

Abstract
In Trypanosoma brucei, most mitochondrial mRNAs undergo internal changes by RNA editing and 3′ end modifications. The temporally separated and functionally distinct modifications are manifested by adenylation prior to editing, and by post‐editing extension of a short A‐tail into a long A/U‐heteropolymer. The A‐tail stabilizes partially and fully edited mRNAs, while the A/U‐tail enables mRNA binding to the ribosome. Here, we identify an essential pentatricopeptide repeat‐containing RNA binding protein, kinetoplast polyadenylation factor 3 (KPAF3), and demonstrate its role in protecting pre‐mRNA against degradation by the processome. We show that KPAF3 recruits KPAP1 poly(A) polymerase to the 3′ terminus, thus leading to pre‐mRNA stabilization, or decay depending on the occurrence and extent of editing. In vitro, KPAF3 stimulates KPAP1 activity and inhibits mRNA uridylation by RET1 TUTase. Our findings indicate that KPAF3 selectively directs pre‐mRNA toward adenylation rather than uridylation, which is a default post‐trimming modification characteristic of ribosomal and guide RNAs. As a quality control mechanism, KPAF3 binding ensures that mRNAs entering the editing pathway are adenylated and, therefore, competent for post‐editing A/U‐tailing and translational activation.
Keywords: pentatricopeptide repeat, polyadenylation, RNA editing, RNA stability, Trypanosoma
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; RNA Biology
Introduction
Parasitic hemoflagellates cause devastating human and animal diseases including African sleeping sickness, Nagana, Chagas disease, and Leishmaniasis. The unique mechanisms of nuclear and mitochondrial gene expression found in these protozoans represent an abundant source of potential therapeutic targets. Some elements of mitochondrial RNA processing are particularly promising because of their derived character, such as mRNA editing (Aphasizheva & Aphasizhev, 2015), or plant‐like features, typified by a multiplicity of pentatricopeptide repeat (35 amino acids, PPR) RNA binding proteins (Aphasizhev & Aphasizheva, 2013; Barkan & Small, 2014). Primary mitochondrial mRNA precursors are transcribed from maxicircle DNA and processed into 12 pre‐edited and six unedited mRNAs; the former require U‐insertion/deletion editing to generate an open reading frame, while the latter contain a translatable coding sequence. Guide RNAs directing the editing reactions are encoded primarily on minicircles. Maxicircle transcripts are thought to be polycistronic, and include rRNAs and mRNAs (Read et al, 1992; Clement et al, 2004). However, the transcription initiation and termination regions, and the mechanism by which mRNAs are released from the precursor remain unknown. The tight packing of individual pre‐mRNAs within a putative precursor, along with monophosphorylated 5′ ends and defined 3′ termini in mature mRNAs, argues for endonuclease involvement. Nonetheless, such activity has not been identified. A plethora of 3′–5′ exonucleases is involved in mRNA editing (Stuart et al, 2005), guide RNA biogenesis (Suematsu et al, 2016), and turnover (Zimmer et al, 2011), but none have been explicitly connected to mRNA processing or decay. The 5′–3′ RNA degradation pathway appears to be absent in kinetoplast mitochondria.
Once liberated from a precursor, 9S and 12S rRNAs are uniformly 3′ uridylated (Adler et al, 1991), while most pre‐mRNAs are adenylated by KPAP1 poly(A) polymerase (Etheridge et al, 2008). The initially added 20–25 nt A‐tail is extended into a 200–300 nt A/U‐heteropolymer by KPAP1 poly(A) polymerase and RET1 TUTase. This reaction also requires a heterodimer of pentatricopeptide repeat‐containing polyadenylation factors, KPAF1 and KPAF2 (Aphasizheva et al, 2011). Importantly, pre‐mRNAs that undergo editing are adenylated prior to internal sequence changes, and receive A/U‐tails upon completion of the editing process, which typically proceeds from the 3′ to the 5′ end. The temporal separation of A‐tailing and A/U‐tailing processes is consistent with the distinct functions of their products: Short A‐tails stabilize partially and fully edited mRNAs, while the long A/U‐tails activate translation by promoting mRNA‐ribosome binding. The developmentally regulated post‐editing A/U‐tailing reaction has been extensively characterized and likely represents a focal point of translational control (Aphasizhev & Aphasizheva, 2013; Ridlon et al, 2013; Aphasizheva et al, 2016a). Conversely, the pre‐editing processing is poorly understood. In this study, we focused on the following questions: (i) how does KPAP1 poly(A) polymerase selectively target mRNAs, but not rRNAs or gRNAs; (ii) what is the mechanism of A‐tail addition by this inefficient enzyme; (iii) whether the A‐tail is required and sufficient for mRNA stabilization; and (iv) is there a quality checkpoint that allows only adenylated mRNAs to proceed through the editing pathway.
It has been demonstrated that the short A‐tail exerts opposite effects on mRNA decay depending on the occurrence and extent of editing. Specifically, pre‐edited mRNAs are moderately destabilized by this cis‐element, while partially and fully edited transcripts depend on the short A‐tail for steady‐state maintenance (Kao & Read, 2005; Etheridge et al, 2008). Hence, the stability rules switch between pre‐edited and edited mRNAs depending on a few initiating editing events, which typically take place at the 3′ region. Furthermore, in organello studies implicated mRNA uridylation in accelerating decay (Militello & Read, 2000; Ryan & Read, 2005), while genetic repression of RET1 TUTase caused accumulation of mRNA precursors (Aphasizheva & Aphasizhev, 2010). To consolidate these observations, we inferred the existence of a trans‐acting mRNA binding factor that recognizes the 3′ region and recruits KPAP1 poly(A) polymerase to distinguish mitochondrial pre‐mRNA from rRNAs and gRNAs. It is plausible that KPAP1 tethering would stimulate mRNA adenylation. We further reasoned that RET1 TUTase's demonstrated roles in mRNA processing and decay implicate the mitochondrial 3′ processome (MPsome) in these processes. This complex of RET1, DSS1 3′–5′ exonuclease, and several structural subunits catalyzes primary uridylation and processive 3′–5′ degradation of guide RNA precursors, and secondary uridylation of mature gRNAs (Suematsu et al, 2016). It follows that the U‐tails characteristic of gRNAs and rRNAs may represent a post‐trimming modification left behind by the MPsome. Likewise, binding of a hypothetical mRNA‐specific trans‐acting factor would channel pre‐mRNAs for A‐tailing, rather than U‐tailing, and enable subsequent RNA editing and A/U‐addition.
Here, we identified an essential pentatricopeptide repeat‐containing (PPR) protein, termed kinetoplast polyadenylation factor 3 (KPAF3), that interacts with mitochondrial poly(A) polymerase KPAP1 complex and binds predominantly to mRNA 3′ regions, but not to rRNAs or gRNAs. KPAF3 knockdown leads to a decline or, in few instances, upregulation of mRNA depending on the occurrence and extent of editing events. Most importantly, KPAF3 stabilizes pre‐edited transcripts irrespective of the A‐tail's presence. In vitro, KPAF3 stimulates KPAP1 polyadenylation activity and inhibits RNA uridylation by RET1 TUTase. We show that the mitochondrial 3′ processome is responsible not only for generating minicircle‐encoded gRNAs, but also for 3′–5′ processing and decay of maxicircle‐encoded rRNA and mRNA precursors. To that end, KPAF3 protects RNA against 3′–5′ degradation by the purified MPsome in vitro. Our findings indicate that KPAF3 recognizes G‐rich octamer sequences and directs pre‐mRNAs toward adenylation rather than uridylation, which appears to be a default 3′ modification introduced by the MPsome‐embedded RET1 TUTase. Conceivably, KPAF3 binding provides a quality control checkpoint to ensure that mRNAs entering the editing pathway are adenylated and, therefore, competent for post‐editing A/U‐tailing and translation.
Results
Identification of the kinetoplast polyadenylation factor 3 (KPAF3)
The nuclear genome of Trypanosoma brucei encodes ~40 pentatricopeptide repeat‐containing RNA binding proteins, most of which are imported into the mitochondrion (Aphasizhev & Aphasizheva, 2013). Previous studies demonstrated that nearly 20 PPRs populate mitochondrial ribosomes (Zikova et al, 2008), and several interact with KPAP1 poly(A) polymerase (Etheridge et al, 2008). Some ribosome‐embedded PPRs are essential for maintaining ribosomal RNAs (Pusnik et al, 2007) and for translational activation of specific transcripts (Aphasizheva et al, 2016a). Conversely, a heterodimer of kinetoplast polyadenylation factors 1 and 2 is required for A/U‐tailing of fully edited and unedited mRNAs in a reaction catalyzed by KPAP1 and RET1 (Aphasizheva et al, 2011). We demonstrated that KPAP1 poly(A) polymerase knockdown leads to accumulation of deadenylated pre‐edited transcripts, while RNAs edited beyond initial sites are efficiently degraded (Etheridge et al, 2008). To identify a potential factor(s) that stabilizes pre‐edited transcripts, we performed a targeted RNAi screen of predicted PPRs (Aphasizhev & Aphasizheva, 2013), and analyzed mitochondrially encoded ribosomal protein S12 (RPS12) mRNA in respective knockdowns (Fig 1A and B). Repression of Tb927.9.12770 caused degradation of the pre‐edited mRNA while leaving 9S and 12S rRNAs unaffected. This 1,022 amino acid protein with 18 predicted PPR repeats (https://toolkit.tuebingen.mpg.de/#/tools/tprpred) was previously detected in the polyadenylation complex (Etheridge et al, 2008). It shows similarity to KPAF1 in repeat organization (Fig 1C) and sequence (32% identity, 55% similarity); this protein was termed kinetoplast polyadenylation factor 3, KPAF3.
Figure 1. KPAF3 is a component of the mitochondrial polyadenylation complex.
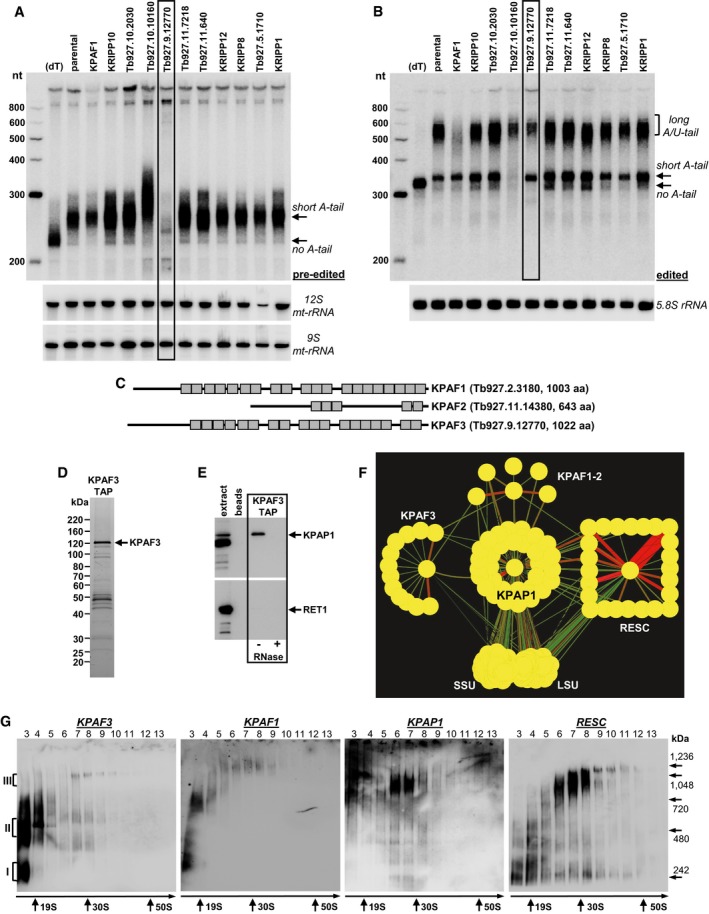
- Representative Northern blotting of pre‐edited RPS12 mRNA and 9S and 12S mt‐rRNAs from targeted RNAi screen of predicted PPR proteins. Knockdown was induced for 72 h, and total RNA was separated on 5% polyacrylamide/8 M urea gel. Gene ID numbers are from http://tritrypdb.org. (dT); RNA was hybridized with 20‐mer oligo(dT) and treated with RNase H to eliminate A‐tails. nt, RNA length in nucleotides. Positions of short A‐tailed and non‐adenylated transcripts are indicated by arrows.
- Same membrane as in (A) was hybridized with probes specific for edited RPS12 mRNA and cytoplasmic 5.8S rRNA (loading control). Edited RNAs with long A/U‐tail, short A‐tail, and non‐adenylated form are indicated.
- Schematic repeat organization of kinetoplast polyadenylation factors 1, 2, and 3.
- Tandem affinity purification of KPAF3. Final fraction was separated on 8–16% SDS gel and stained with Sypro Ruby.
- KPAF3 fractions purified from mock‐ and RNase‐treated mitochondrial extracts were subjected to immunoblotting with antibodies against KPAP1 poly(A) polymerase and RET1 TUTase. Beads, purification from parental cell line.
- Model of interactions between KPAP1 poly(A) polymerase, polyadenylation factors, RNA editing substrate binding complex (RESC), and the ribosome. KPAP1, KPAF2, KPAF3, GRBC1, L3, and S17 proteins were affinity purified from mitochondrial lysates. The network was generated in Cytoscape software from bait–prey pairs in which the prey protein was identified by five or more unique peptides (Table EV1). The edge thickness correlates with normalized spectral abundance factor (NSAF) values.
- Mitochondrial fraction was extracted with detergent, and soluble contents were separated for 5 h at 178,000 g in 10–30% glycerol gradient. Each fraction was further resolved on 3–12% Bis‐Tris native gel. Positions of native protein standards are denoted by arrows. KPAP1 and KPAF3 were visualized by immunoblotting. RNA editing substrate binding complex (RESC) was detected with antibodies against guide RNA binding subunits GRBC1 and GRBC2. Thyroglobulin (19S) and bacterial ribosomal subunits were used as apparent S‐value standards.
Source data are available online for this figure.
To verify KPAF3's association with the polyadenylation complex, the C‐terminally TAP‐tagged polypeptide was expressed in a procyclic (insect) developmental form of T. brucei, and isolated by tandem affinity chromatography (Fig 1D). Purification was also conducted from RNase‐treated mitochondrial lysate, and final fractions were analyzed by immunoblotting and LC‐MS/MS. KPAP1 poly(A) polymerase was readily detectable among proteins co‐purifying with KPAF3, but the association appears to be RNA‐dependent (Fig 1E). Conversely, RET1 TUTase implicated in post‐editing A/U‐tailing was not identified in either sample by immunoblotting or mass spectrometry (Fig 1E and Table EV1). Next, the normalized spectral abundance factors (Neilson et al, 2013) were used to build an interaction network between KPAF3 and established components of the polyadenylation complex, KPAP1 and KPAF1‐2. As shown in Fig 1F, KPAF3 interacts with several hypothetical proteins lacking any functional motifs, and with KPAF1‐2 and KPAP1 complexes. The latter is involved in extensive contacts with the RNA editing substrate binding complex (RESC) and the ribosome, as documented previously (Aphasizheva et al, 2014). Mass spectrometry analysis of RNase‐treated samples indicated that interactions among KPAP1 poly(A) polymerase and KPAF1‐2 and KPAF3 polyadenylation factors are sufficiently stable to withstand two‐step purification, but nonetheless depend entirely on RNA component (Fig EV1A). To assess the size and confirm a critical role of RNA in polyadenylation complex assembly, mock‐ and RNase‐treated mitochondrial lysates were fractionated on glycerol gradient and native gel, and analyzed by immunoblotting. In agreement with proteomic analysis, KPAF3 was found to exist as unassociated protein (I), a distinct complex of ~500 kDa (II), and as a minor fraction bound to ~1 MDa particle (III) (Fig 1G). Conversely, KPAP1 was predominantly detected in ~1 MDa complex, from which it could be effectively released by RNase treatment (Fig EV1B). Remarkably, the ~1 MDa particle closely resembles sedimentation and native gel migration properties of the RESC complex. Based on proteomics and co‐fractionation data, it appears that mitochondrial polyadenylation complex represents a ribonucleoprotein assembly engaged in extensive interactions with RNA editing substrate binding complex.
Figure EV1. Model of RNA‐independent interactions between KPAP1 poly(A) polymerase, KPAF 1‐2, KPAF3, RNA editing substrate binding complex (RESC), and the ribosome.
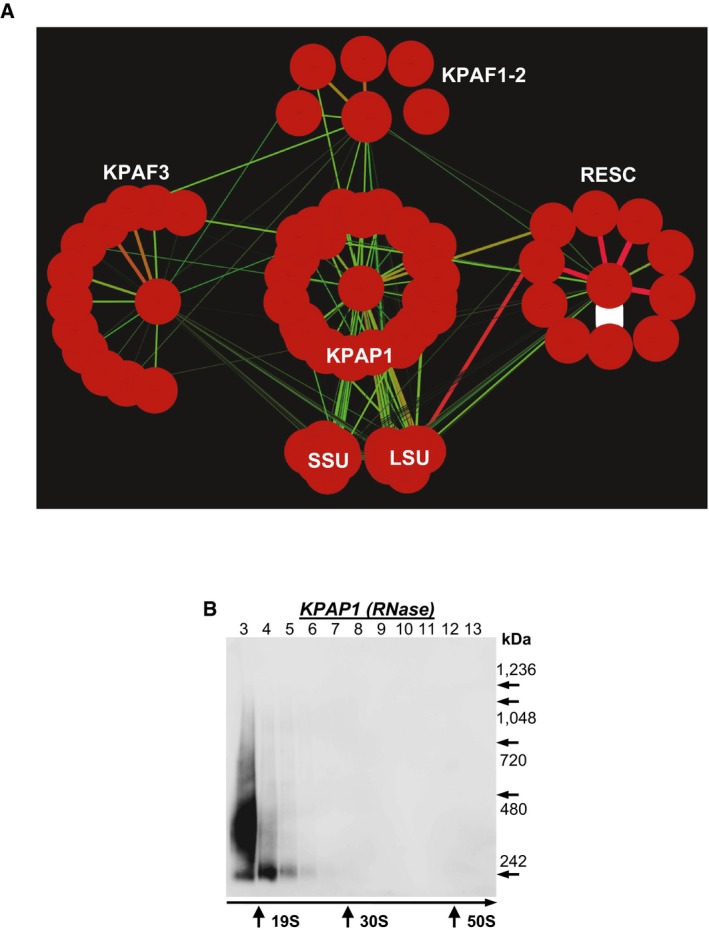
- KPAP1, KPAF2, KPAF3, GRBC1, L3, and S17 proteins were affinity purified from mitochondrial lysates in the presence of RNase A (0.1 mg/ml) and RNase T1 (500 U/ml). The network was generated in Cytoscape software from bait–prey pairs in which the prey protein was identified by five or more unique peptides (Table EV1). The edge thickness correlates with NSAF values.
- Mitochondrial fraction was extracted with detergent in the presence of RNase A (0.1 mg/ml) and RNase T1 (500 U/ml), and soluble contents were separated for 5 h at 178,000 g in 10–30% glycerol gradient. Each fraction was further resolved on 3–12% Bis‐Tris native gel. Positions of native protein standards are denoted by arrows. KPAP1 was visualized by immunoblotting. Thyroglobulin (19S) and bacterial ribosomal subunits were used as apparent S‐value standards.
KPAF3 is essential for mitochondrial mRNA maintenance and parasite viability
The potential role of KPAF3 in mitochondrial RNA processing and cell viability was examined in an actively respiring insect form of T. brucei. Inducible RNAi knockdown triggered rapid and efficient KPAF3 depletion without affecting steady‐state levels of other polyadenylation complex components (KPAP1), or guide RNA processing (RET1 TUTase) and stabilization (GRBC1‐2) factors (Fig 2A). Nonetheless, KPAF3 repression instigated a strong cell growth inhibition phenotype after ~72 h of RNAi induction, and complete cell division arrest and cell death at later time points (Fig 2B). Quantitative RT–PCR of RNA samples isolated at 55 h post‐RNAi induction demonstrated somewhat disparate effects ranging from significant downregulation, such as the case of edited cytochrome b and ND3 transcripts, to moderate upregulation of their respective pre‐edited forms, or pre‐edited mRNA decline (A6, RPS12, CO3; Fig 2C). In agreement with Northern blotting (Fig 1A), mitochondrial ribosomal RNAs remained virtually unaffected. We conclude that KPAF3 is essential for the parasite's viability and functions in an mRNA‐specific processing pathway that differentially affects transcripts depending on their editing status.
Figure 2. KPAF3 knockdown inhibits cell growth and affects mitochondrial mRNAs.
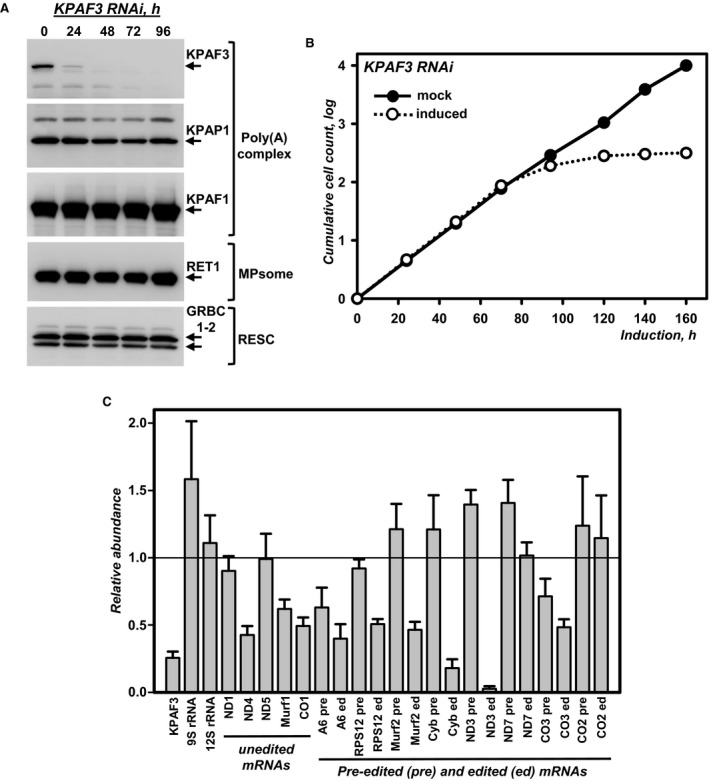
- Cell lysates prepared at indicated time points of KPAF3 RNAi induction were subjected to sequential immunoblotting with antigen‐purified antibodies against KPAF3, KPAP1, KPAF1, GRBC1‐2, and monoclonal antibodies against RET1 TUTase.
- Growth kinetics of procyclic parasite suspension cultures after mock induction and RNAi expression.
- Quantitative real‐time RT–PCR analysis of RNAi‐targeted KPAF3 mRNA, and mitochondrial rRNAs and mRNAs. The assay distinguishes edited and corresponding pre‐edited transcripts, and unedited mRNAs. RNA levels were normalized to β‐tubulin mRNA. RNAi was induced for 55 h. Error bars represent the standard deviation from at least three replicates. The thick line at “1” reflects no change in relative abundance; bars above or below represent an increase or decrease, respectively. Pre, pre‐edited mRNA; ed, edited mRNA.
Source data are available online for this figure.
KPAF3 affects mRNA abundance depending on the occurrence and extent of editing
Changes in relative abundance, as conventionally measured by qRT–PCR, attest to the overall output of a multistep process, but provide limited information about co‐occurrence of RNA editing and 3′ processing events. To integrate KPAF3 interactions with its impact on mRNA adenylation and steady‐state level, we next investigated protein dose‐dependent changes in pan‐edited, moderately edited, and unedited mRNAs. RPS12 mRNA was chosen as a model pan‐edited transcript because it represents a single editing domain, that is, multiple overlapping gRNAs direct editing in a 3′–5′ hierarchical order (Maslov & Simpson, 1992). Total RNA was collected at 24‐h intervals of KPAP1, dual KPAF1‐2 and KPAF3 RNAi induction to reflect changes upon gradual protein depletion. Samples were analyzed by high‐resolution Northern blotting with probes selective for pre‐edited, partially edited (~70% completed), and fully edited mRNAs (Fig 3A). Repressing KPAP1 poly(A) polymerase rapidly eliminated both short A‐ and long A/U‐tailed fully edited mRNAs, while KPAF1/2 knockdown led to a loss of the A/U‐tailed form. In a KPAF3 RNAi cell line, the initial increase in both edited forms was followed by rapid decline after 72 h (Table EV2). The most dramatic differences, however, were detected at the level of pre‐edited mRNA: KPAP1 RNAi caused a loss of the short A‐tail and moderate accumulation of non‐adenylated transcript, while KPAF3 depletion led to a gradual loss of the short A‐tail, followed by rapid and complete degradation of an entire mRNA body after 72 h of RNAi. Consistent with an established KPAF1‐2 role in post‐editing adenylation/uridylation, a simultaneous knockdown of both factors mostly affected A/U‐tailed edited mRNA. To conclude, the loss of A‐tail due to KPAP1 knockdown stabilizes pre‐edited mRNA, but is detrimental to edited form. Conversely, the KPAF3 repression initially leads to decreased adenylation during which mRNA abundance remains mostly unaffected (up to 48 h), but then triggers rapid degradation of an entire pre‐edited mRNA (Table EV2). To that end, the bi‐phasic decay of edited RNA mirrors the loss of a pre‐edited form at later RNAi time points.
Figure 3. KPAF3 repression exerts opposite effects on moderately or massively edited mRNAs.
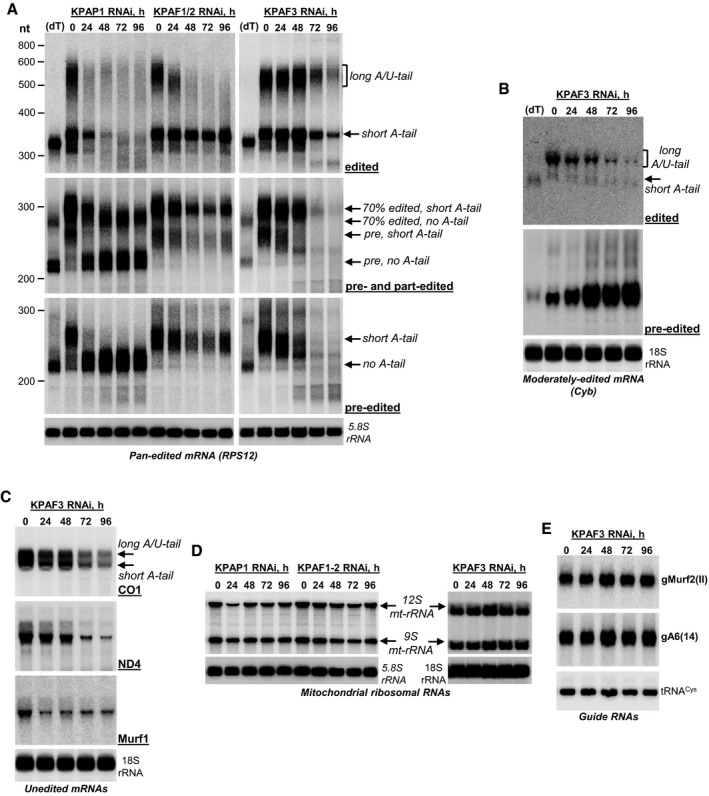
- Northern blotting of pre‐edited, partially edited, and fully edited RPS12 mRNAs. Total RNA was separated on a 5% polyacrylamide/8 M urea gel and sequentially hybridized with radiolabeled single‐stranded DNA probes. Zero time point refers to mock‐induced RNAi cell line. Cytosolic ribosomal RNA (5.8S) was used as loading control. (dT), RNA was hybridized with 20‐mer oligo(dT) and treated with RNase H to eliminate A‐tails. Positions of short A‐tailed and non‐adenylated transcripts are indicated by arrows. Pre, pre‐edited mRNA. Partially edited mRNAs (70% edited) were detected with a probe complementary to positions 33–70 in pre‐edited RPS12 mRNA.
- Northern blotting of moderately edited cyb mRNA. Total RNA was separated on a 1.7% agarose/formaldehyde gel and sequentially hybridized with oligonucleotide probes selective for pre‐edited and fully edited sequences. Cytosolic 18S ribosomal RNA was used as loading control.
- Northern blotting of unedited mRNA. Total RNA was separated on a 1.7% agarose/formaldehyde gel and hybridized with oligonucleotide probes. Cytosolic 18S rRNA was used as loading control.
- Northern blotting of mitochondrial ribosomal RNAs. Total RNA was separated on a 5% polyacrylamide/8 M urea (KPAP1 and KPAF1‐2 RNAi) or 1.8% agarose (KPAF3 RNAi) gels and hybridized with radiolabeled single‐stranded DNA probes specific for 9S and 12S mt‐rRNA. Cytosolic ribosomal RNAs were used as loading controls.
- Guide RNA Northern blotting. Total RNA was separated on a 10% polyacrylamide/8 M urea gel and hybridized with oligonucleotide probes specific for maxicircle‐encoded gMurf2(II) and minicircle‐encoded gA6(14). Mitochondrially localized tRNACys was used as loading control.
In pan‐edited RPS12 mRNA, initiating editing events occur within ~20 nt from the polyadenylation site and expand processively toward the 5′ end to alter the sequence and create an open reading frame (Read et al, 2016). In moderately edited cyb mRNA, 34 uridines are inserted at 13 closely spaced sites located ~1,000 nt away from the 3′ end. In contrast to RPS12 mRNA, pre‐edited cyb transcript accumulated, while the edited form declined during KPAF3 RNAi (Fig 3B). Thus, KPAF3 binding may exert a destabilizing or stabilizing effect depending on the proximity or remoteness, respectively, of editing and polyadenylation events. As expected, this correlative does not apply to unedited mRNAs as these were uniformly downregulated along with KPAF3 depletion (Fig 3C). Finally, the lack of appreciable effects on mitochondrial rRNAs further establishes KPAF3 as an mRNA‐specific factor (Fig 3D). Interestingly, the levels of maxicircle‐encoded Murf2(II) and minicircle‐encoded A6(14) gRNAs increased by ~50% (Fig 3E). This phenomenon is consistent with reported gRNA upregulation upon inhibition of RNA editing (Aphasizheva et al, 2014), which in this case could be caused by depletion of pre‐edited mRNA in KPAF3 RNAi cells.
KPAF3 stabilizes pre‐edited mRNA irrespective of adenylation state
The outcomes of KPAP1 poly(A) polymerase repression suggest that non‐adenylated mRNA remains stable, while in KPAF3 knockdown the initial gradual loss of the short A‐tail is followed by rapid degradation of an entire mRNA body. It is therefore possible that KPAF3 is essential for mRNA polyadenylation by KPAP1, but the mRNA stabilization is achieved by KPAF3 binding rather than by the A‐tail's presence. It follows that non‐adenylated mRNA appearing upon incomplete KPAF3 depletion at early RNAi time points would decay like the adenylated transcript in the mock control. To test this hypothesis, we used an arrested transcription assay (Aphasizheva & Aphasizhev, 2010) to assess mRNA decay rates in a KPAF3 RNAi cell line. The RNAi time points when the short A‐tails is diminished, but pre‐edited mRNA is not yet degraded (36 h), and when most mRNA is degraded (48 h) were selected as starting points. Edited forms were analyzed in the same experiment (Fig 4 and Table EV3). Upon transcriptional arrest with a combination of Actinomycin D and ethidium bromide, mRNA editing proceeds throughout the assay causing multiple internal U‐insertions, as reflected by the lengthening of pre‐edited mRNA. Remarkably, irrespective of differences in mRNA abundance at 36‐ and 48‐h RNAi time points, the decay kinetics of non‐adenylated transcripts closely resembled those of adenylated mRNAs (Fig 4A). As expected, the stability of either short A‐tailed or long A/U‐tailed edited mRNAs was not compromised by KPAF3 knockdown (Fig 4B). In aggregate, the outcomes of dose‐response RNAi experiments and real‐time decay assay suggest that KPAF3 plays an essential role in mRNA polyadenylation, but its stabilizing function does not depend on the A‐tail's presence.
Figure 4. KPAF3 is necessary and sufficient to stabilize pre‐edited mRNAs.
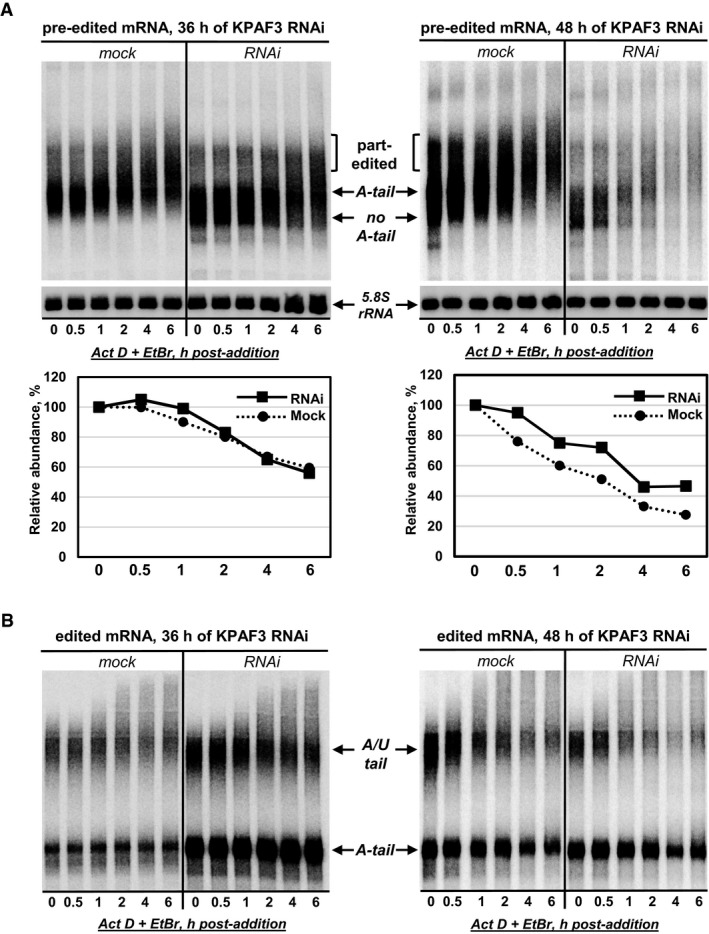
- Pre‐edited mRNA decay in KPAF3 RNAi cells at 36 h (left panel) and 48 h (right panel) post‐induction. After RNAi induction, Actinomycin D and ethidium bromide were added to inhibit transcription. Total RNA was isolated from cells collected at indicated time points after ActD/EtBr bromide addition, separated on denaturing 5% PAGE, and hybridized with DNA probe for pre‐edited RPS12 mRNA. Quantitation was performed in reference to 5.8S rRNA. The graphs below Northern blotting panels represent changes in relative abundance, assuming the mRNA/5.8S rRNA ratio at the time of ActD/EtBr addition as 100%. Contrast was increased in the right panel to reflect RNA loss at 48 h of KPAF3 RNAi.
- Fully edited RPS12 mRNA decay in KPAF3 RNAi cells. Same membrane as in (A) was hybridized with a probe specific for a fully edited RPS12 mRNA.
KPAF3 stimulates KPAP1‐catalyzed RNA adenylation and inhibits uridylation by RET1 TUTase in vitro
Recombinant KPAP1 poly(A) polymerase lacks a pronounced RNA substrate specificity and, adds no more than ~30 adenosines at the highest concentration (300 μM) achievable in vitro (Etheridge et al, 2008). In contrast, RET1 TUTase processively polymerizes hundreds of uridines in vitro, and targets all classes of mitochondrial RNAs by adding U‐tails to rRNAs and gRNAs, and by contributing Us to mRNA long A/U‐tails (Aphasizheva & Aphasizhev, 2010; Aphasizheva et al, 2011). It seems plausible that a protein factor essential for mRNA stability and polyadenylation would stimulate KPAP1 polymerase activity and prevent RET1‐mediated uridylation, which stimulates mRNA decay (Ryan & Read, 2005). Conversely, the KPAF1‐2 heterodimer required for post‐editing A/U‐tailing would not be expected to stimulate KPAP1 activity on pre‐edited mRNA. To investigate whether KPAF3 stimulates KPAP1‐catalyzed RNA adenylation and inhibits uridylation by RET1 TUTase, we have established an in vitro system composed of purified recombinant proteins (Figs 5 and EV2) and synthetic 66 nt RNA resembling a 3′ region of pre‐edited RPS12 mRNA. KPAP1‐ and RET1‐catalyzed reactions in the presence of cognate nucleotide triphosphates and 5′‐radiolabeled RNA produced patterns like those reported previously for generic RNA substrates: distributive addition of ~30 As and processive polymerization of hundreds of Us, respectively (Fig 5A; Etheridge et al, 2008; Rajappa‐Titu et al, 2016). Addition of KPAF3 at increasing concentrations significantly stimulated KPAP1 activity and partially inhibited RNA uridylation by RET1. Combining KPAP1 and RET1 in the same reaction supplied with either ATP, UTP, or both NTPs produced similar outcomes (Fig 5A). The presence of KPAF1‐2 heterodimer caused a moderate concentration‐dependent inhibition of both KPAP1 (Fig 5B) and RET1 (Fig 5C). Nonetheless, introducing KPAF3 in the KPAF1‐2 background rescued polyadenylation activity, but not RNA uridylation.
Figure 5. Reconstitution of KPAF3‐stimulated mRNA adenylation in vitro .

- KPAP1 and RET1 were incubated with 5′‐labeled 66‐mer RNA, ATP, and UTP, as indicated, in the presence of increasing KPAF3 amounts. Protein inclusion in combinatorial reactions is indicated by brackets.
- KPAP1 was incubated with 5′‐labeled 66‐mer RNA and ATP in the presence of increasing KPAF1‐2 and KPAF3 amounts.
- RET1 was incubated with 5′‐labeled 66‐mer RNA and UTP in the presence of increasing KPAF1‐2 and KPAF3 amounts for 20 min.
- KPAP1 was incubated with 5′‐labeled 24‐mer RNAs and ATP in the presence of increasing KPAF3 amounts.
- KPAP1 was incubated with 5′‐labeled 24‐mer [6A] RNA, ATP, and increasing concentrations of indicated homopolymers in the absence (left panel) or presence (right panel) of fixed KPAF3 amount.
- KPAP1 was incubated with 5′‐labeled 24‐mer RNA terminating with 6As, various NTPs, and fixed KPAF3 amount, as indicated.
Figure EV2. Recombinant proteins used in reconstitution reactions.
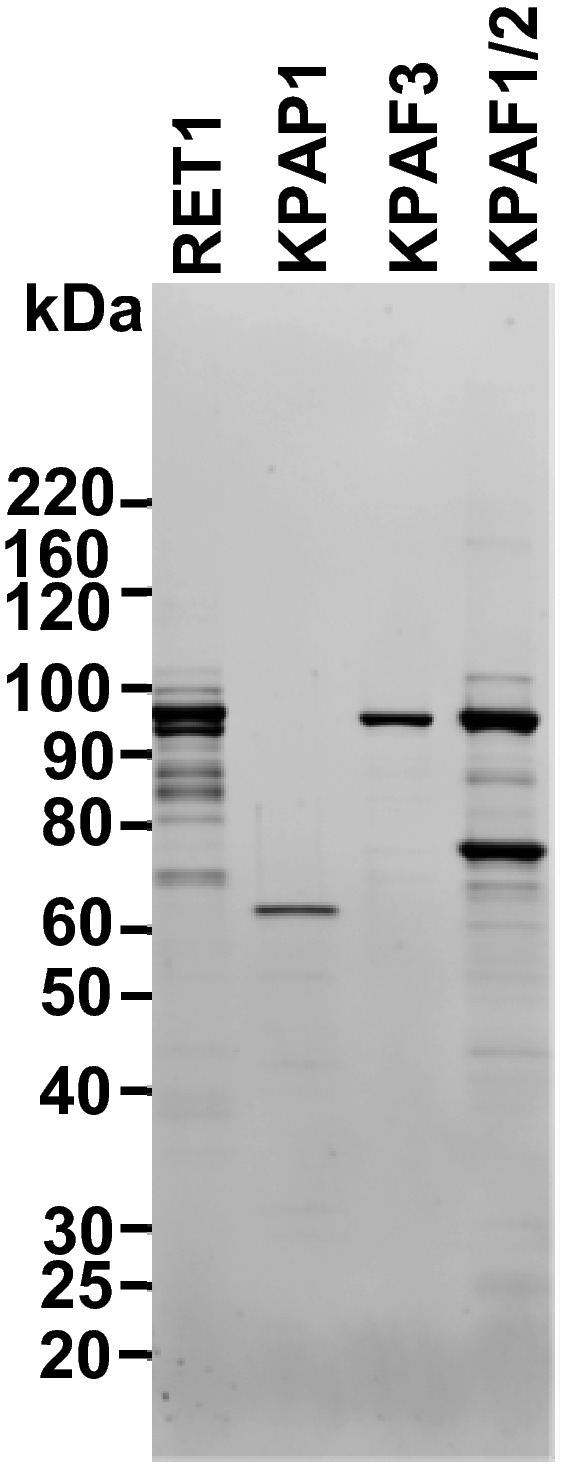
Recombinant proteins were purified from Escherichia coli by sequential metal affinity, ion exchange, and size exclusion chromatographic steps. Final fractions were separated on 8–16% SDS–PAGE and stained with Sypro Ruby. Positions of molecular mass markers are indicated.
Stimulating KPAP1 and inhibiting RET1 by KPAF3 raises the order of events question and stipulates that initial rounds of adenosine or uridine additions by these enzymes may create positive or negative KPAF3 binding determinants, respectively. To assess this possibility, KPAF3‐dependent stimulation was carried out on otherwise identical RNA substrates terminating with different six‐nucleotide stretches. Although the experiment with 6G was uninstructive because of G‐quadruplex formation, equally efficient extensions of 6A, 6U, and 6C substrates demonstrated that KPAP1 has no preference for a specific nucleotide at the 3′ end. The A‐tail therefore does not contribute to KPAF3‐dependent stimulation and is unlikely to constitute the KPAF3‐binding element (Fig 5D). To further confirm this assertion, we conducted competition experiments with poly(A), poly(U), and poly(C) homopolymers and observed minimal inhibition of KPAP1 activity by poly(A) irrespective of KPAF3 presence (Fig 5E). Interestingly, polypyrimidines effectively inhibited KPAP1 activity in both settings. Because auxiliary factors may modulate NTP selectivity of non‐canonical poly(A) polymerases (Rissland & Norbury, 2009), we next inquired whether KPAF3 alters KPAP1 specificity toward UTP, CTP, or GTP (Fig 5F). The nucleotide incorporation patterns, however, indicated general stimulation of KPAP1 activity in the presence of any NTP, which suggests KPAF3's role in increasing KPAP1 affinity for RNA substrate. Collectively, in vitro reconstitution data demonstrate that KPAF3 specifically stimulates mRNA adenylation by KPAP1 and inhibits U‐additions by RET1.
Mitochondrial 3′ processome executes nucleolytic processing of mRNA and rRNA precursors
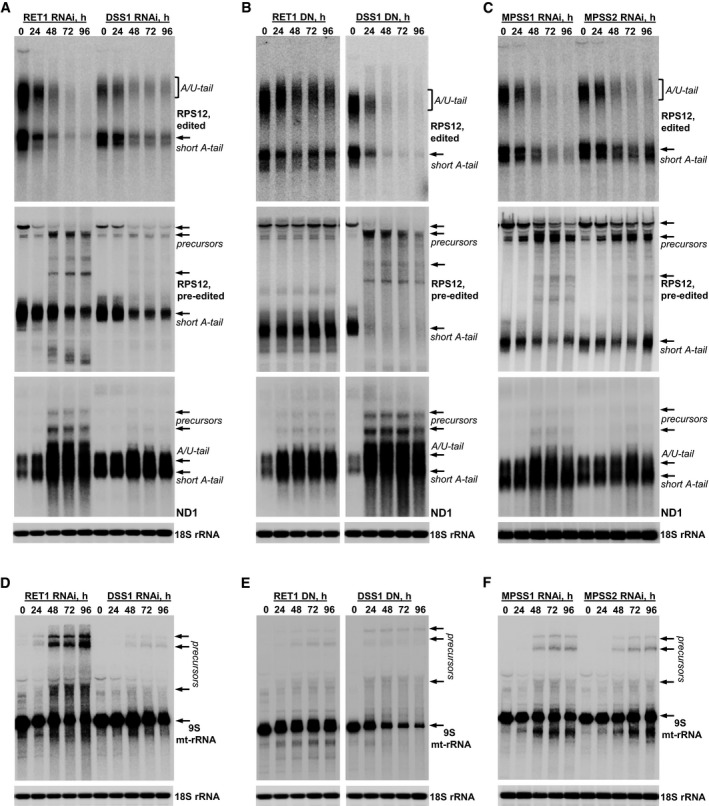
RET1‐catalyzed uridylation reportedly targets all classes of mitochondrial RNAs and stimulates mRNA decay (Ryan & Read, 2005; Aphasizheva & Aphasizhev, 2010). However, a more recent study established RET1 as a subunit of the mitochondrial 3′ processome (MPsome), a stable protein complex that also includes the 3′–5′ exonuclease DSS1, and three structural subunits MPSS1, 2, and 3 (Suematsu et al, 2016). The MPsome generates guide RNAs by uridylating and processively trimming their ~900 nt primary precursors, and then uridylating mature gRNAs. It has been proposed that initial uridylation by RET1 stimulates substrate recognition by DSS1, while secondary uridylation adds a 1–15 nt U‐tail. The latter event likely disengages the MPsome and defines the correctly processed 3′ end. Hence, the short U‐tail may be considered as a hallmark of MPsome‐processed RNA. In this context, it is puzzling why rRNAs indeed terminate with U‐tails while mRNAs are adenylated, even though the corresponding genes are closely positioned in the maxicircle. Because KPAF3 is required for mRNA stabilization and stimulates polyadenylation, we next asked what feature defines the mitochondrial transcript as an mRNA. As a first step, we inquired whether mRNA and rRNA precursors are processed and/or degraded by the MPsome. The steady‐state levels of representative pan‐edited (RPS12) and unedited (ND1) mRNA were analyzed in RET1 TUTase and DSS1 exonuclease knockdowns (Fig 6A). Since either protein is likely to be essential for MPsome integrity, we also assessed the contribution of their enzymatic activities by conditional overexpression of catalytically inactive enzymes (Fig 6B). To verify that the observed effects require the entire MPsome complex, we performed RNAi knockdowns of structural subunits MPSS1 and MPSS2 (Fig 6C). Finally, 9S small ribosomal RNA processing defects were examined in all cell lines described above (Fig 6D–F). Repressing enzymatic and structural MPsome components causes loss of mature gRNAs (Suematsu et al, 2016), and, predictably, production of edited mRNA. Pre‐edited mRNAs were also downregulated, while precursors of diverse sizes accumulated, most prominently upon overexpression of the DSS1 dominant negative variant. Conversely, full‐length unedited ND1 mRNA amassed, along with the appearance of longer ND1‐containing transcripts. This implicates the MPsome not only in mRNA precursor processing, but also in normal decay of mature mRNAs. Finally, 9S rRNA precursors appeared prominently in RET1 RNAi cells, and, to a lesser degree, in all other knockdown cell lines. These results demonstrate that maxicircle‐encoded ribosomal and messenger RNAs are transcribed as 3′ extended precursors and processed by MPsome‐dependent 3′–5′ degradation. The uniform effects of RET1 knockdown suggest that, like in guide RNA processing, uridylation initiates rRNA and mRNA precursor trimming. However, the secondary uridylation event marking the 3′ end of the MPsome‐processed molecule occurs only in rRNAs, while pre‐mRNAs are routed for 3′ adenylation.
Figure 6. Maxicircle‐encoded RNA precursors accumulate in MPsome‐deficient parasites.
-
AEdited and pre‐edited RPS12 mRNA, and unedited ND1 mRNA were analyzed in RET1 and DSS1 RNAi cells. Precursors are indicated by arrows.
-
BEdited and pre‐edited RPS12 mRNA, and unedited ND1 mRNA were analyzed in cell lines overexpressing dominant negative (DN) RET1 and DSS1 variants.
-
CEdited and pre‐edited RPS12 mRNA, and unedited ND1 mRNA were analyzed in MPSS1 and MPSS2 RNAi cells.
-
D–FSame membranes as shown in panels (A–C) were hybridized with radiolabeled oligonucleotide probes specific for 9S mt‐rRNA.
KPAF3 binding inhibits RNA degradation by the MPsome in vitro
Although autonomous DSS1 protein is catalytically inactive, as an MPsome subunit it displays a uridylation‐stimulated processive 3′–5′ exonuclease, and catalysis‐dependent RNA unwinding activities (Suematsu et al, 2016). Our present and published studies suggest that RNA uridylation is coupled with degradation via RET1 and DSS1 association into a stable protein complex. Furthermore, it appears that 3′–5′ degradation represents a major pathway for processing rRNA, mRNA and gRNA precursors, and normal decay of functional molecules. Mature 50–60 nt gRNAs are stabilized by direct binding to specialized subunits of the RNA editing substrate binding complex (RESC), which has no bearing on mRNA or rRNA stability (Weng et al, 2008; Aphasizheva et al, 2014). Therefore, we next tested whether KPAF3 can bind an mRNA precursor in vitro and block its degradation by purified MPsome. The synthetic RNA substrate included a 3′ region of RPS12 mRNA and a downstream sequence consisting of a short intergenic linker, and a 5′ fragment of adjacent ND5 mRNA. At increasing concentrations, recombinant KPAF3 formed three RNA‐protein complexes, which indicates the presence of at least two high affinity binding sites. Increasing the KPAF3 concentration above 50 nM led to a complete RNA sequestration into the largest complex (Fig 7A) and inhibition of the MPsome's 3′–5′ exonuclease activity, which typically degrades RNA into residual 5–7 nt 5′ fragments (Fig 7B). These experiments demonstrate that KPAF3‐bound RNA is resistant to degradation by affinity‐purified MPsome in vitro.
Figure 7. KPAF3‐bound RNA is resistant to 3′–5′ degradation by the MPsome.
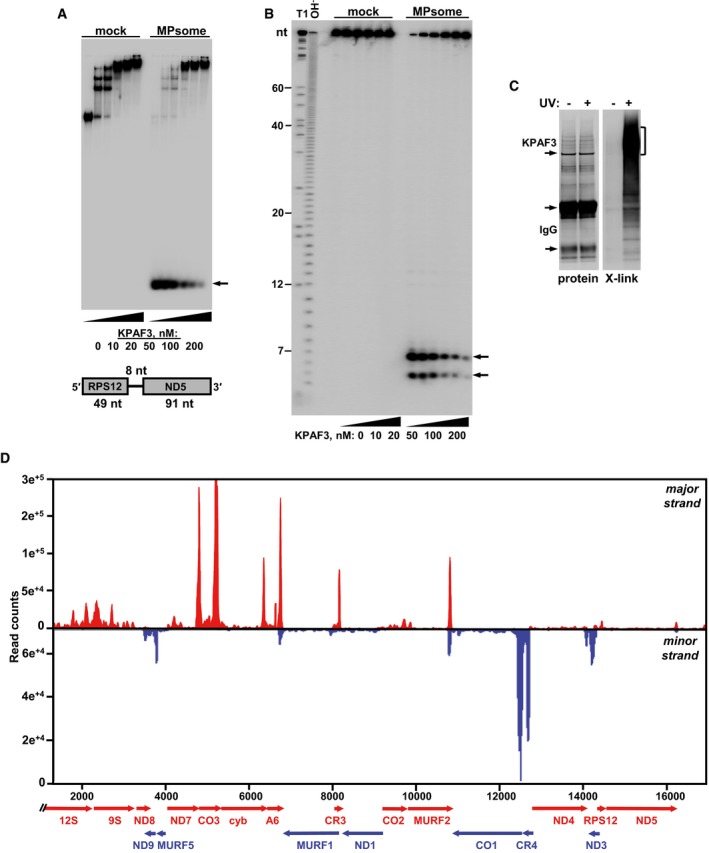
- Electrophoretic mobility shift assay. Increasing amounts of recombinant KPAF3 were incubated with 5′‐radiolabeled RNA in the absence (mock) or presence (MPsome) of tandem affinity‐purified DSS1 exonuclease. Reactions were separated on 7% Tris‐borate native PAGE. Degradation products are indicated by arrows.
- Analysis of RNA degradation patterns. Same reaction products as in (A) were deproteinized and separated on high‐resolution 15% polyacrylamide/8 M urea gel. T1, RNA was digested with G‐specific RNase T1. OH−, alkaline ladder.
- Isolation of in vivo RNA‐KPAF3 crosslinks. KPAF3‐TAP fusion protein was affinity purified from parasites subjected to UV‐irradiation (+) or mock‐treated (−). Final fractions were subjected to partial RNase I digestion, and RNA fragments covalently bound to the protein were radiolabeled. Upon separation on SDS–PAGE, RNA‐protein crosslinks were transferred onto nitrocellulose membrane. Protein patterns were visualized by Sypro Ruby staining (left panel) and RNA‐protein crosslinks by exposure to phosphor storage screen (right panel). RNA was eluted from areas indicated by brackets, converted into barcoded libraries, and sequenced.
- In vivo positioning of KPAF3 binding sites. Crosslinked fragments were mapped to gene‐containing region of the maxicircle. Annotated mitochondrial transcripts encoded on major strand are indicated by red arrows, and those on minor strand are delineated by blue arrows.
Source data are available online for this figure.
KPAF3 determines mRNA identity and 3′ modification status
Upon establishing the coupled uridylation and 3′–5′ degradation as the major mRNA and rRNA processing and decay pathways, and the potential role of KPAF3 in blocking mRNA degradation, we investigated the in vivo positioning of KPAF3 binding sites in respect to mRNA and rRNA 3′ termini. To determine the KPAF3 binding sites, we applied in vivo UV‐crosslinking, partial RNase digestion, and affinity purification of TAP‐tagged polypeptide followed by deep sequencing of crosslinked RNAs (CLAP‐Seq, Fig 7C). We note that many genes in maxicircle DNA are interspersed by very short non‐coding regions, while mature 5′ and 3′ ends of transcripts encoded by adjacent genes often overlap (Clement et al, 2004; Aphasizheva & Aphasizhev, 2010). Mapping of CLAP‐Seq reads to the maxicircle revealed a strong bias in KPAF3 binding toward the 3′ ends of pre‐edited and unedited transcripts encoded on both strands. Conversely, reads derived from abundant 9S and 12S ribosomal RNAs were distributed throughout the respective genes (Fig 7D). A composite plot calculated for all mRNAs with the termination codon set as zero further demonstrates KPAF3's preferential binding to 3′ ends, including untranslated regions (Fig 8A). Because 3′ UTRs of mitochondrial transcripts tend to be short, we next inquired whether KPAF3‐protected fragments include non‐encoded 3′ extensions. As demonstrated by average nucleotide frequency in each position (Fig EV3), the KPAF3 in vivo footprint indeed extends into the A‐tail (Fig 8B).
Figure 8. KPAF3 binding enables polyadenylation of functional 3′ termini.
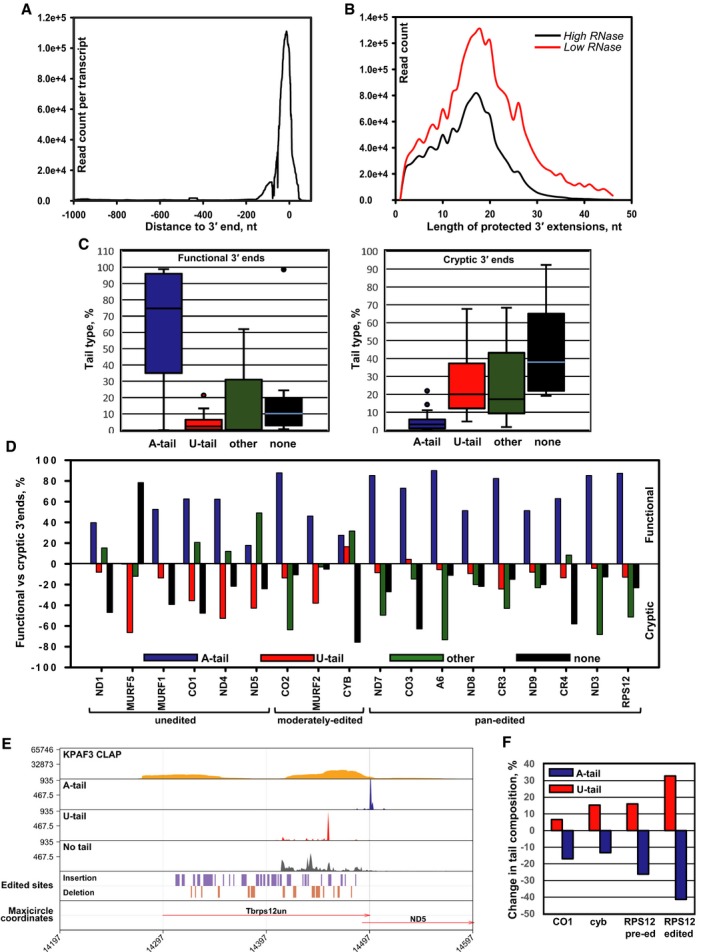
- Overall distribution of KPAF3 binding sites in mitochondrial mRNAs. The CLAP‐Seq reads were aligned to unedited and fully edited sequences. Read counts located 1,000 nt upstream and 100 nt downstream of the 3′ end in each transcript were collected in 1 nt bins. The average coverage across all genes was plotted.
- Length distribution of KPAF3‐protected extensions. The 3′ end sequences that do not align to encoded or edited 3′ ends were considered as 3′ modifications. The length distribution of KPAF3‐protected tail sequences was derived from KPAF3 CLAP‐Seq experiments performed at low and high RNase I concentrations.
- Global modification status of functional and cryptic 3′ termini. Based on 3′ RACE data, the 3′ end patterns were classified as functional (left graph, include stop codon and 3′ UTR) and cryptic (right graph, truncations extend into coding sequence). The percentages of A‐tailed (> 90% As), U‐tailed (> 90% Us), unmodified (lack unmapped 3′ overhangs), and other (those that do not fall into previous three categories) were calculated for all mitochondrial mRNAs. The boxplot distribution of 3′ modification patterns for functional termini in all transcripts is shown on the left panel, and cryptic ends are plotted on the right panel. The horizontal line within each boxplot indicates the median value for all mRNAs. The error bars indicate the 1.5× interquartile range (IQR) distance from median. IQR is defined as the distance between the 25%–75% quantiles. The upper and lower bounds of the box indicate the 75% and 25% quantiles.
- Transcript‐specific differences in modification patterns between functional (upper panel) and cryptic (lower panel) 3′ termini.
- KPAF3 binding correlates with downstream adenylation events. A‐tailed, U‐tailed, and unmodified 3′ ends of pre‐edited RPS12 mRNAs were mapped along with KPAF3 CLAP‐Seq reads and positions of editing events onto maxicircle DNA. Gene positions are shown by arrows.
- Representative unedited (CO1), moderately edited (cyb), and pan‐edited (RPS12) mRNAs were subjected to 3′ RACE in mock‐induced and KPAF3 RNAi knockdown cell line. The percentage of different tail types was calculated for the pre‐edited or edited form (not distinguished in 5′ edited cyb mRNA). The changes in A‐tail and U‐tail percentage at the functional 3′ ends were compared between control and KPAF3 knockdown cells.
Figure EV3. Nucleotide frequency per each position in KPAF3‐CLAP sequences.
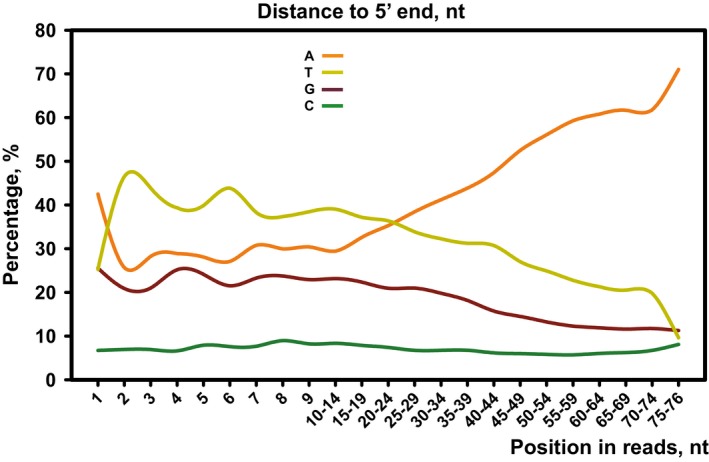
Nucleotide frequency was calculated for each position of KPAF3‐protected RNA sequence.
Considering the specific activation of KPAP1 poly(A) polymerase and inhibition of RET1 TUTase by KPAF3 in vitro (Fig 5), the transcriptome‐wide positioning of this PPR protein supports the like functions in vivo. On the other hand, the MPsome‐catalyzed processing is likely to leave uridylated residues at the 3′ end, as is the case for rRNAs. Hence, we reasoned that KPAF3 stimulates adenylation and blocks uridylation downstream of its binding site. To validate this hypothesis, 3′ ends of annotated mRNAs were analyzed by linker ligation, amplification with gene‐specific primers positioned 150–200 nt upstream of the stop codon, and sequencing (3′ RACE). The modification status was classified as A‐tailed (> 90% As), U‐tailed (> 90% Us), unmodified, and other (no single nucleotide constitutes more than 90%). Unexpectedly, deep sequencing of 3′ regions and their non‐encoded extensions revealed not only mRNAs with predominantly adenylated canonical 3′ UTRs, but also a substantial population of unmodified or uridylated truncated transcripts (Fig 8C). At the individual transcript level, most mRNAs conform to this tenet, but there are a few exceptions. For example, MURF5 encoding a predicted 79 amino acid protein with no discernable motifs is unmodified, while a substantial fraction of cytochrome b mRNA is uridylated (Fig 8D). To clarify the relationship between positioning of KPAF3 binding sites, and the adenylated, uridylated, or unmodified states of adjacent 3′ ends, the CLAP‐Seq reads and 3′ RACE reads were mapped to individual mRNAs. Because 3′ and 5′ ends of mature transcripts encoded by neighboring genes often overlap, 100 nt‐long maxicircle sequences were added to both ends (Fig 8E). Overall, there is a strong correlation between KPAF3 binding upstream of canonical adenylated sites and lack thereof near truncated, mostly uridylated or unmodified 3′ ends. Importantly, truncated termini are virtually absent in edited mRNAs (Appendix Fig S1); this observation provides further support for the A‐tail's critical role in stabilizing edited, but not pre‐edited mRNAs. In other words, the truncated non‐adenylated mRNAs are rapidly degraded once editing is initiated. These findings strongly suggest that the mitochondrial transcript is distinguished as mRNA by KPAF3 binding; this event selects a functional 3′ terminus, stimulates its adenylation, and inhibits uridylation, thereby impeding the MPsome's 3′–5′ exonuclease activity.
If KPAF3 repression prevents mRNA adenylation and allows uridylation, a balance between A‐tailing and U‐tailing processes would be expected to shift toward the latter in the KPAF3 RNAi cell line. To test this prediction, 3′ RACE combined with paired‐end sequencing was performed on total RNA collected at 48 h of RNAi induction (Fig 3A) for representative pre‐edited and edited RPS12 mRNA, and unedited CO1 mRNA, which are predominantly adenylated, and partially uridylated cyb mRNA (Fig 8D). Statistical analysis of modifications at canonical 3′ ends demonstrated a uniform increase in uridylated versus adenylated mRNAs (Fig 8F). These results further assert KPAF3's role in determining the 3′ modification status and, therefore, channeling pre‐mRNA for downstream processing by RNA editing and A/U‐tailing.
KPAF3 binding specificity underlies the adenylation—editing connection
The loss of A‐tail in KPAP1 poly(A) polymerase knockdown stabilizes pre‐edited mRNA, but causes concurrent decay of the edited form. In contrast, KPAF3 repression leads to a loss of the adenylated form, followed by rapid degradation of an entire pre‐edited mRNA (Fig 3A). These observations, along with KPAF3's ability to protect RNA against degradation by the MPsome (Fig 7A and B), suggest that pre‐edited RNA is stabilized by KPAF3, while the short A‐tail is essential for maintaining the edited form. Hence, the stabilization mechanism switches from trans‐ to cis‐mode along with internal changes introduced by editing. It is therefore possible that KPAF3 specifically recognizes pre‐edited, but not edited sequences. Thus, KPAF3 displacement by U‐insertions and deletions ensures that editing proceeds to completion only on adenylated mRNA, which would receive long A/U‐tail upon generation of the open reading frame. Because applying RNA recognition rules developed for plant PPRs (Cheng et al, 2016) to KPAF3 proved of limited value, we derived a consensus KPAF3 binding motif from UV‐crosslinked RNA fragments (Fig 9A), and calculated the number of these motifs per transcript for pre‐edited and corresponding edited mRNAs (Fig 9B). The G‐rich octamers were found in most pre‐edited mRNAs (Table EV4), but were absent or significantly reduced in pan‐edited transcripts. The moderately edited cyb mRNA had the lowest number of KPAF3 binding sites and is apparently not subject to differential KPAF3 binding.
Figure 9. KPAF3 recognizes G‐rich sequences characteristic of pre‐edited mRNAs.
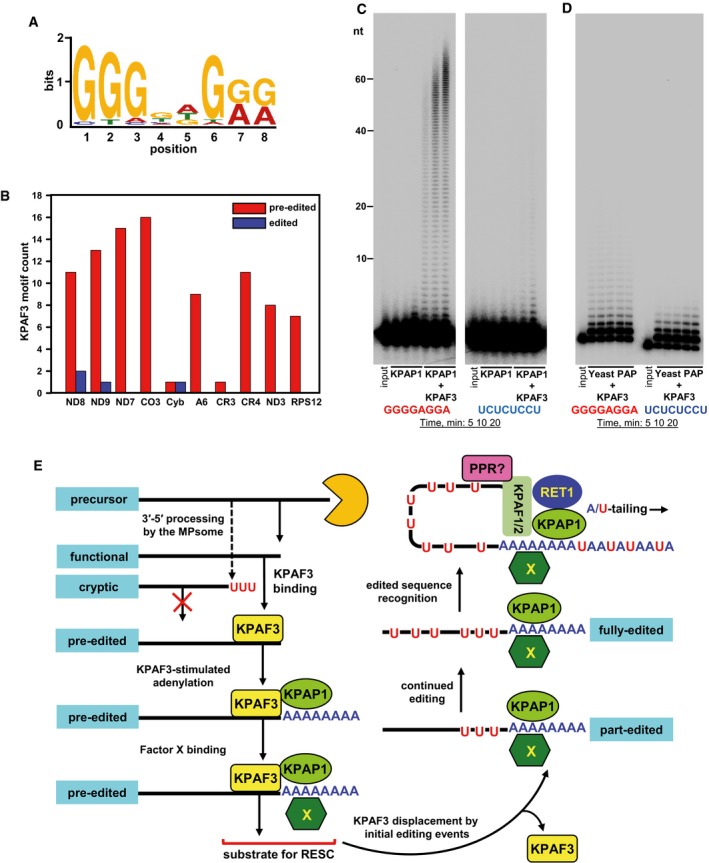
- KPAF3 in vivo binding motif. The MACS algorithm was used to call KPAF3 CLAP‐Seq peaks separately on both maxicircle strands. The significant peaks from samples treated with low and high RNase I concentrations were extended on both sides by 100 nt and used as input; the maxicircle sequences were used as the background model. The MEME algorithm was applied to predict the enriched motif for KPAF3 binding.
- Distribution of KPAF3 binding sites between pre‐edited and edited transcripts. The motif shown in panel (A) was queried against edited and pre‐edited maxicircle transcripts using FIMO algorithm with a P‐value cutoff at 0.001. The number of predicted motifs in pre‐edited and edited maxicircle transcripts was plotted as a bar graph.
- Motif‐dependent stimulation of KPAP1 poly(A) polymerase by KPAF3. Synthetic 5′‐radiolabeled 40‐mers containing either predicted G‐rich motif (left panel), or arbitrary pyrimidine octamer (right panel), was incubated with 100 nM of KPAP1 in the presence or absence of 100 nM of KPAF3. Reactions were performed for 5, 10, 20 min, and products were resolved on 10% polyacrylamide/8 M urea gel.
- Specificity of KPAP1 poly(A) polymerase stimulation by KPAF3. The assay was performed with 14 nM of yeast poly(A) polymerase in the presence of 0, 25, 50, 100, and 200 nM of KPAF3 for 20 min, and products were resolved on 10% polyacrylamide/8 M urea gel.
- Model for functional coupling of primary precursor processing, adenylation, and editing processes. The MPsome‐catalyzed 3′–5′ degradation pauses near the mature 3′ end by a still‐unknown mechanism. Upon pausing, however, two outcomes become feasible depending on the KPAF3 binding site's proximity to the 3′ end: (i) KPAF3 recruits KPAP1 poly(A) polymerase and stimulates short A‐tail addition to downstream terminus; and (ii) lack of bound KPAF3 causes MPsome to dissociate leaving either the unmodified 3′ end, or that with RET1‐added U‐tail. The former modification likely designates the transcript as mRNA, while the latter occurs on rRNAs and truncated mRNA species. A hypothetical factor X is proposed to bind the A‐tail to stabilize edited mRNA once the editing machinery displaces KPAF3 from the 3′ region. Addition of long A/U‐tail to a pre‐existing 3′ A‐tail is triggered upon completion of editing, which typically occurs at the 5′ end. Hence, we hypothesize the existence of a PPR factor that recognizes the RNA sequence created de novo by editing, and recruits KPAF1/2 factors and RET1 TUTase to short A‐tail preloaded with KPAP1 and Factor X. This event likely triggers A/U‐tailing, leading to translational activation (Aphasizheva et al, 2011).
To validate the inferred binding site's functionality, we performed in vitro reconstitution of KPAF3‐stimulated polyadenylation with synthetic 40‐mer derived from pre‐edited A6 mRNA. A parallel experiment replaced the purine‐rich octamer with pyrimidine nucleotides. As shown in Fig 9C, the recombinant KPAP1 added 1–2 adenosines equally inefficiently to both substrates, while in the presence of KPAF3 only the binding site‐containing substrate was effectively polyadenylated. We conclude that KPAF3‐bound RNA is not only resistant to MPsome‐catalyzed degradation, but also represents a competent substrate for KPAP1 poly(A) polymerase. To that end, replacing KPAP1 with yeast poly(A) polymerase produced virtually identical extension patterns with both substrates confirming the specificity of KPAP1‐KPAF3 functional axis (Fig 9D).
Discussion
Functional coupling of primary transcript processing, pre‐editing A‐tailing, internal U‐insertions/deletions, and post‐editing A/U‐tailing is critical for generating translation‐competent mitochondrial mRNAs in the unicellular parasite Trypanosoma brucei. Although the key 3′ modification enzymes, KPAP1 poly(A) polymerase and RET1 TUTase, and KPAF1/2 factors required for the A/U‐tailing have been identified, the pathway leading to formation of mRNA 3′ end prior to modification remained unknown (reviewed in Aphasizheva & Aphasizhev, 2015; Read et al, 2016). Furthermore, it was unclear why 3′ adenylation predominantly targets mRNAs, whereas ribosomal RNAs and guide RNAs are uridylated. Finally, the mechanism by which pre‐edited mRNA stabilization switches from being the A‐tail independent to strictly adenylation‐reliant upon internal sequence changes introduced by editing was puzzling (Kao & Read, 2005; Etheridge et al, 2008).
Here, we identified and characterized the pentatricopeptide repeat‐containing polyadenylation factor KPAF3 as essential for parasite viability, and demonstrated its role in stabilizing pre‐edited and unedited mRNAs. PPR proteins are defined by arrays of 35‐amino acid helix‐turn‐helix motifs (Small & Peeters, 2000) and often recruit RNA modification enzymes to transcripts containing specific binding sites. Each repeat in the array is thought to recognize a single nucleotide via amino acid side chains occupying two cardinal positions (Barkan & Small, 2014). By combining genetic knockdowns, proteomic and RNA analyses, in vivo RNA‐protein crosslinking, real‐time decay assays, deep sequencing of mRNA termini, and in vitro reconstitutions, we show that KPAF3 interacts with polyadenylation complex and selectively stimulates adenylation of mitochondrial mRNAs. Importantly, KPAF3's stabilizing effect is consummated through direct binding to the mRNA 3′ end rather than stimulating short A‐tail addition. It follows that the A‐tail addition is a consequence of KPAF3 binding and ensuing recruitment of KPAP1 poly(A) polymerase to pre‐edited mRNA. The KPAF3 binding site was defined by motif analysis of in vivo crosslinked fragments and in vitro reconstitution as G‐rich octamer. In agreement with KPAF3's role in mRNA adenylation, these motifs are clustered at the 3′ regions of pre‐edited and unedited mRNAs. However, very few binding sites are retained in U‐rich pan‐edited mRNAs. Indeed, mRNAs with truncated unmodified termini are virtually absent in edited mRNAs; this observation suggests that dysfunctional truncated mRNAs are rapidly degraded once editing is initiated. Our findings explain the stability switch phenomenon by selective KPAF3 binding to purine‐rich pre‐edited transcripts, and by the elimination of KPAF3 binding determinants during editing. Although ubiquitous distribution of binding sites in pre‐edited and unedited mRNAs argues for KPAF3's general role in mRNA stabilization and adenylation, it is noted that individual transcripts are affected by KPAF3 knockdown to various degrees. Hence, contribution of transcript‐specific PPRs seems plausible (Aphasizhev & Aphasizheva, 2013).
The extensive RNA‐mediated contacts between polyadenylation complex and the RNA editing substrate binding complex (RESC, Fig 1E) provide a physical basis for functional coupling between adenylation and editing. It is only fitting that the RESC complex, which convenes pre‐edited mRNAs and guide RNAs (Aphasizheva et al, 2014), would recognize the polyadenylation complex‐bound 3′ mRNA region, and outcompete KPAF3 to initiate 3′–5′ editing progression (Maslov & Simpson, 1992). We further speculate that a hypothetical Factor X binds the nascent A‐tail to ensure mRNA stabilization upon KPAF3 displacement from the mRNA 3′ region by the editing process (Fig 9E). Ultimately, this mechanism may serve as a quality checkpoint guaranteeing that only adenylated pre‐mRNA proceeds through the editing pathway and receives the A/U‐tail, which activates translation (Aphasizheva et al, 2011).
It is commonly held that both maxicircle DNA strands are transcribed into polycistronic precursors that undergo endonucleolytic cleavage to produce individual rRNAs and pre‐mRNAs (Hashimi et al, 2013). This notion is supported by the monophosphorylated state of mature mRNA 5′ ends, but the cognate endonuclease remains unknown. To the contrary, we demonstrate that 3′–5′ degradation catalyzed by the mitochondrial processome (MPsome) represents the major processing pathway for primary maxicircle transcripts yielding both mRNAs and rRNAs. This protein complex of RET1 TUTase, DSS1 exonuclease, and three structural proteins has been implicated in uridylation‐induced, antisense RNA‐controlled processing of guide RNA precursors. These findings raise several fundamental questions of which only some are resolved in this study. U‐tails found in mature guide RNAs (Blum & Simpson, 1990) and rRNAs (Adler et al, 1991) denote MPsome‐processed molecules, whereas mRNAs are adenylated (Bhat et al, 1991). In this study, a deeper analysis of mRNA 3′ termini illustrated that 3′–5′ degradation often infringes on the mRNA body, leaving truncated molecules uridylated or unmodified. Crucially, superimposition of KPAF3 CLAP‐Seq reads with 3′ RACE data revealed that termini located downstream of KPAF3 binding sites are mainly adenylated and, therefore, will give rise to functional mRNAs with intact stop codons and 3′ UTRs (Fig 8E and Appendix Fig S1). Thus, KPAF3 defines mRNA identity by routing G‐rich octamer‐containing transcripts toward adenylation by KPAP1 while simultaneously impeding their uridylation by the MPsome‐imbedded RET1 TUTase.
In vitro, KPAF3‐bound RNA is refractory to RET1‐catalyzed uridylation and is resistant to MPsome‐dependent degradation, but competent for A‐tailing by KPAP1 (Figs 5, 7 and 9). To that end, PPR proteins reportedly participate in delimiting the mRNA body in plant organelles by inhibiting exonucleolytic activities (Pfalz et al, 2009). However, we were unable to reproduce precise MPsome pausing or stopping near the predicted KPAF3 binding sites in model RNA substrates (Fig 7 and data not shown). Although only partially conclusive, this outcome suggests that KPAF3 is either insufficient for, or not involved in stopping the MPsome at defined 3′ UTR boundaries. Instead, KPAF3 binds to specific RNA sequences upstream of the 3′ end generated by MPsome dissociation, and recruits KPAP1 poly(A) polymerase. It seems plausible that the antisense RNA‐based mechanism by which MPsome pausing is achieved during guide RNA processing may also be applicable to rRNA and mRNA 3′ end definition (Suematsu et al, 2016). We also note that 3′–5′ degradation is logistically incongruous with liberation of multiple individual pre‐mRNAs from a polycistronic precursor. In contrast to the currently accepted notion of multicistronic precursor processing by a hypothetical endonuclease, our findings advocate the possibility that maxicircle transcripts are synthesized from individual promoters as 3′ extended pre‐mRNAs. In this scenario, the 5′ and 3′ termini would be defined by transcription initiation and MPsome pausing, respectively. In any event, these hypotheses constitute a reasonable ground for future studies.
Materials and Methods
Trypanosome culture, RNAi, protein expression, and RNA analysis
Plasmids for RNAi knockdowns were generated by cloning ~500‐bp gene fragments into p2T7‐177 vector for tetracycline‐inducible expression (Wickstead et al, 2002). Linearized constructs were transfected into a procyclic 29‐13 T. brucei strain (Wirtz et al, 1999). For inducible protein expression, full‐length genes were cloned into pLew‐MHTAP vector (Jensen et al, 2007). RNAi, mitochondrial isolation, glycerol gradient, native gel, total RNA isolation, Northern blotting, qRT–PCR, and tandem affinity purification were performed as described in Aphasizheva et al (2016b). The change in relative abundance was calculated based on qRT–PCR, or Northern blotting, data assuming the ratio between analyzed transcripts and control RNAs in mock‐induced cells as 1 or 100%, respectively. RNA decay assays were described in Aphasizheva and Aphasizhev (2010).
Purification of recombinant proteins and antibody production
Full‐length KPAF3 gene was cloned into pET28c vector (Millipore) to generate C‐terminal 6His‐fusion protein, and transformed into BL21 (DE3) STAR Escherichia coli strain. Bacterial culture was grown in 4 l of 2xYT media supplement with 1% glucose and 50 μg/ml of kanamycin at 37°C until ~0.6 OD600. The temperature was decreased to 18°C, and expression was induced with addition of 0.1 mM IPTG for 3 h. Cells were harvested by centrifugation, washed in PBS, frozen in liquid nitrogen, and cryogenically pulverized in CryoMill (Retsch). Powder was resuspended in 60 ml of lysis buffer [50 mM HEPES pH 8.0, 50 mM NaCl, 10 units/ml of DNase I (Sigma)] supplemented with one tablet of EDTA‐free Protease Inhibitor cocktail (ThermoFisher Scientific). The lysate was sonicated three times at 12 W for 30 s. Sodium chloride was adjusted to 300 mM, and the extract was cleared by centrifugation at 110,000 g in a SW32Ti rotor for 30 min. Cleared extract was loaded onto 5 ml HiTrap Talon column (GE). The column was washed with 80 ml of 50 mM HEPES pH 7.3, 300 mM NaCl, and with 40 ml of the same buffer containing 10 mM imidazole. Protein was eluted with 40 ml of 50 mM HEPES pH 7.3, 300 mM NaCl, and 200 mM imidazole. The peak fraction (~15 ml) was diluted threefold with 25 mM Tris–HCl pH 7.5, 0.1 mM EDTA, and 1 mM DTT and immediately loaded onto 5 ml HiTrap Q column (GE Healthcare) equilibrated with 100 mM KCl, 25 mM Tris–HCl pH 7.5, 0.1 mM EDTA, and 1 mM DTT. Protein was eluted with 80 ml linear gradient of KCl from 100 to 500 mM. The KPAF3‐containing fraction was concentrated to 0.2 ml, and loaded onto Superose 12 10/300GL column (GE) in 25 mM Tris–HCl pH 7.5, 200 mM KCl, 0.1 mM EDTA, 1 mM DTT, and 5% glycerol. KPAF1 and KPAF2 were co‐expressed in pET‐Duet1 vector (Millipore) and purified by the same three‐step procedure. KPAP1 poly(A) polymerase and RET1 TUTase were purified as described (Aphasizhev & Aphasizheva, 2007; Etheridge et al, 2008). For polyclonal antibody production, rabbits were immunized with purified KPAF3. Antibody was purified on solid media‐immobilized recombinant KPAF3. Antibodies against KPAP1 (Etheridge et al, 2008), RET1 (Suematsu et al, 2016), and GRBC1/2 (Aphasizheva et al, 2014) were described previously.
Western blotting
Western blotting was performed with rabbit antigen‐purified rabbit polyclonal antibodies (KPAP1, KPAF1, GRBC1‐2), mouse monoclonal antibody (RET1), or with anti‐CBP antibodies (GenScript) to detect TAP‐tagged proteins. Quantitative chemiluminescent images were acquired with LAS‐4000 (GE Healthcare).
Mass spectrometric analysis by LC‐MS/MS
Affinity‐purified complexes were precipitated by addition of trichloroacetic acid and deoxycholate to 20 and 0.1%, respectively, washed three times with ice‐cold acetone, and digested with LysC peptidase in 8 M urea (1:50 ratio) for 4 h at 37°C. Reaction was diluted fivefold with 50 mM Na‐bicarbonate (pH 7.5) and further digested with trypsin (1:100 ratio) for 16 h. Peptides were purified on Vivapure spin columns (Sartorius). LC‐MS/MS was carried out by nanoflow reversed phase liquid chromatography (RPLC; Eksigent, CA) coupled on‐line to a Linear Ion Trap (LTQ)‐Orbitrap mass spectrometer (Thermo‐Electron Corp). The LC analysis was performed using a capillary column (100 μm ID × 150 mm) with Polaris C18‐A resin (Varian Inc., CA). The peptides were eluted using a linear gradient of 2–35% B in 85 min at a flow of 300 nl/min (solvent A: 100% H2O, 0.1% formic acid; solvent B: 100% acetonitrile, 0.1% formic acid). A cycle of full FT scan mass spectrum (m/z 350–1,800, resolution of 60,000 at m/z 400) followed by 10 data‐dependent MS/MS spectra acquired in the linear ion trap with normalized collision energy (setting of 35%). Target ions already selected for MS/MS were dynamically excluded for 30 s.
Protein identification by database searching
Monoisotopic masses of parent ions and corresponding fragment ions, parent ion charge states, and ion intensities from the tandem mass spectra (MS/MS) were obtained by using in‐house software with Raw_Extract script from Xcalibur v2.4. Following automated data extraction, resultant peak lists for each LC‐MS/MS experiment were submitted to the development version of Protein Prospector (UCSF) for database searching similarly as described (Fang et al, 2012). Each project was searched against a normal form concatenated with the random form of the T. brucei database (www.genedb.org, v5). Trypsin was set as the enzyme with a maximum of two missed cleavage sites. The mass tolerance for parent ion was set as ±20 ppm, whereas ±0.6 Da tolerance was chosen for the fragment ions. Chemical modifications such as protein N‐terminal acetylation, methionine oxidation, N‐terminal pyroglutamine, and deamidation of asparagine were selected as variable modifications during database search. The Search Compare program in Protein Prospector was used for summarization, validation, and comparison of results. Protein identification is based on at least three unique peptides with expectation value ≤ 0.05.
Rapid amplification of 3′ cDNA ends (3′ RACE)
Total RNA was dephosphorylated and ligated with 5′ phosphorylated RNA oligonucleotide RA3, and cDNA was synthesized with RTR primer. PCR was performed in two steps: five cycles with gene‐specific primer and PR1, and additional cycles with PR1 and Index 1 primers (Illumina TrueSeq small RNA library preparation kit). The resultant libraries were sequenced on Illumina MiSeq in 300 nt single‐end or 225 nt paired‐end modes.
Crosslinking–affinity purification and deep sequencing (CLAP‐Seq)
Methods described in Aphasizheva et al (2016b) have been used to perform in vivo crosslinking, affinity purification, and RNA‐Seq library preparation from KPAF3‐bound RNA fragments, with minor modifications. Specifically, total cell lysates were obtained from 5 × 109 parasites and were treated at low (50 U) and high (500 U) RNase I concentration for 5 min at 37°C in 10 ml lysates.
In vitro reconstitution
KPAP1 and RET1 activities in the presence of KPAF1‐2 and KPAF3. RNA (0.1 pmol) was 5′ labeled with 32P, pre‐incubated in 10 μl of reaction buffer (20 mM Tris–HCl pH 7.6, 10 mM MgCl2, 1 mM DTT) with 100 μM of rNTP in the presence of 20, 50, or 200 nM of KPAF3 or KPAF1‐2 for 10 min at 30°C. Reactions were started by adding KPAP1 to 100 nM, or RET1 to 10 nM, incubated for 20 min, and stopped with 15 μl of 95% formamide, 10 mM EDTA, 0.05% xylene cyanol, and 0.05% bromophenol blue, and separated on 10% polyacrylamide/8 M urea gel. Gels were exposed to phosphor storage screens, and images were acquired with Typhoon FLA 7000 scanner (GE Healthcare). For competition experiments, 0.1 pmol of radiolabeled 6A RNA was incubated with 10, 50, 250, 1,000, 5,000 ng of poly(A), poly(U) and poly(C) (Sigma), and 100 nM of KPAF3 in 10 μl of reaction buffer.
Motif‐dependent stimulation of KPAP1 activity by KPAF3. RNA substrates derived from pre‐edited A6 mRNA sequence (positions 324–363) were purchased from Sigma Genosys and purified on 15% polyacrylamide/8 M urea gel.
A6: GAAAGGUUAGGGGGAGGAGAGAAGAAAGGGAAAGUUGUGA
A6 mutated motif: GAAAGGUUAGUCUCUCCUGAGAAGAAAGGGAAAGUUGUGA
Yeast poly(A) polymerase was purchased from Affymetrix. Reactions were performed as above with 100 nM of KPAP1 or 14 nM of yeast poly(A) polymerase, and 100 nM of KPAF3 for 5, 10, and 20 min.
MPsome activity. RPS12 + ND5 RNA substrate (maxicircle positions 14435–14580; GenBank M94286.1)
GGAACCCUUUGUUUUGGUUAAAGAAACAUCGUUUAGAAGAGAUUUUAGAAUAAGAUAUGUUUUUAAUAUUUUUUUUAUUUUUUAUAAUGUUUGGGUUUAUAUCAGGUUCAUUUAUGUUUGGUAGGAAUUUUCUAAGUUUUUGAUUAU was prepared by in vitro transcription, 5′ labeled with 32P, and pre‐incubated with 10, 20, 50, 100, 200 nM of KPAF3 for 10 min in 9 μl mixture containing 50 mM Tris–HCl pH 8.0, 0.05% NP‐40, 1 mM DTT, 50 μM MgCl2. The reaction was started with 1 μl of TAP‐purified DSS1, incubated at 30°C for 15 min, and either diluted with 4 μl of Native PAGE 4× sample buffer (Life Technologies) or stopped with 95% formamide, 10 mM EDTA, 0.05% xylene cyanol, and 0.05% bromophenol blue. Products were analyzed on native 7% Tris‐borate or denaturing 15% polyacrylamide gels, respectively.
See Appendix for detailed protocols and RNA sequences. DNA oligonucleotide sequences are provided in Appendix Table S1.
Accession numbers
The protein‐coding sequence of the KPAF3 gene has been deposited in GenBank under accession code KY645970. Deep sequencing data have been deposited into the Sequence Read Archive (SRA) under accession code SRP100492.
Author contributions
LZ and TY performed bioinformatics analysis with advice from SM and IA. FMS conducted 3′RACE and characterized native complexes. TS carried out in vitro reconstitutions. LH directed protein identification by mass spectrometry. RA and IA performed genetic knockdowns, affinity purifications, RNA analysis, and CLAP experiments. IA and RA wrote the manuscript. All authors contributed to the final version of the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 7
Acknowledgements
We thank members of our laboratories for stimulating discussions and Mikhail Mesitov for technical assistance. This work was supported by NIH grants AI113157 to IA, AI091914, and AI101057 to RA, and GM074830 and GM106003 to LH.
The EMBO Journal (2017) 36: 2435–2454
References
- Adler BK, Harris ME, Bertrand KI, Hajduk SL (1991) Modification of Trypanosoma brucei mitochondrial rRNA by posttranscriptional 3′ polyuridine tail formation. Mol Cell Biol 11: 5878–5884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R, Aphasizheva I (2007) RNA editing uridylyltransferases of trypanosomatids. Methods Enzymol 424: 51–67 [DOI] [PubMed] [Google Scholar]
- Aphasizheva I, Aphasizhev R (2010) RET1‐catalyzed uridylylation shapes the mitochondrial transcriptome in Trypanosoma brucei . Mol Cell Biol 30: 1555–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Maslov D, Wang X, Huang L, Aphasizhev R (2011) Pentatricopeptide repeat proteins stimulate mRNA adenylation/uridylation to activate mitochondrial translation in trypanosomes. Mol Cell 42: 106–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R, Aphasizheva I (2013) Emerging roles of PPR proteins in trypanosomes: switches, blocks, and triggers. RNA Biol 10: 1495–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Zhang L, Wang X, Kaake RM, Huang L, Monti S, Aphasizhev R (2014) RNA binding and core complexes constitute the U‐insertion/deletion editosome. Mol Cell Biol 34: 4329–4342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Aphasizhev R (2015) U‐insertion/deletion mRNA‐editing holoenzyme: definition in sight. Trends Parasitol 13: 1078–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Maslov DA, Qian Y, Huang L, Wang Q, Costello CE, Aphasizhev R (2016a) Ribosome‐associated pentatricopeptide repeat proteins function as translational activators in mitochondria of trypanosomes. Mol Microbiol 99: 1043–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Zhang L, Aphasizhev R (2016b) Investigating RNA editing factors from trypanosome mitochondria. Methods 107: 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan A, Small I (2014) Pentatricopeptide repeat proteins in plants. Annu Rev Plant Biol 65: 415–442 [DOI] [PubMed] [Google Scholar]
- Bhat GJ, Myler PJ, Stuart K (1991) The two ATPase 6 mRNAs of Leishmania tarentolae differ at their 3′ ends. Mol Biochem Parasitol 48: 139–150 [DOI] [PubMed] [Google Scholar]
- Blum B, Simpson L (1990) Guide RNAs in kinetoplastid mitochondria have a nonencoded 3′ oligo‐(U) tail involved in recognition of the pre‐edited region. Cell 62: 391–397 [DOI] [PubMed] [Google Scholar]
- Cheng S, Gutmann B, Zhong X, Ye Y, Fisher MF, Bai F, Castleden I, Song Y, Song B, Huang J, Liu X, Xu X, Lim BL, Bond CS, Yiu SM, Small I (2016) Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J 85: 532–547 [DOI] [PubMed] [Google Scholar]
- Clement SL, Mingler MK, Koslowsky DJ (2004) An intragenic guide RNA location suggests a complex mechanism for mitochondrial gene expression in Trypanosoma brucei . Eukaryot Cell 3: 862–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etheridge RD, Aphasizheva I, Gershon PD, Aphasizhev R (2008) 3′ adenylation determines mRNA abundance and monitors completion of RNA editing in T. brucei mitochondria. EMBO J 27: 1596–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Kaake RM, Patel VR, Yang Y, Baldi P, Huang L (2012) Mapping the protein interaction network of the human COP9 signalosome complex using a label‐free QTAX strategy. Mol Cell Proteomics 11: 138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimi H, Zimmer SL, Ammerman ML, Read LK, Lukes J (2013) Dual core processing: MRB1 is an emerging kinetoplast RNA editing complex. Trends Parasitol 29: 91–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BC, Kifer CT, Brekken DL, Randall AC, Wang Q, Drees BL, Parsons M (2007) Characterization of protein kinase CK2 from Trypanosoma brucei . Mol Biochem Parasitol 151: 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CY, Read LK (2005) Opposing effects of polyadenylation on the stability of edited and unedited mitochondrial RNAs in Trypanosoma brucei . Mol Cell Biol 25: 1634–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov DA, Simpson L (1992) The polarity of editing within a multiple gRNA‐mediated domain is due to formation of anchors for upstream gRNAs by downstream editing. Cell 70: 459–467 [DOI] [PubMed] [Google Scholar]
- Militello KT, Read LK (2000) UTP‐dependent and ‐independent pathways of mRNA turnover in Trypanosoma brucei mitochondria. Mol Cell Biol 20: 2308–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilson KA, Keighley T, Pascovici D, Cooke B, Haynes PA (2013) Label‐free quantitative shotgun proteomics using normalized spectral abundance factors. Methods Mol Biol 1002: 205–222 [DOI] [PubMed] [Google Scholar]
- Pfalz J, Bayraktar OA, Prikryl J, Barkan A (2009) Site‐specific binding of a PPR protein defines and stabilizes 5′ and 3′ mRNA termini in chloroplasts. EMBO J 28: 2042–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusnik M, Small I, Read LK, Fabbro T, Schneider A (2007) Pentatricopeptide repeat proteins in Trypanosoma brucei function in mitochondrial ribosomes. Mol Cell Biol 27: 6876–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajappa‐Titu L, Suematsu T, Munoz‐Tello P, Long M, Demir O, Cheng KJ, Stagno JR, Luecke H, Amaro RE, Aphasizheva I, Aphasizhev R, Thore S (2016) RNA Editing TUTase 1: structural foundation of substrate recognition, complex interactions and drug targeting. Nucleic Acids Res 44: 10862–10878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read LK, Myler PJ, Stuart K (1992) Extensive editing of both processed and preprocessed maxicircle CR6 transcripts in Trypanosoma brucei . J Biol Chem 267: 1123–1128 [PubMed] [Google Scholar]
- Read LK, Lukes J, Hashimi H (2016) Trypanosome RNA editing: the complexity of getting U in and taking U out. Wiley Interdiscip Rev RNA 7: 33–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon L, Skodova I, Pan S, Lukes J, Maslov DA (2013) The importance of the 45S ribosomal small subunit‐related complex for mitochondrial translation in Trypanosoma brucei . J Biol Chem 288: 32963–32978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Norbury CJ (2009) Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat Struct Mol Biol 16: 616–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CM, Read LK (2005) UTP‐dependent turnover of Trypanosoma brucei mitochondrial mRNA requires UTP polymerization and involves the RET1 TUTase. RNA 11: 763–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small ID, Peeters N (2000) The PPR motif ‐ a TPR‐related motif prevalent in plant organellar proteins. Trends Biochem Sci 25: 46–47 [DOI] [PubMed] [Google Scholar]
- Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK (2005) Complex management: RNA editing in trypanosomes. Trends Biochem Sci 30: 97–105 [DOI] [PubMed] [Google Scholar]
- Suematsu T, Zhang L, Aphasizheva I, Monti S, Huang L, Wang Q, Costello CE, Aphasizhev R (2016) Antisense transcripts Delimit Exonucleolytic activity of the mitochondrial 3′ processome to generate guide RNAs. Mol Cell 61: 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Aphasizheva I, Etheridge RD, Huang L, Wang X, Falick AM, Aphasizhev R (2008) Guide RNA‐binding complex from mitochondria of trypanosomatids. Mol Cell 32: 198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B, Ersfeld K, Gull K (2002) Targeting of a tetracycline‐inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei . Mol Biochem Parasitol 125: 211–216 [DOI] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross GA (1999) A tightly regulated inducible expression system for conditional gene knock‐outs and dominant‐negative genetics in Trypanosoma brucei . Mol Biochem Parasitol 99: 89–101 [DOI] [PubMed] [Google Scholar]
- Zikova A, Panigrahi AK, Dalley RA, Acestor N, Anupama A, Ogata Y, Myler PJ, Stuart K (2008) Trypanosoma brucei mitochondrial ribosomes: affinity purification and component identification by mass spectrometry. Mol Cell Proteomics 7: 1286–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer SL, McEvoy SM, Li J, Qu J, Read LK (2011) A novel member of the RNase D exoribonuclease family functions in mitochondrial guide RNA metabolism in Trypanosoma brucei . J Biol Chem 286: 10329–10340 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 7