

Abstract
Type 2 inflammation is a defining feature of infection with parasitic worms (helminths), as well as being responsible for widespread suffering in allergies. However, the precise mechanisms involved in T helper (Th) 2 polarization by dendritic cells (DCs) are currently unclear. We have identified a previously unrecognized role for type I IFN (IFN‐I) in enabling this process. An IFN‐I signature was evident in DCs responding to the helminth Schistosoma mansoni or the allergen house dust mite (HDM). Further, IFN‐I signaling was required for optimal DC phenotypic activation in response to helminth antigen (Ag), and efficient migration to, and localization with, T cells in the draining lymph node (dLN). Importantly, DCs generated from Ifnar1 −/− mice were incapable of initiating Th2 responses in vivo. These data demonstrate for the first time that the influence of IFN‐I is not limited to antiviral or bacterial settings but also has a central role to play in DC initiation of Th2 responses.
Keywords: dendritic cell, interferon, priming, Th2
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Introduction
Although both helminth infection and allergies exert a devastating global impact and lack effective vaccines or refined therapeutics, basic understanding of the key cell types and mediators that initiate and control type 2 immunity is limited. Despite the potential for innate immune cells such as group 2 innate lymphoid cells to influence type 2 responses (McKenzie et al, 2014), it is DCs that are critical for activation and polarization of Th2 immunity (Hammad et al, 2010; Phythian‐Adams et al, 2010; McKenzie et al, 2014). However, the signals that DCs provide to facilitate Th2 polarization remain unclear (MacDonald & Maizels, 2008; Bouchery et al, 2014).
IFN‐I is most well known for its pro‐inflammatory role in antiviral immunity (Hoffmann et al, 2015). However, this large family of cytokines (including 14 IFNα subtypes in mice, as well as IFNβ) exert diverse effects in a range of infection settings (Bouchery et al, 2014; McNab et al, 2015). All IFN‐I subtypes bind to a common receptor expressed on immune, stromal, and epithelial cells, and studies using IFN‐I receptor‐deficient mice (Ifnar1 −/−) indicate that IFN‐I signaling can either enhance or impair the inflammatory response during viral and bacterial infection, depending on context (Ivashkiv & Donlin, 2014; McNab et al, 2015). Many IFN‐I effects are mediated by a direct impact on DC phenotype and functionality. For example, IFN‐I responsiveness controls the ability of CD8α+ cDC1s to cross‐present viral and tumor Ag to CD8+ T cells (Diamond et al, 2011; Pinto et al, 2011; Ivashkiv & Donlin, 2014) and also influences DC activation, migration, and T cell priming in vitro (Parlato et al, 2001; Montoya, 2002; Mattei et al, 2009; Diamond et al, 2011; Pinto et al, 2011). Although it has been suggested that IFN‐I may be produced by DCs exposed to live Schistosoma mansoni eggs (Trottein et al, 2004), the role of IFN‐I in type 2 inflammation, and how DC function may be modulated by this cytokine family during the orchestration of Th2 responses, is unknown.
We have investigated the key factors involved in DC activation and function during Th2 induction using in vitro‐generated murine Flt3‐L BMDCs (FLDCs), which reflect the heterogeneity and complexity of DC subsets in vivo (Naik et al, 2005), and using in vivo models of Th2 priming. These studies indicate that in addition to producing IFN‐I in response to helminth Ag and allergens, DC‐intrinsic IFN‐I signaling is required for their effective migration, localization, and Th2 induction in vivo. For the first time, we establish a central role for IFN‐I as an early positive regulator of Th2 immunity and demonstrate that IFN‐I signaling is essential for optimal DC function during type 2 priming.
Results
An IFN‐I signature and Th2 induction by FLDCs
To date, studies have failed to identify a defined DC phenotypic activation profile in response to Th2‐polarizing helminths (MacDonald & Maizels, 2008; Bouchery et al, 2014). Previous work in this area has predominantly been carried out using murine GM‐CSF‐derived BMDCs (GMDCs) or cell lines (MacDonald et al, 2001; Trottein et al, 2004). However, FLDCs better represent DC subsets in vivo (Naik et al, 2005; Helft et al, 2015), generating CD24hi equivalents of CD8α+ cDC1s, CD11b+ cDC2s, and plasmacytoid DCs (pDCs; Fig 1A; Brasel et al, 2000; Gilliet et al, 2002; Guilliams et al, 2014). FLDCs were cultured overnight with the potent Th2‐inducing soluble egg Ag from S. mansoni (SEA; MacDonald & Maizels, 2008), or heat‐killed Salmonella typhimurium (St) as a Th1/17 control (Perona‐Wright et al, 2012; Cook et al, 2015). Surprisingly, FLDCs secreted significant levels of IFN‐I in response to SEA, including IFNα3 and IFNβ, in excess of that induced by St (Fig 1B). IFN‐I production by FLDCs responding to Th2‐Ag was not restricted to SEA, but was also evident following their exposure to the allergen house dust mite (HDM; Fig 1C). Despite significant IFN‐I induction, SEA failed to stimulate production of the inflammatory mediators IL‐1β, IL‐6, IL‐12p40, IL‐12p70, or TNFα, or regulatory IL‐10, from FLDCs (Fig 1D), as reported previously by our group and others using GMDCs or human PBMC DCs (MacDonald et al, 2001; de Jong et al, 2002). In stark contrast, St induced high‐level production of these cytokines (Fig 1D). Although it has recently been suggested that DC‐derived IL‐10 and IL‐33 are important inducers of Th2 responses (Williams et al, 2013), we were unable to detect significant levels of either IL‐10 (Fig 1D) or IL‐33 (Fig EV1A) following FLDC exposure to SEA.
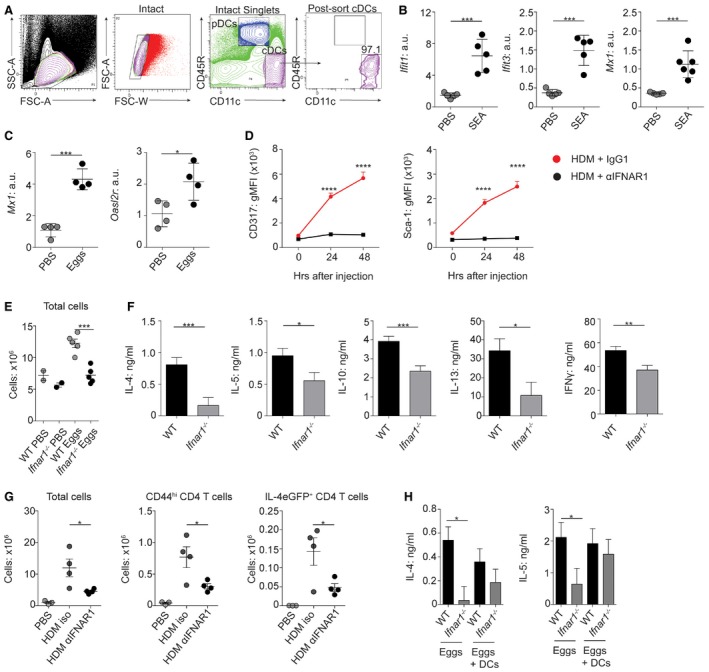
Figure 1. FLDCs produce IFN‐I in response to Th2 Ag.
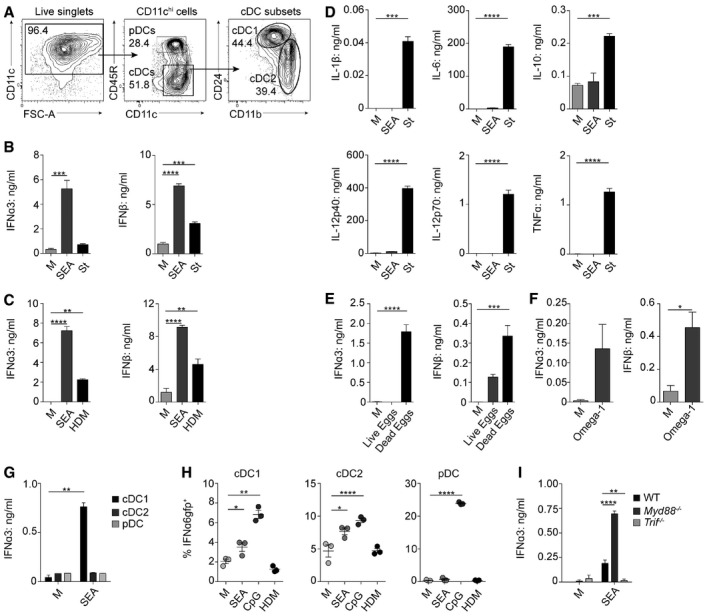
-
AFLDC subsets were defined by expression of CD11c, CD45R (B220), CD11b, and CD24. pDCs were identified as CD45R+ CD11clo; cDCs are CD11c+ CD45R− with cDC1 and cDC2 subsets.
-
B, CIFN‐I production as measured by ELISA on supernatants from bulk cultures cultured in medium alone (M) or stimulated with 25 μg/ml soluble egg Ag from Schistosoma mansoni (SEA) (B, C), 5 μg/ml Salmonella typhimurium (St) (B), or 50 μg/ml house dust mite (HDM) extract (C).
-
DProduction of DC cytokines from bulk cultures following exposure to SEA or St.
-
E–GIFN‐I production as measured by ELISA on supernatants from bulk cultures cultured in medium alone (M) or stimulated with 1:500 live or dead whole S. mansoni eggs (E), 0.5 μg/ml recombinant omega‐1 (F), or FACS‐isolated subsets after stimulation with SEA (G).
-
HPercentage of IFNα6GFP+ DCs after stimulation with SEA, 1 μg/ml CpG, or HDM.
-
IIFNα3 production by WT, Myd88 −/−, and Trif −/− bulk FLDCs after exposure to SEA.
Figure EV1. FLDCs’ responses to Ag.
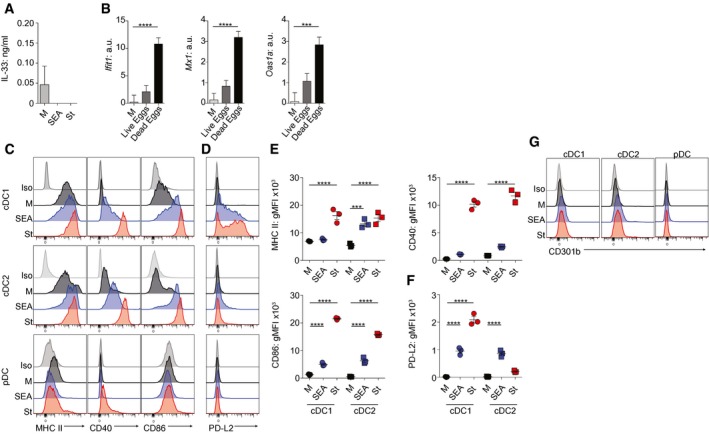
-
AIL‐33 production by FLDCs cultured in medium alone (M) or with Ag.
-
BWT FLDCs were cultured at a ratio of 1 live or dead egg: 500 DCs, or in medium alone (M), for 6 h for gene expression analysis (normalized against Gapdh, a.u.).
-
C, DDC phenotype following overnight culture in medium alone (M, black), SEA (blue), or St (red). Isotype control (Iso, gray‐shaded).
-
E, FgMFI of surface markers for cDC1 and cDC2 FLDC subsets.
-
GCD301b expression of FLDC subsets following 18‐h culture in the presence or absence of Ag.
In order to assess whether IFN‐I induction by the soluble extract SEA was also evident with whole S. mansoni eggs, we cultured FLDCs with live or dead eggs (Fig 1E). This demonstrated significant induction of both IFNα3 and IFNβ following FLDC exposure to dead eggs, as well as a clear IFN‐I signature in the form of upregulated IFN‐stimulated genes (ISGs), including Ifit1, Mx1, and Oas1a (Fig EV1B). Co‐culture of FLDCs with live eggs showed only a trend toward IFN‐I secretion and ISG induction, suggesting that the components responsible for DC IFN‐I production are predominantly released from dead, or dying, eggs. In addition, IFN‐I production was also detected in FLDCs exposed to the T2 ribonuclease (RNase) omega‐1 (Everts et al, 2009; Wilbers et al, 2017), which is the major Th2 immunostimulatory factor in, and one of the most abundant components of, S. mansoni egg secretions (Cass et al, 2007; Fig 1F).
To identify which FLDC subset(s) produced IFN‐I in response to Th2‐inducing Ag, cells were sorted prior to SEA stimulation, demonstrating that the primary source was cDC1s (Fig 1G). This was unexpected, given that pDCs are a key source of this cytokine, particularly in early viral infection (Swiecki & Colonna, 2010). As a complementary approach, we cultured FLDCs generated from mice that express GFP when IFNα6 is expressed (Kumagai et al, 2007) with SEA, CpG, or HDM (Fig 1H). These IFNα6 reporter cells confirmed that in addition to cDC1‐restricted production of IFNα3 (Fig 1G), both cDC1s and cDC2s had the capacity to produce IFNα6 in response to SEA. They also demonstrated that pDCs did not respond to the Th2‐associated Ags SEA or HDM with upregulation of IFN‐I production and that, although HDM promoted FLDC IFNα3 and IFNβ (Fig 1C), no IFNα6 was induced in either cDCs or pDCs by this allergen. As expected, CpG was the most effective IFNα6 stimulus in all three FLDC subsets. Together, this shows that cDCs, and particularly cDC1s, can produce IFN‐I in response to both helminth Ag and allergens, as has been reported for antiviral settings (Diebold et al, 2003; Kato et al, 2005).
cDC1s in vivo produce IFN‐I in response to TLR3‐TRIF agonists such as polyI:C (Miyake et al, 2009), while pDCs primarily produce IFN‐I following TLR7 or TLR9 stimulation in a MyD88‐dependent manner (McNab et al, 2015). In agreement with this, we found that FLDC production of IFN‐I in response to SEA was TRIF‐dependent, not MyD88‐dependent (Fig 1I). In fact, IFN‐I induction was negatively regulated by MyD88 (Fig 1I), a phenomenon reported for TRIF‐dependent cytokine responses in macrophages (Johnson et al, 2008), but not previously identified as a mechanism of regulating TRIF‐dependent IFN‐I production in DCs.
In addition to their impact on DC cytokine secretion, SEA and St upregulated expression of surface molecules associated with Ag presentation and costimulation (MHC II, CD40, and CD86) in both cDC1s and cDC2s (Fig EV1C and E), while pDC phenotypic activation was not dramatically altered by either Ag (Fig EV1C). It has recently been suggested that expression of PD‐L2 and CD301b may be typical of DCs activated with Th2‐inducing Ag (Gao et al, 2013; Kumamoto et al, 2013). Although upregulation of PD‐L2 was observed on cDCs responding to SEA, increased expression of this marker was not restricted to Th2 Ag, but was also evident on DCs responding to St (Fig EV1D and F). Neither Ag influenced the low level of CD301b expressed by any FLDC subset (Fig EV1G). Notably, in most cases, the degree to which activation molecules were upregulated was less striking with SEA than St (Fig EV1). This muted cDC surface activation following exposure to SEA is in keeping with previous reports of GMDCs or human PBMC DCs exposed to SEA in vitro, or splenic and hepatic DCs from S. mansoni‐infected mice, and may reflect an alternative, and more restricted, DC activation phenotype that is common to Th2 settings (MacDonald et al, 2001; de Jong et al, 2002; Straw et al, 2003; MacDonald & Maizels, 2008; Lundie et al, 2016).
In order to act as effective Ag‐presenting cells (APCs), DCs must be able to migrate to the dLN (Alvarez et al, 2008). Since the role of the different DC subsets in Th2 priming is currently unclear (MacDonald & Maizels, 2008; Bouchery et al, 2014), and FLDC ability to generate Th2 responses has not yet been addressed, we next assessed their capacity to migrate effectively, and to initiate and polarize immune responses after adoptive transfer into naïve mice. Following injection of dsRed+ FLDCs, cDCs capably trafficked to the dLN (Fig 2A and B). Transferred CD45R+ pDCs could not be detected in the dLN (Fig 2A), in agreement with previous work demonstrating that pDCs can only gain entry to a LN via the blood through inflamed high endothelial venules and do not routinely migrate via the lymphatics (Diacovo, 2005). While the proportions of cDC1s and cDC2s were approximately 50:50 in cultures prior to transfer (as in Fig 1A), the majority of DCs reaching the dLN were cDC2s (Fig 2A and C), consistent with the suggestion that cDC2s are the primary DC subset that polarizes Th2 responses in vivo (Tussiwand et al, 2015). Interestingly, even though SEA‐exposed cDCs maintained a muted activation phenotype post‐transfer (Fig 2D), a characteristic that has also been identified in vivo during S. mansoni infection (Straw et al, 2003), they competently migrated to the dLN T cell zones (Fig 2E). In addition, despite their limited activation profile, and significant IFN‐I production, SEA‐exposed FLDCs effectively promoted Th2 polarization following transfer, with clear induction of IL‐4, IL‐5, IL‐10, and IL‐13 in dLN restimulations (Fig 2F). As in many Th2 settings (Pearce & MacDonald, 2002), transferred FLDCs also induced SEA‐specific IFNγ. It is possible that the minor population of cDC1s present in the dLN were responsible for the low‐level IFNγ response that was seen in this setting, as has been reported with the in vivo equivalents of this subtype (Everts et al, 2016). In keeping with their inflammatory phenotype, St‐exposed FLDCs capably induced recipient IL‐10, IL‐17, and IFNγ production (Fig 2G). Consistent with previous reports, no IL‐17 was detectable following SEA‐activated DC transfer (Larkin et al, 2012; Cook et al, 2015) and no Th2 cytokines were evident following St‐activated DC transfer.
Figure 2. FLDCs migrate to the dLN and induce Ag‐specific responses following adoptive transfer.
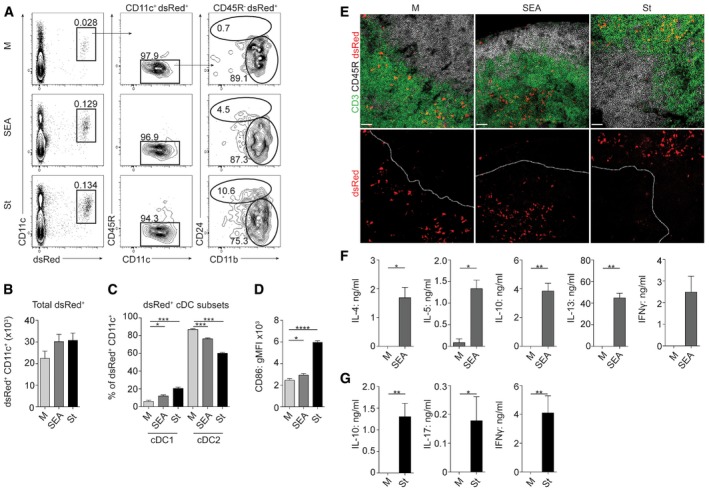
-
AdsRed+ CD11c+ FLDCs were gated as pDCs (CD45R+), cDC1s (CD45R− CD24+), or cDC2s (CD45R− CD11b+).
-
BAbsolute numbers of dsRed+ CD11c+ FLDCs in the dLN calculated from flow analysis and cell counts.
-
CPercentage of transferred dsRed+ CD11c+ cells that were cDC1s or cDC2s.
-
DCD86 expression on transferred dsRed+ CD11c+ FLDCs.
-
EConfocal microscopy of dLN sections after FLDC transfer: Upper row depicts overlay of CD3 (green), CD45R (gray) and dsRed (red); bottom row depicts dsRed (red) alone. White dashed line represents division between T cell (CD3+) and B cell zones (CD45R+). Scale bars represent 38 μm.
-
F, GSeven days after transfer, dLN cells were restimulated for 72 h with 15 μg/ml SEA (F), 1 μg/ml St (G), or medium alone (M) and cytokine production assessed by ELISA.
FLDCs depend on IFN‐I signaling for Th2 induction
Having established that SEA triggered FLDC IFN‐I secretion (Fig 1), while also conferring Th2‐initiation ability (Fig 2), we next addressed the importance of IFN‐I production in the Th2 induction process. Given that DCs themselves can be a target of IFN‐I (Montoya, 2002; Mattei et al, 2009), we first determined whether IFN‐I‐responsiveness was required for optimal DC function following SEA stimulation. To this end, we used Ifnar1 −/− mice to generate FLDCs lacking the IFNAR1 subunit of the IFN‐I receptor, thus rendering them unresponsive to IFN‐I (Hwang et al, 1995). Ifnar1 −/− FLDCs displayed significantly reduced expression of the ISGs Ifit1 and Mx1 in response to SEA (Fig 3A), and markedly impaired secretion of IFN‐I (Fig 3B), consistent with previous reports using Ifnar1 −/− fibroblasts in non‐Th2 settings (Marié et al, 1998). Additional aspects of SEA‐induced activation of WT cDC1s and cDC2s were dramatically impaired in Ifnar1 −/− FLDCs, with reduced expression of all markers measured (Fig 3C). IFN‐I signaling has been implicated in controlling many aspects of DC function, not limited to surface activation and cytokine production, but also regulating their survival and turnover (Mattei et al, 2009). However, we did not identify any defect in either differentiation, or survival, of Ifnar1 −/− FL‐cDCs (Fig EV2). Most strikingly, in the absence of IFNAR1, the ability of FLDCs to promote SEA‐specific Th2 responses in vivo was completely ablated, while IFNγ induction by these DCs was not significantly affected (Fig 3D). Further, using IL‐4 reporter mice (Mohrs et al, 2005) as recipients, we found that Ifnar1 −/− SEA‐activated FLDCs displayed impaired ability to prime CD4+ T cell IL‐4 secretion (huCD2) or mRNA expression (eGFP) in vivo (Fig 3E). Together, our data demonstrate for the first time that IFN‐I responsiveness is essential for optimal DC activation by Th2‐inducing Ag and is a key factor governing DC Th2 priming ability in vivo.
Figure 3. FLDCs depend on IFN‐I signaling for optimal activation and Th2 induction.
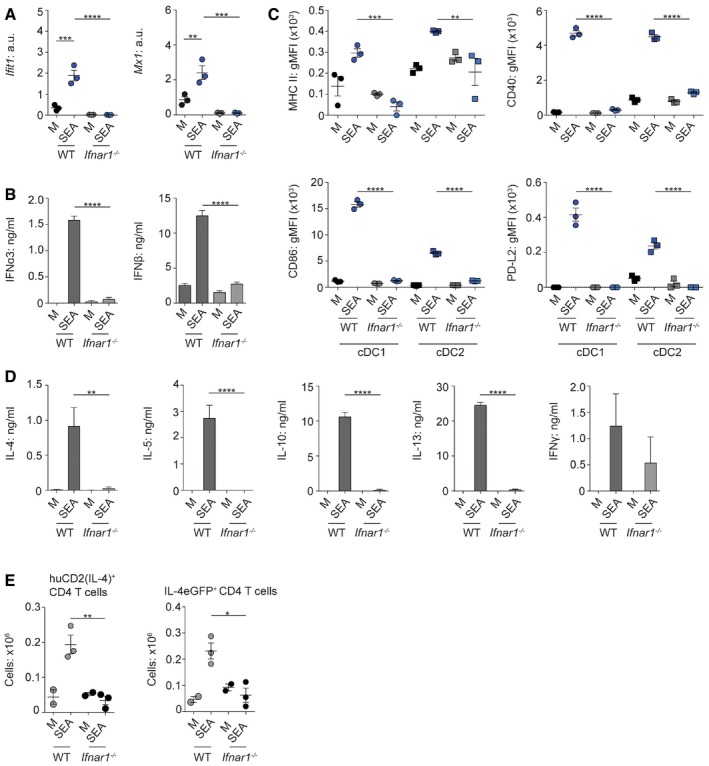
-
A–DWT or Ifnar1 −/− FLDCs were cultured with 25 μg/ml SEA or in medium alone (M) for 6 h for gene expression analysis (A) (normalized against Gapdh, a.u.) or 18 h for analysis of IFN‐I production (B), surface phenotype (C), or injection s.c. into naïve WT mice (D). (D) Seven days after transfer, dLN cells were restimulated for 72 h with 15 μg/ml SEA or medium alone and cytokine production assessed by ELISA (medium‐stimulation‐alone values subtracted).
-
EWT or Ifnar1 −/− FLDCs were transferred s.c. into KN2xIL‐4eGFP mice; 7 days later, the presence of IL‐4+ (huCD2+, IL‐4 protein) and IL‐4eGFP+ (IL‐4 mRNA) CD4+ T cells was assessed by flow cytometry.
Figure EV2. Ifnar1 −/− FLDCs display normal development and viability.
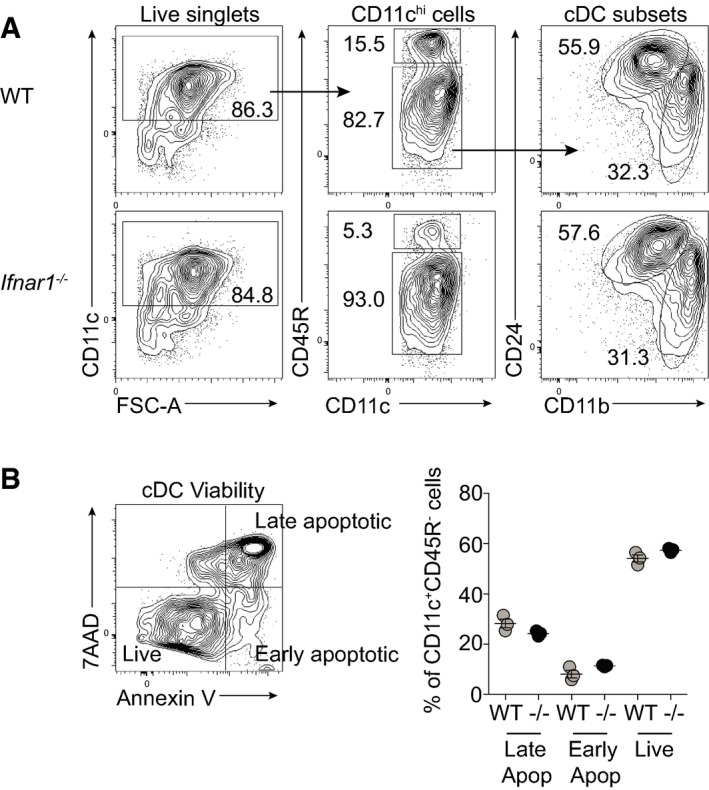
- FLDCs were cultured for 18 h in medium alone and analyzed for subsets by flow cytometry.
- Following 18‐h culture, FLDCs were stained with 7AAD and Annexin V to assess viability of cDC populations. 7AAD+ Annexin V+ were classed as late apoptotic, 7AAD− Annexin V+ as early apoptotic, and 7AAD− Annexin V− as live cells. Results are mean ± SEM.
cDCs depend on IFN‐I signaling for effective migration to, and localization within, the dLN
To determine why Ifnar1 −/− FLDCs were impaired in their Th2 induction ability, we first assessed their capacity to process and present Ag to OVA‐specific OT‐II CD4+ T cells. T cell proliferation was comparable in co‐cultures containing WT or Ifnar1 −/− cDCs with either OVA protein (OVA) or peptide (pOVA) indicating that Ifnar1 −/− cDCs had no defect in their capacity to capture, process, or present Ag in vitro (Fig 4A and B). Supporting unimpaired Ag uptake and processing in the absence of IFNAR responsiveness, Ifnar1 −/− cDCs internalized and processed DQ‐OVA as effectively as WT (Fig 4C). Further demonstrating that Ifnar1 −/− cDCs displayed no fundamental deficiency in their ability to polarize CD4+ T cells in vitro, both WT and Ifnar1 −/− cDCs co‐cultured with IL‐4, IL‐13, or IL‐10 reporter CD4+ T cells (Mohrs et al, 2005; Kamanaka et al, 2006; Neill et al, 2010; Cook et al, 2015) effectively induced T cell expression of huCD2 (IL‐4; Fig 4D), IL‐10eGFP (Fig 4E), and IL‐13eGFP (Fig 4F) under polarizing conditions. These in vitro experiments indicate that DC Ag uptake, processing, and presentation were not significantly impaired in the absence of IFNAR1 signaling.
Figure 4. Ifnar1 −/− FL‐cDC APC function is comparable to WT FL‐cDC in vitro .
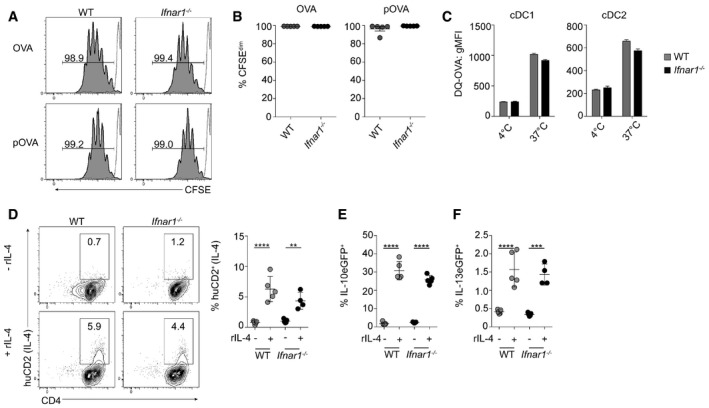
-
A, BFL‐cDCs were sorted and co‐cultured with CFSE‐labeled OT‐II T cells, with either OVA protein (OVA) or peptide (pOVA), for 96 h. Proliferation was determined by flow cytometric analysis of CFSE dilution. The gray dashed line represents non‐proliferating OT‐II T cell controls.
-
CTo assess Ag uptake and processing, WT or Ifnar1 −/− FLDCs were cultured with DQ‐OVA for 2 h at 37°C or 4°C and uptake assessed by flow cytometry.
-
D–FSorted FL‐cDCs were cultured with eGFP− CD4+ T cells from KN2xIL‐13eGFP or KN2xIL‐10eGFP animals, with anti‐CD3, in the presence (+) or absence (−) of 20 ng/ml rIL‐4. IL‐4 (huCD2, D), IL‐10eGFP (E), or IL‐13eGFP (F) expression on CD4+ T cells was assessed by flow cytometry after 72 h of culture. Positive cells were expressed as a percentage of all CD4+ T cells.
The ability of Ifnar1 −/− cDCs to activate and polarize Th2 cells in vitro (Fig 4), but not in vivo (Fig 3), strongly suggested that IFN‐I responsiveness may be important for effective migration of Th2‐promoting DCs. As DC migration through the lymphatic system, and within the dLN, is governed by the chemokine receptor CCR7 and its ligands CCL21 and CCL19 (Braun et al, 2011), we next assessed FL‐cDC subset expression of CCR7 (Fig 5A). Both cDC subsets expressed low levels of CCR7 on their surface in medium‐alone conditions, which may account for the steady‐state migration that was evident following adoptive transfer of these unstimulated cells (Fig 2B). cDC1s and cDC2s upregulated CCR7 following SEA exposure, and this was significantly reduced in the absence of IFNAR1 (Fig 5A). However, we saw little expression of CXCR5 on WT or Ifnar1 −/− cDCs (Fig 5B), despite effective staining of this chemokine receptor on other cell types, including St‐stimulated GMDCs (Fig EV3A) and T follicular helper cells (Fig EV3B), and in contrast to previous work suggesting that CXCR5 is required for DC induction of Th2 responses (León et al, 2012).
Figure 5. FL‐cDC migration and localization in vivo is deficient in the absence of IFNAR signaling.
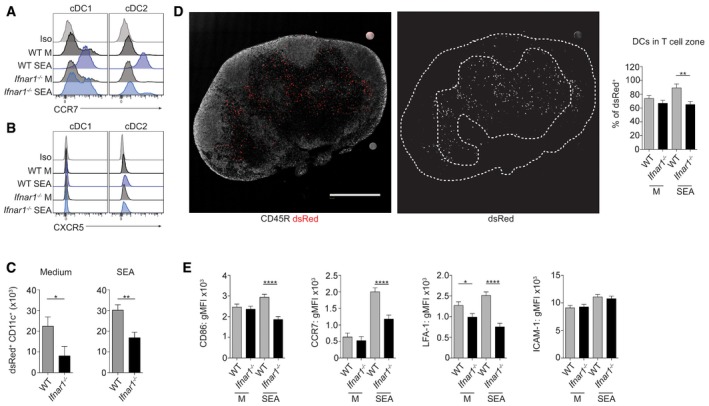
-
A, BWT or Ifnar1 −/− FLDCs were cultured for 18 h with 25 μg/ml SEA, or in medium alone (M) and CCR7 (A) and CXCR5 (B) expression assessed by flow cytometry.
-
C–EWT or Ifnar1 −/− dsRed+ FLDCs were injected s.c. into WT mice, and dLNs were harvested 48 h later and analyzed by flow cytometry (C) or confocal microscopy (D) to identify dsRed+ cells. (C) The total number of dsRed+ cDCs in the dLN was calculated from flow data and cell counts. WT data are from the same pooled experiments as Fig 2B. (D) Left image shows an overlay of CD45R+ B cells (gray) and dsRed+ cells (red). Scale bar represents 1 mm. Right image shows dsRed alone (gray), white dashed line represents B cell zone of LN, defined by CD45R staining in left image. Images shown are of dLN following transfer of WT FLDCs cultured in medium alone. The percentage of WT and Ifnar1 −/− dsRed+ cells within the T cell zone was calculated for each condition. (E) The surface phenotype of transferred WT and Ifnar1 −/− dsRed+ CD11c+ cells was assessed by flow cytometry.
Figure EV3. St elicits a strong upregulation of CXCR5 on the surface of GMDCs.
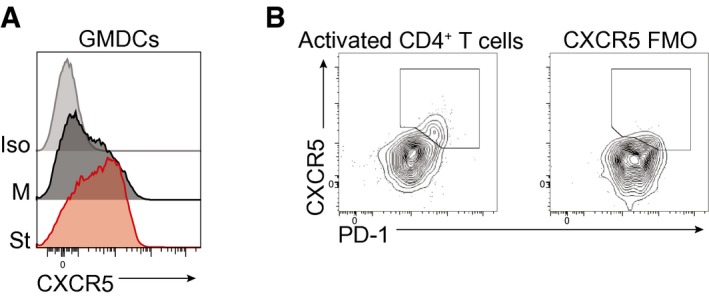
- GMDCs were cultured for 18 h in medium alone (M) or in the presence of St, and the expression of CXCR5 was analyzed by flow cytometry and compared to an isotype control (Iso).
- Expression of CXCR5 and PD‐1 on activated (CD44hi CD62Llo) CD4+ Foxp3− T cells from hepatic LN of Schistosoma mansoni‐infected mouse on day 42 of infection. FMO (fluorescence minus one) of CXCR5 expression.
Our tracking experiments using WT dsRed+ FLDCs had demonstrated efficient migration of cDCs to dLN T cell zones following adoptive transfer (Fig 2A–C). However, in the absence of IFNAR1 expression, cDC numbers reaching the dLN after injection were reduced by ~50% (Fig 5C). Since CCR7 is required for effective migration of cDCs to, and appropriate localization within, dLN (Braun et al, 2011), confocal microscopy was used to determine the location of transferred Ifnar1 −/− DCs (Fig 5D). While the majority of WT SEA‐exposed dsRed+ FLDCs homed to the dLN T cell zones, there was a significant reduction in the proportion of Ifnar1 −/− SEA‐exposed DCs found within this region (Fig 5D). In agreement with our phenotypic studies prior to transfer (Figs 3C and 5A), Ifnar1 −/− SEA‐exposed cDCs expressed lower levels of CD86 and CCR7 on their surface post‐transfer, compared to WT (Fig 5E). Similarly, LFA‐1 expression was lower on Ifnar1 −/− DCs compared to WT post‐transfer, while ICAM‐1 levels were equivalent (Fig 5E).
IFN‐I involvement in Th2 development in vivo is not restricted to FLDCs
Having identified that IFNAR1 plays a key role in regulating activation of FLDCs responding to Th2 Ag in vitro (Fig 1), and their ability to initiate Th2 responses in vivo (Fig 3), we next assessed the broader role of this signaling pathway in Th2 priming in vivo.
We first investigated the expression of ISGs in splenic cDCs purified by FACS from mice challenged intravenously (i.v.) with SEA (Fig 6A). This demonstrated significant upregulation of a range of ISGs by cDCs responding to SEA in vivo, including Ifit1, Ifit3, and Mx1 (Fig 6B). ISG upregulation was not restricted to the spleen, as mice that had been sensitized intraperitoneally (i.p.) and challenged i.v. with dead S. mansoni eggs (Wynn et al, 1997) showed significant expression of the ISGs Mx1 and Oasl2r in the lung (Fig 6C). Further, when mice were sensitized intradermally (i.d.) with HDM, cDC expression of the IFN‐responsive surface markers CD317 (Blasius et al, 2006) and Sca‐1 (Essers et al, 2009) was significantly upregulated compared to expression on cDCs from mice treated with the IFNAR1‐blocking antibody MAR1‐5A3 (Sheehan et al, 2006), indicating that expression of these markers was IFN‐I‐driven (Fig 6D). Together, these data demonstrate that SEA, S. mansoni eggs, and HDM stimulate an IFN‐I signature in vivo and that this is not restricted to FLDCs in vitro.
Figure 6. IFN‐I is required for Th2 induction in vivo .
-
ASplenic DCs were FACS‐isolated following 12‐h exposure to 50 μg SEA administered i.v. Cells were gated as intact singlets and cDCs identified as CD45R− CD11c+ cells sorted to > 95% purity.
-
BmRNA expression of ISGs by splenic sorted cDCs was assessed by qPCR (normalized against Gapdh, a.u.) following in vivo exposure to SEA.
-
CmRNA expression of ISGs in whole lung following pulmonary challenge with Schistosoma mansoni eggs (normalized against beta‐actin, a.u.).
-
DMice were challenged i.d. with 100 μg HDM in conjunction with 200 μg of the IFNAR1‐blocking Ab MAR1‐5A3 or an isotype control. Twenty‐four or 48 h later, auricular LNs were harvested and CD317 and Sca‐1 expression on cDCs analyzed.
-
E, FA total of 2,500 S. mansoni eggs were injected s.c. per foot into WT or Ifnar1 −/− mice, dLNs were harvested 7 days later, and cell counts were performed (E). (F) Cells were cultured with anti‐CD3 for 72 h and then cytokines measured by ELISA (medium background subtracted).
-
GIL‐4eGFP mice were challenged i.d. with 100 μg HDM in conjunction with 200 μg of the IFNAR1‐blocking Ab MAR1‐5A3 or an isotype control. Mice were given a second dose of Ab i.p. 48 h later. On day 7, dLNs were harvested and total cell numbers, CD44hi CD4+ T cell numbers, and IL‐4eGFP+ T cell numbers assessed.
-
HA total of 2.5 × 103 S. mansoni eggs were injected s.c. with (+DC) or without 1 × 106 WT FLDCs into WT or Ifnar1 −/− recipients. On day 7, dLNs were harvested, cells were stimulated with anti‐CD3 for 72 h, and cytokines were measured by ELISA (medium background subtracted).
We next assessed the importance of IFN‐I responsiveness for Th2 development after direct in vivo challenge with either dead S. mansoni eggs or HDM. While WT mice displayed significantly increased cellularity in the dLN following S. mansoni egg injection, this was not evident in similarly challenged Ifnar1 −/− mice (Fig 6E). Importantly, there was a significant defect in Th2 polarization and IFNγ induction in the dLN of Ifnar1 −/− mice after egg injection (Fig 6F). This was not just a feature of helminth Th2 immunity, or of Ifnar1 −/− mice, as administration of the IFNAR1‐neutralizing antibody MAR1‐5A3 (Sheehan et al, 2006) significantly impaired cellularity, and numbers of dLN CD44hi effector and IL‐4‐producing CD4+ T cells, in the dLNs of mice sensitized i.d. with HDM (Fig 6G). Finally, this impairment in Th2 induction in Ifnar1 −/− mice after egg injection was “rescued” by co‐transfer of WT FLDCs (Fig 6H), indicating that IFN‐I responsiveness in DCs alone is sufficient for Th2 initiation in an Ifnar1 −/− environment. Together, these data establish the importance of IFNAR1 signaling in DCs for optimal Th2 polarization against either helminths or allergens in vivo.
Discussion
We have identified IFN‐I production by DCs responding to Th2 Ag (Fig 1) and demonstrated for the first time that IFN‐I responsiveness is essential for optimal DC activation and effective Th2 response induction in vivo (Figs 3 and EV4). This extends our fundamental understanding of the context‐dependent role for IFN‐I during immune response development, building upon previous studies in which it has been implicated as an important regulator of basal DC activation (Montoya, 2002), and function during viral infection (Pinto et al, 2011).
Figure EV4. A TRIF‐dependent IFN‐I signature is induced in response to Schistosoma mansoni egg Ags and is required for optimal DC activation, migration, and Th2 priming capacity.
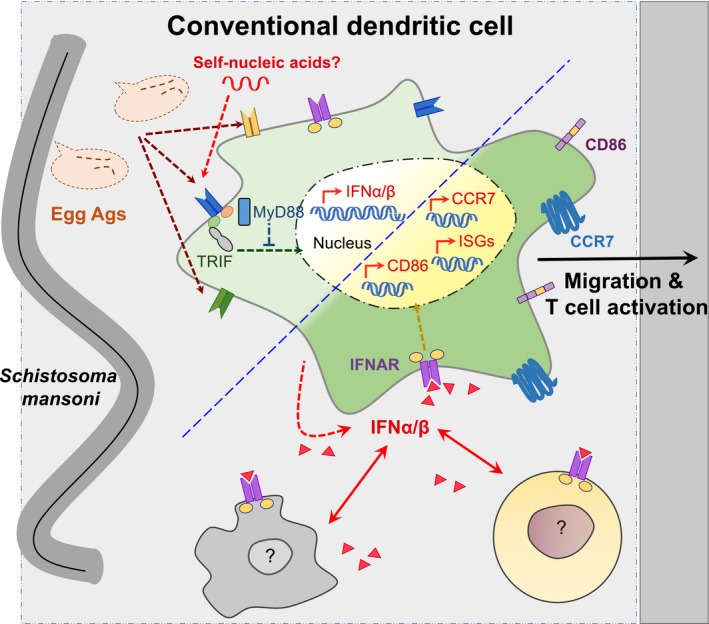
Th2‐inducing Ags, such as those released from S. mansoni eggs, bind to a range of Ag receptors on the surface of cDCs. Ags such as omega‐1, which binds the mannose receptor, likely act in concert with parasite‐ and/or host‐derived nucleic acid ligands that bind TRIF‐dependent receptors and activate the transcription of IFN‐I genes. This leads to the secretion of IFN‐I, including IFNα subtypes and IFNβ. cDCs themselves express the IFN‐I receptor IFNAR and can therefore respond to IFN‐I produced from a range of sources including DCs and other immune cells. This stimulates the activation of IFN‐dependent signaling pathways which leads to transcription of a range of IFN‐dependent genes, including ISGs, and upregulation of surface markers such as CCR7 and CD86. In this way, IFN‐I signaling enhances the activation, migration, and antigen presentation capabilities of cDCs during type 2 inflammation.
A key aspect of DC activity controlled by IFN‐I signaling, which is essential for APC function, is effective migration (Kapsenberg, 2003; Alvarez et al, 2008). Studies using in vitro migration assays have previously suggested that IFN‐I is required for effective transmigration (Mattei et al, 2009; Rouzaut et al, 2010). Our study is the first to demonstrate the importance of IFN‐I in promoting DC migration in vivo—from the tissue site, via the lymphatics, to the dLN—in response to pathogen‐associated Ag (Fig 5).
Perhaps surprisingly, there is very little work currently published that has focused on the migratory capacity of DCs in a Th2 setting. While it is widely accepted that CCR7 and its ligands CCL19 and CCL21 are the key chemokine receptor–ligand pairs that control DC migration under most circumstances (Alvarez et al, 2008), León et al (2012) suggested that during helminth infection, the essential chemokine that governs DC Th2 induction is CXCR5 (León et al, 2012). CXCR5 interacts with CXCL13 and is required for the formation of germinal centers (Crotty, 2011). CCR7 was expressed to some degree on cDCs cultured in medium alone (Fig 5), perhaps accounting for the homeostatic migration of unstimulated DCs that is evident in Fig 2. Although we identified SEA‐specific upregulation of CCR7 expression on cDCs, significant CXCR5 expression was not seen under any circumstances (Fig 5). Further, our data demonstrate an integral role for signaling via IFNAR1 in effective DC Th2 priming and LN T cell zone localization in vivo, strongly indicating that DC induction of Th2 responses does not require localization to the fringe of the B cell zone, as has previously been proposed (León et al, 2012). It is possible that at later stages of immune activation during helminth infection, CXCR5 becomes more important for T cell polarization and for T follicular helper cell differentiation (Fairfax et al, 2015), while in the early stages of Th2 induction CCR7 plays a more dominant role. Further work is required to determine whether distinct chemokine receptor–ligand pairs are key for DC function at different stages of type 2 differentiation.
As reported by others in non‐Th2 settings (Mattei et al, 2009), we found that CCR7 upregulation on SEA‐activated cDCs was controlled by IFNAR1 signaling, particularly on cDC2s (Fig 5), the primary DC subset responsible for Th2 priming. Additionally, by phenotypic analysis of DCs post‐transfer, we identified that optimal expression of the integrin LFA‐1 also depended on IFNAR1 (Fig 5). It has previously been shown using human PBMC DCs that LFA‐1 facilitates transmigration of DCs across endothelium and is upregulated on DCs by IFN‐I treatment (Rouzaut et al, 2010). Further, disruption of Rap1, which is required for cell surface stabilization of LFA‐1 (Hogg et al, 2011), leads to decreased GMDC ability to localize to T cell zones of skin dLN (Katagiri et al, 2004). As it has been shown that Rap1 activation is also controlled by IFNAR (Platanias, 2005), this adds further weight to our hypothesis that IFN‐I may be influencing cDC migration by facilitating the upregulation of important chemokine receptors and integrins on the surface of cDCs. Thus, it is likely that the combined impact of reduced CCR7 and LFA‐1 accounts for defective migration and homing of Ifnar1 −/− FLDCs in vivo. In addition, the significant reduction in cDC expression of MHC II, CD40, CD86, and PD‐L2 (Fig 3C), all thought to be involved in Th2 induction (MacDonald et al, 2001, 2002; Straw et al, 2003; Gao et al, 2013), likely contributes to the striking inability of Ifnar1 −/− FLDCs to effectively prime Th2 responses in vivo. CD40 in particular has been shown to be essential for this process (MacDonald et al, 2002), and the significant impairment in CD40 expression is likely to contribute to the phenotype we see in vivo.
In our work, despite lower levels of activation (Fig 3), Ifnar1 −/− cDC APC ability was not significantly impaired in vitro (Fig 4), contrasting previously published reports that suggested a reduced priming capacity of Ifnar1 −/− cDCs in OVA‐specific T cell priming models in vitro (Montoya, 2002; Diamond & Farzan, 2013). This discrepancy could be due to technical differences, for example, Ag availability or the ratio of DCs:T cells present, that might alter the importance of cDC IFN‐I responsiveness during T cell activation in vitro. Conceivably, the strikingly impaired Th2 priming by Ifnar1 −/− DCs that we have identified following their adoptive transfer into recipient mice (Fig 3) may in part be due to restricted Ag availability and a relatively small Ag‐specific T cell pool in vivo in WT mice.
An important aspect of DC function in type 2 settings that remains poorly understood is the pattern recognition receptors (PRRs) and pathways used by DCs to respond to Th2‐associated Ag, and how these might influence Th2 priming. Here, we demonstrate that the FLDC IFN‐I response to SEA is dependent on the adaptor protein TRIF, while MyD88 acts as a negative regulator of this pathway (Figs 1 and EV4). Identification of TRIF as an important adaptor protein for DC activation by SEA suggests a role for the TRIF‐dependent PRRs in sensing IFN‐I inducing ligands following exposure to Th2 Ag (Fig EV4). Such PRRs are primarily nucleic acid sensing and include TLR3, TLR4, and the cytoplasmic receptor complex DDX1–DDX21–DDX36 (Broz & Monack, 2013; McNab et al, 2015). The role of MyD88 as a negative regulator of PRR signaling has not previously been reported in a Th2 setting, although it is known that Th2 induction is not impaired in the absence of splenic DC, GMDC, or global MyD88 expression (Layland et al, 2005; Jankovic et al, 2002, 2004; Marshall & Pearce, 2008). Our data demonstrate a requirement for TRIF in optimal FLDC activation in response to a Th2 Ag and highlight the need for further study in this area to address the impact of TRIF deficiency on DC Th2 induction. Additionally, our demonstration of FLDC IFN‐I induction by the key immunostimulatory molecule in SEA omega‐1 (Fig 1) suggests that glycoproteins and C‐type lectin receptors such as the mannose receptor, which has been shown to recognize omega‐1 (Everts et al, 2012), may be involved in this process.
Having identified that there was a degree of differential expression of the IFN‐I subtypes IFNα3, IFNα6, and IFNβ by cDCs responding to Th2 Ag (Fig 1), it would be of interest to characterize cDC expression of additional IFN‐I subtypes in response to different Th2 Ags. Whether distinct subtypes are more or less responsible for conferring effective Th2 induction ability on cDCs remains to be shown, although current understanding is that the diverse IFN‐I subtypes exert similar biological function in viral infection (van Pesch et al, 2004).
The question of why IFN‐I induction, normally associated with intracellular infections, has evolved against a helminth parasite, and whether this IFN‐I signal benefits the host and/or the parasite, remains. IFN‐I has previously been suggested to act as a damage signal that enhances early innate immune activation (Gregorio et al, 2010). Many of the PRRs that induce IFN‐I production sense nucleic acid ligands (McNab et al, 2015). Our work comparing the IFN‐I signature induced by live or dead eggs (Figs 1 and EV1) suggests that the molecules responsible for IFN‐I induction are primarily components released by dead or dying eggs. This would include omega‐1, as well as implicating ligands such as nucleic acids that are not normally secreted by live intact eggs. In addition, omega‐1 is a RNase that degrades messenger RNAs (Everts et al, 2012), likely leading to the production of self‐nucleic acid ligands. This RNase activity is essential for the Th2‐polarizing capacity of omega‐1 (Everts et al, 2012). Thus, it is possible that one way that Th2 Ag enhances polarization of Th2 cells may be via the stimulation of IFN‐I production, a previously unrecognized role for this damage‐associated signal. It has also been shown that IFN‐I can limit IL‐12 production upon secondary stimulation with TLRs (McNab et al, 2015); thus, IFN‐I may also promote Th2 polarization by blocking differentiation down the Th1 route.
Our identification of this central role for IFN‐I in DC Th2 promotion raises many new questions about the broader impact of IFN‐I during type 2 inflammation. Importantly, the DC focus of our work does not exclude additional roles for IFN‐I in the wider Th2 setting, and how IFN‐I may influence effector functions of other immune cells during helminth infection remains to be determined. Similarly, our identification of a DC‐derived source of IFN‐I does not rule out its production by additional cell types exposed to helminth products or allergens in vivo (Fig EV4). Discovery of a key role for IFN‐I in DC Th2 ability has implications not only for development of therapeutics aimed at modulating type 2 responses, but also in the context of helminth co‐infection with bacterial or viral pathogens, where IFN‐I is known to impact dramatically on disease outcome (McNab et al, 2015). In future work, it will be important to define whether IFN‐I induced in response to helminth Ag is a long‐term cost, or benefit, to pathogen and host.
Together, our work emphasizes the pleiotropic nature of IFN‐I and the importance of considering the previously unappreciated role of this family of cytokines, ordinarily associated with antiviral immunity, in promotion of type 2 inflammation.
Materials and Methods
Mice
C57BL/6xIL‐4eGFP, dsRed, dsRedxIfnar1 −/−, Ifnar1 −/−, Myd88 −/−, IFNα6GFP, KN2xIL‐4eGFP, KN2xIL‐10eGFP, KN2xIL‐13eGFP, OT‐IIxLy5.1, and Trif −/− mice, all on the C57BL/6 background, were bred and maintained at the University of Edinburgh, the University of Manchester, or the Malaghan Institute of Medical Research, under specific pathogen‐free conditions (Hwang et al, 1995; Adachi et al, 1998; Barnden et al, 1998; Yamamoto, 2003; Vintersten et al, 2004; Mohrs et al, 2005; Kamanaka et al, 2006; Kumagai et al, 2007; Neill et al, 2010). Control C57BL/6 (WT) mice were either bred and maintained in‐house or obtained commercially from Harlan or the Jackson Laboratory. Experiments were conducted under a license granted by the Home Office (UK), in accordance with local guidelines, or under approval from the Victoria University Animal Ethics Committee, and performed according to Malaghan Institute guidelines (HDM challenge in IL‐4eGFP mice), or under approval from the ethics committee of the University of Liege (S. mansoni egg lung challenge model).
Cell culture
For FLDC generation, BM cells were cultured with 200 ng/ml Flt3‐L (PeproTech), according to published methods (Naik et al, 2010). Briefly, BM cells were flushed from femurs and tibia of adult mice and resuspended for counting after red blood cell (RBC) lysis. Cells were then cultured at 37°C 5% CO2 at 1.5 × 106 cells/ml in RPMI 1640 medium (Sigma) containing Flt3‐L, 10% fetal calf serum (Sigma), 2 mM L‐glutamine, 50 mM 2‐mercaptoethanol (Invitrogen), 50 U/ml penicillin, and 50 μg/ml streptomycin (Sigma). FLDCs were harvested on d8 and replated at 2 × 106/ml for 18 h in the presence or absence of Ag. For gene expression analyses, cells were cultured for 6 h in medium alone or with Ag. Cells were exposed to 25 μg/ml SEA, 5 μg/ml St, 50 μg/ml HDM (Dermatophagoides pteronyssinus; Greer Laboratories), 1:500 live S. mansoni eggs (purified from the livers of C57BL/6 mice at approximately day 49 of S. mansoni infection), or dead eggs (freeze/thawed) or 500 ng/ml recombinant omega‐1 (generated in Nicotiana benthamiana and purified from the leaf extracellular space using POROS 50 cation resin (Life Technologies) (Wilbers et al, 2017). Endotoxin‐free SEA from S. mansoni eggs was prepared in‐house as previously described (MacDonald et al, 2001). In brief, S. mansoni eggs were homogenized using a Tenbroeck tissue grinder in PBS, and supernatant was collected, centrifuged, filtered (0.45 μm), and stored at −70°C. St, strain SL3261, was provided by Dr. Maurice Gallagher (University of Edinburgh) and was heat‐killed by incubation at 80°C for 30 min and stored in PBS at 4°C until use.
FLDC sorting and flow cytometry
For some experiments, FLDC bulk cultures were sorted by FACS into separate subset populations prior to Ag stimulation. FLDCs were harvested on d8 of culture, and following FcR‐Block (2.4G2), cells were surface‐stained with mAbs against CD11c, CD45R (B220), CD11b, and CD24. Sorted or unsorted DC populations were replated at 2 × 106/ml and stimulated with Ag for 18 h as described above. Following Ag stimulation, DC surface activation was assessed by flow cytometry. Cells were first stained with LIVE/DEAD Fixable Aqua or UV (Life Technologies) and, following FcR‐Block, were stained with combinations of the following mAbs: CCR7, CD11b, CD11c, CD24, CD40, CD45R, CD301b, CXCR5, CD86, MHC II, and PD‐L2. For analysis of cell viability, cells were stained with Annexin V and 7‐AAD. All antibodies for flow cytometry were purchased from BD, eBioscience, or BioLegend. IFNα6GFP FLDCs were also stained with the above antibodies and their expression of GFP analyzed by flow cytometry. To assess antigen uptake, 2 × 105 FLDCs were incubated with 10 μg DQ‐OVA (Life Technologies) for 2 h at 37°C or 4°C, before flow cytometric analysis. Samples were acquired on a FACSCanto II or LSR‐Fortessa flow cytometer using BD FACSDiva Software and analyzed with FlowJo software (Tree Star, Inc.) to ascertain proportional expression and geometric mean fluorescence intensity (gMFI) for surface marker expression.
Cell tracking and transfer
For cell tracking and transfer experiments, WT, dsRed+, Ifnar1 −/−, or Ifnar1 −/− dsRed+ FLDCs were cultured as above for 6 or 18 h, with SEA or St or in medium alone. For tracking of dsRed+ cells, FLDCs were injected subcutaneously (s.c.) into WT recipients (1 × 106/foot). Following dsRed+ cell transfer, dLNs were harvested at 48 h post‐injection. Popliteal LNs (pLNs) were digested at 37°C for 30 min with 0.4 U/ml Liberase TL (Roche) and 80 U/ml DNase I type IV (Sigma). Single‐cell suspensions were prepared by mechanical disruption of LNs through a 70‐μm filter. LN suspensions were then analyzed by flow cytometry for the presence of dsRed+ CD11c+ cells. dLN cells were first stained with LIVE/DEAD Fixable Aqua or UV, and following FcR‐Block, cells were stained with mAb against CD11b, CD11c, CD24, CD45R, CD86, CCR7, ICAM‐1, and LFA‐1. Identification of transferred cells was performed following exclusion of Lin+ cells using CD3, CD19, CD49b, Ly6G, and NK1.1. Monocytes (Ly6C+ CD11b+) and macrophages (CD11c− F4/80+) were also excluded. For cell transfer experiments, FLDCs were injected into WT recipients (0.25 × 106/foot) and dLNs harvested 7 days later. Single‐cell suspensions of LN cells (5 × 106 cells/ml) were cultured in X‐VIVO 15 medium (Lonza) containing 2 mM L‐glutamine and 50 μM 2‐ME (Life Technologies) in 96‐well plates at 37°C 5% CO2 and stimulated with SEA (15 μg/ml) or St (1 μg/ml). Supernatants were harvested after 72 h and cytokine production assessed by ELISA.
Immunohistochemistry
Following dsRed+ cell transfer, dLNs from recipient mice were fixed in 4% paraformaldehyde (PFA; Sigma), incubated sequentially in 15 and 30% sucrose (Sigma) solutions overnight to quench residual PFA, and then mounted in Optimal Cutting Temperature (O.C.T.) embedding medium (Sakura). Thin‐section confocal microscopy was performed on 20‐μm cryostat sections, which were fixed in ice‐cold acetone for 10 min, and then incubated for 1 h with mAb against CD3 conjugated to 488 (eBioscience, UK), and CD45R conjugated to Alexa647 (BD Biosciences, UK). Slides were washed extensively in PBS–Tween‐20, PBS, and then water before nuclei were stained with DAPI‐supplemented ProLong Fade Gold (Life Technologies) mounting media. Samples were imaged at room temperature (~23°C) using TCS SP5 II or TCS SP8 CW microscopes (Leica Microsystems), with laser lines with emission wavelengths of 405 (DAPI), 488 (Alexa Fluor 488), 543 (dsRed), and 633 and 647 (Alexa Fluor 647) nm, with 20× (HC PL APO; N.A. = 0.75), 40× (HCX PL APO CS; N.A. = 1.25), and 63× (HCX PL APO; N.A. = 1.4) objectives with immersion oil (Type F, Leica), using LAS AP or LAS X software (Leica). Fluorophore emission light was collected using PMT or Hybrid Detectors (HyD). Data were rendered and analyzed using Volocity (Improvision) or ImageJ (NIH) software. To analyze the localization of dsRed+ cells in specific dLN regions, the total LN area was measured on a binary image of the DAPI staining, the B cell zone area identified using CD45R staining, and the T cell zone as the CD45R− region. A threshold of 65‐255 (the mean intensity of background in uninjected samples) was applied to the dsRed channel and the total area of dsRed ascertained. dsRed+ cells within the T cell zone were then calculated as a percentage of all dsRed+ cells within the LN.
Immunizations and in vivo treatments
To assess the IFN‐I signature of in vivo DCs responding to Th2 Ag, mice were injected intravenously with PBS or 50 μg SEA. Eighteen hours later, spleens were digested (as pLN, above) for 15 min at 37°C, single‐cell suspensions prepared by mechanical disruption through 70‐μm filters and low‐density cells enriched using NycoPrep (1.077 g/ml; Axis‐Shield). For purification of splenic DCs, non‐DC lineage cells were removed using biotinylated mAbs against murine CD2, CD3ε, CD49b, mIgM, and erythrocytes (Ter‐119) and MyOne Streptavidin Dynabeads (Invitrogen). Cells were then stained with antibodies against CD11c and CD45R and cDCs sorted as CD11chi CD45R− directly into RNALater (Ambion) for subsequent RNA extraction. To assess Th2 priming in vivo, 2.5 × 103 dead S. mansoni eggs were injected s.c. per foot into recipient mice. In some experiments, 1 × 106 WT FLDCs were co‐transferred with S. mansoni eggs. Draining pLNs were harvested 7 days later, and restimulated with plate‐bound αCD3 (0.5 μg/well) for 72 h. An alternative model of in vivo priming involved intradermal (i.d.) challenge into the ear pinnae with 100 μg HDM whole bodies. Auricular dLNs were collected 24 h, 48 h, or 7 days later. Flow cytometry was used to analyze CD45R− cDC expression of CD317 and Sca‐1 at 24 h or 48 h, and to assess the number of IL‐4eGFP+ T cells at day 7. In these experiments, mice were either treated with an anti‐IFNAR1 antibody (MAR1‐5A3) or an isotype control (MOPC‐21; both from BioXcell), with 200 μg Ab mixed with HDM given i.d. Mice received a second Ab dose 48 h later i.p. In order to address induction of ISG expression following dead S. mansoni egg challenge of the lung, mice were injected with 5,000 dead S. mansoni eggs i.p. On day 14 mice were challenged with 5,000 dead eggs i.v. Lungs were then collected on day 22 and a portion reserved for gene expression analysis.
ELISA
ELISAs were performed on culture supernatants using paired mAb and recombinant cytokine standards, or DuoSets (BioLegend, eBioscience, BD, R&D Systems, and PeproTech). For restimulation data, medium‐alone values were subtracted from Ag restimulated cytokine levels for each sample.
RNA extraction and qPCR analysis
RNA was recovered from FLDCs or splenic DCs using TRIzol reagent (Life Technologies) or RNALater, respectively. Lung tissue was prepared using the mirVana miRNA isolation kit (Life Technologies). RNA was translated into cDNA using Superscript III Reverse Transcriptase and Oligo (dT; Life Technologies). Quantitative RT–PCR was performed using a LightCycler 480 II Real‐Time PCR machine (Roche) and LightCycler‐DNA master SYBR Green I (Roche) and compared to a serially diluted standard of pooled cDNA. The relative amount of mRNA for genes of interest was normalized to GAPDH or beta‐actin. The following primers were used: β‐actin: 5′‐AGAGGGAAATCGTGCGTGAC‐3′, 5′‐ACGGCCAGGTCATCACTATTG‐3′; Gapdh: 5′‐AATGTGTCCGTCGTGGATCT‐3′, 5′‐CCCAGCTCTCCCCATACATA‐3′; Ifit1: 5′‐TCTAAACAGGGCCTTGCAG‐3′, 5′‐GCAGAGCCCTTTTTGATAATGT‐3′; Ifit3: 5′‐TGAACTGCTCAGCCCACA‐3′, 5′‐TCCCGGTTGACCTCACTC‐3′; Mx1: 5′‐TTCAAGGATCACTCATACTTCAGC‐3′, 5′‐GGGAGGTGAGCTCCTCAGT‐3′; Oas1a: 5′‐GCTGCCAGCCTTTGATGT‐3′, 5′‐TGGCATAGATTGTGGGATCA‐3′; and Oasl2r: 5′‐GGATGCCTGGGAGAGAATCG‐3′, 5′‐CAGTTTCGAAGAGCAGGCGA‐3′.
DC: T cell co‐culture assays
For CFSE dilution assays, splenic and LN CD4+ OT‐II T cells were purified using CD4+ Dynabeads (Life Technologies) following the manufacturer's protocol. T cells were labeled with 5 μM CFSE (Life Technologies) for 15 min at 37°C, prior to culture with 5 × 104 WT or Ifnar1 −/− FLDCs in the presence of 0.01 μg/ml OVA323–339 peptide (CRB) or 5 μg/ml OVA protein (Sigma) which had been endotoxin depleted in‐house. Cultures were incubated at 37°C for 4 days prior to assessment of CFSE dilution by flow cytometry. In vitro co‐culture polarization experiments were performed with 5 × 104 KN2xIL‐13eGFP− CD4+ or KN2xIL‐10eGFP− CD4+ T cells which were cultured in 96‐well plates for 4 days with 2,500 WT or Ifnar1 −/− FLDCs, 1 μg/ml soluble anti‐CD3, and with or without rIL‐4 (20 ng/ml; PeproTech; Cook et al, 2015). T cell expression of huCD2 (IL‐4; Life Technologies) or eGFP (IL‐10 or IL‐13) was then assessed by flow cytometry.
Statistical analysis
Statistical analyses were carried out using GraphPad Prism 5 or JMP (SAS Institute). The data were checked to confirm normality and that groups had equal variance. One‐way analysis of variance (ANOVA) with Tukey's multiple comparison tests was employed to determine significant differences between sample groups. Results from these tests were reported as significant if P ≤ 0.05, with results from these tests shown as mean ± SEM. For some experiments, statistical analysis was carried out using JMP, in which case data were analyzed using three‐way full‐factorial fit models to assess effects such as “genotype”, “treatment”, and “experiment” on the response variable of interest. This allowed the interaction between effects to be taken into account in addition to their impact on the response variable, which enabled experimental repeats to be pooled, increasing the power of the analysis. The least squares mean results table from the three‐way full‐factorial analysis was used to test the contrast between specific experimental groups using a joint F‐test. A difference between experimental groups was taken to be significant if the P‐value (Prob > F) was less than or equal to 0.05, with results in graphs shown as least squares mean ± SEM.
Author contributions
LMW and RJL designed the project and coordinated and carried out the experimental work. JGB and ANRC helped design the project and carried out the microscopy work. SLB, AMD, LMC, LHJ‐J, PCC, and ATP‐A carried out the experimental work. PCC, LHJ‐J, and ATP‐A contributed to the design of in vitro and in vivo assays. RHPW contributed recombinant omega‐1. DMD provided some equipment and supervised some of the research. CJ provided IFNα6GFP mice. BGD and FR supervised some of the research. ASM conceived and designed the project and supervised the research. LMW, RJL, and ASM wrote the manuscript, with valuable input from all the other authors.
Conflict of interest
F.R., A.S.M., and L.M.C. are listed as inventors on a provisional patent application concerned with the subject matter of this paper. The MCCIR is a joint venture between the University of Manchester, GSK, and AstraZeneca.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was supported by the MRC (G0701437 to A.S.M.) and the Wellcome Trust (WT086628MA). R.J.L. is the recipient of a National Health and Medical Research Council of Australia (NHMRC) Early Career Fellowship. Funding was also provided to F.R. by the Health Research Council of New Zealand. B.G.D. is a senior researcher from the F.R.S‐FNRS. A.M.D. was funded by the ULg‐Marie Curie COFUND Program. C.J. is supported by the MRC (G0800311). The authors thank John Grainger, James Hewitson, and Mark Travis for critical reading of the manuscript, Rinku Rajan for technical support, Richard Preziosi for statistical advice, Martin Waterfall and Gareth Howell for cell sorting and assistance with flow cytometry, and David Gray, Caetano Reis e Sousa, Andrew McKenzie, Markus Mohrs, and Shizuo Akira for provision of OT‐II × Ly5.1, Myd88 −/−, Trif −/−, IL‐10eGFP, Ifnar1 −/−, IL‐13eGFP, KN2, and IFNα6GFP mice. Biomphalaria glabrata snails used to generate eggs and SEA for this research were supplied by the N.I.A.I.D. Schistosomiasis Resource Center at the Biomedical Research Institute (Rockville, MD), through Contract HHSN272201000005I for distribution through BEI Resources, with the help of Mark Wilson.
The EMBO Journal (2017) 36: 2404–2418
References
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S (1998) Targeted disruption of the MyD88 gene results in loss of IL‐1‐ and IL‐18‐mediated function. Immunity 9: 143–150 [DOI] [PubMed] [Google Scholar]
- Alvarez D, Vollmann EH, von Andrian UH (2008) Mechanisms and consequences of dendritic cell migration. Immunity 29: 325–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnden MJ, Allison J, Heath WR, Carbone FR (1998) Defective TCR expression in transgenic mice constructed using cDNA‐based α‐and β‐chain genes under the control of heterologous regulatory elements. Immunol Cell Biol 76: 34–40 [DOI] [PubMed] [Google Scholar]
- Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M (2006) Bone marrow stromal cell antigen 2 is a specific marker of type I IFN‐producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol 177: 3260–3265 [DOI] [PubMed] [Google Scholar]
- Bouchery T, Kyle R, Ronchese F, Le Gros G (2014) The differentiation of CD4(+) T‐helper cell subsets in the context of helminth parasite infection. Front Immunol 5: 487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasel K, De Smedt T, Smith JL, Maliszewski CR (2000) Generation of murine dendritic cells from flt3‐ligand‐supplemented bone marrow cultures. Blood 96: 3029–3039 [PubMed] [Google Scholar]
- Braun A, Worbs T, Moschovakis GL, Halle S, Hoffmann K, Bölter J, Münk A, Förster R (2011) Afferent lymph‐derived T cells and DCs use different chemokine receptor CCR7‐dependent routes for entry into the lymph node and intranodal migration. Nat Immunol 12: 879–887 [DOI] [PubMed] [Google Scholar]
- Broz P, Monack DM (2013) Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol 13: 551–565 [DOI] [PubMed] [Google Scholar]
- Cass CL, Johnson JR, Califf LL, Xu T, Hernandez HJ, Stadecker MJ, Yates JR, Williams DL (2007) Proteomic analysis of Schistosoma mansoni egg secretions. Mol Biochem Parasitol 155: 84–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PC, Owen H, Deaton AM, Borger JG, Brown SL, Clouaire T, Jones G‐R, Jones LH, Lundie RJ, Marley AK, Morrison VL, Phythian‐Adams AT, Wachter E, Webb LM, Sutherland TE, Thomas GD, Grainger JR, Selfridge J, McKenzie ANJ, Allen JE et al (2015) A dominant role for the methyl‐CpG‐binding protein Mbd2 in controlling Th2 induction by dendritic cells. Nat Commun 6: 6920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S (2011) Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29: 621–663 [DOI] [PubMed] [Google Scholar]
- Diacovo TG (2005) Adhesive mechanisms governing interferon‐producing cell recruitment into lymph nodes. J Exp Med 202: 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD (2011) Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208: 1989–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Farzan M (2013) The broad‐spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 13: 46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al‐Shamkhani A, Flavell R, Borrow P, Reis e Sousa C (2003) Viral infection switches non‐plasmacytoid dendritic cells into high interferon producers. Nature 424: 324–328 [DOI] [PubMed] [Google Scholar]
- Essers MAG, Offner S, Blanco‐Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A (2009) IFNα activates dormant haematopoietic stem cells in vivo . Nature 458: 904–908 [DOI] [PubMed] [Google Scholar]
- Everts B, Perona‐Wright G, Smits HH, Hokke CH, van der Ham AJ, Fitzsimmons CM, Doenhoff MJ, van der Bosch J, Mohrs K, Haas H, Mohrs M, Yazdanbakhsh M, Schramm G (2009) Omega‐1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J Exp Med 206: 1673–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Hussaarts L, Driessen NN, Meevissen MHJ, Schramm G, van der Ham AJ, van der Hoeven B, Scholzen T, Burgdorf S, Mohrs M, Pearce EJ, Hokke CH, Haas H, Smits HH, Yazdanbakhsh M (2012) Schistosome‐derived omega‐1 drives Th2 polarization by suppressing protein synthesis following internalization by the mannose receptor. J Exp Med 209: 1753–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Tussiwand R, Dreesen L, Fairfax KC, Huang SC‐C, Smith AM, O'Neill CM, Lam WY, Edelson BT, Urban JF, Murphy KM, Pearce EJ (2016) Migratory CD103+ dendritic cells suppress helminth‐driven type 2 immunity through constitutive expression of IL‐12. J Exp Med 213: 35–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfax KC, Everts B, Amiel E, Smith AM, Schramm G, Haas H, Randolph GJ, Taylor JJ, Pearce EJ (2015) IL‐4‐secreting secondary T follicular helper (Tfh) cells arise from memory T cells, not persisting Tfh cells, through a B cell‐dependent mechanism. J Immunol 199: 2999–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Nish SA, Jiang R, Hou L, Licona‐Limón P, Weinstein JS, Zhao H, Medzhitov R (2013) Control of T helper 2 responses by transcription factor IRF4‐dependent dendritic cells. Immunity 39: 722–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliet M, Boonstra A, Paturel C, Antonenko S, Xu X‐L, Trinchieri G, O'Garra A, Liu Y‐J (2002) The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3‐ligand and granulocyte/macrophage colony‐stimulating factor. J Exp Med 195: 953–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, Arai N, Gallo RL, Digiovanni J, Gilliet M (2010) Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med 207: 2921–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S (2014) Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14: 571–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MAM, Kool M, Muskens F, Lambrecht BN (2010) Inflammatory dendritic cells–not basophils–are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med 207: 2097–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, Sousa CRE (2015) GM‐CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+MHC II+ macrophages and dendritic cells. Immunity 42: 1197–1211 [DOI] [PubMed] [Google Scholar]
- Hoffmann H‐H, Schneider WM, Rice CM (2015) Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol 36: 124–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg N, Patzak I, Willenbrock F (2011) The insider's guide to leukocyte integrin signalling and function. Nat Rev Immunol 11: 416–426 [DOI] [PubMed] [Google Scholar]
- Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, Kola I (1995) A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci USA 92: 11284–11288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14: 36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic D, Kullberg MC, Hieny S, Caspar P, Collazo CM, Sher A (2002) In the absence of IL‐12, CD4(+) T cell responses to intracellular pathogens fail to default to a Th2 pattern and are host protective in an IL‐10(−/−) setting. Immunity 16: 429–439 [DOI] [PubMed] [Google Scholar]
- Jankovic D, Kullberg MC, Caspar P, Sher A (2004) Parasite‐induced Th2 polarization is associated with down‐regulated dendritic cell responsiveness to Th1 stimuli and a transient delay in T lymphocyte cycling. J Immunol 173: 2419–2427 [DOI] [PubMed] [Google Scholar]
- Johnson AC, Li X, Pearlman E (2008) MyD88 functions as a negative regulator of TLR3/TRIF‐induced corneal inflammation by inhibiting activation of c‐Jun N‐terminal kinase. J Biol Chem 283: 3988–3996 [DOI] [PubMed] [Google Scholar]
- de Jong EC, Vieira PL, Kalinski P, Schuitemaker JHN, Tanaka Y, Wierenga EA, Yazdanbakhsh M, Kapsenberg ML (2002) Microbial compounds selectively induce Th1 cell‐promoting or Th2 cell‐promoting dendritic cells in vitro with diverse th cell‐polarizing signals. J Immunol 168: 1704–1709 [DOI] [PubMed] [Google Scholar]
- Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara‐Tejero M, Galán JE, Harhaj E, Flavell RA (2006) Expression of interleukin‐10 in intestinal lymphocytes detected by an interleukin‐10 reporter knockin tiger mouse. Immunity 25: 941–952 [DOI] [PubMed] [Google Scholar]
- Kapsenberg M (2003) Dendritic‐cell control of pathogen‐driven T‐cell polarization. Nat Rev Immunol 3: 984–993 [DOI] [PubMed] [Google Scholar]
- Katagiri K, Ohnishi N, Kabashima K, Iyoda T, Takeda N, Shinkai Y, Inaba K, Kinashi T (2004) Crucial functions of the Rap1 effector molecule RAPL in lymphocyte and dendritic cell trafficking. Nat Immunol 5: 1045–1051 [DOI] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S (2005) Cell type‐specific involvement of RIG‐I in antiviral response. Immunity 23: 19–28 [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S (2007) Alveolar macrophages are the primary interferon‐α producer in pulmonary infection with RNA viruses. Immunity 27: 240–252 [DOI] [PubMed] [Google Scholar]
- Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A (2013) CD301b+ dermal dendritic cells drive T helper 2 cell‐mediated immunity. Immunity 39: 733–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin BM, Smith PM, Ponichtera HE, Shainheit MG, Rutitzky LI, Stadecker MJ (2012) Induction and regulation of pathogenic Th17 cell responses in schistosomiasis. Semin Immunopathol 34: 873–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layland LE, Wagner H, Da Costa CUP (2005) Lack of antigen‐specific Th1 response alters granuloma formation and composition in Schistosoma mansoni‐infected MyD88−/− mice. Eur J Immunol 35: 3248–3257 [DOI] [PubMed] [Google Scholar]
- León B, Ballesteros‐Tato A, Browning JL, Dunn R, Randall TD, Lund FE (2012) Regulation of TH2 development by CXCR5+ dendritic cells and lymphotoxin‐expressing B cells. Nat Immunol 13: 681–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundie RJ, Webb LM, Marley AK, Phythian‐Adams AT, Cook PC, Jackson‐Jones LH, Brown S, Maizels RM, Boon L, O'Keeffe M, MacDonald AS (2016) A central role for hepatic conventional dendritic cells in supporting Th2 responses during helminth infection. Immunol Cell Biol 94: 400–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald AS, Straw AD, Bauman B, Pearce EJ (2001) CD8‐ dendritic cell activation status plays an integral role in influencing Th2 response development. J Immunol 167: 1982–1988 [DOI] [PubMed] [Google Scholar]
- MacDonald AS, Straw AD, Dalton NM, Pearce EJ (2002) Cutting edge: Th2 response induction by dendritic cells: a role for CD40. J Immunol 168: 537–540 [DOI] [PubMed] [Google Scholar]
- MacDonald AS, Maizels RM (2008) Alarming dendritic cells for Th2 induction. J Exp Med 205: 13–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marié I, Durbin JE, Levy DE (1998) Differential viral induction of distinct interferon‐alpha genes by positive feedback through interferon regulatory factor‐7. EMBO J 17: 6660–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall F, Pearce E (2008) Uncoupling of induced protein processing from maturation in dendritic cells exposed to a highly antigenic preparation from a helminth parasite. J Immunol 181: 7562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattei F, Bracci L, Tough DF, Belardelli F, Schiavoni G (2009) Type I IFN regulate DC turnover in vivo . Eur J Immunol 39: 1807–1818 [DOI] [PubMed] [Google Scholar]
- McKenzie ANJ, Spits H, Eberl G (2014) Innate lymphoid cells in inflammation and immunity. Immunity 41: 366–374 [DOI] [PubMed] [Google Scholar]
- McNab F, Mayer‐Barber K, Sher A, Wack A, O'Garra A (2015) Type I interferons in infectious disease. Nat Rev Immunol 15: 87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake T, Kumagai Y, Kato H, Guo Z, Matsushita K, Satoh T, Kawagoe T, Kumar H, Jang MH, Kawai T, Tani T, Takeuchi O, Akira S (2009) Poly I: C‐induced activation of NK cells by CD8+ dendritic cells via the IPS‐1 and TRIF‐dependent pathways. J Immunol 183: 2522–2528 [DOI] [PubMed] [Google Scholar]
- Mohrs K, Wakil AE, Killeen N, Locksley RM, Mohrs M (2005) A two‐step process for cytokine production revealed by IL‐4 dual‐reporter mice. Immunity 23: 419–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya M (2002) Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 99: 3263–3271 [DOI] [PubMed] [Google Scholar]
- Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, Lahoud MH, O'Keeffe M, Shao Q‐X, Chen W‐F, Villadangos JA, Shortman K, Wu L (2005) Cutting edge: generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms‐like tyrosine kinase 3 ligand bone marrow cultures. J Immunol 174: 6592–6597 [DOI] [PubMed] [Google Scholar]
- Naik SH, O'Keeffe M, Proietto A, Shortman HHK, Wu L (2010) CD8+, CD8−, and plasmacytoid dendritic cell generation in vitro using flt3 ligand. Methods Mol Biol 595: 167–176 [DOI] [PubMed] [Google Scholar]
- Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie ANJ (2010) Nuocytes represent a new innate effector leukocyte that mediates type‐2 immunity. Nature 464: 1367–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlato S, Santini SM, Lapenta C, Di Pucchio T, Logozzi M, Spada M, Giammarioli AM, Malorni W, Fais S, Belardelli F (2001) Expression of CCR‐7, MIP‐3beta, and Th‐1 chemokines in type I IFN‐induced monocyte‐derived dendritic cells: importance for the rapid acquisition of potent migratory and functional activities. Blood 98: 3022–3029 [DOI] [PubMed] [Google Scholar]
- Pearce EJ, MacDonald AS (2002) The immunobiology of schistosomiasis. Nat Rev Immunol 2: 499–511 [DOI] [PubMed] [Google Scholar]
- Perona‐Wright G, Lundie RJ, Jenkins SJ, Webb LM, Grencis RK, MacDonald AS (2012) Concurrent bacterial stimulation alters the function of helminth‐activated dendritic cells, resulting in IL‐17 induction. J Immunol 188: 2350–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Pesch V, Lanaya H, Renauld J‐C, Michiels T (2004) Characterization of the murine alpha interferon gene family. J Virol 78: 8219–8228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phythian‐Adams AT, Cook PC, Lundie RJ, Jones LH, Smith KA, Barr TA, Hochweller K, Anderton SM, Hammerling GJ, Maizels RM, MacDonald AS (2010) CD11c depletion severely disrupts Th2 induction and development in vivo . J Exp Med 207: 2089–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AK, Daffis S, Brien JD, Gainey MD, Yokoyama WM, Sheehan KCF, Murphy KM, Schreiber RD, Diamond MS (2011) A temporal role of type I interferon signaling in CD8+ T cell maturation during acute West Nile virus infection. PLoS Pathog 7: e1002407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC (2005) Mechanisms of type‐I‐ and type‐II‐interferon‐mediated signalling. Nat Rev Immunol 5: 375–386 [DOI] [PubMed] [Google Scholar]
- Rouzaut A, Garasa S, Teijeira Á, González I, Martinez‐Forero I, Suarez N, Larrea E, Alfaro C, Palazón A, Dubrot J, Hervás‐Stubbs S, Melero I (2010) Dendritic cells adhere to and transmigrate across lymphatic endothelium in response to IFN‐α. Eur J Immunol 40: 3054–3063 [DOI] [PubMed] [Google Scholar]
- Sheehan KCF, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo‐Arthur C, Carrero JA, White JM, Hertzog PJ, Schreiber RD (2006) Blocking monoclonal antibodies specific for mouse IFN‐alpha/beta receptor subunit 1 (IFNAR‐1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res 26: 804–819 [DOI] [PubMed] [Google Scholar]
- Straw AD, MacDonald AS, Denkers EY, Pearce EJ (2003) CD154 plays a central role in regulating dendritic cell activation during infections that induce Th1 or Th2 responses. J Immunol 170: 727–734 [DOI] [PubMed] [Google Scholar]
- Swiecki M, Colonna M (2010) Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol Rev 234: 142–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trottein F, Pavelka N, Vizzardelli C, Angeli V, Zouain CS, Pelizzola M, Capozzoli M, Urbano M, Capron M, Belardelli F, Granucci F, Ricciardi‐Castagnoli P (2004) A type I IFN‐dependent pathway induced by Schistosoma mansoni eggs in mouse myeloid dendritic cells generates an inflammatory signature. J Immunol 172: 3011–3017 [DOI] [PubMed] [Google Scholar]
- Tussiwand R, Everts B, Grajales‐Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, Wu X, Wong R, Anderson DA, Murphy TL, Pearce EJ, Murphy KM (2015) Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 42: 916–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vintersten K, Monetti C, Gertsenstein M, Zhang P, Laszlo L, Biechele S, Nagy A (2004) Mouse in red: Red fluorescent protein expression in mouse ES cells, embryos, and adult animals. Genesis 40: 241–246 [DOI] [PubMed] [Google Scholar]
- Wilbers RHP, Westerhof LB, van Noort K, Obieglo K, Driessen NN, Everts B, Gringhuis SI, Schramm G, Goverse A, Smant G, Bakker J, Smits HH, Yazdanbakhsh M, Schots A, Hokke CH (2017) Production and glyco‐engineering of immunomodulatory helminth glycoproteins in plants. Sci Rep 7: 45910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, Decker DC, Blaine KM, Fixsen BR, Singh H, Sciammas R, Sperling AI (2013) Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun 4: 2990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Morawetz R, Scharton‐Kersten T, Hieny S, Morse HC, Kühn R, Müller W, Cheever AW, Sher A (1997) Analysis of granuloma formation in double cytokine‐deficient mice reveals a central role for IL‐10 in polarizing both T helper cell 1‐ and T helper cell 2‐type cytokine responses in vivo . J Immunol 159: 5014–5023 [PubMed] [Google Scholar]
- Yamamoto M (2003) Role of adaptor TRIF in the MyD88‐independent toll‐like receptor signaling pathway. Science 301: 640–643 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File