S‐T, Hershko A (2014) Disassembly of mitotic checkpoint complexes by the joint action of the AAA‐ATPase TRIP13 and p31(comet). Proc Natl Acad Sci USA 111: 12019–12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faesen AC, Thanasoula M, Maffini S, Breit C, Müller F, van Gerwen S, Bange T, Musacchio A (2017) Basis of catalytic assembly of the mitotic checkpoint complex. Nature 542: 498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraschini R, Beretta A, Sironi L, Musacchio A, Lucchini G, Piatti S (2001) Bub3 interaction with Mad2, Mad3 and Cdc20 is mediated by WD40 repeats and does not require intact kinetochores. EMBO J 20: 6648–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Daniel K, Wojtasz L, Toth A, Höög C (2010) A novel mammalian HORMA domain‐containing protein, HORMAD1, preferentially associates with unsynapsed meiotic chromosomes. Exp Cell Res 316: 158–171 [DOI] [PubMed] [Google Scholar]
- Habu T, Kim SH, Weinstein J, Matsumoto T (2002) Identification of a MAD2‐binding protein, CMT2, and its role in mitosis. EMBO J 21: 6419–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Holland AJ, Fachinetti D, Kulukian A, Cetin B, Cleveland DW (2013) Catalytic assembly of the mitotic checkpoint inhibitor BubR1‐Cdc20 by a Mad2‐induced functional switch in Cdc20. Mol Cell 51: 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Hashimoto H, Murakumo Y, Kobayashi S, Kogame T, Unzai S, Akashi S, Takeda S, Shimizu T, Sato M (2010) Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase zeta and REV1. J Biol Chem 285: 12299–12307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG, Johnston RC, Smith DL, Murray AW (2000) MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J Cell Biol 148: 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, Leitner A, Beck M, Hartl F‐U, Ban N, Malmström L, Aebersold R (2012) Structural probing of a protein phosphatase 2A network by chemical cross‐linking and mass spectrometry. Science 337: 1348–1352 [DOI] [PubMed] [Google Scholar]
- Ho H‐C, Burgess SM (2011) Pch2 acts through Xrs2 and Tel1/ATM to modulate interhomolog bias and checkpoint function during meiosis. PLoS Genet 7: e1002351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW (2010) Polo‐like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol 188: 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth NM, Goetsch L, Byers B (1990) The HOP1 gene encodes a meiosis‐specific component of yeast chromosomes. Cell 61: 73–84 [DOI] [PubMed] [Google Scholar]
- Hoofnagle AN, Resing KA, Ahn NG (2003) Protein analysis by hydrogen exchange mass spectrometry. Annu Rev Biophys Biomol Struct 32: 1–25 [DOI] [PubMed] [Google Scholar]
- Howell BJ, Moree B, Farrar EM, Stewart S, Fang G, Salmon ED (2004) Spindle checkpoint protein dynamics at kinetochores in living cells. Curr Biol 14: 953–964 [DOI] [PubMed] [Google Scholar]
- Inoue M, Kamikubo H, Kataoka M, Kato R, Yoshimori T, Wakatsuki S, Kawasaki M (2008) Nucleotide‐dependent conformational changes and assembly of the AAA ATPase SKD1/VPS4B. Traffic 9: 2180–2189 [DOI] [PubMed] [Google Scholar]
- Jao CC, Ragusa MJ, Stanley RE, Hurley JH (2013) A HORMA domain in Atg13 mediates PI 3‐kinase recruitment in autophagy. Proc Natl Acad Sci USA 110: 5486–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Tang D, Shen Y, Xue Z, Wang H, Shi W, Zhang C, Du G, Li Y, Cheng Z (2016) P31comet, a member of the synaptonemal complex, participates in meiotic DSB formation in rice. Proc Natl Acad Sci USA 113: 10577–10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N, Barot A, Jamison C, Börner GV (2009) Pch2 links chromosome axis remodeling at future crossover sites and crossover distribution during yeast meiosis. PLoS Genet 5: e1000557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapust RB, Tözsér J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS (2001) Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild‐type catalytic proficiency. Protein Eng 14: 993–1000 [DOI] [PubMed] [Google Scholar]
- Keeney S, Lange J, Mohibullah N (2014) Self‐organization of meiotic recombination initiation: general principles and molecular pathways. Annu Rev Genet 48: 187–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, Hara K, Shimizu T, Sato M, Hashimoto H (2012) Structural basis of recruitment of DNA polymerase ζ by interaction between REV1 and REV7 proteins. J Biol Chem 287: 33847–33852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Rosenberg SC, Kugel CL, Kostow N, Rog O, Davydov V, Su TY, Dernburg AF, Corbett KD (2014) The chromosome axis controls meiotic events through a hierarchical assembly of HORMA domain proteins. Dev Cell 31: 487–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulukian A, Han JS, Cleveland DW (2009) Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell 16: 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambing C, Osman K, Nuntasoontorn K, West A, Higgins JD, Copenhaver GP, Yang J, Armstrong SJ, Mechtler K, Roitinger E, Franklin FCH (2015) Arabidopsis PCH2 mediates meiotic chromosome remodeling and maturation of crossovers. PLoS Genet 11: e1005372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara‐Gonzalez P, Westhorpe FG, Taylor SS (2012) The spindle assembly checkpoint. Curr Biol 22: R966–R980 [DOI] [PubMed] [Google Scholar]
- Luo X, Fang G, Coldiron M, Lin Y, Yu H, Kirschner MW, Wagner G (2000) Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat Struct Biol 7: 224–229 [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Rizo J, Yu H (2002) The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell 9: 59–71 [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H (2004) The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol 11: 338–345 [DOI] [PubMed] [Google Scholar]
- Ma HT, Poon RYC (2016) TRIP13 regulates both the activation and inactivation of the spindle‐assembly checkpoint. Cell Rep 14: 1086–1099 [DOI] [PubMed] [Google Scholar]
- Mapelli M, Filipp FV, Rancati G, Massimiliano L, Nezi L, Stier G, Hagan RS, Confalonieri S, Piatti S, Sattler M, Musacchio A (2006) Determinants of conformational dimerization of Mad2 and its inhibition by p31comet. EMBO J 25: 1273–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli M, Massimiliano L, Santaguida S, Musacchio A (2007) The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell 131: 730–743 [DOI] [PubMed] [Google Scholar]
- Mapelli M, Musacchio A (2007) MAD contortions: conformational dimerization boosts spindle checkpoint signaling. Curr Opin Struct Biol 17: 716–725 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miniowitz‐Shemtov S, Eytan E, Kaisari S, Sitry‐Shevah D, Hershko A (2015) Mode of interaction of TRIP13 AAA‐ATPase with the Mad2‐binding protein p31comet and with mitotic checkpoint complexes. Proc Natl Acad Sci USA 112: 11536–11540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe N, Han H, Gonciarz MD, Eckert DM, Karren MA, Whitby FG, Sundquist WI, Hill CP (2014) The oligomeric state of the active Vps4 AAA ATPase. J Mol Biol 426: 510–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED (2007) The spindle‐assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8: 379–393 [DOI] [PubMed] [Google Scholar]
- Musacchio A (2015) The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol 25: R1002–R1018 [DOI] [PubMed] [Google Scholar]
- Nelson CR, Hwang T, Chen PH, Bhalla N (2015) TRIP13/PCH‐2 promotes Mad2 localization to unattached kinetochores in the spindle checkpoint response. J Cell Biol 211: 503–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Lin Z, Li W, Lu J, Shen Y, Wang C (2013) Structural insights into the unusually strong ATPase activity of the AAA domain of the Caenorhabditis elegans fidgetin‐like 1 (FIGL‐1) protein. J Biol Chem 288: 29305–29312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AH, Plug AW, van Vugt MJ, de Boer P (1997) A drying‐down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res 5: 66–68 [DOI] [PubMed] [Google Scholar]
- Primorac I, Musacchio A (2013) Panta rhei: the APC/C at steady state. J Cell Biol 201: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi S, Kim DJ, Stjepanovic G, Hurley JH (2015) Structure of the human Atg13‐Atg101 HORMA heterodimer: an interaction hub within the ULK1 complex. Structure 23: 1848–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R (2008) Identification of cross‐linked peptides from large sequence databases. Nat Methods 5: 315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig I, Dowdle JA, Toth A, de Rooij DG, Jasin M, Keeney S (2010) Mouse TRIP13/PCH2 is required for recombination and normal higher‐order chromosome structure during meiosis. PLoS Genet 6: E1001062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll‐Mecak A, Vale RD (2008) Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 451: 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SC, Corbett KD (2015) The multifaceted roles of the HORMA domain in cellular signaling. J Cell Biol 211: 745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San‐Segundo PA, Roeder GS (1999) Pch2 links chromatin silencing to meiotic checkpoint control. Cell 97: 313–324 [DOI] [PubMed] [Google Scholar]
- Scott A, Chung H‐Y, Gonciarz‐Swiatek M, Hill GC, Whitby FG, Gaspar J, Holton JM, Viswanathan R, Ghaffarian S, Hill CP, Sundquist WI (2005) Structural and mechanistic studies of VPS4 proteins. EMBO J 24: 3658–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JV, Botvinick E, Bonday Z, Furnari F, Berns MW, Cleveland DW (2004) Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr Biol 14: 942–952 [DOI] [PubMed] [Google Scholar]
- Shibuya H, Morimoto A, Watanabe Y (2014) The dissection of meiotic chromosome movement in mice using an in vivo electroporation technique. PLoS Genet 10: e1004821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M, Chuma S, Yoshida K, Morita T, Nakatsuji N (2005) RNA interference during spermatogenesis in mice. Dev Biol 282: 524–534 [DOI] [PubMed] [Google Scholar]
- Simonetta M, Manzoni R, Mosca R, Mapelli M, Massimiliano L, Vink M, Novak B, Musacchio A, Ciliberto A (2009) The influence of catalysis on mad2 activation dynamics. PLoS Biol 7: e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi L, Melixetian M, Faretta M, Prosperini E, Helin K, Musacchio A (2001) Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. EMBO J 20: 6371–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi L, Mapelli M, Knapp S, de Antoni A, Jeang K‐T, Musacchio A (2002) Crystal structure of the tetrameric Mad1‐Mad2 core complex: implications of a ‘safety belt’ binding mechanism for the spindle checkpoint. EMBO J 21: 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner JJ, Lim WK, Bédard S, Black BE, Englander SW (2012) Protein dynamics viewed by hydrogen exchange. Protein Sci 21: 996–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanzione M, Baumann M, Papanikos F, Dereli I, Lange J, Ramlal A, Tränkner D, Shibuya H, de Massy B, Watanabe Y, Jasin M, Keeney S, Toth A (2016) Meiotic DNA break formation requires the unsynapsed chromosome axis‐binding protein IHO1 (CCDC36) in mice. Nat Cell Biol 18: 1208–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 154: 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki SW, Yamamoto H, Oikawa Y, Kondo‐Kakuta C, Kimura Y, Hirano H, Ohsumi Y (2015) Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci USA 112: 3350–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JL, White SR, Lauring B, Kull FJ (2012) Crystal structure of the human spastin AAA domain. J Struct Biol 179: 133–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichner A, Eytan E, Sitry‐Shevah D, Miniowitz‐Shemtov S, Dumin E, Gromis J, Hershko A (2011) p31comet Promotes disassembly of the mitotic checkpoint complex in an ATP‐dependent process. Proc Natl Acad Sci USA 108: 3187–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker D, Mohibullah N, Zhu X, Keeney S (2014) Homologue engagement controls meiotic DNA break number and distribution. Nature 510: 241–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe A, Johnson VL, Taylor SS (2004) Truncating APC mutations have dominant effects on proliferation, spindle checkpoint control, survival and chromosome stability. J Cell Sci 117: 6339–6353 [DOI] [PubMed] [Google Scholar]
- Tipton AR, Wang K, Oladimeji P, Sufi S, Gu Z, Liu S‐T (2012) Identification of novel mitosis regulators through data mining with human centromere/kinetochore proteins as group queries. BMC Cell Biol 13: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G, Blitzblau HG, Tame MA, Falk JE, Curtin L, Hochwagen A (2011) Protection of repetitive DNA borders from self‐induced meiotic instability. Nature 477: 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G (2015) Pch2(TRIP13): controlling cell division through regulation of HORMA domains. Chromosoma 124: 333–339 [DOI] [PubMed] [Google Scholar]
- Venkatesan K, Rual J‐F, Vazquez A, Stelzl U, Lemmens I, Hirozane‐Kishikawa T, Hao T, Zenkner M, Xin X, Goh K‐I, Yildirim MA, Simonis N, Heinzmann K, Gebreab F, Sahalie JM, Cevik S, Simon C, de Smet A‐S, Dann E, Smolyar A et al (2009) An empirical framework for binary interactome mapping. Nat Methods 6: 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzthoeni T, Joachimiak LA, Rosenberger G, Röst HL, Malmström L, Leitner A, Frydman J, Aebersold R (2015) xTract: software for characterizing conformational changes of protein complexes by quantitative cross‐linking mass spectrometry. Nat Methods 12: 1185–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Sturt‐Gillespie B, Hittle JC, Macdonald D, Chan GK, Yen TJ, Liu S‐T (2014) Thyroid hormone receptor interacting protein 13 (TRIP13) AAA‐ATPase is a novel mitotic checkpoint‐silencing protein. J Biol Chem 289: 23928–23937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhorpe FG, Tighe A, Lara‐Gonzalez P, Taylor SS (2011) p31comet‐mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci 124: 3905–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtasz L, Daniel K, Roig I, Bolcun‐Filas E, Xu H, Boonsanay V, Eckmann CR, Cooke HJ, Jasin M, Keeney S, McKay MJ, Toth A (2009) Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA‐ATPase. PLoS Genet 5: e1000702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtasz L, Cloutier JM, Baumann M, Daniel K, Varga J, Fu J, Anastassiadis K, Stewart AF, Reményi A, Turner JMA, Toth A (2012) Meiotic DNA double‐strand breaks and chromosome asynapsis in mice are monitored by distinct HORMAD2‐independent and ‐dependent mechanisms. Genes Dev 26: 958–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woltering D, Baumgartner B, Bagchi S, Larkin B, Loidl J, de los Santos T, Hollingsworth NM (2000) Meiotic segregation, synapsis, and recombination checkpoint functions require physical interaction between the chromosomal proteins Red1p and Hop1p. Mol Cell Biol 20: 6646–6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H (2004) Conformation‐specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. EMBO J 23: 3133–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Li B, Tomchick DR, Machius M, Rizo J, Yu H, Luo X (2007) p31comet blocks Mad2 activation through structural mimicry. Cell 131: 744–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Li B, Liu C‐J, Tomchick DR, Machius M, Rizo J, Yu H, Luo X (2008) Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol 6: e50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Stjepanovic G, Shen Q, Martin A, Hurley JH (2015) Vps4 disassembles an ESCRT‐III filament by global unfolding and processive translocation. Nat Struct Mol Biol 22: 492–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Rosenberg SC, Moeller A, Speir JA, Su TY, Corbett KD (2015) TRIP13 is a protein‐remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife 4: e07367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Wu S, Zhou Q, Vivona S, Cipriano DJ, Cheng Y, Brunger AT (2015) Mechanistic insights into the recycling machine of the SNARE complex. Nature 518: 61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File
Data Availability Statement
The crystal structures reported here are available at the Protein Data Bank (http://www.rcsb.org) under the accession codes 5VQ9 (TRIP13E253Q Apo; https://doi.org/10.2210/pdb5vq9/pdb) and 5VQA (TRIP13E253Q·ATP; https://doi.org/10.2210/pdb5vqa/pdb). Diffraction datasets for both structures are available at the SBGrid Data Bank (https://data.sbgrid.org) under the accession codes 409 (TRIP13E253Q Apo; https://doi.org/10.15785/sbgrid/409) and 410 (TRIP13E253Q·ATP; https://doi.org/10.15785/sbgrid/410).